

Abstract
The dystonias are a group of disorders defined by sustained or intermittent muscle contractions that result in involuntary posturing or repetitive movements. There are many different clinical manifestations and causes. Although they traditionally have been ascribed to dysfunction of the basal ganglia, recent evidence has suggested dysfunction may originate from other regions, particularly the cerebellum. This recent evidence has led to an emerging view that dystonia is a network disorder that involves multiple brain regions. The new network model for the pathogenesis of dystonia has raised many questions, particularly regarding the role of the cerebellum. For example, if dystonia may arise from cerebellar dysfunction, then why are there no cerebellar signs in dystonia? Why are focal cerebellar lesions or degenerative cerebellar disorders more commonly associated with ataxia rather than dystonia? Why is dystonia more commonly associated with basal ganglia lesions rather than cerebellar lesions? Can answers obtained from animals be extrapolated to humans? Is there any evidence that the cerebellum is not involved? Finally, what is the practical value of this new model of pathogenesis for the neuroscientist and clinician? This article explores potential answers to these questions.
Keywords: dystonia, cerebellum, network model
INTRODUCTION
The dystonias are a heterogeneous collection of disorders defined by sustained or intermittent muscle contractions that result in involuntary posturing or repetitive movements (Albanese et al., 2013). The defining features of the dystonias involve the nature of abnormal movements, not the molecular or neuropathological causes. The abnormal movements of the dystonias have varied clinical expressions which are classified according to four dimensions that include the body region involved, the age at onset, temporal aspects of symptoms, and whether or not there are associated neurological features (Table 1).
Table 1.
Classification of Clinical Manifestations of Dystonia
| Age at onset |
| Infancy (birth to 2 years) |
| Childhood (3–12 years) |
| Adolescence (13–20 years) |
| Early adulthood (21–40 years) |
| Late adulthood (>40 years) |
| Body distribution |
| Focal (one region of the body) |
| Segmental (two or more contiguous regions) |
| Multifocal (two or more non-contiguous regions) |
| Generalized (trunk plus 2 other regions) |
| Hemidystonia (several sites on one side of the body) |
| Temporal pattern |
| Disease course |
| Static |
| Progressive |
| Variability |
| Persistent |
| Action-specific |
| Diurnal |
| Paroxysmal |
| Associated features |
| Isolated or combined with another movement disorder |
| Occurrence of other neurological or systemic manifestations |
The varied clinical manifestations of dystonia also have many different causes (Tables 2–3). These causes are classified according to two dimensions, including evidence for heritability and/or structural pathology of the nervous system (Albanese et al., 2013, Fung et al., 2013). Many genes have been identified, and each is associated with a distinct clinical phenotype (Tanabe et al., 2009, Gasser and Asmus, 2010, Bragg et al., 2011, LeDoux, 2012, LeDoux et al., 2013, Lohmann and Klein, 2013). Structural pathology causing dystonia also varies considerably. Lesions may be inherited or acquired, focal or diffuse, obvious enough to be seen on routine imaging studies, or sufficiently subtle to require functional imaging or detailed histological assessments (Neychev et al., 2011).
Table 2.
Some known inherited causes of dystonia
| Amino acid metabolism |
| glutaric academia |
| GAMT deficiency |
| Hartnup disease |
| homocystinuria |
| methylmalonic acidemia |
| propionic acidemia |
| sulfite oxidase deficiency |
| Neurotransmitter metabolism |
| AADC deficiency |
| dihydropterin reductase deficiency |
| GTP cyclohydrolase deficiency |
| PTPS deficiency |
| tyrosine hydroxylase deficiency |
| Lipid metabolism/storage |
| GM1 or GM2 gangliosidosis |
| Krabbe disease |
| metachromatic leukodystrophy |
| neuronal ceroid lipofuscinosis |
| Niemann-Pick disease, type C |
| Pelizaeus-Merzbacher disease |
| Ion/metal homeostasis |
| aceruloplasminemia |
| Cav2.1 calcium channel defects |
| Fahr disease |
| neuroferritinopathy |
| rapid-onset dystonia-Parkinsonism |
| Wilson disease |
| Polyglutamine expansions |
| dentato-rubral-pallidoluysian atrophy |
| Huntington disease |
| spinocerebellar ataxias (1, 2, 3, 6, 7, 17) |
| DNA handling/transcription |
| ataxia-oculomotor apraxia |
| ataxia telangiectasia |
| Cockayne syndrome |
| Lubag |
| Rett syndrome |
| xeroderma pigmentosum |
| Mitochondrial function |
| deafness-dystonia syndrome |
| fumarase deficiency |
| Leber hereditary optic neuropathy |
| Leigh disease |
| MELAS |
| MERRF |
| pyruvate dehydrogenase deficiency |
| Other |
| ataxia with vitamin E deficiency |
| biotin responsive basal ganglia disease |
| frontotemporal dementias |
| Lesch-Nyhan disease |
| myoclonus dystonia |
| neuroacanthocytosis |
| neuronal intranuclear inclusion disease |
| Oppenheim dystonia |
| pantothenate kinase neurodegeneration |
| triosephosphate isomerase |
| whispering dysphonia |
Abbreviations: AADC, aromatic amino acid decarboxylase; Cav2.1, P/Q-type voltage regulated calcium channel; GAMT, guanidinoacetate methyltransferase; GTP, gunosine triphosphate; MELAS, mitochondrial encephalopathy with lactic acidosis and stroke-like episodes; MERRF, mitochondrial encephalopathy with ragged red fibers; PTPS, pyruvoyltetrahydropterin synthase.
Table 3.
Some known acquired causes of dystonia
| Medications |
| carbamazepine |
| cinnarizine |
| dopamine antagonists/agonists |
| fenfluramine |
| flunarizine |
| levodopa |
| phenytoin |
| prochlorperazine |
| metaclopramide |
| serotonin uptake inhibitors |
| tiagabine |
| Toxins |
| 3-nitropropionic acid |
| bilirubin (kernicterus) |
| carbon disulfide |
| carbon monoxide |
| cyanide |
| disulfiram |
| manganese |
| methanol |
| Vascular |
| stroke (hemmorhagic or ischemic) |
| vascular malformation |
| vasculitis |
| Infection |
| bacterial |
| fungal |
| prion |
| protozoan |
| viral |
| Autoimmune |
| anti-phospholipid syndrome |
| dystonia gravidarum |
| hymenoptera stings |
| multiple sclerosis |
| Sjögren’s syndrome |
| systemic lupus erythematosis |
| subacute sclerosis panencephalitis |
| Trauma |
| head |
| nerve |
| spine |
| Structural |
| abscess |
| Arnold-Chiari malformation |
| atlanto-axial subluxation |
| syringomyelia |
| tumors (brain, spine) |
| Other |
| cerebral palsy |
| corticobasal ganglionic degeneration |
| hypoparathyroidism |
| multiple system atrophy |
| Parkinson’s disease |
| progressive supranuclear palsy |
| tic disorders |
The marked heterogeneity of clinical syndromes and biological causes raises the obvious question regarding why the dystonias are grouped together as a family. Despite the heterogeneity, they share clinical features of involuntary over-activity of multiple muscles (Albanese et al., 2013). They also share certain biological features (Defazio et al., 2007, Bragg et al., 2011, Jinnah et al., 2013, LeDoux et al., 2013). At the genetic level, for example, one mutation can cause several different clinical phenotypes affecting different body parts at different ages and with different temporal evolution. This observation demonstrates that a single etiology can cause multiple different but related phenotypes. At the physiological level, a common finding across many etiologically unrelated dystonias is an alteration in the normal balance between inhibitory and excitatory processes (Quartarone and Hallett, 2013). This observation suggests that there may be shared systems-level abnormalities, despite differences in the original trigger. The differences and similarities are conceptualized by a model similar to those for other neurological diseases, where multiple different etiologies ultimately share some final common pathway(s) to produce the final outcome (Figure 1). In this model, all types of dystonia do not follow the same pathways of pathogenesis. Instead, specific subgroups of dystonia may share certain biological defects at the molecular, cellular, physiological, or anatomical levels.
Figure 1.

Conceptual model for accommodating differences and similarities among the many different types of dystonia. The basic concept for pathogenesis involves a series of events at the molecular, cellular, anatomic, and/or systems levels (A). In dystonia, pathological changes associated with an array of initial insults may converge in shared downstream pathways to produce the final syndrome. Some may converge at the molecular or biochemical levels, for example by affecting the same neurotransmitter system (B). Others may converge at the cellular level, by affecting the same type of cell to cause dystonia (C). Still others may converge at higher biological levels, such as physiological or anatomical pathways. Presumably, of these pathways converge at some final common pathway that produced excessive involuntary muscle contractions common to all dystonias. However this is a conceptual model only, and it remains feasible that there are multiple entirely independent pathways that produce a similar final phenotype.
At the anatomical level, dystonia traditionally has been ascribed to dysfunction of the basal ganglia. However, more recent evidence has pointed also to the cerebellum (Neychev et al., 2011, Avanzino and Abbruzzese, 2012, Sadnicka et al., 2012, Filip et al., 2013, Lehericy et al., 2013), challenging traditional concepts that the basal ganglia always play the dominant role. Since the role of the basal ganglia has been addressed many times already by others (Berardelli et al., 1998, Perlmutter and Mink, 2004, Hallett, 2006, Breakefield et al., 2008, Peterson et al., 2010), the current review focuses on the emerging evidence for the cerebellum, and how this emerging evidence might be accommodated with evidence already accumulated for the basal ganglia.
Evidence for Involvement of the Basal Ganglia
Dystonia historically has been attributed to dysfunction of the basal ganglia and its connections. The evidence supporting a role for the basal ganglia is strong and has been reviewed many times (Berardelli et al., 1998, Perlmutter and Mink, 2004, Hallett, 2006, Breakefield et al., 2008, Peterson et al., 2010). Herz was among the first to catalog autopsy findings in various acquired forms of dystonia. Although overt lesions are uncommon in most forms of dystonia, they are most common in the putamen, caudate, and pallidum (Herz, 1944). These autopsy findings later were corroborated by others (Hedreen et al., 1988, McGeer and McGeer, 1995). The autopsy findings also were confirmed by clinical neuroimaging surveys, where focal lesions frequently were found in the basal ganglia or its connections (Marsden et al., 1985, Pettigrew and Jankovic, 1985, Obeso and Gimenez-Roldan, 1988, Bhatia and Marsden, 1994).
Even when obvious lesions were not evident with histopathological or clinical imaging methods, basal ganglia abnormalities frequently were found via other imaging methods including positron emission tomography (PET) studies of regional metabolic activity detected with [18F]-fluorodeoxyglucose uptake or regional blood flow, voxel-based morphometry (VBM), functional MRI (fMRI), and diffusion tensor imaging (DTI). These studies also have been reviewed in detail (Niethammer et al., 2010, Neychev et al., 2011, Zoons et al., 2011, Lehericy et al., 2013). The basal ganglia also are implicated by neurosurgical studies showing that dystonia improves in patients following lesions or deep brain stimulation of the internal segment of the globus pallidus or pallidotomy (Gross, 2008, Vidailhet et al., 2012, Moro et al., 2013). Finally, animal studies implicate the basal ganglia in dystonia, since manipulations of various portions of the basal ganglia can induce dystonia (Jinnah et al., 2005, Guehl et al., 2009). Thus there is strong convergent evidence from many sources that the basal ganglia play an important role in dystonia. However, the basal ganglia may not be the cause for all types of dystonia.
Evidence for Involvement of the Cerebellum
The evidence pointing to the basal ganglia as a cause for dystonia is strong, but there has been increasing appreciation that other brain regions are involved, particularly the cerebellum. This evidence also has been reviewed (Neychev et al., 2011, Avanzino and Abbruzzese, 2012, Sadnicka et al., 2012, Filip et al., 2013). Similar to the case for the basal ganglia, autopsy and clinical imaging studies have linked some subtypes of dystonia with focal lesions of cerebellar circuits (Neychev et al., 2011, Zoons and Tijssen, 2013), and recent studies have shown subtle defects in cerebellar Purkinje cells in autopsy specimens from patients with cervical dystonia (Ma et al., 2012, Prudente et al., 2012). The same functional imaging studies that revealed abnormalities in the basal ganglia very frequently simultaneously revealed abnormalities in the cerebellum, and in some cases the cerebellar changes were greater than those in the basal ganglia (Odergren et al., 1998, Hutchinson et al., 2000, Delmaire et al., 2007, Thobois et al., 2008, Argyelan et al., 2009). These cerebellar abnormalities originally were interpreted as secondary compensatory changes to causal pathology in the basal ganglia, but more recent re-interpretations suggest the cerebellar abnormalities may be causal (Argyelan et al., 2009, Carbon and Eidelberg, 2009).
Human physiological studies also have revealed subclinical abnormalities of eyeblink conditioning in focal dystonia (Teo et al., 2009) and saccadic adaptation in DYT11 dystonia (Hubsch et al., 2011). These functions are intrinsic to the cerebellum, and they cannot be readily ascribed to basal ganglia dysfunction (Avanzino and Abbruzzese, 2012, Sadnicka et al., 2012). It also has been proposed that cerebellar dysfunction underlies defects in sensorimotor integration or maladaptive plasticity (Neychev et al., 2011, Quartarone and Hallett, 2013). Moreover, cerebellar modulation of motor cortex excitability is reduced in patients with focal hand dystonia (Brighina et al., 2009). Some surgical studies also suggest involvement of the cerebellum, since dentatectomy (Hitchcock, 1973, Davis, 2000) or deep brain stimulation of regions of the thalamus receiving cerebellar afferents can relieve dystonia in some cases (Fukaya et al., 2007, Goto et al., 2008, Morishita et al., 2010, Hedera et al., 2013). Finally, there is strong evidence from animal models that abnormal activity of the cerebellum can cause dystonia (LeDoux et al., 1993, LeDoux et al., 1995, LeDoux et al., 1998, Pizoli et al., 2002, Xiao and Ledoux, 2005, Neychev et al., 2008, Calderon et al., 2011, Alvarez-Fischer et al., 2012, Fan et al., 2012, Raike et al., 2012). Similar to results for the basal ganglia, there is strong convergent evidence that the cerebellum plays an important role in dystonia.
Top Ten Questions Regarding A Role for the Cerebellum in Dystonia
The emerging evidence regarding a role for the cerebellum has led to the view that dystonia is a network disorder that involves both the basal ganglia and cerebellum, and perhaps other regions too. This new model for the anatomical pathogenesis of dystonia has raised many questions. The purpose of this section is to address the most obvious questions aimed at relatively newer concepts incorporating the cerebellum in a model for the pathogenesis of dystonia.
1. If dystonia arises from cerebellar dysfunction, then why are there no cerebellar signs in dystonia?
If dystonia arises from cerebellar dysfunction, then concomitant “cerebellar signs” might be expected in patients with dystonia. In clinical neurology, the most commonly recognized cerebellar signs include ataxia and ocular nystagmus. These signs may occur in patients with various types of dystonia, but they are uncommon. The paucity of overt cerebellar signs questions the existence of cerebellar dysfunction underlying dystonia.
On the other hand, action-induced tremors are very common in dystonia. In fact, tremor is so common that it is the only neurological sign that is permitted in the current definition of “isolated dystonia” (Albanese et al., 2013). The frequency of tremor varies from 5–60%, depending on how it is defined and assessed (Quinn et al., 2011, Elble, 2013). Tremors can occur within the same body region affected by dystonia, or in another body part. Tremors can be irregular and jerky, or more regular and sinusoidal. The relationship between tremor and dystonia currently is a controversial matter, with some evidence suggesting they are two distinct but commonly co-morbid disorders and other evidence suggesting they are different manifestations of the same disorder (Hedera et al., 2010, Quinn et al., 2011, Schiebler et al., 2011, Elble, 2013, Pita Lobo et al., 2013). In either case, many studies have linked action-induced tremors with dysfunction of circuits involving the cerebellum and inferior olives (Raethjen and Deuschl, 2012). If tremor is a cerebellar sign, then cerebellar signs are common in dystonia.
Other cerebellar signs also occur in dystonia, although they are not overtly apparent in traditional clinical examinations. One of these is eye blink conditioning, a phenomenon that is reliant solely on cerebellar networks (Hopp and Fuchs, 2004, Gerwig et al., 2007, Thompson and Steinmetz, 2009). This phenomenon is elicited by applying a puff of air to the cornea (unconditioned stimulus) to induce a reflexive blink. The puff is paired repeatedly with an auditory tone (conditioned stimulus). After repeated pairings, the tone elicits a blink without the puff. Eye blink conditioning is less robust among patients with cervical and focal hand dystonia, suggesting an intrinsic cerebellar defect (Teo et al., 2009, Hoffland et al., 2013).
Another phenomenon mediated exclusively by the cerebellum that is abnormal in dystonia is saccadic adaptation (Hopp and Fuchs, 2004, Gerwig et al., 2007). In a saccadic adaptation task, a subject is asked to make a saccade towards a target to one side. However, the target is moved slightly during the saccade, resulting in the eye missing the target. The individual then must make a second corrective saccade to reach the target. With repeated trials, the individual learns to adjust the first saccade so that a second one is no longer necessary. Saccadic adaptation is abnormal in patients with the myoclonus dystonia syndrome, revealing another cerebellar sign in dystonia (Hubsch et al., 2011). In summary, some cerebellar signs are common in dystonia (tremor, and motor learning involving eye blink conditioning and saccadic adaptation), while others are uncommon (ataxia and ocular nystagmus).
2. If dystonia arises from cerebellar dysfunction, then why do focal cerebellar lesions more commonly cause ataxia, not dystonia?
Focal lesions of the cerebellum or its outflow pathways are most often associated with ataxia, not dystonia. One explanation for why cerebellar lesions cause ataxia and not dystonia may involve the nature of the lesion and its consequences (Figure 2).
Figure 2.
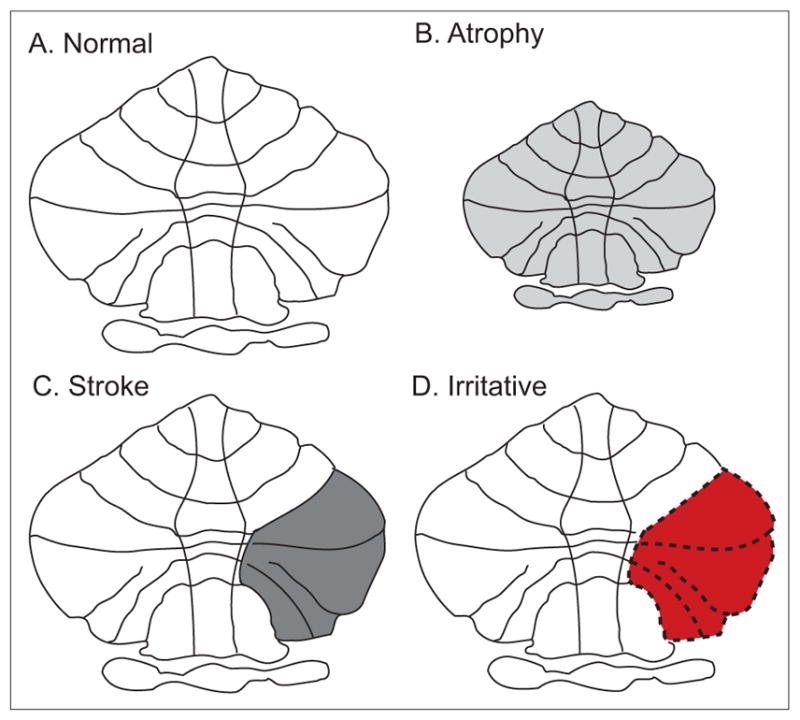
Different consequences of cerebellar lesions. Lesions of the cerebellum may cause different phenotypes depending on the nature of the lesion and its consequences. Normal cerebellar structure and output from the cerebellum result in normal patterns of movements (A). Decreased cerebellar output due to cerebellar atrophy (B) or stroke (C) may cause ataxia. In contrast, irritative lesions causing distorted cerebellar output (represented in red) may lead to dystonia (D).
A simple and well-documented illustration of this point involves the consequences of lesions in the primary motor region of the cerebral cortex. Destructive lesions with loss of cortical function result in weakness or paralysis. On the other hand, irritative lesions causing distorted cortical output cause epileptic seizures. Irritative lesions often are not visible with routine clinical imaging studies. In some cases the responsible lesions can be revealed with detailed histological studies. In other cases, the “lesion” is based on a functional defect. Thus lesions of the motor cortex can cause different motor phenotypes, depending on the nature of the lesion and its consequences (Figure 2). This phenomenon is not unique to the cerebral cortex, but also is known to occur in most other regions of the nervous system.
Overt lesions that are most obvious in imaging studies are those that involve loss of cerebellar tissue, such as stroke, and they usually cause ataxia. But what might be the outcome of lesions that are irritative and distort cerebellar output instead? We have proposed that dystonia can result from lesions of the cerebellum that distort, rather than reduce, cerebellar output (Neychev et al., 2011). Such lesions may result from local irritation, such as hemorrhage, or functional derangements due to compressive effects of mass lesions. Such “lesions” may also be primarily functional, with no overt anatomical abnormality, analogous to epileptic seizures.
This hypothesis regarding a distortion or increase of cerebellar output is consistent with prior literature indicating that cerebellar lesions occasionally may cause dystonia (Neychev et al., 2011). Such lesions are more likely to be space-occupying lesions such as tumors that compress and distort surrounding cerebellar functions, or hemorrhages that cause a local irritative focus. This hypothesis also is consistent with observations that the vast majority of function-based imaging studies of human dystonia, such as PET studies of fluorodeoxyglucose or blood flow, have revealed increases in cerebellar activity, not decreases (Neychev et al., 2011, Zoons et al., 2011). Furthermore, the hypothesis is consistent with the strong relationship between dystonia and tremor, because tremor also is viewed as functional distortion of cerebellar processing. Finally, the distinction between lesions that cause distorted function versus loss of function is consistent with multiple animal studies showing that dystonia arises from distorted cerebellar output, such as an abnormal increase in Purkinje neuron firing or abnormal bursting patterns, rather than loss of output (LeDoux et al., 1993, LeDoux et al., 1995, LeDoux et al., 1998, Pizoli et al., 2002, Xiao and Ledoux, 2005, Neychev et al., 2008, Calderon et al., 2011, Alvarez-Fischer et al., 2012, Fan et al., 2012, Raike et al., 2012). This hypothesis should be more directly addressed by examining the physiological properties of cerebellar neurons in humans with dystonia.
3. If dystonia arises from cerebellar dysfunction, then why are degenerative cerebellar syndromes associated with ataxia, not dystonia?
Degenerative processes involving the cerebellum, such as the spinocerebellar ataxias (SCAs), are most often associated with ataxia, not dystonia. The explanation is similar to that regarding focal cerebellar lesions. Degenerative loss of cerebellar neurons results in ataxia because of the loss of normal cerebellar function.
However, some degenerative disorders of the cerebellum are associated with dystonia. Dystonia is most prominent in SCA3 (Machado-Joseph disease) but may also occur in SCA1, SCA2, SCA6, ataxia telangiectasia, Freidreich’s ataxia, and others (Kuoppamaki et al., 2003, Le Ber et al., 2006, Neychev et al., 2011). In most such cases, dystonia and ataxia occur together, but in some cases, dystonia may be the dominant feature. These observations raise a related question: how can one disease process cause two different motor phenotypes at the same time? One potential explanation is that ataxia results from pathology in the cerebellum and dystonia results from concomitant pathology of the basal ganglia. Another possible explanation is that these diseases simultaneously cause both destructive and irritative lesions in the cerebellum that are separated in time or space.
Most cerebellar degenerations are caused by slowly progressive dysfunction of cerebellar neurons, with the final outcome being degenerative loss of these neurons. However, very little is known about what these neurons are doing before they die and disappear. If they produce distorted output in their pre-agonal state, as often is the case with diseased neurons, it is possible that they may cause dystonia before they disappear and contribute to development of ataxia. If this is the case, then longitudinal studies of such cases might reveal dystonia early in the disease, with progressively more ataxia as the disease progresses.
Additionally, the cerebellum is a large structure, which is somatotopically organized and structurally compartmentalized. In many cerebellar degenerations, different compartments are more seriously affected than others (Manni and Petrosini, 2004, Mottolese et al., 2012). This relatively selective vulnerability of different cerebellar compartments could result in degenerative loss of cells in a particularly vulnerable region, simultaneous with abnormal function of surviving cells in a less vulnerable region. If this is the case, then cerebellar dysfunction in a single individual might cause ataxia and dystonia in different body regions, and the relative expression of each will be dependent on how far the disease has progressed within the relevant somatotopically associated regions of the cerebellum. This hypothesis could be explored by careful clinico-pathological or clinico-radiological correlation studies in affected individuals with varying degrees of ataxia and dystonia.
4. Why is dystonia more commonly associated with lesions of the basal ganglia, not the cerebellum?
Dystonia traditionally has been associated with overt lesions of the basal ganglia or its connections in autopsy and imaging studies. One of the earliest studies addressing this question involved patients with hemidystonia and it demonstrated that the majority of lesions evident from histopathology or structural imaging studies were in the putamen (Marsden et al., 1985). This study had an enduring effect, with the conclusions being somewhat uncritically extrapolated to other forms of dystonia. However, the etiological heterogeneity of dystonia is now better appreciated, and more recent studies have shown that the distribution of lesions is not the same for all types of dystonia. For example, the majority of lesions among patients with cervical or craniofacial dystonia involves the cerebellum or brainstem (LeDoux and Brady, 2003, Khooshnoodi et al., 2013). The concept that dystonia is more commonly associated with lesions of the basal ganglia needs refinement with reference to the type of dystonia in question.
The nature of the lesion and its consequences also must be considered. As emphasized earlier, lesions that cause loss of function have consequences that differ from lesions that cause irritation or distortion of function (Figure 2). Most types of lesions detected by clinical histopathological methods and clinical imaging studies of dystonia are biased towards loss of function such as stroke or degenerative atrophy. Such methods are insensitive to microstructural defects or functional disturbances.
Finally, it is important to acknowledge an important phenomenon in studies that attempt to link focal lesions with dystonia. Following an acute insult to the nervous system, dystonia rarely emerges immediately. Instead, it typically emerges after a delay of several weeks or years (Saint Hilaire et al., 1991, Ghika et al., 1994, He et al., 1995, Scott and Jankovic, 1996, Palfi et al., 2000, Kim, 2001). This delay argues that the lesion itself cannot be causing dystonia by loss of function from the damaged region. Instead, dystonia must arise from some secondary adaptive response to the lesion. The location of this adaptive response is unknown. It may occur in nearby undamaged structures, or it may occur in more remote regions. Thus the lesion method, while used extensively to establish structure-function relationships in clinical neurology, has an unexplained handicap in studies of dystonia. However, studies addressing potential adaptive changes in the brain that occur with the development of dystonia could provide powerful clues to the real source of the problem.
5. Basal ganglia, cerebellum or both?
The new network model has generated some novel questions not anticipated by models focusing exclusively on the basal ganglia (Jinnah and Hess, 2006). Can dystonia arise from causal pathology in a single node of the network? Is the causal node always the same, or are different nodes responsible for different types of dystonia? Does one node serve as a “final common pathway” leading to dystonia? Does dystonia require combined dysfunction of more than one node, in a “two-hit” model for pathogenesis? Can dystonia arise instead from aberrant communication between nodes (Figure 3)?
Figure 3.
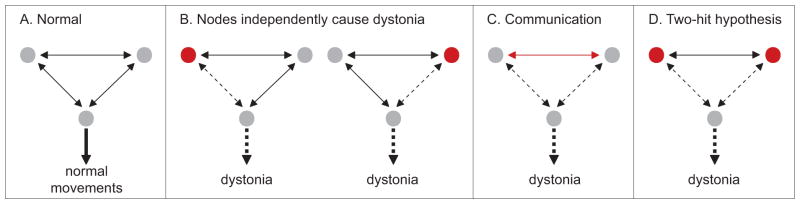
The network model for dystonia. Normal movements are known to require combined action of distinct motor systems including the motor cortex, basal ganglia, cerebellum, and brainstem (A). Dystonia may arise from dysfunction of one, or another node in the network (B). Dystonia may alternatively require abnormal communication between nodes (C), or dysfunction of both nodes as proposed by the two-hit hypothesis (D). In the diagram above, dysfunction in any part of the network is represented in red.
There is some experimental evidence for each of these scenarios. For example, there are multiple clinical and animal studies suggesting that dystonia may arise from focal lesions or manipulation of the basal ganglia or cerebellum. These observations imply that dystonia may arise from independent pathology in a single node, which is not always the same. Indeed, there are multiple studies that have suggested different types of dystonia do not share the same neuroanatomical pathogenesis (Carbon et al., 2004, Carbon and Eidelberg, 2009, Neychev et al., 2011, Sadnicka et al., 2013).
There also is evidence for a two-hit process for pathogenesis. For example, partial interruption of sensory afferents from peripheral nerves combined with dysfunction of the nigrostriatal pathway can result in dystonia (Schicatano et al., 1997). Similarly, subclinical lesions of the basal ganglia exacerbate dystonia in rodent models where dystonia is triggered by cerebellar dysfunction (Neychev et al., 2008, Calderon et al., 2011). Recent studies demonstrating subcortical connections between the basal ganglia and cerebellum have raised the possibility that defective communication may also cause dystonia in some circumstances (Hoshi et al., 2005, Bostan et al., 2010, Bostan and Strick, 2010). Further studies will be needed to discriminate among these possibilities, and it seems feasible that the many different types of dystonia will not all share the same mechanisms of anatomical pathogenesis.
6. Are animal models useful for exploring abnormal cerebellar function in dystonia?
The vast majority of our knowledge regarding the anatomical substrates responsible for dystonia in humans comes from indirect evidence. Human clinico-pathological and clinico-radiological studies linking lesions with dystonia are necessarily correlational, and they cannot establish causal links (Hedreen et al., 1988, McGeer and McGeer, 1988). The unexplained delays between lesion and symptom development imply adaptive changes that may be far from the original lesion. Human functional imaging studies also are largely correlational, and it rarely is possible to discriminate the regions causing dystonia from downstream compensatory changes (Neychev et al., 2011, Zoons et al., 2011). Even results of neurosurgical interventions cannot establish causation, because the effects of local stimulation or lesions of one region are transmitted to other regions (Thobois et al., 2008), and because of the unexplained delay between intervention and improvement in dystonia (Vidailhet et al., 2012, Moro et al., 2013). To prove a causal link between specific brain regions and dystonia, it would be necessary to experimentally manipulate the regions to cause dystonia in otherwise normal individuals. Conversely, it would be necessary to target these same regions to suppress dystonia in affected patients. Such studies cannot be methodically conducted in humans for obvious ethical reasons.
However, such studies can be conducted in animals. Multiple studies in normal primates have shown that dystonic movements can be provoked by various manipulations of different brain regions including the basal ganglia, cerebellum, midbrain, and other regions (Magoun et al., 1935, Kemberling et al., 1952, Foltz et al., 1959, Klier et al., 2002, Guehl et al., 2009). Multiple studies in normal rodents also have shown dystonic movements to be induced by manipulating a variety of brain regions (Jinnah et al., 2005, Wilson and Hess, 2013). For example, inducing distorted cerebellar output in otherwise normal mice or rats produces dystonia (Pizoli et al., 2002, Neychev et al., 2008, Calderon et al., 2011, Alvarez-Fischer et al., 2012, Raike et al., 2012). Conversely, in rodent models with inherited genetic defects, it also has been possible to demonstrate alleviation of dystonia by manipulation of specific brain regions. For instance, chronic generalized dystonia in the Dt rat can be eliminated by surgical ablation of the cerebellum, or targeted ablation of specific cerebellar output nuclei (LeDoux et al., 1993, LeDoux et al., 1995). Paroxysmal dystonic movements in tottering mutant mice can be eliminated by surgical ablation of the cerebellum, or by deletion of abnormally functioning Purkinje neurons (Campbell et al., 1999, Neychev et al., 2008, Raike et al., 2012). In both cases, dystonic movements are replaced by ataxic movements, an observation consistent with concepts regarding distorted function versus loss of function of the cerebellum in dystonia versus ataxia. Although these animal studies provide strong evidence that abnormal output from the cerebellum causes dystonia, an obvious question is whether they are relevant to human dystonia.
The cerebellum demonstrates obvious differences across species, most notably its gross structural appearance (Glickstein and Voogd, 1995). However, there are more similarities than differences, particularly among mammals. The cerebellum is a phylogenetically very old structure with highly conserved similarities across many species including afferent and efferent connectivity, intrinsic laminar structure, physiological properties of cerebellar neurons, and chemical transmitters. At present, there is no compelling evidence that information learned from animals cannot be extrapolated to humans. Therefore, rather than dismiss the evidence from animal studies, it seems more fruitful to take advantage of the unique insights they can provide, and follow up with more targeted studies of humans to either validate or refute the findings (Figure 4, A and B).
Figure 4.

Contributions of human and animal studies. Both human and animal studies are required to provide a complete view of the pathogenesis of dystonia. Clinical observations and results from human studies can be used to guide experimental questions and hypotheses when designing animal studies (A and B). On the other hand, findings from animal studies can be applied in human research to validate or refute conceptual models as well as to aid in the development of more targeted interventions (A and B). To avoid potential problems related to cross-species translation of results, we propose an iterative approach, in which answers to experimental questions obtained from simple models are verified in more advanced animal models, and subsequently validated in human studies (B). For example, results from rodents or fruit fly studies can be verified first in nonhuman primates before validation in humans. Similarly, findings from cell culture models can be tested in rodents before application to human studies.
7. Are results from non-human primates regarding cerebellar involvement in dystonia more trustworthy than results from other animals?
Non-human primates are valuable for disease research because they most closely resemble humans. However, broad use of primates is limited by relatively high expenses, the need for specialized centers for maintenance and testing, and ethical issues related to the application of certain types of experimental manipulations in primates. In addition, genetically modified primate models are not widely available. Because of these difficulties, the use of primates for dystonia research has been limited (Guehl et al., 2009). Although a variety of pharmacological or surgical manipulations have been reported to induce dystonic movements in non-human primates, most have been reported from a single laboratory, and none of them has been fully validated as a model for dystonia.
In comparison, research involving rodents is less expensive and less encumbered by ethical questions. Rodent research also is more amenable to the expertise available in many laboratories, and genetic manipulations. As a result, the vast majority of research in animals has focussed on rodents (Jinnah et al., 2005, Oleas et al., 2013, Wilson and Hess, 2013). Several rodent models have been rigorously validated for dystonia research, and the most compelling results pointing to the cerebellum have come from rodents. Unfortunately, there are many well-documented examples where research in rodents has led to results that could not be extrapolated to humans (Jinnah et al., 2008). These examples have led to some well-deserved scepticism for rodent models, especially among clinicians. To avoid potential problems related to cross-species translation of results, we have advocated an iterative approach, in which answers to experimental questions obtained from animals are subsequently validated in human studies (Figure 4, A and B). For example, the concepts regarding a causal role for distorted cerebellar functioning as a cause for dystonia originated from studies of rodents (LeDoux et al., 1993, Campbell and Hess, 1998, LeDoux and Lorden, 1998, Campbell et al., 1999), but they have now been at least partly validated through a variety of human studies including neuroimaging of affected patients (Neychev et al., 2011, Zoons et al., 2011), non-invasive physiological studies (Brighina et al., 2009, Teo et al., 2009, Hubsch et al., 2011, Hoffland et al., 2013), and histopathological studies of brains collected at autopsy (Prudente et al., 2012). More precise and direct validation studies in humans often are not feasible, so the further development of non-human primates models of dystonia seems valuable to help bridge the gap between rodents and humans (Figure 4B).
8. Is there evidence against a role for the cerebellum?
Several types of evidence have been cited as evidence against a role for the cerebellum in dystonia. Examples often cited include studies showing that focal basal ganglia lesions cause dystonia, the ability of dopamine-related drugs to cause dystonia in some circumstances, and disorders such as dopa-responsive dystonia. In these cases, the basal ganglia clearly play a central role in the pathogenesis of dystonia, and there seems little reason to suspect cerebellar involvement. While this evidence provides strong support for a role for the basal ganglia for specific subtypes of dystonia, it does not provide evidence against a role for the cerebellum for others. By the same token, evidence suggesting a role for the cerebellum does not refute a role for the basal ganglia. These two regions are not mutually exclusive in the network model.
9. What is the practical relevance of the network model for neuroscientists?
Most of the evidence from human studies supporting a role for the basal ganglia or the cerebellum in dystonia is correlational, and proof of a causal link is limited. More direct evidence is needed to validate concepts derived from indirect human observations (Figure 4). Often, the most direct evidence comes from studies of surrogate models, such as animal or tissue culture models.
Traditional concepts regarding a central role for the basal ganglia in dystonia have led to this region being the target of many scientific studies of animal and cell models. In comparison, there are far fewer studies of the cerebellum. For example, there are many electrophysiological studies of the properties of basal ganglia neurons in animal models for dystonia, especially cholinergic and dopaminergic neurons (Centonze et al., 2003, Pisani et al., 2006, Pisani et al., 2007, Martella et al., 2009, Grundmann et al., 2012, Sciamanna et al., 2012a, Sciamanna et al., 2012b, Oleas et al., 2013). There are far fewer studies of cerebellar neurons in dystonia models (LeDoux et al., 1998, Chen et al., 2009). Similarly, there are many pharmacological and biochemical studies of basal ganglia pathways in animal models (Jinnah et al., 1992, Jinnah et al., 1994, Balcioglu et al., 2007, Grundmann et al., 2007, Zhao et al., 2008, Hewett et al., 2010, Page et al., 2010, Lange et al., 2011, Dang et al., 2012, Song et al., 2012, Song et al., 2013), but comparatively few for cerebellar pathways (Fureman et al., 2002, Fureman and Hess, 2005, Fan et al., 2012).
The ideal strategic development of a tissue culture model for dystonia obviously involves the type of cell selected as the model. To date, most culture models have aimed at non-neuronal cells, neuroblastoma lines, or dopamine-like neurons (Gonzalez-Alegre et al., 2005, Hewett et al., 2006, Breakefield et al., 2008, Granata et al., 2009). There are no culture models relevant to dystonia based on cerebellar neurons. Considering that the pathogenic effects of gene defects and other triggers for dystonia are likely to be specific to selected neuronal populations, the development of more targeted tissue culture models seems important. In general, the paucity of information regarding the cerebellum and its neurons in animal and cell models of dystonia provides fruitful grounds for further basic studies.
10. What is the practical relevance of the network model for clinical scientists?
Current treatments of dystonia are mostly symptomatic and not always effective. More definitively identifying the source of the problem is useful for the development of new treatment strategies that control or eliminate symptoms more completely. For instance, deep brain stimulation of the internal segment of the globus pallidus works for some, but not all patients, with dystonia, and the reason for the different outcomes remains unclear. Perhaps better results in some cases might be achieved by targeting other circuits. Indeed, several studies have suggested that targeting the cerebellar receiving zone in the thalamus may be particularly effective for certain types of dystonia (Fukaya et al., 2007, Goto et al., 2008, Morishita et al., 2010, Hedera et al., 2013). Additionally, non-invasive transcranial stimulation of the cerebellum has been reported to temporarily attenuate the severity of dystonia (Brighina et al., 2009, Bradnam et al., 2013). Whether different stimulation paradigms might have a more potent and enduring effect remains to be explored.
Novel pharmacological interventions also depend on the neurochemical functionality of the putative brain region where abnormalities arise. While there are remarkably effective drugs for specific types of dystonia arising from basal ganglia dysfunction, such as levodopa in dopa-responsive dystonia, there has been limited success with drugs for most other types of dystonia (Jinnah and Hess, 2008). A broader view that considers the neurochemical properties of cerebellar or other regions may point to alternative pharmacological targets for dystonia. In summary, the development of novel surgical and pharmacological treatment strategies for dystonia both rely on conceptual models regarding its anatomical substrates. The new network model opens the door to rational design of multiple new therapeutic strategies that would never be considered with traditional models of pathogenesis that focus only on the basal ganglia.
Looking ahead
Decades of research focussing on the basal ganglia have generated a wealth of information regarding the pathogenesis of dystonia. The network model involving the cerebellum is a relatively new concept that is still in evolution, and it has generated more questions than answers. In fact, some investigators have criticized the model as being inconsistent with traditional knowledge, or questioned the need for a new model (Sadnicka et al., 2012).
Any good conceptual model should be compatible with existing knowledge. As summarized above, the new network model is consistent with traditional knowledge. Indeed, it provides a broad conceptual paradigm that accommodates results from prior clinical and animal studies that have pointed either to the basal ganglia, cerebellum or both (Figure 3). Future efforts to understand the pathogenesis of dystonia are not well served by over-simplistic conceptual models that view a complex problem through a lens that is too narrowly focussed on a single brain region.
Any good conceptual model also should be useful to scientists and clinicians. As noted above, the new model is useful in guiding novel experimental questions and pointing towards novel therapeutic strategies. Perhaps most importantly, any novel model should be testable and should stimulate novel ideas. Indeed, the new model generates a number of testable hypotheses that can be the target of future research.
Perhaps the last question that needs to be addressed is whether or not we are ready for clinical interventions targeting the cerebellum. In view of the available evidence regarding the role of the cerebellum in dystonia, combined with the paucity of effective treatments for many types of dystonia, it is tempting to consider whether interventions more directly targeting the cerebellum might be worthwhile. Enthusiasm for human studies must be tempered by the fact that the most compelling evidence implicating the cerebellum in dystonia so far has come from rodent studies. There are no results from non-human primates addressing the value of cerebellar interventions in dystonia, and the available studies of humans are largely anecdotal. All of the evidence from humans pointing to the cerebellar receiving zone in the thalamus for certain types of dystonia comes from individual case reports or small retrospective series of cases (Fukaya et al., 2007, Goto et al., 2008, Morishita et al., 2010, Hedera et al., 2013). Additionally, reports of improvement in cervical dystonia following removal of a posterior fossa meningioma are anecdotal (Krauss et al., 1997). The report of resolution of upper limb dystonia following treatment of a tuberculous mass of the cerebellum in one case is similarly anecdotal (Alarcon et al., 2001).
Although the available evidence is provocative, it is quite limited when considering humans and non-human primates, so interventions targeting the cerebellum as a treatment for patients with dystonia seem premature. However, it is clear that more methodical studies in non-human primates or carefully designed clinical trials in select patient populations with dystonia must be considered.
Highlights.
There is increasing evidence that dystonia is a network disorder involving multiple brain regions
Regions involved include the basal ganglia, cerebellum, thalamus and cortex
Cerebellar involvement likely derives from abnormal output, not loss of output
The network model leads to several specific and testable hypotheses
The network model has direct implications for new therapies aimed at specific anatomical targets
Acknowledgments
This summary was supported in part by U54 NS065701 from the Office of Rare Diseases Research in the National Center for Advancing Translational Sciences and the National Institute of Neurological Disorders and Stroke at the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Cecilia N. Prudente, Email: cpruden@emory.edu.
Ellen J. Hess, Email: ellen.hess@emory.edu.
H. A. Jinnah, Email: hjinnah@emory.edu.
References
- Alarcon F, Tolosa E, Munoz E. Focal limb dystonia in a patient with a cerebellar mass. Arch Neurol. 2001;58:1125–1127. doi: 10.1001/archneur.58.7.1125. [DOI] [PubMed] [Google Scholar]
- Albanese A, Bhatia K, Bressman SB, DeLong MR, Fahn S, Fung VSC, Hallett M, Jankovic J, Jinnah HA, Klein C, Lang AE, Mink JW, Teller JK. Phenomenology and classification of dystonia: A consensus update. Mov Disord. 2013;28:863–873. doi: 10.1002/mds.25475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Fischer D, Grundmann M, Lu L, Samans B, Fritsch B, Moller JC, Schaefer MK, Hartmann A, Oertel WH, Bandmann O. Prolonged generalized dystonia after chronic cerebellar application of kainic acid. Brain Res. 2012;1464:82–88. doi: 10.1016/j.brainres.2012.05.007. [DOI] [PubMed] [Google Scholar]
- Argyelan M, Carbon M, Niethammer M, Ulug AM, Voss HU, Bressman SB, Dhawan V, Eidelberg D. Cerebellothalamocortical connectivity regulates penetrance in dystonia. J Neurosci. 2009;29:9740–9747. doi: 10.1523/JNEUROSCI.2300-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanzino L, Abbruzzese G. How does the cerebellum contribute to the pathophysiology of dystonia. Basal Ganglia. 2012;2:231–235. [Google Scholar]
- Balcioglu A, Kim MO, Sharma N, Cha JH, Breakefield XO, Standaert DG. Dopamine release is impaired in a mouse model of DYT1 dystonia. J Neurochem. 2007;102:783–788. doi: 10.1111/j.1471-4159.2007.04590.x. [DOI] [PubMed] [Google Scholar]
- Berardelli A, Rothwell JC, Hallett M, Thompson PD, Manfredi M, Marsden CD. The pathophysiology of primary dystonia. Brain. 1998;121:1195–1212. doi: 10.1093/brain/121.7.1195. [DOI] [PubMed] [Google Scholar]
- Bhatia KP, Marsden CD. The behavioral and motor consequences of focal lesions of the basal ganglia in man. Brain. 1994;117:859–876. doi: 10.1093/brain/117.4.859. [DOI] [PubMed] [Google Scholar]
- Bostan AC, Dum RP, Strick PL. The basal ganglia communicate with the cerebellum. Proc Natl Acad Sci U S A. 2010;107:8452–8456. doi: 10.1073/pnas.1000496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostan AC, Strick PL. The cerebellum and basal ganglia are interconnected. Neuropsychol Rev. 2010;20:261–270. doi: 10.1007/s11065-010-9143-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradnam, Ridding, McDonnell Non-invasive cerebellar stimulation for treatment of focal dystonia. Second International Congress on Treatment of Dystonia; Hannover Germany. 2013. [Google Scholar]
- Bragg DC, Armata IA, Nery FC, Breakefield XO, Sharma N. Molecular pathways in dystonia. Neurobiol Dis. 2011;42:136–147. doi: 10.1016/j.nbd.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nat Rev Neurosci. 2008;9:222–234. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- Brighina F, Romano M, Giglia G, Saia V, Puma A, Giglia F, Fierro B. Effects of cerebellar TMS on motor cortex of patients with focal dystonia: a preliminary report. Exp Brain Res. 2009;192:651–656. doi: 10.1007/s00221-008-1572-9. [DOI] [PubMed] [Google Scholar]
- Calderon DP, Fremont R, Kraenzlin F, Khodakhah K. The neural substrates of rapid-onset dystonia-parkinsonism. Nat Neurosci. 2011;14:357–365. doi: 10.1038/nn.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DB, Hess EJ. Cerebellar circuitry is activated during convulsive episodes in the tottering (tg/tg) mutant mouse. Neurosci. 1998;85:773–783. doi: 10.1016/s0306-4522(97)00672-6. [DOI] [PubMed] [Google Scholar]
- Campbell DB, North JB, Hess EJ. Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp Neurol. 1999;160:268–278. doi: 10.1006/exnr.1999.7171. [DOI] [PubMed] [Google Scholar]
- Carbon M, Eidelberg D. Abnormal structure-function relationships in hereditary dystonia. Neuroscience. 2009;164:220–229. doi: 10.1016/j.neuroscience.2008.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon M, Su S, Dhawan V, Raymond D, Bressman S, Eidelberg D. Regional metabolism in primary torsion dystonia: effects of penetrance and genotype. Neurology. 2004;62:1384–1390. doi: 10.1212/01.wnl.0000120541.97467.fe. [DOI] [PubMed] [Google Scholar]
- Centonze D, Grande C, Saulle E, Martin AB, Gubellini P, Pavon N, Pisani A, Bernardi G, Moratalla R, Calabresi P. Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. J Neurosci. 2003;23:8506–8512. doi: 10.1523/JNEUROSCI.23-24-08506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Popa LS, Wang X, Gao W, Barnes J, Hendrix CM, Hess EJ, Ebner TJ. Low-frequency oscillations in the cerebellar cortex of the tottering mouse. J Neurophysiol. 2009;101:234–245. doi: 10.1152/jn.90829.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Cheetham CC, Lu J, Vo V, Lovinger DM, Li Y. An anticholinergic reverses motor control and corticostriatal LTD deficits in Dyt1 DeltaGAG knock-in mice. Behav Brain Res. 2012;226:465–472. doi: 10.1016/j.bbr.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R. Cerebellar stimulation for cerebral palsy spasticity, function, and seizures. Arch Med Res. 2000;31:290–299. doi: 10.1016/s0188-4409(00)00065-5. [DOI] [PubMed] [Google Scholar]
- Defazio G, Berardelli A, Hallett M. Do primary adult-onset focal dystonias share aetiological factors? Brain. 2007;130:1183–1193. doi: 10.1093/brain/awl355. [DOI] [PubMed] [Google Scholar]
- Delmaire C, Vidailhet M, Elbaz A, Bourdain F, Bleton JP, Sangla S, Meunier S, Terrier A, Lehericy S. Structural abnormalities in the cerebellum and sensorimotor circuit in writer’s cramp. Neurology. 2007;69:376–380. doi: 10.1212/01.wnl.0000266591.49624.1a. [DOI] [PubMed] [Google Scholar]
- Elble RJ. Defining dystonic tremor. Curr Neuropharmacol. 2013;11:48–52. doi: 10.2174/157015913804999478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Hughes KE, Jinnah HA, Hess EJ. Selective and sustained AMPA receptor activation in cerebellum induces dystonia in mice. J Pharmacol Exp Ther. 2012;340:733–741. doi: 10.1124/jpet.111.190082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filip P, Lungu OV, Bares M. Dystonia and the cerebellum: A new field of interest in movement disorders? Clin Neurophysiol. 2013;124:1269–1274. doi: 10.1016/j.clinph.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Foltz EL, Knopp LM, Ward AA. Experimental spasmodic torticollis. J Neurosurg. 1959;16:55–67. doi: 10.3171/jns.1959.16.1.0055. [DOI] [PubMed] [Google Scholar]
- Fukaya C, Katayama Y, Kano T, Nagaoka T, Kobayashi K, Oshima H, Yamamoto T. Thalamic deep brain stimulation for writer’s cramp. J Neurosurg. 2007;107:977–982. doi: 10.3171/JNS-07/11/0977. [DOI] [PubMed] [Google Scholar]
- Fung VS, Jinnah HA, Bhatia K, Vidailhet M. Assessment of the patient with dystonia: An update on dystonia syndromes. Mov Disord. 2013;28:889–898. doi: 10.1002/mds.25549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fureman BE, Hess EJ. Noradrenergic blockade prevents attacks in a model of episodic dysfunction caused by a channelopathy. Neurobiol Dis. 2005;20:227–232. doi: 10.1016/j.nbd.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Fureman BE, Jinnah HA, Hess EJ. Triggers of paroxysmal dyskinesias in the calcium channel mouse mutant tottering. Pharmacol Biochem Behav. 2002;73:631–637. doi: 10.1016/s0091-3057(02)00854-7. [DOI] [PubMed] [Google Scholar]
- Gasser T, Asmus F. Dystonia-plus syndromes. Eur J Neurol. 2010;17 (Suppl 1):37–45. doi: 10.1111/j.1468-1331.2010.03049.x. [DOI] [PubMed] [Google Scholar]
- Gerwig M, Kolb FP, Timmann D. The involvement of the human cerebellum in eyeblink conditioning. Cerebellum. 2007;6:38–57. doi: 10.1080/14734220701225904. [DOI] [PubMed] [Google Scholar]
- Ghika J, Bogousslavsky J, Henderson J, Maeder P, Regli F. The “jerky dystonic unsteady hand”: a delayed motor syndrome in posterior thalamic infarctions. J Neurol. 1994;241:537–542. doi: 10.1007/BF00873516. [DOI] [PubMed] [Google Scholar]
- Glickstein M, Voogd J. Lodewijk Bolk and the comparative anatomy of the cerebellum. Trends Neurosci. 1995;18:206–210. doi: 10.1016/0166-2236(95)93903-b. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Alegre P, Bode N, Davidson BL, Paulson HL. Silencing primary dystonia: lentiviralmediated RNA interference therapy for DYT1 dystonia. J Neurosci. 2005;25:10502–10509. doi: 10.1523/JNEUROSCI.3016-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto S, Shimazu H, Matsuzaki K, Tamura T, Murase N, Nagahiro S, Kaji R. Thalamic Vo-complex vs pallidal deep brain stimulation for focal hand dystonia. Neurology. 2008;70:1500–1501. doi: 10.1212/01.wnl.0000310430.00743.11. [DOI] [PubMed] [Google Scholar]
- Granata A, Schiavo G, Warner TT. TorsinA and dystonia: from nuclear envelope to synapse. J Neurochem. 2009;109:1596–1609. doi: 10.1111/j.1471-4159.2009.06095.x. [DOI] [PubMed] [Google Scholar]
- Gross R. What happened to posteroventral pallidotomy for Parkinson’s disease and dystonia? NeuroRx. 2008;5:281–293. doi: 10.1016/j.nurt.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann K, Glockle N, Martella G, Sciamanna G, Hauser TK, Yu L, Castaneda S, Pichler B, Fehrenbacher B, Schaller M, Nuscher B, Haass C, Hettich J, Yue Z, Nguyen HP, Pisani A, Riess O, Ott T. Generation of a novel rodent model for DYT1 dystonia. Neurobiol Dis. 2012;47:61–74. doi: 10.1016/j.nbd.2012.03.024. [DOI] [PubMed] [Google Scholar]
- Grundmann M, Reischmann B, Vanhoutte G, Hubener J, Teismann P, Hauser TK, Bonin M, Wilbertz J, Horn S, Nguyen HP, Kuhn M, Chanarat S, Wolburg H, Van der Linden A, Riess O. Overexpression of human wildtype torsinA and human deltaGAG torsinA in a transgenic mouse model causes phenotypic abnormalities. Neurobiol Dis. 2007;27:190–206. doi: 10.1016/j.nbd.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Guehl D, Cuny E, Ghorayeb I, Michelet T, Bioulac B, Burbaud P. Primate models of dystonia. Prog Neurobiol. 2009;87:118–131. doi: 10.1016/j.pneurobio.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Hallett M. Pathophysiology of dystonia. J Neural Transm Suppl. 2006;70:485–488. doi: 10.1007/978-3-211-45295-0_72. [DOI] [PubMed] [Google Scholar]
- He F, Zhang S, Qian G, Zhang C. Delayed dystonia with striatal CT lucencies induced by a mycotoxin (3-nitropropionic acid) Neurology. 1995;45:2178–2183. doi: 10.1212/wnl.45.12.2178. [DOI] [PubMed] [Google Scholar]
- Hedera P, Phibbs FT, Dolhun R, Charles PD, Konrad PE, Neimat JS, Davis TL. Surgical targets for dystonic tremor: Considerations between the globus pallidus and ventral intermediate thalamic nucleus. Parkinsonism Relat Disord. 2013;19:684–686. doi: 10.1016/j.parkreldis.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Hedera P, Phibbs FT, Fang JY, Cooper MK, Charles PD, Davis TL. Clustering of dystonia in some pedigrees with autosomal dominant essential tremor suggests the existence of a distinct subtype of essential tremor. BMC Neurol. 2010;10:66. doi: 10.1186/1471-2377-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedreen JC, Zweig RM, DeLong MR, Whitehouse PJ, Price DL. Primary dystonias: A review of the pathology and suggestions for new directions of study. In: Fahn S, et al., editors. Advances in Neurology Dystonia 2. Vol. 50. 1988. pp. 123–132. [PubMed] [Google Scholar]
- Herz E. Dystonias, part 3: pathology and conclusions. Arch Neurol Psychiatr. 1944;52:20. [Google Scholar]
- Hewett J, Johanson P, Sharma N, Standaert D, Balcioglu A. Function of dopamine transporter is compromised in DYT1 transgenic animal model in vivo. J Neurochem. 2010;113:228–235. doi: 10.1111/j.1471-4159.2010.06590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewett JW, Zeng J, Niland BP, Bragg DC, Breakefield XO. Dystonia-causing mutant torsinA inhibits cell adhesion and neurite extension through interference with cytoskeletal dynamics. Neurobiol Dis. 2006;22:98–111. doi: 10.1016/j.nbd.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Hitchcock E. Dentate lesions for involuntary movement. Proc Royal Soc Med. 1973;66:877–879. doi: 10.1177/003591577306600923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffland BS, Kassavetis P, Bologna M, Teo JT, Bhatia KP, Rothwell JC, Edwards MJ, van de Warrenburg BP. Cerebellum-dependent associative learning deficits in primary dystonia are normalized by rTMS and practice. Eur J Neurosci. 2013;38:2166–2177. doi: 10.1111/ejn.12186. [DOI] [PubMed] [Google Scholar]
- Hopp JJ, Fuchs AF. The characteristics and neuronal substrate of saccadic eye movement plasticity. Prog Neurobiol. 2004;72:27–53. doi: 10.1016/j.pneurobio.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Hoshi E, Tremblay L, Feger J, Carras PL, Strick PL. The cerebellum communicates with the basal ganglia. Nature Neurosci. 2005;8:1491–1493. doi: 10.1038/nn1544. [DOI] [PubMed] [Google Scholar]
- Hubsch C, Vidailhet M, Rivaud-Pechoux S, Pouget P, Brochard V, Degos B, Pelisson D, Golmard JL, Gaymard B, Roze E. Impaired saccadic adaptation in DYT11 dystonia. J Neurol Neurosurg Psychiatry. 2011;82:1103–1106. doi: 10.1136/jnnp.2010.232793. [DOI] [PubMed] [Google Scholar]
- Hutchinson M, Nakamura T, Moeller JR, Antonini A, Belakhlef A, Dhawan V, Eidelberg D. The metabolic topography of esential blepharospasm. Neurology. 2000;55:673–677. doi: 10.1212/wnl.55.5.673. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Berardelli A, Comella C, Defazio G, DeLong M, Factor S, Hallett M, Galpern W, Ludlow CL, Perlmutter JS, Rosen A. The focal dystonias: Current views and challenges for future research. Mov Disord. 2013;7:926–943. doi: 10.1002/mds.25567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnah HA, Hess EJ. A new twist on the anatomy of dystonia: the basal ganglia and the cerebellum. Neurology. 2006;67:1740–1741. doi: 10.1212/01.wnl.0000246112.19504.61. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Hess EJ. Experimental therapeutics for dystonia. Neurotherapeutics. 2008;5:198–209. doi: 10.1016/j.nurt.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnah HA, Hess EJ, LeDoux MS, Sharma N, Baxter MG, DeLong MR. Rodent models for dystonia research: characteristics, evaluation, and utility. Mov Disord. 2005;20:283–292. doi: 10.1002/mds.20364. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Langlais PJ, Friedmann T. Functional analysis of brain dopamine systems in a genetic mouse model of Lesch-Nyhan syndrome. J Pharmacol Exp Ther. 1992;263:596–607. [PubMed] [Google Scholar]
- Jinnah HA, Richter A, Mink JW, Caldwell GA, Caldwell KA, Gonzalez-Alegre P, Cookson MR, Breakefield XO, Delong MR, Hess EJ. Animal models for drug discovery in dystonia. Expert Opin Drug Discovery. 2008;3:83–97. doi: 10.1517/17460441.3.1.83. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Wojcik BE, Hunt MA, Narang N, Lee KY, Goldstein M, Wamsley JK, Langlais PJ, Friedmann T. Dopamine deficiency in a genetic mouse model of Lesch-Nyhan disease. J Neurosci. 1994;14:1164–1175. doi: 10.1523/JNEUROSCI.14-03-01164.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemberling SR, Baird HW, Spiegel EA. Experimental torticollis of rhombencephalic origin. J Neuropathol Exp Neurol. 1952;11:184–191. doi: 10.1097/00005072-195204000-00006. [DOI] [PubMed] [Google Scholar]
- Khooshnoodi MA, Factor SA, Jinnah HA. Secondary blepharospasm associated with structural lesions of the brain. J Neurol Sci. 2013;331:98–101. doi: 10.1016/j.jns.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS. Delayed onset mixed involuntary movements after thalamic stroke. Brain. 2001;124:299–309. doi: 10.1093/brain/124.2.299. [DOI] [PubMed] [Google Scholar]
- Klier EM, Wang H, Constantin AG, Crawford JD. Midbrain control of three-dimensional head orientation. Science. 2002;295:1314–1316. doi: 10.1126/science.1067300. [DOI] [PubMed] [Google Scholar]
- Krauss JK, Seeger W, Jankovic J. Cervical dystonia associated with tumors of the posterior fossa. Mov Disord. 1997;12:443–447. doi: 10.1002/mds.870120329. [DOI] [PubMed] [Google Scholar]
- Kuoppamaki MPG, Quinn N, Wood NW, Bhatia KP. Slowly progressive cerebellar ataxia and cervical dystonia: clinical presentation of a new form of spinocerebellar ataxia? Mov Disord. 2003;18:200–206. doi: 10.1002/mds.10308. [DOI] [PubMed] [Google Scholar]
- Lange N, Hamann M, Shashidharan P, Richter A. Behavioural and pharmacological examinations in a transgenic mouse model of early-onset torsion dystonia. Pharmacol Biochem Behav. 2011;97:647–655. doi: 10.1016/j.pbb.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Le Ber I, Clot F, Vercueil L, Camzuat A, Viemont M, Benamar N, De Liege P, Ouvrard-Hernandez AM, Pollak P, Stevanin G, Brice A, Durr A. Predominant dystonia with marked cerebellar atrophy: a rare phenotype in familial dystonia. Neurology. 2006;67:1769–1773. doi: 10.1212/01.wnl.0000244484.60489.50. [DOI] [PubMed] [Google Scholar]
- LeDoux MS. The genetics of dystonias. Adv Genet. 2012;79:35–85. doi: 10.1016/B978-0-12-394395-8.00002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux MS, Brady KA. Secondary cervical dystonia associated with structural lesions of the central nervous system. Mov Disord. 2003;18:60–69. doi: 10.1002/mds.10301. [DOI] [PubMed] [Google Scholar]
- LeDoux MS, Dauer WT, Warner T. Emerging molecular pathways for dystonia. Mov Disord. 2013 doi: 10.1002/mds.25547. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux MS, Hurst DC, Lorden JF. Single-unit activity of cerebellar nuclear cells in the awake genetically dystonic rat. Neurosci. 1998;86:533–545. doi: 10.1016/s0306-4522(98)00007-4. [DOI] [PubMed] [Google Scholar]
- LeDoux MS, Lorden JF. Abnormal cerebellar output in the genetically dystonic rat. In: Fahn S, et al., editors. Dystonia 3. Vol. 78. Philadelphia: Lippincott-Raven Publishers; 1998. pp. 63–78. [PubMed] [Google Scholar]
- LeDoux MS, Lorden JF, Ervin JM. Cerebellectomy eliminates the motor syndrome of the genetically dystonic rat. Exp Neurol. 1993;120:302–310. doi: 10.1006/exnr.1993.1064. [DOI] [PubMed] [Google Scholar]
- LeDoux MS, Lorden JF, Meinzen-Derr J. Selective elimination of cerebellar output in the genetically dystonic rat. Brain Res. 1995;697:91–103. doi: 10.1016/0006-8993(95)00792-o. [DOI] [PubMed] [Google Scholar]
- Lehericy S, Tijssen MA, Vidailhet M, Kaji R, Meunier S. The anatomical basis of dystonia: Current view using neuroimaging. Mov Disord. 2013;28:944–957. doi: 10.1002/mds.25527. [DOI] [PubMed] [Google Scholar]
- Lohmann K, Klein C. Genetics of dystonia: What’s known? What’s new? What’s next? Mov Disord. 2013;28:899–905. doi: 10.1002/mds.25536. [DOI] [PubMed] [Google Scholar]
- Ma K, Babij R, Cortes E, Vonsattel JP, Louis ED. Cerebellar pathology of a dual clinical diagnosis: Patients with essential tremor and dystonia. Tremor Other Hyperkinet Mov. 2012;2:1–6. doi: 10.7916/D8JD4VJ5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoun HW, Hare WK, Ranson SW. Electrical stimulation of the interior of the cerebellum in the monkey. Am J Physiol. 1935;112:329–339. [Google Scholar]
- Manni E, Petrosini L. A century of cerebellar somatotopy: a debated representation. Nat Rev Neurosci. 2004;5:241–249. doi: 10.1038/nrn1347. [DOI] [PubMed] [Google Scholar]
- Marsden CD, Obeso JA, Zarranz JJ. The anatomical basis of symptomatic dystonia. Brain. 1985;108:463–483. doi: 10.1093/brain/108.2.463. [DOI] [PubMed] [Google Scholar]
- Martella G, Tassone A, Sciamanna G, Platania P, Cuomo D, Viscomi MT, Bonsi P, Cacci E, Biagioni S, Usiello A, Bernardi G, Sharma N, Standaert DG, Pisani A. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain. 2009;132:2336–2349. doi: 10.1093/brain/awp194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL. The dystonias. Can J Neurol Sci. 1988;15:447–483. [PubMed] [Google Scholar]
- McGeer EG, McGeer PL. Pathology of dystonias. In: Tsui JKC, Calne DB, editors. Handbook of dystonia. New York: Marcel Dekker; 1995. pp. 77–102. [Google Scholar]
- Morishita T, Foote KD, Haq IU, Zeilman P, Jacobson CE, Okun MS. Should we consider Vim thalamic deep brain stimulation for select cases of severe refractory dystonic tremor. Stereotact Funct Neurosurg. 2010;88:98–104. doi: 10.1159/000289354. [DOI] [PubMed] [Google Scholar]
- Moro E, Gross RE, Krauss JK. What’s new in surgical treatments for dystonia? Mov Disord. 2013;28:1013–1020. doi: 10.1002/mds.25550. [DOI] [PubMed] [Google Scholar]
- Mottolese C, Richard N, Harquel S, Szathmari A, Sirigu A, Desmurget M. Mapping motor representations in the human cerebellum. Brain. 2012;136:330–342. doi: 10.1093/brain/aws186. [DOI] [PubMed] [Google Scholar]
- Neychev V, Fan X, Mitev VI, Hess EJ, Jinnah HA. The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain. 2008;131:2499–2509. doi: 10.1093/brain/awn168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neychev VK, Gross R, Lehericy S, Hess EJ, Jinnah HA. The functional neuroanatomy of dystonia. Neurobiol Dis. 2011;42:185–201. doi: 10.1016/j.nbd.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer M, Carbon-Correll M, Argyelan M, Eidelberg D. Hereditary dystonia as a neurodevelopmental circuit disorder: Evidence from neuroimaging. Neurobiol Dis. 2010;42:202–209. doi: 10.1016/j.nbd.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeso JA, Gimenez-Roldan S. Clinicopathologic correlation in symptomatic dystonia. Adv Neurol. 1988;50:113–122. [PubMed] [Google Scholar]
- Odergren T, Stone-Elander S, Ingvar M. Cerebral and cerebellar activation in correlation to the action-induced dystonia in writer’s cramp. Mov Disord. 1998;13:497–508. doi: 10.1002/mds.870130321. [DOI] [PubMed] [Google Scholar]
- Oleas J, Yokoi F, MPD, Pisani A, Li Y. Engineering animal models for dystonia: What have we learned? Mov Disord. 2013;28:990–1000. doi: 10.1002/mds.25583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page ME, Bao L, Andre P, Pelta-Heller J, Sluzas E, Gonzalez-Alegre P, Bogush A, Khan LE, Iacovitti L, Rice ME, Ehrlich ME. Cell-autonomous alteration of dopaminergic transmission by wild type and mutant (DeltaE) TorsinA in transgenic mice. Neurobiol Dis. 2010;39:318–326. doi: 10.1016/j.nbd.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfi S, Leventhal L, Goetz CG, Hantraye T, Roitberg BZ, Sramek J, Emborg M, Kordower JH. Delayed onset of progressive dystonia following subacute 3-nitropropionic acid treatment in Cebus apella monkeys. Mov Disord. 2000;15:524–530. [PubMed] [Google Scholar]
- Perlmutter JS, Mink JW. Dysfunction of dopaminergic pathways in dystonia. Adv Neurol. 2004;94:163–170. [PubMed] [Google Scholar]
- Peterson DA, Sejnowski TJ, Poizner H. Convergent evidence for abnormal striatal synaptic plasticity in dystonia. Neurobiol Dis. 2010;37:558–573. doi: 10.1016/j.nbd.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettigrew LC, Jankovic J. Hemidystonia: a report of 22 patients and a review of the literature. J Neurol Neurosurg Psychiatry. 1985;48:650–657. doi: 10.1136/jnnp.48.7.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Bernardi G, Ding J, Surmeier DJ. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007;30:545–553. doi: 10.1016/j.tins.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Pisani A, Martella G, Tscherter A, Bonsi P, Sharma N, Bernadi G, Standaert DG. Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol Dis. 2006;24:318–325. doi: 10.1016/j.nbd.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Pita Lobo P, Quattrocchi G, Jutras MF, Sangla S, Apartis E, Vidailhet M, Grabli D. Primary writing tremor and writer’s cramp: Two faces of a same coin? Mov Disord. 2013 doi: 10.1002/mds.25340. [DOI] [PubMed] [Google Scholar]
- Pizoli CE, Jinnah HA, Billingsley ML, Hess EJ. Abnormal cerebellar signaling induces dystonia in mice. J Neurosci. 2002;22:7825–7833. doi: 10.1523/JNEUROSCI.22-17-07825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudente CN, Pardo CA, Xiao J, Hanfelt J, Hess EJ, LeDoux MS, Jinnah HA. Neuropathology of cervical dystonia. Exp Neurol. 2012;241:95–104. doi: 10.1016/j.expneurol.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartarone A, Hallett M. Emerging concepts in the physiological basis of dystonia. Mov Disord. 2013;28:958–967. doi: 10.1002/mds.25532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn NP, Schneider SA, Schwingenschuh P, Bhatia KP. Tremor - some controversial aspects. Mov Disord. 2011;26:18–23. doi: 10.1002/mds.23289. [DOI] [PubMed] [Google Scholar]
- Raethjen J, Deuschl G. The oscillating central network of essential tremor. Clin Neurophysiol. 2012;123:61–64. doi: 10.1016/j.clinph.2011.09.024. [DOI] [PubMed] [Google Scholar]
- Raike RS, Pizoli CE, Weisz C, van den Maagdenberg AM, Jinnah HA, Hess EJ. Limited regional cerebellar dysfunction induces focal dystonia in mice. Neurobiol Dis. 2012;49:200–210. doi: 10.1016/j.nbd.2012.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadnicka A, Hoffland BS, Bhatia KP, van de Warrenburg BP, Edwards MJ. The cerebellum in dystonia - Help or hindrance? Clin Neurophysiol. 2012;123:65–70. doi: 10.1016/j.clinph.2011.04.027. [DOI] [PubMed] [Google Scholar]
- Sadnicka A, Teo J, Kojovic M, Kassavetis P, Saifee T, Isabel P, Schwingenschuh P, Rothwell J, Bhatia K, Edwards M. Genotype specific cerebellar involvement in dyt1 and dyt6 dystonia? J Neurol Neurosurg Psychiatry. 2013;84:e2. [Google Scholar]
- Saint Hilaire MH, Burke RE, Bressman SB, Brin MF, Fahn S. Delayed-onset dystonia due to perinatal or early childhood asphyxia. Neurology. 1991;41:216–222. doi: 10.1212/wnl.41.2_part_1.216. [DOI] [PubMed] [Google Scholar]
- Schicatano EJ, Basso MA, Evinger C. Animal model explains the origins of the cranial dystonia benign essential blepharospasm. J Neurophysiol. 1997;77:2842–2846. doi: 10.1152/jn.1997.77.5.2842. [DOI] [PubMed] [Google Scholar]
- Schiebler S, Schmidt A, Zittel S, Baumer T, Gerloff C, Klein C, Munchau A. Arm tremor in cervical dystonia-Is it a manifestation of dystonia or essential tremor? Mov Disord. 2011;26:1789–1792. doi: 10.1002/mds.23837. [DOI] [PubMed] [Google Scholar]
- Sciamanna G, Hollis R, Ball C, Martella G, Tassone A, Marshall A, Parsons D, Li X, Yokoi F, Zhang L, Li Y, Pisani A, Standaert DG. Cholinergic dysregulation produced by selective inactivation of the dystonia-associated protein torsinA. Neurobiol Dis. 2012a;47:416–427. doi: 10.1016/j.nbd.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G, Tassone A, Mandolesi G, Puglisi F, Ponterio G, Martella G, Madeo G, Bernardi G, Standaert DG, Bonsi P, Pisani A. Cholinergic dysfunction alters synaptic integration between thalamostriatal and corticostriatal inputs in DYT1 dystonia. J Neurosci. 2012b;32:11991–12004. doi: 10.1523/JNEUROSCI.0041-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott BL, Jankovic J. Delayed-onset progressive movement disorders after static brain lesions. Neurology. 1996;46:68–74. doi: 10.1212/wnl.46.1.68. [DOI] [PubMed] [Google Scholar]
- Song CH, Bernhard D, Bolarinwa C, Hess EJ, Smith Y, Jinnah HA. Subtle microstructural changes of the striatum in a DYT1 knock-in mouse model of dystonia. Neurobiol Dis. 2013;54:362–371. doi: 10.1016/j.nbd.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CH, Fan X, Exeter CJ, Hess EJ, Jinnah HA. Functional analysis of dopaminergic systems in a DYT1 knock-in mouse model of dystonia. Neurobiol Dis. 2012;48:66–78. doi: 10.1016/j.nbd.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe LM, Kim CE, Alagem N, Dauer WT. Primary dystonia: molecules and mechanisms. Nat Rev Neurol. 2009;5:598–609. doi: 10.1038/nrneurol.2009.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo JT, van de Warrenburg BP, Schneider SA, Rothwell JC, Bhatia KP. Neurophysiological evidence for cerebellar dysfunction in primary focal dystonia. J Neurol Neurosurg Psychiatry. 2009;80:80–83. doi: 10.1136/jnnp.2008.144626. [DOI] [PubMed] [Google Scholar]
- Thobois S, Ballanger B, Xie-Brustolin J, Damier P, Durif F, Azulay JP, Derost P, Witjas T, Raoul S, Le Bars D, Broussolle E. Globus pallidus stimulation reduces frontal hyperactivity in tardive dystonia. J Cereb Blood Flow Metab. 2008;28:1127–1138. doi: 10.1038/sj.jcbfm.9600610. [DOI] [PubMed] [Google Scholar]
- Thompson RF, Steinmetz JE. The role of the cerebellum in classical conditioning of discrete behavioral responses. Neuroscience. 2009;162:732–755. doi: 10.1016/j.neuroscience.2009.01.041. [DOI] [PubMed] [Google Scholar]
- Vidailhet M, Jutras MF, Grabli D, Roze E. Deep brain stimulation for dystonia. J Neurol Neurosurg Psychiatry. 2012;84:1029–1040. doi: 10.1136/jnnp-2011-301714. [DOI] [PubMed] [Google Scholar]
- Wilson B, Hess EJ. Animal models for dystonia. Mov Disord. 2013;28:982–989. doi: 10.1002/mds.25526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Ledoux MS. Caytaxin deficiency causes generalized dystonia in rats. Brain Res Mol Brain Res. 2005;141:181–192. doi: 10.1016/j.molbrainres.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Zhao Y, DeCuypere M, LeDoux MS. Abnormal motor function and dopamine neurotransmission in DYT1 DeltaGAG transgenic mice. Exp Neurol. 2008;210:719–730. doi: 10.1016/j.expneurol.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoons E, Booij J, Nederveen AJ, Dijk JM, Tijssen MA. Structural, functional and molecular imaging of the brain in primary focal dystonia--a review. Neuroimage. 2011;56:1011–1020. doi: 10.1016/j.neuroimage.2011.02.045. [DOI] [PubMed] [Google Scholar]
- Zoons E, Tijssen MAJ. Pathological changes in the brain in cervical dystonia pre- and post-mortem: a commentary with a special focus on the cerebellum. Exp Neurol. 2013;247:130–133. doi: 10.1016/j.expneurol.2013.04.005. [DOI] [PubMed] [Google Scholar]