

Abstract
Age associated decline of the immune system continues to be a major health concern. All components of innate and adaptive immunity are adversely affected to lesser or greater extent by ageing resulting in an overall decline of immunocompetence. As a result in the aged population, there is increased susceptibility to infection, poor responses to vaccination, and increased incidence of autoreactivity. There is an increasing focus on the role of T cells during ageing because of their impact on the overall immune responses. A steady decline in the production of fresh naïve T cells, more restricted T cell receptor (TCR) repertoire and weak activation of T cells are some of the effects of ageing. In this review we summarize our present understanding of the effects of ageing on naïve CD4 T cells and potential approaches for therapeutic interventions to restore protective immunity in the aged population.
Keywords: Adaptive immunity, ageing, haematopoietic stem cells, innate immunity, TCR repertoire, T cells, thymic involution, transcription factors
Introduction
Higher organisms have evolved an elaborate defense mechanism against a variety of pathogenic organisms. The first line of such defense is termed as the innate immune response as it is evolutionarily older, found in many more organisms, is non-specific in nature and depends upon recognition of highly conserved pathogen associated molecular patterns (PAMPs) by a series of germline encoded pattern recognition receptors (PRRs). These receptors are expressed on a range of cells which respond to pathogenic threats by a number of means such as engulfment of pathogen, production of antimicrobial peptides and acid hydrolases, autophagy, production of highly reactive and oxidizing molecules like free radicals or reactive oxygen species (ROS). Together these mechanisms mediate the killing and clearance of organisms detrimental to the host1. A second defense mechanism evolved in jawed vertebrates and is now shared in humans. It is characterized by the presence of a large array of anticipatory receptors that can recognize practically any kind of antigen2. These receptors are generated by rearrangement of genes and are expressed on specialized cells called lymphocytes2. This kind of immune response is termed the adaptive immune response. Specificity to a particular antigen, generation of long-lived memory and tolerance to self-components are hallmarks of adaptive immune responses. The primary components of adaptive immunity are T and B lymphocytes, which originate in thymus and bone marrow, respectively and enter in circulation after maturation. The distinction of innate and adaptive response is not an absolute bifurcation of immunity as both these branches constantly cross communicate; naive T cells are primed by dendritic cells (DCs) which in turn secrete cytokines required for the recruitment and activation of innate immune cells like macrophages. Both these immune responses work together to protect the host from an invading pathogen.
The immune system in humans develops very early at the embryonic stage itself with the appearance of the first haematopoietic stem cells (HSC) in embryonic yolk sac during the first week of development, next migrating to liver and spleen, arriving in bone marrow eventually, which serves as primary centre of haematopoesis at birth and throughout the adult life of an individual3. Precursors of T lymphocytes migrate from the bone marrow and colonize the thymus. All the important events, such as gene rearrangement for generating diversity, development of functional lymphocyte and tolerisation to self-antigens take place in the bone marrow for B cells and in the thymus for T cells resulting in a fully competent lymphocyte repertoire. These lymphocytes stay in circulation throughout the adult life sensing non-self entities and mounting appropriate immune responses. As efficient as the immune system is in its multilayered approach in fighting pathogens, its efficacy decreases with ageing. It has been observed that intrinsic defects in cells of the immune system accumulate over a period of time manifesting themselves in the inability of the aged to mount an effective immune response against infectious disease and a heightened risk of developing autoimmune responses4. Age related immune dysfunctions are often described as ‘immunesenescence’ which can be a whole organismal effect or an effect on individual cells and its causes are wide ranging. From intrinsic defects such as thymic involution with age that leads to reduced thymic output in terms of naïve T cell numbers, reduction of B cell progenitors from bone marrow, oligoclonal expansion and accumulation of T cells because of chronic viral infections to an overall decline of regenerative capacity of the HSC with age or shortening of telomeres with successive cell division, many can be listed. All these factors contribute towards the phenomenon of immunesenescence5,6. Though age related changes on lymphocytes are well documented7, innate immune responses are also compromised in aged individuals8.
Effect of ageing on innate immunity
Many cell types of the innate immune system such as macrophages, DCs and neutrophils are adversely affected with advancing age. Macrophages/monocytes are present in all tissues and are the first cells to encounter particulate matter apart from neutrophils. It has been observed that macrophages from aged individuals have lower expression of surface molecules like major histocompatibility complex II (MHC-II)9 and toll like receptors (TLRs) which could adversely impact their antigen presentation, produce poor levels of interleukin (IL)-6 and tumour necrosis factor (TNF)-α when stimulated with known agonists of TLR as compared to macrophages from young mice10. Other receptor driven functions like chemotaxis, phagocytosis and respiratory burst, essential for containing the pathogens are also compromised in the elderly11. Dendritic cells are another major component of the innate immune responses that are present in all tissues; these are characterized by their high phagocytic and antigen processing and presentation capabilities. After antigen acquisition, DCs mature and migrate to the nearest lymph node by upregulating C-C chemokine receptor type 7 (CCR7) receptors. Once inside the lymph node these start priming naïve T cells. Cytokine secretion from DCs also determines to a significant extent what differentiation pathway helper T cells would be directed to. Apart from macrophages, DCs are another cell type affected by ageing. Langerhans cells are DCs present in skin and their numbers decrease with increasing age12. DCs in aged individuals also have lower numbers of co-stimulatory molecules and lower production of IL-12, though these retain their ability of antigen presentation13, indicating that some of the age related dysfunction observed in T cells could be related to changes in the effectiveness of the DC population in the aged. After an infection the first cell type to infiltrate the infected sites are neutrophils which are armed with a range of antimicrobial arsenal like superoxide, reactive nitrogen intermediates, antimicrobial peptides and degradative enzymes. These very short lived cells rapidly initiate an effective antimicrobial defense before other specialized cells, e.g., the lymphocyte can react to the threat. No change in their numbers has been observed in the aged versus the young14 but all functional aspects of neutrophils such as chemotaxis, production of superoxide and their ability to respond to survival signals from granulocyte macrophage colony-stimulating factor (GM-CSF) are compromised leading to more apoptotic cells at the site of infection15. The presence of these apoptotic cells at the site of infection could delay the resolution of infection resulting in persistent inflammation. Natural killer (NK) cells originate from common lymphoid progenitors that give rise to T and B lymphocytes; however, these do not express unique antigen receptors as expressed by either B cells or T cells. They mediate MHC-independent direct and rapid killing of virus-infected cells or tumour cells by the release of perforin and granzymes, and are identified by the expression of CD56 and CD16. It has been observed that overall NK cell numbers increase with age, evident from an increase in the cytotoxic CD56dim population; however, their killer activity on a per cell basis decreases in the aged individuals and so does their ability to produce cytokines such as IL-8 and chemokines such as regulated on activation normal T-cell expressed and secreted (RANTES) and macrophage inflammatory protein (MIP)-1α, which might lead to a greater susceptibility of aged towards infection16.
Effect of ageing on adaptive immunity
B lymphocytes are crucial for humoral immunity. These cells originate in foetal liver or adult bone marrow from HSCs that give rise to common lymphoid precursors (CLPs). Some cells from CLPs differentiate into B cells under the influence of the bone marrow microenvironment by differential expression of transcription factors and cytokine receptors. The developing B cells undergo a series of stages characterized by the presence of cell surface markers and immunoglobulin gene recombination and are termed as pro-B cells, pre-B cells, immature-B cells, transitional-B cells and mature-B cells17. B cell development proceeds to the next stage only in the case of successful gene rearrangement of immunoglobulin genes; otherwise these are deleted. Their interaction with bone marrow stromal cells is very crucial for their development. Self-reactive immature-B cells are eliminated resulting in central tolerance of remaining B cells. B cells at immature-B cell stage expressing IgM on their surface migrate to the secondary lymphoid organs such as the lymph nodes or spleen. After encountering an antigen, either the B cells migrate to the medulla of lymph nodes, complete their differentiation and undergo class switching, becoming antibody secreting plasma cells or migrate into a nearby follicle, forming a germinal centre by proliferating rapidly and undergoing somatic hypermutation, resulting in the selection of cells expressing immunoglobulins with an even higher affinity for previously encountered antigen (Figure A). Ageing affects B cell responses quantitatively, indicated by reduced antibody production and qualitatively, evident in the production of lower-affinity antibodies18. The B cell pool in the aged decreases possibly because there is an overall decline in bone marrow output and reduction in numbers of HSCs committing to B cell lineage, resulting in a decrease of early B cell precursors in the aged mice (Figure B). For successfully clearing an infection, B cells undergo class switching, brought about by the help provided by T cells, whereby these can switch the production of surface IgM to IgG, IgE or IgA. Class switching produces essentially an antibody with the same specificity but with different effector function. It has been reported that both, the enzyme for class switching, activation-induced cytidine deaminase (AID) and E47, the transcription factor that controls its expression, are downregulated in aged murine B cells due to decreased mRNA stability19. Age related changes affect B cell population partly because of T cell dysfunction in the elderly but also because of the above-mentioned intrinsic defects in B cells and translate at a functional level in the compromised response of the elderly to vaccination and with the production of auto-reactive and lower affinity antibodies.
Fig.
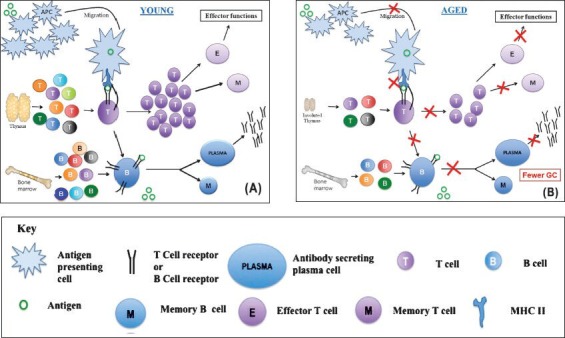
Effect of ageing on adaptive immunity. (A) Broad outline of the adaptive immune responses in the young host. Antigen presenting cells (APCs) pick up invading organisms by phagocytosis and migrate to regional lymph nodes. Here they encounter diverse population of naïve T cells which are produced continuously by a well-functioning thymus. On encounter with the antigen presented as peptide major histocompatibility complex (pMHC) complex by professional APCs such as dendritic cells (DCs), T cells get activated and multiply. The expanded population further differentiates into the effector and memory T cells. Naïve B cells are also generated steadily by the bone marrow in the young. On encounter with antigen these get activated and differentiate to produce memory B cells and antibody secreting plasma cells. In the germinal centre antigen-specific T cells provide help to B cells for class switching. (B) Broad outline of the adaptive immune responses in the aged host. As compared to the young hosts, efficiency of APC migration and making stable contact with T cells (immunological synapse) decreases. This results in qualitatively and quantitatively poor activation and differentiation of T cells. The thymic output also decreases in the aged host further compromising the immune response. B cell output from the bone marrow is also adversely affected with age. T-B interactions happen less efficiently with fewer germinal centres (ac) getting formed and poor differentiation process leading to poor antibody responses. The red crosses indicate a reduction in the respective activity.
The effects of ageing on T cells are more researched and documented than for any other cell populations, primarily because these are the effectors and regulators of the immune response and because of thymic involution. This natural phenomenon reduces the output numbers of naïve T cells with increasing age. T cell precursors from bone marrow routinely seed the thymus where they undergo maturation and enter the periphery. Like B cells, T lymphocytes also undergo gene rearrangement for antigen receptors and extensive selection process where self-reactive T cells are deleted. Apart from the conspicuous effect of thymic involution on naïve T cells, it has been observed that ageing results in other functional defects as well: decreased T cell repertoire, decreased interleukin (IL)-2 production, and an increased memory T cell population due to low grade viral infection (Figure B)20. All these factors have a significant negative impact on the immunity of the aged individuals.
Generation and activation of T cells
T cell precursors arise from HSCs in bone marrow of adult or foetal liver. Thymic seeding progenitors (TSPs) arrive in the thymus in small numbers where upon their interaction with the thymic epithelium give rise to the earliest thymic progenitors (ETPs). ETPs develop gradually through successive stages of differentiation from CD4-CD8- double negatives (DNs), to CD4+CD8+ double positives (DPs), to either CD4+ or CD8+ single positive (SPs) T cells restricting their lineage options at each stage. The development and schooling of T lymphocytes takes place in the thymus. The thymus is differentiated into an outer cortical region and an inner medullary region. One of the most important factors governing T cell development is the thymic microenvironment and the interaction of the T cell progenitors with thymic stromal cells. It is in the thymus that multipotent T cell progenitors lose alternative lineage differentiation capacity to become fully committed T cells.
Developmental stages of T cell progenitors is determined by thymic microenvironment
Depending on their developmental stage, T cell precursors can be found in different thymic regions. ETPs enter the thymus from blood vessels near the cortico-medullary junction and under the influence of CCR7 and CCR9 settle into the thymus21. These early ETPs are characterized as CD4-CD8-CD3-CD44+CD25- double negative (DN1) cells. These DN1 cells carry multi-lineage potential; and still retain the capability to differentiate into B cells, NK cells, DCs and T cells. A crucial factor that pushes DN1 cells exclusively towards a T cell fate is Notch signaling22. DN1 cells express the molecule, Notch1 and the corresponding ligand, delta-like ligand 4 (DLL4) is expressed by thymic epithelial cells. Induced deletion of Notch1 leads to complete block of T cell development leading to the ectopic development of immature B cells in the thymus23, indicating a central role of this molecule in T cell lineage commitment. However, little is known about the molecular mechanism by which Notch signaling induces this commitment. The cells now migrate deeper into the cortex, interaction with thymic epithelial cells (TECs) and fibroblasts leads to their differentiation into DN2 stage characterized byCD4-CD8-CD3-CD44+CD25+ expression and the beginning of rearrangement of T cell receptor (TCR) gene locus24,25. DN2 cells carry the potential to differentiate into NK cells and DCs apart from T cell potential; however, these do not have any B cell potential left. Depending on IL-7 receptor expression these DN2 cells could be further subdivided as IL-7Rhi and IL-7Rlo that have the capacity to differentiate into γδ or αβ T cells, respectively26. DN2 cells go through a transition from an early DN2a (CD4-CD8-CD44+CD25+CD117hi) stage having the potential to give rise to DCs and NK cells to a late DN2b (CD4-CD8-CD44+CD25+CD117int) stage, completely losing the potential to differentiate into DCs and only retaining alternative NK cell lineage potential27. Cells now migrate to the sub-capsular zone (SCZ) of the thymus maturing into DN3 cells defined as, CD4-CD8-CD3-CD44loCD25+. At this stage, final commitment to T cell lineage occurs with no alternative developmental pathway left to explore. DN3 cells begin to rearrange β-chain locus. A pre-T cell receptor is expressed at this stage on the cell surface comprising a TCRβ chain, a surrogate chain called pTα (pre T cell receptor α chain) and components of CD3 chains28. Cells that fail to productively rearrange their β-chain locus die. DN3 cells now migrate back towards the medullary cortex losing the expression of CD25 and becoming DN4 cells characterized by CD4-CD8-CD3-CD44-CD25- expression. Signaling through the pre-TCR leads to the expression of CD4 and CD8 both on the surface of cells resulting into CD4+CD8+ double positive (DP) cells29. Rearrangement of the α-chain locus begins at this stage and DP cells start expressing low levels of the TCR complex. DP cells undergo positive or negative selection by interacting with self-peptide MHC (pMHC) complex expressed on fibroblasts, DCs and TECs. Most cells fail to make it through, and die in the process. Relatively very few cells with intermediate avidity to self-pMHC survive that finally become CD4-CD8+ or CD4+CD8- T cells30.
Transcription factors
T cell lineage commitment is a sequential process where at each stage, T cells lose the potential to differentiate into alternate lineages. Several transcription factors are activated and silenced during this process. One of these is BCL11b (B cell lymphoma 11b), a zinc finger transcription factor that is expressed at the DN2 stage and maintained throughout thymocyte development. BCL11b suppresses NK cell and myeloid lineages. Its deletion at the DN3 stage where thymocytes have committed to the T cell lineage lose expression of T cell lineage genes and start expressing NK cell lineage genes. This indicates the crucial importance of BCL11b transcription factor in the lineage commitment of thymocytes to a T cell fate31. Another essential transcription factor for T cell development is GATA3. Its role in initiating a Th2 effector programme is well established; however, GATA3 is also required for early T cell development in the thymus. It is expressed in ETPs and the expression continues until the DN3 stage after which it subsides, again becoming upregulated in single positive CD4+ T cells. GATA3 deficient foetal liver cells give rise to very few ETPs, and no downstream T cell lineage progenitors. GATA3 deficiency has no effect on survival or proliferation, indicating that GATA3 most likely regulates differentiation of early T cell progenitors32. Expression of both these transcription factors BCL11b and GATA3 is induced by another transcription factor, T cell factor 1 (TCF-1) which is expressed in ETPs and is strongly upregulated by Notch signaling. TCF-1 deficiency results in decreased thymic cellularity. Overexpression of TCF-1 leads to the induction of T cell lineage programme even in the absence of Notch signaling indicating its important role in early thymic development of T cell progenitors33.
Chemokines
As evident from the above description, T cell development within the thymus is location specific. T cell progenitors are recruited to the thymus depending on the expression of chemokine receptors where they migrate from the cortico-medullary junction to interior parts of the cortex to the sub-capsular zone and then traverse back into the cortex. These intrathymic movements are controlled by the expression of chemokine receptors on T cell progenitors and their ligands expressed by thymic epithelial cells. The chemokine receptors crucial for settling of T cell progenitors into the thymus are CCR7 and CCR9. The ligands for CCR7 and CCDR9 are CCL19, CCL21, and CCL25, respectively, all of which are secreted in abundance by the thymic stromal cells. Adult mice lacking both CCR7 and CCR9 fail to recruit any T cell progenitors to the thymus21,34. Once inside the thymus, DN cells migrate towards the outer cortex, which is controlled by CXC chemokine receptor CXCR4 expression and its ligand CXC chemokine ligand CXCL12. CXCR4 deficiency leads to the blockage of migration of thymocytes from the cortico-medullary junction to the cortex and, therefore, these fail to progress from DN1 stage to later stages of development35. Thymocytes undergo positive selection by their interactions with self-pMHC, which also results in the upregulation of CCR9 and CCR7 resulting in their migration back into the medulla. Chemokines play a very important role in localizing thymocytes in the different microenvironments within the thymus leading to their appropriate development into functional T cells.
Homeostatic maintenance of naïve T cell pool
Most of the thymocytes are deleted owing to their ability to bind self pMHC complex either too strongly or too weakly, thereby significantly decreasing the chances for autoimmune responses later in their life time. The small pool of naïve T cells that survive are the cells with optimal binding ability to MHC. This T cell pool egresses the thymus and stays in constant circulation in the periphery searching for the presence of an appropriate ligand in the form of a peptide loaded MHC molecule. This naïve T cell pool stays constant through adult life despite thymic involution and transition of naïve T cells into memory T cells due to activation36. The survival and expansion of this naïve T cell pool in this stage is controlled by a number of factors, primary among these is their interaction with self-peptide loaded MHC and the presence of IL-7. The role of self-pMHC in maintenance of the naïve T cell pool has been reported in several studies. One such study has shown that CD4+ T cells engrafted into recombinant activating gene Rag-2-/- MHC II-/- recipient mice gradually decline over a period of 6 months. This suggests that low level interactions of self-pMHC with TCR play a significant role in long term survival and maintenance of the naïve T cell pool, probably by rescuing the cells from death37. Another study38 found a different requirement for maintenance and proliferation of naïve and memory CD8+T cell population. Naïve T cells appear fastidious in their requirement for survival and proliferation on MHC I molecules, while memory CD8 T cells expand to a significant extent even in the absence of all MHC I molecules. In contrast, Dorfman et al39 have shown that naïve CD4 and CD8 T cells that interact with self-pMHC show activation of the TCR as evident by the partial phosphorylation of the TCRζ chain and the phosphorylation diminishes in absence of self-pMHC. However, such interactions were not found to be necessary for naïve T cell survival as evident by their survival in MHC deficient hosts for almost a month39. Despite contradictory initial claims, it is now well established that homeostatic proliferation requires low threshold of TCR activation that is provided by self-pMHC complex and full gain of effector function in such a situation will be detrimental for self. Thus, suboptimal induction of genes normally associated with TCR activation is sufficient for homeostatic proliferation of naïve T cells, a unique gene pattern is neither observed nor seems to be required for such proliferation in naïve CD8 T cells40. Another study indicated that when monoclonal transgenic CD4+ T cells were transferred at low frequency, they were better able to survive than when transferred at higher frequencies establishing a role for intraclonal competition for self-pMHC II in maintenance of the naïve T cell pool in the periphery41. Naïve CD4+T cells cannot survive in the absence of γc, a receptor that is utilized by many cytokines such as IL-2, IL-4, IL-7, IL-9, and IL-1542. Among all γc cytokines, IL-7 was demonstrated to be critically involved in the proliferation and survival of naïve T cells as adoptively transferred naïve T cells failed to proliferate in IL-7-/- mouse and their numbers declined within a month indicating death in the absence of IL-743. IL-7 is also required for proliferation of naïve CD4+T cells in a lymphopenic condition where it acts by upregulating b cell lymphoma Bcl-2, an anti-apoptotic protein in response to IL-7R engagement44. IL-7 mediated survival of CD4+T cells has been shown to be a result of higher telomerase activity in these cells as compared to memory T cells45. Stromal and epithelial cells in the bone marrow and thymus produce IL-7 for lymphopoiesis and it is also produced in secondary lymphoid organs for homeostasis of mature T cells. Both these factors, self-pMHC and IL-7, are crucial for maintaining a constant naïve T cell pool with broad specificity throughout the life of an individual.
T cell activation
Naïve T cells after completing their development, exit the thymus and stay in circulation patrolling secondary lymphoid organs such as lymph nodes and spleen, for potential pathogenic or non-self entities. A successful immune response is dependent on the engagement and activation of the T cell receptor with a defined specificity with a foreign peptide loaded MHC molecule in a sea of self-pMHC complexes present on the surface of cells. This engagement occurs in the T cell zone of secondary lymphoid organs. Such a TCR-pMHC interaction leads to the formation of an immunological synapse, which is a localized accumulation of the TCR complex and associated intracellular signaling molecules46. Binding of the TCR complex with pMHC leads to the phosphorylation of immunoreceptor-Tyrosine based activation motifs (ITAMs) in the ζ chain of CD3 molecules by Lck and Fyn, kinases of the Src family. This creates a binding site for zeta-chain-associated protein kinase (ZAP)-70, which is then phosphorylated by Lck. After activation, phosphorylated ZAP-70 phosphorylates scaffold proteins linker of activation of T cells (LAT) and SH2 domain containing leukocyte protein (SLP)-76. Phosphorylation of both these molecules leads to the recruitment of other proteins, Vav (a guanine nucleotide exchange factor), the adaptor proteins non-catalytic region of tyrosine kinase adaptor protein (NCK) and Grb2-related adaptor downstream of Shc (GADS), and an inducible T cell kinase interleukin-2-inducible T-cell kinase (ITK). ITK phosphorylates phospholipase Cγ1 (PLCγ1) resulting in the generation of the second messengers, diacylglycerol (DAG) and inositoltrisphosphate (IP3), by hydrolyzing phosphatidylinositol 4,5-bisphosphate (PIP2). DAG activates PKCθ and mitogen-activated protein kinase/extracellular signal-regulated kinase (MAP/Erk) pathways, which leads to the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF)-κB while IP3 causes the release of intracellular calcium leading to the activation of calcineurin, which further activates nuclear factor of activated T cells (NFAT). Both these transcription factors drive the expression of several genes required for the activation and proliferation of T cells, including IL-247.
Naïve T cells, in addition to the primary signal by the TCR complex, also require co-stimulatory signals. In the absence of co-stimulation, T cells become unresponsive which is described as ‘anergy’. These co-stimulatory signals are conveyed independently of the TCR through CD28, a molecule expressed on T cells, via its engagement with either CD80 or CD86 on antigen presenting cells. In the absence of CD28 mediated signaling, T cells show reduced proliferation and poor help to B cells seen as inhibition in germinal centre formation and immunoglobulin class switching. CD28 signaling primarily works by activating phosphoinositide-3 kinase pathway (PI3K). PI3K leads to the activation of Akt, which phosphorylates many proteins. Some of these proteins further lead to the nuclear transport of NF-κB and NFAT. These transcription factors aid in the transcription of prosurvival proteins like Bcl-xl with IL-2 contributing to the overall signals generated by TCR signaling. CD28 signaling does not seem to provide a qualitatively different signal but only helps in amplifying the signaling response generated by the TCR complex48. Apart from CD28, other signaling molecules also provide co-stimulatory signals. Key among these is inducible co-stimulator (ICOS), which is expressed on the activated T cells and shares many structural similarities with CD28. Similar to CD28, ICOS enhances T cell differentiation, cytokine production and immunoglobulin production. ICOS stimulation, however, does not lead to the production of IL-2, therefore, endowing T cells in the periphery with effector functions without providing them with the ability to further expand. Members of the tumor necrosis factor receptor (TNFR) family CD27, CD40, OX40 and 4-1BB are other important co-stimulatory molecules, which enhance and sustain an immune response and are important for memory T cell generation. Apart from co-stimulatory signals that enhance the TCR pMHC signals, there are inhibitory signals also generated that control this primary signal; key among such molecules is a homologue of CD28, cytotoxic T-lymphocyte associated antigen-4 (CTLA-4) and programmed death-1. Like CD28, CTLA-4 can also bind to CD80 and CD86. However, it does not enhance the primary signal generated by TCR-pMHC, inhibiting instead IL-2 production and progression to the cell cycle, thus limiting the scale of the T cell response and preventing autoimmunity49,50. The result of TCR stimulation with pMHC and co-stimulatory signals is the proliferation and differentiation of T cells into effector populations. IL-2 is the primary cytokine driving T cell proliferation. It is a 15.5 kDa secretory molecule produced primarily by activated T cells and acts in an autocrine and paracrine manner via IL-2 receptor. IL-2R can be dimeric, composed of a common γ-chain and IL-2Rβ, which binds to IL-2 with weak affinity or it can be trimeric composed of an additional IL-2Rα chain and binds with a high affinity. Signaling by IL-2R leads to the activation of Akt, MAP kinase and JAK-STAT pathways which mediate cell differentiation, growth, survival and activation induced cell death51,52. The cytokine milieu and expression of specific transcription factors determine the outcome of an infection by guiding CD4 T cells toward distinct effector populations. A combination of IL-12 with STAT-4 and T-bet helps in the differentiation of the T helper (Th)-1 cells lineage that secretes interferon (IFN)-γ and is important for the clearance of intracellular pathogens. IL-4 along with GATA-3 and STAT-6 is required for the development of the Th2 lineage that secretes IL-4, IL-5 and IL-13 and is required to fight extracellular pathogens. Transforming growth factor (TGF)-β and IL-6 along with RORγt are required for the differentiation of the Th17 lineage which is essential for antibacterial and antifungal responses while TGF-β along with IL-2 promotes the differentiation of T regulatory (Treg) cells that help in preventing autoimmunity and generation of tolerance52.
After initial TCR stimulation in lymphoid tissues, T cells undergo a series of phases. A proliferation or expansion phase where antigen activated T cells clonally expand rapidly and differentiate into effector populations (Figure A) to control the infection. This is followed by a contraction phase where more than 90 per cent of cells die to maintain homeostasis. Lastly, a memory phase where a small number of these T cells is maintained in case of a future antigenic challenge. The memory T cell pool, in case of successive antigenic challenge, rapidly proliferates and results in an immune response, which is faster and stronger than the primary response and clears up the infection relatively sooner. Memory T cells are able to mount a vigorous response against a secondary challenge due to permanent changes in chromatin structure and transcription factors leading to elevated messenger RNA levels for effector molecules like IFN-γ, perforin and granzyme. Memory T cells could be further subdivided based on the expression of surface markers as central memory T cells (TCM) (CD62L+CCR7+) and effector memory T cells (TEM) (CD62L- CCR7-). TCM cells home to secondary lymphoid organs and produce IL-2, undergoing regeneration to maintain a constant pool and are involved in secondary challenges and maintenance of long-lived memory. In contrast, TEM migrate to inflamed peripheral tissues and on TCR stimulation rapidly produce inflammatory cytokines providing protection from ongoing infection53.
Effect of ageing on T cells
Effects of ageing on HSCs
A very small population of HSCs residing in the bone marrow generates all the cells of the haematopoietic lineage; B cells, T cells, NK cells, DCs, macrophages, granulocytes, platelets and erythrocytes. All these cells have a definite life span after which these die off and are cleared from the system, being replaced by fresh cells generated by the stem cells in bone marrow. HSCs retain the capacity to regenerate themselves and also give rise to daughter cells that could follow a myeloid or lymphoid pathway, thus maintaining a constant pool of effector cells in the system. The process of ageing affects all the cells including HSCs, despite their self-renewal capabilities. One of the most conspicuous effects is a shift in balance from the lymphoid lineage in young adults to the myeloid lineage in the elderly54, manifested as a reduced number of memory B cells and naïve T cells in the elderly, adversely affecting the immune response. Oxidative stress and reduced telomerase activity are other two factors that affect the functioning of HSCs in the aged population55,56. With increasing age there is an accumulation of ROS induced DNA damage, along with a decline in the activity of DNA repair gene expression, resulting in decreased activity of the HSCs. Telomeres are found at the end of eukaryotic chromosomes that prevent chromosomes from fusing with each other. After successive divisions, telomere length decreases which is restored by the activity of telomerase57. HSCs of aged show decreased telomerase activity, compromising genomic integrity and restricting the proliferative capacity of HSCs. The effect of ageing on immune and blood cells could be traced back to general defects accumulated in HSCs over a life time in the aged.
Thymic involution
One of the most significant contributors to the phenomenon of ageing is the process of thymic involution, a gradual decline in the structural integrity of the thymus with age, resulting in an atrophied thymus (Figure B). This structural waning of the thymus results in a steady decline in the production of naïve T cell numbers leading to a restricted TCR repertoire. As more naïve T cells turn into memory cells owing to constant antigenic stimulation, there is an increased risk of severe infection in the elderly due to a paucity of naïve T cells. The thymus represents a major component of the immune system, where T cell progenitors undergo differentiation, generate TCR diversity, become self-restricted and undergo positive and negative selection to avoid immunodeficiency or autoimmunity later in life58. The thymus develops at the foetal stage and gains its maximum functioning around the perinatal period after which it declines gradually, a process that continues throughout adult life59. An aged thymus is characterized by reduction in overall size along with an increase in nonfunctional, non-thymopoietic perivascular space and a decrease in functional, thymic epithelial space. This is accompanied by a loss of organized thymic architecture and an increased deposition of adipocytes within the thymus60. Several possible hypotheses for age associated thymic involution have been proposed such as, the paucity of T cell progenitors from bone marrow, effect of circulating hormones and cytokines, loss of thymic architecture with increasing age and changes in productive TCR rearrangement; yet none of these convincingly explain why thymic function declines with age given that naïve T cells are required throughout the adult life. A common mechanism proposed for thymic involution is puberty and evidence comes from the fact that chemical or surgical castration of male mice leading to sex steroid ablation, reverses age related thymic decline and restores thymic function61. In contrast, administration of androgens and estrogens in uncastrated mice results in decreased thymopoiesis62. Interpreting these data of thymic involution in mice as a result of puberty is slightly more challenging in humans because the human thymus gains its full functionality right after birth and starts declining within a year, long before attaining sexual maturity. In mice, however, the time between weaning and sexual maturity is not that far apart and coincides with the beginning of thymic involution. Also, androgen levels decrease in humans with age, yet there is no observable reversal of thymic involution with age. These arguments point to the fact that levels of sex hormones might influence the process of thymic involution and rebound but these are not the only factors responsible for such a phenomenon; other contributing factors must be involved. T cell progenitors and thymic epithelial cells constantly cross-communicate and are mutually inter-dependent for survival and maintenance. There is a qualitative decline in the production of ETPs with age. HSCs and ETPs from aged mice are not very efficient in foetal thymus reconstitution assays63. Bone marrow precursor transplant from a young mouse into an aged does not fully restore thymic function64, indicating that intrinsic defects within the ETPs and intrathymic microenvironment together contribute to the overall phenomenon of thymic involution. The levels of transcription factors are also altered in the aged thymus. The expression of E2A, a transcription factor required for early T cell development declines as much as 18-fold by 7 months of age while the expression of its negative regulator LMO2 increases with increasing age. The expression of Foxn1, a key transcription factor required for the development of thymic epithelium, shows a 16-fold reduction at 12 months of age in BALB/c mice65. Several cytokines of the IL-6 family are secreted by the thymus and have a potent effect on naïve T cells and are associated with ageing. Leukemia inhibitory factor (LIF), IL-6, Oncostatin M (OSM), and stem cell factor (SCF) are expressed at slightly higher levels in aged individuals but the levels of IL-7, a cytokine crucial for maturation and maintenance of thymocytes, stays constant66. Furthermore, exogenous treatment of mice with IL-6 family cytokines induces rapid and acute thymic involution66. These studies suggest that levels of intrathymic cytokines change over a period of time contributing to the overall decline of thymic function. Despite the lack of a clear consensus on what exactly are the initiating and driving factors of thymic involution, it is clear that the decline of naïve T cell numbers in the periphery is directly associated with this phenomenon. More investigation needs to be done to understand the cellular and molecular mechanism of thymic involution.
Effect of ageing on homeostasis of naïve CD4 T cell pool and TCR diversity
Naïve CD4 T cell production and homeostasis are tightly regulated to maintain a constant pool of cells. After leaving the thymus, recent thymic immigrants (RTEs) are exported to secondary lymphoid organs where these receive low level but critical survival signals from self-pMHC and IL-7. Under homeostatic conditions, naïve cells undergo a very low level of spontaneous proliferation; however, these can expand rapidly under lymphopenic conditions probably by sensing an excess of a survival signal like IL-7. With the onset of thymic involution this constant source of RTEs declines leading to a decrease in fresh naïve T cell number in the periphery and the homeostatic mechanisms adjust accordingly to maintain a constant pool of cells67. In a young mouse the naïve T cell pool is very dynamic. In response to cognate antigens, some naïve T cells get activated and move out of the pool68, whereas some never encounter their cognate antigen and hence keep recirculating through the secondary lymphoid organs or die to make space for the newly emerging cells from the thymus. Thus, the naïve T cell pool contains cells of varying age. In the aged individual this balance is disturbed with the decline of RTEs. The naïve T cell pool, however, is maintained at a constant level indicating that the T cell pool in the aged contains naïve CD4 T cells of either longer chronological age or progeny of such cells, which in itself is sufficient to accumulate age related defects. Together these events- reduced thymic output, conversion of naïve T cells into memory phenotype cells and accumulation of age related defects in naïve CD4 T cells shift the balance of the total T cell pool towards the memory phenotype. Another effect of age related decline in the number of naïve T cells is oligoclonal expansion of the memory pool. TCR diversity and clonal expansion are the hallmarks of T cell mediated response. The size and diversity of the TCR repertoire determines the successful elimination of pathogens from the system. With increasing age the composition of the total T cell pool, which contains RTEs, older naïve CD4 T cells and memory cells, is disturbed. To compensate for the loss and avoid lymphopenia, fewer naïve T cells die and more memory T cells expand leading to expansion of an oligoclonal repertoire. TCR repertoire diversity is maintained until the sixth decade of life in humans after which it declines substantially. A decline in diversity accompanied with oligoclonal expansion leads to a restricted T cell repertoire that has severe consequences for the immune response of the elderly68.
Effect of ageing on naïve CD4 T cell function
Most likely to compensate for reduced thymic output, naïve CD4 T cells survive for longer periods of time in the periphery, a phenomenon attributed to reduced Bim expression, a proapoptotic Bcl family member69. New naïve CD4 T cells generated from aged stem cells show normal functioning. Similarly, naïve CD4 T cells from aged mice transferred into CD4 depleted mice show normal proliferation and IL-2 production indicating the age related defects in T cells are acquired in the periphery70,71. Due to the longer post-thymic chronological age of naïve CD4 T cells in the periphery, they accrue intrinsic defects that reduce their ability to respond to antigenic stimulus. Evidence suggests that such defects start at the very beginning, during the initiation of TCR signaling itself. The number of aged naïve CD4 T cells forming an immunological synapse with antigen presenting cells (APCs) falls to half as compared to cells from a young mouse72. Furthermore, non-responsive cells express surface P-glycoprotein, a multi-drug resistance pump that is expressed on hyporesponsive CD4 T cells in mice73,74. Even for the cells that form the synapse the quality of signal generated could be compromised as these show an altered cytoskeletal rearrangement and cholesterol levels in lipid raft formation compared to young naïve CD4 T cells75. The recruitment of crucial signaling molecules downstream of TCR, e.g., Lck and LAT, in their phosphorylated forms was also delayed to lipid rafts75. Together these defects adversely influence the strength of signal generated by TCR and are manifested downstream of the initial signaling event. Naïve CD4 T cells were also shown to be impaired in their functionality both in vitro and in vivo conditions. Data from our laboratory show that the frequency of naïve CD4 T cells from aged mice undergoing apoptotic death after activation with anti-CD3 and anti-CD28 antibodies was more as compared to naïve CD4 T cells from young mice76. Loss of mitochondrial membrane potential early post-activation in naïve CD4 T cells from aged mice suggested a mitochondrial origin of this death pathway as opposed to Fas-FasL interaction. Naïve CD4 T cells from aged mice were also found to be compromised in their ability to meet metabolic demands after activation as indicated by either lower lactate production or poor autophagy response as compared to their counterparts from young mice76. Naïve CD4 T cells from aged mice were also found to be more susceptible to DNA damage post-activation or after exposure to gamma-irradiation. Interestingly, those T cells from aged mice that have undergone and survived first few rounds of cell cycle post-activation did not show higher death as compared to their younger counterparts. This possibly suggested that after activation, T cells showing abnormalities, for example, higher death susceptibility or DNA damage, were lost and only those capable of generating a normal response survived76. Some phenotypic features of naïve cells from young and aged mice were also different. A higher frequency of naïve CD4 T cells from aged mice showed decreased CD4 expression and were found to be smaller in size with lower mitochondrial mass while the expression of inhibitory molecules like CD5 and PD-1 were higher as compared to cells from the young mice. These observations suggested that decline of co-receptor expression could be an indicator of global changes associated with age associated dysfunction of naïve CD4 T cells and could serve as an effective marker for activation potential in disease conditions (unpublished data). One of the most significant outcomes of TCR stimulation of naïve CD4 T cells is the production of IL-2, a key cytokine required for survival and proliferation of T cells. Naïve CD4 T cells from the aged show reduced production of IL-2 and proliferation when stimulated with APCs. This could explain the poor generation of either a Th1 or Th2 response, which subsequently could be overcome by providing exogenous IL-2, indicating that the cells still retain the capacity to respond to IL-277. Effectors generated from aged naïve CD4 T cells show reduced expression of differentiation and activation markers like CD25 and CD62L. Unlike newly generated T cells from the stem cells from the aged mouse, adoptively transferred naïve CD4 T cells from the aged mouse into a young host proliferated to a lesser extent than their young counterparts and on stimulation ex vivo showed reduced production of IL-2 indicating the effect of environment contributing to the defects which could not be overcome by providing a younger milieu to the cells. The inability of aged naïve CD4 T cells to activate, produce IL-2 and proliferate also affects their ability to provide cognate help for the generation of the B cell response (Figure B). Naïve CD4 T cells from aged mice when transferred to CD4 deficient young host localized similarly to germinal centres and B cell follicles after immunization but failed to generate robust humoral response. The quantity and quality of such responses are impaired with low IgG titres and reduced frequencies of somatic hypermutation in heavy chain genes. Reduced expression of CD154, an essential molecule for providing cognate help, could be the reason for weak antibody responses in the aged78,79. The defects such as reduced proliferation, cytokine secretion and impaired cognate help were also observed in memory cells generated from aged naïve CD4 T cells indicating impairment in the generation of functional memory (Figure B)76, which could not be overcome even by exogenous IL-2 treatment80.
Translational implications of age related changes
Age associated changes in the immune system adversely affect the ability to fight infections and show decreased efficacy to immunization. The elderlies show increased susceptibility towards established (e.g. Influenza, pneumococcus, etc.) and emerging (e.g. severe acute respiratory syndrome associated corona virus) pathogens81. Among the prophylactic measures available vaccination is the most potent method; however, the aged population fails to generate effective long-lasting memory in response to vaccination. Our data in mice also show that while aged mice respond to protein immunization, their responses wane much faster than those in the young mice76. Influenza vaccine is only 40-60 per cent efficient in the aged population82. Response to other vaccines like hepatitis, tetanus, tick borne encephalitis and Streptococcus pneumonia is also diminished in the older population. Intrinsic defects accumulate in naïve T cells due to their individual longevity in the periphery and leads to defective activation and impaired generation of memory83. Vaccination at an early age continues to protect the individual throughout the adult life but vaccination in the aged does not generate competent memory reflecting possibly the presence of impaired naïve CD4 T cells in the aged population. Deficiencies in the vaccine response of the elderly could be countered by passive immunization with pathogen specific antibodies that is not dependent on the naïve CD4 T cell population. Another approach is to use better adjuvants during vaccination to maximize the immune response. MF59 (oil in water emulsion) is such an adjuvant, which is given in combination with influenza vaccine to stimulate antibody production. Another strong adjuvant is unmethylated CpG containing oligodeoxynucleotide (CpG-ODN), which stimulates TLRs and enhances antibody production to immunization84. Since thymic productivity is the most important factor associated with ageing that negatively influences quality and quantity of naïve CD4 T cells, several therapeutic interventions have been suggested to rectify this decline. Growth hormones, keratinocyte growth factor (KGF) and IL-7 show promising results in inducing thymopoiesis and reversing age related thymic involution. Growth hormones are produced at lower levels in the aged as compared to the young. Growth hormone treatment has been shown to increase thymic cellularity and an increase thymocyte response to mitogen stimulation85,86. KGF is required for the differentiation and proliferation of epithelial cells including thymic epithelial cells. Thymic involution is characterized by a loss of epithelial cells that mediate thymopoiesis by providing microenvironmental niches to developing thymocytes. KGF treatment in aged mice results in enhanced thymic architecture leading to improved thymocyte numbers and peripheral T cell functions. Antibody responses following immunization also increase in KGF treated aged mice87. IL-7, a γ chain receptor cytokine, required for normal homeostasis of naïve and memory T cells has shown promising results in phase I clinical trials. Administration of a non-glycosylated form of IL-7 results in expansion of naïïve CD4 population and T cell immunity88,89. T cells seem to be exquisitely sensitive to the effects of ageing. Our best new insights into ageing are at the cellular and molecular levels with the hope of enhancing the T cell responses by renewing the cells that are present and/or by designing vaccines and adjuvants specifically tuned to generate strong responses in the elderly.
References
- 1.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 2.Pancer Z, Cooper MD. The evolution of adaptive immunity. Annu Rev Immunol. 2006;24:497–518. doi: 10.1146/annurev.immunol.24.021605.090542. [DOI] [PubMed] [Google Scholar]
- 3.Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, Beilhack GF, et al. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol. 2003;21:759–806. doi: 10.1146/annurev.immunol.21.120601.141007. [DOI] [PubMed] [Google Scholar]
- 4.Globerson A, Effros RB. Ageing of lymphocytes and lymphocytes in the aged. Immunol Today. 2000;21:515–21. doi: 10.1016/s0167-5699(00)01714-x. [DOI] [PubMed] [Google Scholar]
- 5.Desai A, Grolleau-Julius A, Yung R. Leukocyte function in the aging immune system. J Leukoc Biol. 2010;87:1001–9. doi: 10.1189/jlb.0809542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ponnappan S, Ponnappan U. Aging and immune function: molecular mechanisms to interventions. Antioxid Redox Signal. 2011;14:1551–85. doi: 10.1089/ars.2010.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weng NP. Aging of the immune system: how much can the adaptive immune system adapt? Immunity. 2006;24:495–9. doi: 10.1016/j.immuni.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solana R, Pawelec G, Tarazona R. Aging and innate immunity. Immunity. 2006;24:491–4. doi: 10.1016/j.immuni.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Plowden J, Renshaw-Hoelscher M, Engleman C, Katz J, Sambhara S. Innate immunity in aging: impact on macrophage function. Aging Cell. 2004;3:161–7. doi: 10.1111/j.1474-9728.2004.00102.x. [DOI] [PubMed] [Google Scholar]
- 10.Renshaw M, Rockwell J, Engleman C, Gewirtz A, Katz J, Sambhara S. Cutting edge: impaired Toll-like receptor expression and function in aging. J Immunol. 2002;169:4697–701. doi: 10.4049/jimmunol.169.9.4697. [DOI] [PubMed] [Google Scholar]
- 11.Gomez CR, Boehmer ED, Kovacs EJ. The aging innate immune system. Curr Opin Immunol. 2005;17:457–62. doi: 10.1016/j.coi.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 12.Cumberbatch M, Dearman RJ, Kimber I. Influence of ageing on Langerhans cell migration in mice: identification of a putative deficiency of epidermal interleukin-1beta. Immunology. 2002;105:466–77. doi: 10.1046/j.1365-2567.2002.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uyemura K, Castle SC, Makinodan T. The frail elderly: role of dendritic cells in the susceptibility of infection. Mech Ageing Dev. 2002;123:955–62. doi: 10.1016/s0047-6374(02)00033-7. [DOI] [PubMed] [Google Scholar]
- 14.Chatta GS, Andrews RG, Rodger E, Schrag M, Hammond WP, Dale DC. Hematopoietic progenitors and aging: alterations in granulocytic precursors and responsiveness to recombinant human G-CSF, GM-CSF, and IL-3. J Gerontol. 1993;48:M207–12. doi: 10.1093/geronj/48.5.m207. [DOI] [PubMed] [Google Scholar]
- 15.Schroder AK, Rink L. Neutrophil immunity of the elderly. Mech Ageing Dev. 2003;124:419–25. doi: 10.1016/s0047-6374(03)00017-4. [DOI] [PubMed] [Google Scholar]
- 16.Solana R, Mariani E. NK and NK/T cells in human senescence. Vaccine. 2000;18:1613–20. doi: 10.1016/s0264-410x(99)00495-8. [DOI] [PubMed] [Google Scholar]
- 17.LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112:1570–80. doi: 10.1182/blood-2008-02-078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frasca D, Blomberg BB. Aging affects human B cell responses. J Clin Immunol. 2011;31:430–5. doi: 10.1007/s10875-010-9501-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frasca D, Van der Put E, Riley RL, Blomberg BB. Reduced Ig class switch in aged mice correlates with decreased E47 and activation-induced cytidine deaminase. J Immunol. 2004;172:2155–62. doi: 10.4049/jimmunol.172.4.2155. [DOI] [PubMed] [Google Scholar]
- 20.Haynes L, Maue AC. Effects of aging on T cell function. Curr Opin Immunol. 2009;21:414–7. doi: 10.1016/j.coi.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krueger A, Willenzon S, Lyszkiewicz M, Kremmer E, Forster R. CC chemokine receptor 7 and 9 double-deficient hematopoietic progenitors are severely impaired in seeding the adult thymus. Blood. 2010;115:1906–12. doi: 10.1182/blood-2009-07-235721. [DOI] [PubMed] [Google Scholar]
- 22.Sambandam A, Maillard I, Zediak VP, Xu L, Gerstein RM, Aster JC, et al. Notch signaling controls the generation and differentiation of early T lineage progenitors. Nat Immunol. 2005;6:663–70. doi: 10.1038/ni1216. [DOI] [PubMed] [Google Scholar]
- 23.Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 1999;10:547–58. doi: 10.1016/s1074-7613(00)80054-0. [DOI] [PubMed] [Google Scholar]
- 24.Godfrey DI, Kennedy J, Mombaerts P, Tonegawa S, Zlotnik A. Onset of TCR-beta gene rearrangement and role of TCR-beta expression during CD3-CD4-CD8- thymocyte differentiation. J Immunol. 1994;152:4783–92. [PubMed] [Google Scholar]
- 25.Takahama Y. Journey through the thymus: stromal guides for T-cell development and selection. Nat Rev Immunol. 2006;6:127–35. doi: 10.1038/nri1781. [DOI] [PubMed] [Google Scholar]
- 26.Kang J, Volkmann A, Raulet DH. Evidence that gammadelta versus alphabeta T cell fate determination is initiated independently of T cell receptor signaling. J Exp Med. 2001;193:689–98. doi: 10.1084/jem.193.6.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masuda K, Kakugawa K, Nakayama T, Minato N, Katsura Y, Kawamoto H. T cell lineage determination precedes the initiation of TCR beta gene rearrangement. J Immunol. 2007;179:3699–706. doi: 10.4049/jimmunol.179.6.3699. [DOI] [PubMed] [Google Scholar]
- 28.Von Boehmer H. Unique features of the pre-T-cell receptor alpha-chain: not just a surrogate. Nat Rev Immunol. 2005;5:571–7. doi: 10.1038/nri1636. [DOI] [PubMed] [Google Scholar]
- 29.Porritt HE, Gordon K, Petrie HT. Kinetics of steady-state differentiation and mapping of intrathymic-signaling environments by stem cell transplantation in nonirradiated mice. J Exp Med. 2003;198:957–62. doi: 10.1084/jem.20030837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klein L, Hinterberger M, Wirnsberger G, Kyewski B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat Rev Immunol. 2009;9:833–44. doi: 10.1038/nri2669. [DOI] [PubMed] [Google Scholar]
- 31.Ashworth TD, Pear WS, Chiang MY, Blacklow SC, Mastio J, Xu L, et al. Deletion-based mechanisms of Notch1 activation in T-ALL: key roles for RAG recombinase and a conserved internal translational start site in Notch1. Blood. 2010;116:5455–64. doi: 10.1182/blood-2010-05-286328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hosoya T, Kuroha T, Moriguchi T, Cummings D, Maillard I, Lim KC, et al. GATA-3 is required for early T lineage progenitor development. J Exp Med. 2009;206:2987–3000. doi: 10.1084/jem.20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weber BN, Chi AW, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, et al. Critical role for TCF-1 in T-lineage specification and differentiation. Nature. 2011;476:63–8. doi: 10.1038/nature10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zlotoff DA, Sambandam A, Logan TD, Bell JJ, Schwarz BA, Bhandoola A. CCR7 and CCR9 together recruit hematopoietic progenitors to the adult thymus. Blood. 2010;115:1897–905. doi: 10.1182/blood-2009-08-237784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plotkin J, Prockop SE, Lepique A, Petrie HT. Critical role for CXCR4 signaling in progenitor localization and T cell differentiation in the postnatal thymus. J Immunol. 2003;171:4521–7. doi: 10.4049/jimmunol.171.9.4521. [DOI] [PubMed] [Google Scholar]
- 36.Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29:848–62. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 37.Takeda S, Rodewald HR, Arakawa H, Bluethmann H, Shimizu T. MHC class II molecules are not required for survival of newly generated CD4+ T cells, but affect their long-term life span. Immunity. 1996;5:217–28. doi: 10.1016/s1074-7613(00)80317-9. [DOI] [PubMed] [Google Scholar]
- 38.Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 1997;276:2057–62. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- 39.Dorfman JR, Stefanova I, Yasutomo K, Germain RN. CD4+ T cell survival is not directly linked to self-MHC-induced TCR signaling. Nat Immunol. 2000;1:329–35. doi: 10.1038/79783. [DOI] [PubMed] [Google Scholar]
- 40.Goldrath AW, Luckey CJ, Park R, Benoist C, Mathis D. The molecular program induced in T cells undergoing homeostatic proliferation. Proc Natl Acad Sci USA. 2004;101:16885–90. doi: 10.1073/pnas.0407417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hataye J, Moon JJ, Khoruts A, Reilly C, Jenkins MK. Naive and memory CD4+ T cell survival controlled by clonal abundance. Science. 2006;312:114–6. doi: 10.1126/science.1124228. [DOI] [PubMed] [Google Scholar]
- 42.Lantz O, Grandjean I, Matzinger P, Di Santo JP. Gamma chain required for naive CD4+ T cell survival but not for antigen proliferation. Nat Immunol. 2000;1:54–8. doi: 10.1038/76917. [DOI] [PubMed] [Google Scholar]
- 43.Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, et al. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci USA. 2001;98:8732–7. doi: 10.1073/pnas.161126098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–32. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 45.Yang Y, An J, Weng NP. Telomerase is involved in IL-7-mediated differential survival of naive and memory CD4+ T cells. J Immunol. 2008;180:3775–81. doi: 10.4049/jimmunol.180.6.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fooksman DR, Vardhana S, Vasiliver-Shamis G, Liese J, Blair DA, Waite J, et al. Functional anatomy of T cell activation and synapse formation. Annu Rev Immunol. 2010;28:79–105. doi: 10.1146/annurev-immunol-030409-101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003;3:939–51. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 49.Watts TH, DeBenedette MA. T cell co-stimulatory molecules other than CD28. Curr Opin Immunol. 1999;11:286–93. doi: 10.1016/s0952-7915(99)80046-6. [DOI] [PubMed] [Google Scholar]
- 50.Riley JL, June CH. The CD28 family: a T-cell rheostat for therapeutic control of T-cell activation. Blood. 2005;105:13–21. doi: 10.1182/blood-2004-04-1596. [DOI] [PubMed] [Google Scholar]
- 51.Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–79. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 52.Liao W, Lin JX, Leonard WJ. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol. 2011;23:598–604. doi: 10.1016/j.coi.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 54.Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci USA. 2005;102:9194–9. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–51. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 56.Rossi DJ, Bryder D, Weissman IL. Hematopoietic stem cell aging: mechanism and consequence. Exp Gerontol. 2007;42:385–90. doi: 10.1016/j.exger.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–9. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 58.Ciofani M, Zuniga-Pflucker JC. The thymus as an inductive site for T lymphopoiesis. Annu Rev Cell Dev Biol. 2007;23:463–93. doi: 10.1146/annurev.cellbio.23.090506.123547. [DOI] [PubMed] [Google Scholar]
- 59.Steinmann GG, Klaus B, Muller-Hermelink HK. The involution of the ageing human thymic epithelium is independent of puberty. A morphometric study. Scand J Immunol. 1985;22:563–75. doi: 10.1111/j.1365-3083.1985.tb01916.x. [DOI] [PubMed] [Google Scholar]
- 60.Flores KG, Li J, Sempowski GD, Haynes BF, Hale LP. Analysis of the human thymic perivascular space during aging. J Clin Invest. 1999;104:1031–9. doi: 10.1172/JCI7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fitzpatrick FT, Kendall MD, Wheeler MJ, Adcock IM, Greenstein BD. Reappearance of thymus of ageing rats after orchidectomy. J Endocrinol. 1985;106:R17–9. doi: 10.1677/joe.0.106r017. [DOI] [PubMed] [Google Scholar]
- 62.Olsen NJ, Olson G, Viselli SM, Gu X, Kovacs WJ. Androgen receptors in thymic epithelium modulate thymus size and thymocyte development. Endocrinology. 2001;142:1278–83. doi: 10.1210/endo.142.3.8032. [DOI] [PubMed] [Google Scholar]
- 63.Min H, Montecino-Rodriguez E, Dorshkind K. Reduction in the developmental potential of intrathymic T cell progenitors with age. J Immunol. 2004;173:245–50. doi: 10.4049/jimmunol.173.1.245. [DOI] [PubMed] [Google Scholar]
- 64.Mackall CL, Punt JA, Morgan P, Farr AG, Gress RE. Thymic function in young/old chimeras: substantial thymic T cell regenerative capacity despite irreversible age-associated thymic involution. Eur J Immunol. 1998;28:1886–93. doi: 10.1002/(SICI)1521-4141(199806)28:06<1886::AID-IMMU1886>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 65.Ortman CL, Dittmar KA, Witte PL, Le PT. Molecular characterization of the mouse involuted thymus: aberrations in expression of transcription regulators in thymocyte and epithelial compartments. Int Immunol. 2002;14:813–22. doi: 10.1093/intimm/dxf042. [DOI] [PubMed] [Google Scholar]
- 66.Sempowski GD, Hale LP, Sundy JS, Massey JM, Koup RA, Douek DC, et al. Leukemia inhibitory factor, oncostatin M, IL-6, and stem cell factor mRNA expression in human thymus increases with age and is associated with thymic atrophy. J Immunol. 2000;164:2180–7. doi: 10.4049/jimmunol.164.4.2180. [DOI] [PubMed] [Google Scholar]
- 67.Sempowski GD, Gooding ME, Liao HX, Le PT, Haynes BF. T cell receptor excision circle assessment of thymopoiesis in aging mice. Mol Immunol. 2002;38:841–8. doi: 10.1016/s0161-5890(01)00122-5. [DOI] [PubMed] [Google Scholar]
- 68.Nikolich-Zugich J. Ageing and life-long maintenance of T-cell subsets in the face of latent persistent infections. Nat Rev Immunol. 2008;8:512–22. doi: 10.1038/nri2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsukamoto H, Huston GE, Dibble J, Duso DK, Swain SL. Bim dictates naive CD4 T cell lifespan and the development of age-associated functional defects. J Immunol. 2010;185:4535–44. doi: 10.4049/jimmunol.1001668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Haynes L, Eaton SM, Burns EM, Randall TD, Swain SL. Newly generated CD4 T cells in aged animals do not exhibit age-related defects in response to antigen. J Exp Med. 2005;201:845–51. doi: 10.1084/jem.20041933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haynes L, Maue AC. Effects of aging on T cell function. Curr Opin Immunol. 2009;21:414–7. doi: 10.1016/j.coi.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garcia GG, Miller RA. Single-cell analyses reveal two defects in peptide-specific activation of naive T cells from aged mice. J Immunol. 2001;166:3151–7. doi: 10.4049/jimmunol.166.5.3151. [DOI] [PubMed] [Google Scholar]
- 73.Eisenbraun MD, Tamir A, Miller RA. Altered composition of the immunological synapse in an anergic, age-dependent memory T cell subset. J Immunol. 2000;164:6105–12. doi: 10.4049/jimmunol.164.12.6105. [DOI] [PubMed] [Google Scholar]
- 74.Bining N, Miller RA. Cytokine production by subsets of CD4 memory T cells differing in P-glycoprotein expression: effects of aging. J Gerontol A Biol Sci Med Sci. 1997;52:B137–45. doi: 10.1093/gerona/52a.3.b137. [DOI] [PubMed] [Google Scholar]
- 75.Garcia GG, Miller RA. Age-dependent defects in TCR-triggered cytoskeletal rearrangement in CD4+ T cells. J Immunol. 2002;169:5021–7. doi: 10.4049/jimmunol.169.9.5021. [DOI] [PubMed] [Google Scholar]
- 76.Mattoo H, Faulkner M, Kandpal U, Das R, Lewis V, George A, et al. Naive CD4 T cells from aged mice show enhanced death upon primary activation. Int Immunol. 2009;21:1277–89. doi: 10.1093/intimm/dxp094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haynes L, Linton PJ, Eaton SM, Tonkonogy SL, Swain SL. Interleukin 2, but not other common gamma chain-binding cytokines, can reverse the defect in generation of CD4 effector T cells from naive T cells of aged mice. J Exp Med. 1999;190:1013–24. doi: 10.1084/jem.190.7.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eaton SM, Burns EM, Kusser K, Randall TD, Haynes L. Age-related defects in CD4 T cell cognate helper function lead to reductions in humoral responses. J Exp Med. 2004;200:1613–22. doi: 10.1084/jem.20041395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maue AC, Eaton SM, Lanthier PA, Sweet KB, Blumerman SL, Haynes L. Proinflammatory adjuvants enhance the cognate helper activity of aged CD4 T cells. J Immunol. 2009;182:6129–35. doi: 10.4049/jimmunol.0804226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Haynes L, Eaton SM, Burns EM, Randall TD, Swain SL. CD4 T cell memory derived from young naive cells functions well into old age, but memory generated from aged naive cells functions poorly. Proc Natl Acad Sci USA. 2003;100:15053–8. doi: 10.1073/pnas.2433717100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen Q, Liang WN, Liu GF, Liu M, Xie XQ, Wu J, et al. Case fatality rate of severe acute respiratory syndromes in Beijing. Biomed Environ Sci. 2005;18:220–6. [PubMed] [Google Scholar]
- 82.Vu T, Farish S, Jenkins M, Kelly H. A meta-analysis of effectiveness of influenza vaccine in persons aged 65 years and over living in the community. Vaccine. 2002;20:1831–6. doi: 10.1016/s0264-410x(02)00041-5. [DOI] [PubMed] [Google Scholar]
- 83.Vrisekoop N, den Braber I, de Boer AB, Ruiter AF, Ackermans MT, van der Crabben SN, et al. Sparse production but preferential incorporation of recently produced naive T cells in the human peripheral pool. Proc Natl Acad Sci USA. 2008;105:6115–20. doi: 10.1073/pnas.0709713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maletto BA, Ropolo AS, Liscovsky MV, Alignani DO, Glocker M, Pistoresi-Palencia MC. CpG oligodeoxynucleotide functions as an effective adjuvant in aged BALB/c mice. Clin Immunol. 2005;117:251–61. doi: 10.1016/j.clim.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 85.Chen BJ, Cui X, Sempowski GD, Chao NJ. Growth hormone accelerates immune recovery following allogeneic T-cell-depleted bone marrow transplantation in mice. Exp Hematol. 2003;31:953–8. doi: 10.1016/s0301-472x(03)00196-6. [DOI] [PubMed] [Google Scholar]
- 86.Tian ZG, Woody MA, Sun R, Welniak LA, Raziuddin A, Funakoshi S, et al. Recombinant human growth hormone promotes hematopoietic reconstitution after syngeneic bone marrow transplantation in mice. Stem Cells. 1998;16:193–9. doi: 10.1002/stem.160193. [DOI] [PubMed] [Google Scholar]
- 87.Alpdogan O, Hubbard VM, Smith OM, Patel N, Lu S, Goldberg GL, et al. Keratinocyte growth factor (KGF) is required for postnatal thymic regeneration. Blood. 2006;107:2453–60. doi: 10.1182/blood-2005-07-2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Alpdogan O, Muriglan SJ, Eng JM, Willis LM, Greenberg AS, Kappel BJ, et al. IL-7 enhances peripheral T cell reconstitution after allogeneic hematopoietic stem cell transplantation. J Clin Invest. 2003;112:1095–107. doi: 10.1172/JCI17865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mackall CL, Fry TJ, Bare C, Morgan P, Galbraith A, Gress RE. IL-7 increases both thymic-dependent and thymic-independent T-cell regeneration after bone marrow transplantation. Blood. 2001;97:1491–7. doi: 10.1182/blood.v97.5.1491. [DOI] [PubMed] [Google Scholar]