

Abstract
HIV continues to be a major health problem worldwide even today. Owing to the intricate nature of its interactions with the immune system, HIV has remained an enigma that cleverly utilizes the host machinery to survive. Its ability to evade the host immune system, at both levels, innate and adaptive, allows the pathogen to replicate and transmit from one host to another. It has been shown that HIV has multipronged effects especially on the adaptive immunity, with CD4+ T cells being the worst affected T cell populations. Various analyses have revealed that the exposure to HIV results in clonal expansion and excessive activation of the immune system. Also, an abnormal process of differentiation has been observed suggestive of an alteration and blocks in the maturation of various T cell subsets. Additionally, HIV has shown to accelerate immunosenescence and exhaustion of the overtly activated T cells. Apart from causing phenotypic changes, HIV has adverse effects on the functional aspect of the immune system, with evidences implicating it in the loss of the capacity of T cells to secrete various antiviral cytokines and chemokines. However, there continues to be many aspects of the immunopathogenesis of HIV that are still unknown and thus require further research to convert the malaise of HIV into a manageable epidemic.
Keywords: Differentiation, HIV, immune activation, immune exhaustion, polyfunctionality, proliferation, T cells
Introduction
T cells are one of the most potent cell populations against invading pathogens and ironically, these are also the cells that make the body vulnerable to HIV. Dalgleish et al1 were the first to arrive at this conclusion and provided the evidence that CD4+ T lymphocytes were potential target cells of HIV. Although initial studies by Chun et al2 proposed the resting CD4+ T cells to be the stable latent reservoir for HIV, but recent latency models suggest other cells such as macrophages and monocytes to be involved in viral latency3. However, the subject remains debatable to date. In recent years, the most predominant theory about HIV immunopathogenesis is that of HIV associated chronic immune activation. This theory suggests hyper and sustained systemic immune activation to be the trigger for perturbations in T cell dynamics and function. Brenchley et al4 proposed that the translocation of microbial products such as lipopolysaccharide (LPS), from leaky gut mucosa could be a cause for the persistent activated state of the immune system. While considerable evidence has been gathered, the most alarming questions of viral replication, viral reservoir, correlates of disease progression, CD4 depletion and vaccine development are still to be answered. One of the major leads gathered so far is the role of adaptive immune response against HIV. In the quest for all these outstanding questions, researchers have found that adaptive immune response could be the connecting link which could provide considerable clues to solve the HIV conundrum. Being at the forefront of adaptive immune response, T cells and more specifically, their activation, proliferation, differentiation, polyfunctionality and exhaustion have been the focus of research efforts to delineate mechanisms leading to HIV/AIDS and effective vaccine strategies. The two recent HIV vaccine efficacy trials, namely the Merck STEP trial and the Thai RV144 trial5 are testament to not only the varying degrees of success but also the setbacks faced in the field of HIV vaccine development. The STEP trial was conducted keeping in mind that the MRKAd5 HIV-1 Gag Pol Nef vaccine would elicit an effective immune response in HIV uninfected individuals, at the initial phase of the trial the vaccine was found to be highly immunogenic. But as the study progressed further to the test of concept trial, the vaccine failed to provide protection as compared to the placebo. The trial also pointed out the fact that interferon gamma (IFN-γ) alone cannot be assumed to be the definite correlate of immune protection5.
In contrast, the Thai RV144 test of concept trial evaluated both the humoral as well as the cell mediated arm of the immune system. Although negligible cell mediated immune response was observed in the trial, still the vaccine was found to be 31.2 per cent efficacious in preventing HIV infection. While the vaccine showed moderate protection, the lack of effective cytotoxic T lymphocyte (CTL) response remained unexplained5. The trials brought out the differences in approach as well as underscored our inadequacy to conclusively identify correlates of protection.
The studies and trials so far have employed non-human primate models (NHP) to decipher the mechanisms of immunological imbalance and optimization of vaccination strategies. Furthermore, the comparative analysis of cellular immune responses generated by vaccine candidates in humans and macaques underscores the importance of NHP models. However, the recent vaccine trials based on initial NHP studies failed to achieve the desired protective threshold in humans, and thus a critical re-evaluation of the used NHP models and the generation of new suitable models that are able to precisely imitate HIV-1 infection in humans is the need of the hour6.
Taking all this into consideration, this review attempts to recapitulate the past achievements and also sheds light on the questions that are road blocks on the way to an effective vaccine. In recent years immune activation has emerged as a linchpin in HIV immunopathogenesis; however, evidence implicating this phenomenon in T cell dysfunction and exhaustion remains elusive. Thus, to enlighten these aspects of HIV and the host, this enduring tale has been discussed in two phases: acute viral infections and chronic HIV infection. It draws up a character sketch of T cells, their strength and weaknesses, and their journey during the course of HIV infection.
T cells and their types
It is now a well established fact that the T cells are a key component of the immune system with numerous studies elucidating their role in thwarting infections and keeping the body disease free. Within the T cell compartment, there are two well characterized subpopulations namely, T helper (CD4) and T cytotoxic cells (CD8)7. Mosmann et al8 further characterized CD4+ T cells into two distinct subsets based on their ability to produce signature cytokines, TH1 subset secreting IFN-γ and TH2 subset secreting interleukin IL-4 and IL-13. In addition to their secretory capacity, Th1 and Th2 cells promote the cell mediated and humoral branch of the immune system respectively8. Recent discovery of a few more subsets of T cells namely Th17 and Treg (regulatory T cells) has further complicated the whole picture. Th17 cells as their name suggests, are active producers of IL-179 and their role against extracellular bacteria and fungi was first reported by Milner et al10 in individuals lacking IL-17 production who were susceptible to bacterial and fungal infections13. Apart from IL-17 production, studies have also reported secretion of other pro-inflammatory cytokines by TH17 cells such as tumour necrosis factor TNF-α, IL-1, IL-2, IL-21 and IL-22. Being localized in the gastrointestinal tract11, these play a significant role in mucosal immunity and are involved in regeneration of the epithelial cells. In contrast to the above mentioned protective T helper populations, the Tregs (CD25+FoxP3+) are suppressive in nature and regulate T cell hyperactivation, proliferation and cytokine production12. Like Th17 cells, these have also been reported in autoimmunity and other disorders12. Initial description of Tregs was reported by Gershon and Kondo in 197013 and later on, these were fully characterized by Sakaguchi et al14. Recent reports suggest that although Th17 and Tregs have a common lineage, but their differentiation pathways are inversely modulated in a number of immune disorders. The common developmental link between these two populations is transforming growth factor (TGF-β). In the Th17 developmental pathway, TGF-β upregulates the master transcription factor retinoic acid receptor-related orphan receptor-γt (RORc gene encoding ROR-γt,)15. But the same TGF-β also induces the Treg developmental pathway by activating the transcription factor Fox P315. Bettelli et al16 reported that IL-6 along with TGF-β can favour the development of Tregs over Th17 cells. These evidences indicate a “see-saw” equation between Th17 and Treg population.
Apart from this, several new models of T cell lineage have been proposed which include TFH (Follicular helper T cell), Th3, Tr1, Th917. The precise role of all these newly identified subset is still under investigation9.
Immune response against acute infections
T cell development: The journey of T cells begins in the bone marrow where haematopoiesis results in the production of progenitor T cells which then migrate to thymus for maturation and differentiation to develop into functionally distinct populations. Post negative and positive selection, CD4+ and CD8+ T cells exit the thymus and enter the circulatory system as “Naïve” (CD45RA+CCR7+) resting T cells having little transcriptional activity and accumulate in the lymphoid tissues, which are also the sites of accumulation of infected cells and pathogens7. Along with functional differences, these subsets exhibit a modified characteristic phenotype. Initial studies using flowcytometry techniques allowed the identification of the specific phenotypes using specific markers such as CD45RA, unprimed T cell marker and CD45RO, a primed memory T cell marker18. Further characterization of naïve T cell subset was made possible using CD27 and CD28, co-stimulatory molecules.
On exposure to antigens, CD4+ and CD8+ T cells undergo changes at the cellular as well as the sub-cellular levels and get activated. During their activated state, T cells undergo a number of phenotypic and functional changes resulting in the upregulated expression of stimulatory and co-stimulatory molecules such as CD28. Also, one of the activation markers which has recently come into limelight is CD38, a glycoprotein that is highly expressed on early T cell precursors19, is downregulated upon maturation and finally in the presence of antigenic stimulus, its expression gets upregulated again19.
The activated CD4 T cells secrete IL-2, a growth factor that acts on the antigen activated lymphocytes and stimulates their proliferation resulting in clonal expansion20. At the phenotypic level receptors such as CC-chemokine receptor 7 (CCR7), the lymph-node homing receptor CD62 ligand (CD62L) and the co-stimulatory receptors CD28 and CD27 are initially expressed on naïve T cells but once primed, there expression is downregulated20. The model developed by Hamann et al21 allowed for simultaneous use of CD45RA and CD27 to define 3 distinct subsets of T cells: naive (N; RA+27+), effector (E; RA+27-), and memory (M; RA-27+) CD8+ subpopulations. These differentiated effector cells exhibit polyfunctional capacity, i.e. the ability to produce a plethora of cytokines such as IFN-γ, TNF-α, IL-2 etc. In addition to this, CD8 T cells upregulate the expression of granzyme and perforin, thus making them cytolytic in nature21.
While both CD4 and CD8 T cells differentiate simultaneously, CD8+ T cells proliferate at a much faster rate thereby forming a larger pool of effector cells as compared to CD4+ T cells. Also, at the genotypic level, the daughter CD8+ effector population is programmed such that no repeated stimulation is required for further proliferation and differentiation making them independent of the antigenic stimulus22. Subsequently, irrespective of the antigen clearance, the expansion phase is followed by a contraction of the effector population where 90-95 per cent of the activated CD8+ effector cells undergo programmed cell death primarily through Fas-mediated apoptosis20. The transition from the death to memory phase is not specific to a predetermined population. A number of factors such as changes in gene expression, function and phenotype decide the fate of effector cells to become memory cells. Now, this newly formed memory compartment is characterized by low granzyme B production and high expression of Bcl-2 (B-cell lymphoma-2) i.e. upregulated by the IL-7/IL-7R pathway23. IL-7 is one of the cytokines involved in T cell homeostasis, maturation, differentiation and long term T cell survival. It is actively produced by the thymic epithelium, stroma and bone marrow. In addition to these changes, a peculiar feature of this memory compartment is its ability to sustain itself for much longer period of time24. This is achieved through slow division of memory CD8 T cells which ensures a steady homeostatic turnover24. Further investigation into this compartment revealed that memory cells residing in lymph nodes were phenotypically and functionally different from those present in blood and spleen. The cells present in the non lymphoid locations were found to be CD62LLo CCR7Lo and could produce IFN-γ and TNF-α efficiently whereas memory cells in lymphoid zones expressed CD62LHi CCR7Hi, and produced IFN-γ, TNF- α along with IL-2. Thus based on their homing and functional capacity, these were categorized as T effector memory (TEM) and T central memory cells (TCM), respectively43. Recent studies have observed a high degree of heterogeneity in the effector memory (EM; CD45RA-CCR7-) subset with differential expression of CD28 and CD27 molecules. Thus Romero et al25 proposed the categorization of this section as well, the resultant being 4 phenotypically distinct subpopulations called EM1(CD27+CD28+), EM2(CD27+CD28-), EM3(CD27-CD28-) and EM4(CD27-CD28+). Of these, three subsets are functionally distinct, with EM1 being memory like, EM2 exhibiting partial replicative history and effector functions and EM3 being effector like. The findings so far have not only enabled researchers to understand the multidimensional process of T cell differentiation but can also be considered as an archetype of immune response in the presence of acute infections.
In this way sufficient numbers of CD4 and CD8 T cells are maintained throughout the life of an individual by slow and steady homeostatic process.
Immune response in chronic infection: In the event of a chronic infection like HIV, where the virus persists in the body in both latent and replicative forms, T cell dynamics play a significant role in deciding the susceptibility to a pathogen. The initial weeks post HIV infection witness a sharp rise in the viral titre along with a depletion of CD4+ T cells most prominently observed in the gastrointestinal tract associated lymphoid tissue26. However, a partial retaliation by the immune system is observed when the decline in viraemia coincides with the appearance of HIV specific CD8+ T cells. It must be noted here, that this initial CD8 response is limited to a few viral epitopes27. Ordinarily, this decline in viraemia plateaus and the virus establishes latent viral reservoirs in the lymph nodes and eludes the cytotoxic response of the immune system and thus the chronic phase of HIV sets in. But investigators have studied a few exceptions to this trend. In the Nairobi cohort, commercial sex workers who were repeatedly exposed to HIV remained uninfected exhibited HIV specific CTL responses in the vaginal mucosa. They also found that the epitopes recognized by CTLs were different from those recognized in HIV infected individuals28. Additionally, studies on long term non progressors have reported stronger CTL responses and control of viral replication29. These studies contradictory to prevalent beliefs prove that CTLs could be a part of the solution.
Chronic phase
The lull before the storm: One of the hallmarks of the chronic phase of HIV infection is the depletion of CD4 T cell pool. While the classic notion of HIV mediated CD4 depletion points to the direct killing of CD4 infected T cells, recent developments have challenged this theory and given rise to newer paradigms. Recent findings suggest that majority of the CD4+ T cells that undergo cell death are in fact not infected with HIV. This loss of uninfected bystander T cells indicates the existence of an alternate mechanism of CD4 T cell depletion independent of productive HIV infection30.
One of the mechanisms that has gained popularity is that of chronic immune activation. Giorgi and colleagues31 demonstrated that CD38 expression on CD8+ T cells correlates strongly with HIV disease progression in HIV infected than HIV uninfected individuals. Further support came from the studies in Simian immunodeficiency virus (SIV) infected primates. Rhesus macaques infected with SIV showed high levels of immune activation, suffered progressive CD4+ T cell depletion and exhibited high rates of viral replication. On the other hand, Sooty mangabey, the natural hosts of SIV, registered low immune activation levels and maintained normal reservoir of CD4 T cell population without developing any immunodeficiency despite high viral replication32. When translated into the pathogenesis of HIV/AIDS in humans, primate studies provide strong support to the hypothesis: Immune activation is the leading cause of bystander CD4 T cell death33. However, the major paradox of immune activation is that HIV causes immunodeficiency but at the same it is responsible for chronic immune activation. The answer lies in the complexity of the immune response against HIV. Although the exact cause of immune activation is still unknown, studies over the past few decades have gathered evidence to the theory that there is no single cause for chronic immune activation33. Rather it is a resultant of a multitude of factors which are still debatable.
(i) Chronic innate immune activation: Innate being the foremost line of defence encounters HIV first and is activated within hours. The first cells to come across HIV are the plasmacytoid dendritirc cells (pDCs)34. pDCs through their CD4 surface molecule interact with HIV and get activated. The persistent stimulation of pDCs by HIV beyond the acute early phase causes continued secretion of IFN-α and indoleamine 2, 3 dioxygenase (IDO)35. In vitro studies have shown IFN-α to be closely associated with increased expression of CD38 on CD8+T cells36. In contrast to this, IFN-α has also been reported to induce apoptosis in CD4+T cells in HIV infected and SIV infected macaques but not in non-human primates with non-pathogenic infection35,36. Thus uncontrolled innate immune activation may lead to dysregulated adaptive immune response. This finding suggests a link between players of activation in innate and adaptive immunity. Also IDO which is required for degradation of tryptophan to kynurenine37 has suppressive effect on T cell proliferation. Two evidences supporting this were murine models where inhibition of HIV induced IDO enhanced the clearance of HIV-infected macrophages38 and in vitro studies which demonstrated the improvement in CD4 T cell proliferation on blocking of HIV-induced IDO39. Thus, the ripples of chronic immune activation in the innate arm of immunity can be felt in the form of immune activation as well as deficiency in adaptive immunity.
(ii) Direct stimulation by HIV proteins: The most obvious mechanism is the direct stimulation of the immune system by the virus itself. Activation sets right in the acute phase of HIV where its presence triggers a cascade of signals. In vitro studies have reported that HIV gene encoded products can directly stimulate the immune system without direct infection40. HIV proteins such as gp-120 through their interaction with CD4 and co-receptors have been shown to activate lymphocytes and macrophages through production of pro-inflammatory cytokines like TNF-α which in turn boosts viral replication41. Two other important proteins that induce hyperactivation of monocytes and macrophages are Nef and Vpr. The Nef and Vpr proteins partially mimic the TNF receptor signalling in these cells and stimulate NFk-β leading to HIV LTR (long terminal repeat) activation and subsequent HIV replication42. However, at the same time, pro-inflammatory cytokines and chemokines production is blocked by Vpr protein43 thereby favouring the recruitment of T cells, monocytes and macrophages44.
In other words, these viral proteins by fooling the immune system ensure a continuous secretion of TNF-α thereby creating an environment of constant inflammation and viral replication. These events ensure a closed loop for immune activation as well as HIV-1 replication thereby creating a vicious cycle.
(iii) Microbial translocation: The human gut has vast surface area that allows it to absorb nutrients and water without infiltration of the gut flora and thereby restricts the translocation of microbial products. Contrary to this notion, the presences of histological abnormalities and lymphocytes in the gut associated lymphoid tissue (GALT) of chronically HIV infected individuals have been reported7,45 highlighting a possible association between mucosal immunity and HIV disease progression. Since then, substantial data have been gathered and different theories have been proposed as the underlying mechanisms. Some researchers endorse the most apparent mechanism of virus mediated toxicity in enterocytes46. This could be due to the inhibitory role of Tat, an HIV protein, on uptake of glucose by enterocytes. Also, gp120 has shown to increase the concentration of calcium in enterocytes leading to ionic imbalance47. Thus, these changes affect the integrity of the mucosa lining of the gastrointestinal tract leading to mobilization of microbial products such as LPS, sCD14 and EndoCAb. To prove this hypothesis, Brenchley et al48 showed that LPS, a common component of bacterial cell wall was increased in plasma of chronically infected HIV individuals and SIV-infected macaques and correlated with immune activation. Supporting evidence was provided by measurement of soluble CD14 (sCD14) in HIV infected individuals and significantly higher levels of plasma CD14 were found in the acute and chronic HIV infected individuals as compared to the HIV seronegative individuals49. Apart from this, they also reported that LPS levels strongly correlated with plasma IFN-α level which led to their hypothesis that certain factors in the intestinal flora could also induce IFN-α production. Thus, they concluded that the microbial products translocated across a leaky gut, may result in systemic hyperactivation of the innate immune system and may further impair the adaptive immune responses4 (Figure). Other microbial products such as flagellin, bacterial 16sDNA and peptidoglycans have also been shown to translocate across the mucosal barrier. These microbial products through their interaction with Toll-like receptors (TLRs) activate the innate immune system. Boasso and Shearer39 proved this hypothesis through their work on murine models and reported that in mice lacking the IFN-α receptor showed milder physiological and functional alterations in their lymphoid organs on treatment with CpG oligodeoxynucleotides (ODN) as compared to those with IFN-γ receptor. Therefore, they propounded the theory that the administration of CpG ODN activates pDC through TLR9 and results in alterations50 which were similar to those seen in HIV infection39.
Fig.
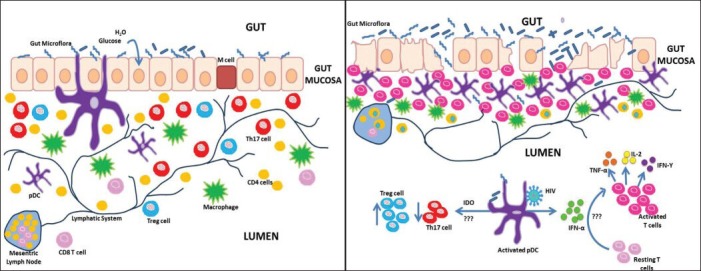
Hypothetical model of gastrointestinal tract (A) Normal Healthy state and (B) chronic HIV infection. (A) In a normal healthy state, the epithelial and M cells lining the mucosal barrier of the gastrointestinal tract forms an effective barrier against invading pathogens. The lamina propria contains loose clusters of T cells, macrophages, plasmacytoid dendritirc cells (pDCs) and follicles. (B) During chronic HIV infection, the mucosal barrier lining the gastrointestinal tract becomes leaky facilitating translocation of microbial products. This results in increased infiltration of inflammatory cells into the lamina propria leading to more proinflammatory cytokine production. Lipopolysaccharide has been reported to activate IFN-α and Indoleamine 2, 3 dioxygenase (IDO) production by (pDCs) promoting immune activation and a shift in the balance from Th17 to Treg population by unknown mechanisms.
The investigators have also identified a preferential loss of Th17 subsets from the GI tract51. Further studies comparing the pathogenic (pigtailed macaques) with the non pathogenic SIV models (African green monkey) revealed loss of Th17 cells with a concomitant increase in Treg7,52 in the pigtail macaques. The loss of Th17/Treg balance was associated with sustained systemic immune activation52. Our group53 has also observed a gradual loss of Th17 cells in the peripheral blood of HIV-1 infected Indian individuals which revealed that this loss of Th17 cells during late stage of HIV-1 infection could render them more prone to opportunistic infection. Furthermore, an enhancement in the proportion of Treg cells was noted during the HIV disease progression. This indicative of progressive HIV infection was associated with the loss of Th17/Treg immune balance. Therefore, our group speculated that the relative balance between Th17 and Treg cellular subsets rather than the function of either alone was critical for immunopathogenesis of HIV infection53.
Consistent with our study, others have also reported a loss of Th17 cells in blood and rectosigmoid mucosal biopsies51 and was found to be associated with sustained immune activation. However, the reason behind this selective loss of Th17 cells remains unclear. One possible mechanism involves the role of IDO metabolism. As IDO catabolises tryptophan to kynurenine and in the case of HIV, an excess of IDO production deprives the cell of the tryptophan pool resulting in the accumulation of catabolites37. These catabolites, on the one hand, prove toxic54 for the cell and induce apoptosis and on the other hand, due to the ability to stimulate FoxP3 expression and downregulate RORc expression, these promote a shift in the balance from Th17 to Treg population55.
Thus constant damage of the gastrointestinal track, decline in Th17 cell population and translocation of microbial products set up an enduring cycle of immune activation and viral replication leading to poor immune response.
Co-infections: A number of opportunistic infections can also contribute to immune activation. Among bacterial infections in vivo studies have revealed tuberculosis (TB) to be a driving factor for HIV replication. Pro-inflammatory cytokines such as TNF-α produced against TB bind to the cell receptors leading to the secretion of active nuclear factor (NF)-kB in large quantities56. NF-kB activates transcription of a number of host genes including HIV-1 LTR sequences subsequently enhancing viral replication57 which in turn maintains the systemic immune activation. Evidence in support of this came from co-infected Ugandan patients whose pleural fluid samples recorded four times higher amount of HIV-1 load than in plasma samples. High levels of TNF-α, IL-6 and other soluble markers were found to be strongly correlated with HIV-1 viral load in the pleural space58.
Looking at this scenario, it appears that it is the innate immune system which initiates the process of immune activation but it is the adaptive immunity that sustains it and gets affected in the process. HIV through immune activation is able to generate new targets for infection and propagation. While these events have been labelled as causes of immune activation, these along with other factors play an important contributory role in immune deficiency. Whether these causes are linked through an unknown network or are a series of events occurring simultaneously still remains to be determined. Also, other governing factors that are contributing to this phenomenon need to be explored.
After-effects of immune activation
I CD4 T cell depletion
(i) Direct virus mediated killing: Brenchley et al59 reported that GI tract harbours the majority of CD4+ T cells followed by lymph nodes and other lymphatic tissues rather than peripheral blood. Most of CD4+ T cells are CCR5+ activated memory cells making them the susceptible targets for HIV. Also as compared to the periphery productive infection rate is higher in mucosal sites. Massive depletion of these cells occurs during the early phase of infection followed by a slow and stable decline of the remaining cells in the chronic phase. In addition to the mucosa, a marked loss in CD4 T cells was also noted in the periphery by many research groups including ours60. We observed significant negative correlations of higher magnitude between CD4+ T cell percentages and plasma HIV-1 RNA levels when adjusted for the effects of the immune activation markers. These data support the idea that viral replication leads to immune activation, which in turn drives CD4+ T cell depletion. However, a lucid mechanism explaining the loss of CD4 T cells in HIV infection has yet to be defined60.
Several mechanisms have been proposed to explain the depletion of CD4 T cells. Studies have shown an increase in CTL responses during the acute phase of infection which coincided with the drop in plasma viraemia. From this it is evident that CTLs try and control the infection by directly killing infected CD4 T cells by cytolysis. Thus, this virus mediated immune response directly results in the preferential loss of CD4 T cells. However, as the error prone reverse transcriptase enzyme generates variants of viral epitopes and the virus evolves constantly, the existing CTLs no longer recognize the mutant epitopes and the immune system fails to generate new cytolytic response against the variant epitopes thereby allowing the virus to escape. In addition to this, HIV encoded protein Nef downregulates the expression of MHC I and prevents recognition by CTLs and thus avoid CTL mediated killing of infected cells61. Hence, the killing effects of CTLs get shadowed by continues evolution of HIV and its proteins. However, this clearly indicates the presence of other factors that are involved in depletion of CD4 T cells.
(ii) Activation induced cell death (AICD): Cell signalling studies have shown that immune activation shares molecular steps that are also involved in apoptosis giving rise to the notion that activation and apoptosis may be two sides of the same coin61. AICD is a mechanism through which the body keeps immune activation in check by directing the activated cell to undergo apoptosis61. During the chronic phase of infection, pro-inflammatory cytokines such as TNF and C-C chemokines are produced in significant amounts by the infected cells62. Studies have shown that TNF family of cytokines can induce apoptosis in a direct manner by inducing the appropriate (or inappropriate) apoptotic signals63. However, their precise role in inducing apoptosis is still under investigation. While, these mechanisms describe the cytopathic role of immune activation through cytokine production, these could not explain apoptosis in bystander cells.
It has been reported that apoptosis occurs predominantly in non-infected bystander cells as compared to the infected cells in HIV infected children and SIV infected macaques64. During the hyperactivated state of HIV infection which is also associated with high viral replication, the expression of host factors such as TNF superfamily ligands and receptors (Fas/Fas-L along with TRAIL-DR5) and viral factors such as Tat, Nef, Vpu, Vpr are upregulated65. These simultaneous events result in depletion of CD4 T cells leading to acquired immunodeficiency. Studies have shown higher expression of both CD95 (Fas) and FasL on peripheral blood mononuclear cells (PBMCs) of HIV infected individuals and have also reported that both CD4 and CD8 T cell populations are vulnerable to Fas mediated apoptosis66. This mechanism was suggested to be the reason for bystander death by AICD. Moreover, Tat has also been reported to induce the production of reactive oxygen intermediates, activation of caspases and other transcription factors such as NF-kB. These signalling events ensure HIV production by NF-kb activation but at the same time, these prepare the cell for apoptosis by caspase activation. The Tat protein in plasma induces apoptosis in infected as well as uninfected cells through its activation of FOXO3a (Forkhead box transcription factor O class 3a) which controls the expression of several proapoptotic genes, including FasL, Bim and TRAIL67, Env (another HIV protein), binds to CD4 molecule and sensitizes the cell to Fas-mediated killing68 with an increase in caspase activity69. Both the secreted and the membrane bound form of Env are able to induce apoptosis in uninfected cells. When these cross-linked cells encounter antigen presenting cells (APCs), stimulatory signals through T cell receptor (TCR) leads to apoptosis in uninfected cells and these negative signals are more enhanced during chronic immune activation resulting in AICD70. This env mediated apoptosis is significantly higher in lymphoid tissues of HIV infected individuals30.
Unlike Env and Tat, Vpr (Virion associated protein) is involved in cell cycle arrest71. By inhibiting the activation of p34cdc2-cyclin B which is required by the cell to enter mitotic phase, Vpr arrests both infected and uninfected cells in G2 phase72. Studies73 have demonstrated that the carboxy-terminal truncation of Vpr induces apoptosis via G1 arrest of the cell cycle. On one hand, Vpr benefits HIV by enhancing viral replication and on the other hand induces cell death73,74.
Like Tat, Nef, another potent HIV protein induces apoptosis in bystander cells through upregulation of Fas and FasL or directly engaging CXCR4 on infected cells75. In addition to this, free Nef in plasma can be present in the form of microvesicles expressing CD45 which are known to cause AICD in resting CD4 T cells75,76.
However, HIV being a true opportunist delays cell death for its sustenance. It can modulate the activity of Nef protein, depending on the stage of infection, to inhibit apoptosis and thus promotes continuous production of progeny virions. In such a scenario, Nef can switch roles and be anti-apoptotic in nature77.
II. Loss of homeostasis
The adverse effects of chronic immune activation in HIV are not restricted to CD4 T cells depletion alone as the expansion of CD4 and CD8 T cells is also seen as an equally detrimental effect. The indiscriminate and chronic immune activation results in the creation of a pool of rapidly proliferating T cells with effector functions which subsequently undergo apoptosis33. This may represent compensatory homeostasis in response to cells killed either directly or indirectly by the virus. In contrary to this, studies monitoring immunological changes with the initiation of HAART have shown that in comparison to the rapid decline in viral titres, the restoration of CD4 T cell counts takes more time with therapy78. Detailed analysis of T cell subsets and their response to HAART has revealed that the normalization of CD4 T cell counts is preceded by an initial drop in the proliferation rates of all the subsets of CD4 and CD8 T cells, i.e. naïve, effector and memory79. These findings clearly indicate that once the viraemia is contained, antigenic stimulus decreases and opportunity for immune activation reduces suggesting that immune activation plays a crucial role in heightened T cell proliferation and dysregulates a normal homeostatic balance. The unremitting rounds of activation and expansion exert a negative pressure on naïve T cell pool. This would not only result in the exhaustion of the T cell compartment but also weaken the immune response to newer infections38,80.
Many studies have proposed two different pathways, thymic-dependent (thymopoiesis) and thymic-independent (homeostatic proliferation of existing cells) for the observed dysregulation in homeostasis81.
(i) Thymic-independent mechanisms: Studies have reported elevated levels of IL-7 during HIV infection which may be produced in response to the depleted pool of T cells to restore homeostasis. Decreased levels of CD127 expression in HIV infected individuals on both the CD4+ and CD8+ T cell compartments have been shown82. Similar findings have also been reported by our group, wherein a decreased expression of CD127 was observed and this decrease was found to be more pronounced on CD8 T cell subsets than CD4 T cells. Additionally, on further investigations, the CD127 expression was observed to be less in naïve, effector, and memory T cell subsets and was more marked for the central and effector memory cell subsets83.
As signaling through CD127-IL-7 receptor complex is fundamental to T cell survival, a depletion of CD127 from these memory cells signifies a functional aberration of these cells that can result in loss of these cells eventually in the course of infection. Reduced expression of CD127 disables the cell to respond to high levels of IL-7 which are mostly seen in HIV infected individuals84. Studies have also shown reduced expression of Bcl-2 in case of HIV infection, suggesting that the reduced expression of CD127 and Bcl-2 might predispose the cell to apoptosis raising the question of a possible association between correlates of disease progression and CD12784,85. In this context, studies were conducted to measure correlation between markers of immune activation and CD127 and have been found to be strongly correlated82. Investigations have also revealed strong correlation between central memory pool affected by immune activation and reduced CD127 expression86. This is further supported by evidences in elite controllers, non-pathogenic retroviral infection models, HIV-2 infection and controlled HIV infection models. Elite controllers are able to maintain stable CD4 counts with an undetectable viral load for long period of time in the absence of therapy. Reports have suggested that these HIV infected individuals show a conserved CD4 central memory cells expressing high levels of CD127. Also it was found that immune activation was restricted to CD4+ TEM compartment which had decreased expression of CD12786. In comparison to the pathogenic HIV infection, SIV infected sooty mangabeys with low levels of immune activation showed stable expression of CD127 on CD4+T cell population which correlated with preserved T cell count87. In another scenario of low immune activation, elevated levels of CD127 were observed in HIV-2 infected individuals88. In addition to this, patients who display recovery of T cell numbers on initiation of antiretroviral therapy have also showed upregulated expression of CD127 on the T cell compartment. However, individuals who despite therapy are not able to control the viral loads recorded poor restoration of CD127 expression85,89.
These findings support the theory that the immune activation and not the viral load, is an independent regulator of CD127 expression.
(ii) Thymic dependent mechanisms: It is apparent that immune activation is one of the major mechanisms for CD4 depletion. However, thymic impairment has also been frequently attributed to the progressive loss of CD4+ T cells90. In support of this, several studies have highlighted the role and effect of HIV on the thymus and to its functions. First line of evidence comes from a study of aborted foetuses from HIV-infected mothers wherein a loss of HIV infected thymocytes has been observed. Furthermore, similar results were seen in thymus biopsies of HIV-infected neonates91, in post mortem thymic tissue from HIV infected individuals92, and in SIV-infected rhesus macaques93. However, till date the precise contribution of HIV in thymic impairment and its possible role in CD4 depletion remain ambiguous. In 1998, Douek et al94 proposed the use of T cell receptor (TCR) excision circle (TREC) as a mean to quantify thymic output. TRECs are extra-chromosomal DNA fragments which are produced during TCR gene rearrangement. These are relatively stable, do not replicate, and have been proposed to be surrogate markers for thymic output95.
To further understand the role of thymus better, studies involving the removal of the thymus were undertaken. A group HIV infected individuals thymectomized before the infection, were studied and it was found that the thymectomy did not exclude long term survival. Also, thymectomized persons on HAART recorded an increase in CD4 T cells which was similar to that observed in non-thymectomized HIV infected individuals92. To directly measure the impairment of thymic output in SIV infection, Arron et al96 infected a group of thymectomized and group of sham operated juvenile rhesus macaques with the assumption that if SIV infection had a negative effect on the thymic output, then in thymectomized SIV infected macaque models, the condition should not deteriorate. On the contrary, they reported that the sham-operated, SIV infected macaques displayed a slower decline in CD4+ T cells and total TRECs in the CD4+ T cell population than the thymectomized, SIV-infected macaques. Also, when compared with the healthy uninfected thymectomized macaques, the SIV infected thymectomized macaques showed a major loss of CD4 T cells and TRECs. When translated into HIV infection, perhaps the decline in CD4 and TREC content could be due to the immune activation and increased proliferation resulting in dilution of TRECs96.
These findings point to the fact that (i) the impairment of thymus due to SIV does not result in the decline of CD4 T cells and TRECs, rather the peripheral effects of SIV should be taken into account, and (ii) TREC numbers are strongly dependent on T cell division rather than thymic dysfunction and thus these cannot be a marker for thymic function.
III. Maturational and differentiation defects
With growing evidence of HIV induced disturbance of the T cell differentiation and its impact on the immunopathogenesis, Clerici et al97 questioned the possibility of immune abnormalities occurring in foetuses of HIV infected pregnant women. Abnormal maturation of the T cell naïve and memory compartments were observed in infected aborted foetuses as well as uninfected children of HIV infected mothers leading the group to emphasize on the persistent effects HIV had on the immune system even after termination of the antigen stimulus. It also raised various concerns of the impaired immune system making the children susceptible to autoimmune diseases98. While the consequences of HIV infection of the immune system were becoming less ambiguous, Appay et al99 set out to understand the dynamics of T cell responses specifically in the acute and chronic phases of HIV infection. They pointed out that during the acute phase of the infection, the T cell compartment mostly comprises naοve T cells (CD27+CD28+) which underwent maturation by differentiating rapidly (within 2-4 wk) into intermediately differentiated cells (CD27-CD28+) and as the infection progresses to the chronic stage, the T population displayed a quantum shift towards the CD27-CD28- subsets. Although the study highlighted the shift in the phenotype, it cautioned against the oversimplified classifications of T cell subsets as a clear distinction between the CD28- and CD28+ population, suggesting the need for further scrutiny. However, the subsequent study by Tussey et al100 challenged this conclusion by stating that well before seroconversion, the CD8 population may exhibit a mixed expression of CD28 indicating a period of priming where the expression of CD28 is downregulated. They further argue that there is a flux between the maturation subsets in response to the antigenic stimulus, i.e. when the antigen load is low, the cells mature to the memory stage but during recrudescence, the memory subsets are recruited back to their previous state101.
To understand how the continuous antigenic stimulus disturbs the differentiation process better, Younes et al102 examined the effect of HIV viraemia on the differentiation of T cells. They found that persistent viraemia resulted in the alteration of the differentiation pathway by blocking the formation of stable long term central memory T cells. Instead, the continuous stimulation caused further differentiation of the effector T cells (Tem1) to form a functionally limited effector T cell population (Tem2). Furthermore, their data supported the previous findings that chronic progressive HIV-1 infection is related to a maturation block from effector memory phenotype (EM, CD45RA/CCR7) to full effector phenotype (E, CD45RA+/CCR7)103.
These conclusions also raised critical questions regarding the possible role of terminally differentiated HIV specific effector cells in mediating viral control. Our group also investigated quantitative alterations in the naive (CD45RA+CD62L+), memory/effector (CD45RO+) and activated (HLA-DR+CD38+) T lymphocyte subpopulations in treatment naive, HIV-1 infected Indian patients. Significant correlation obtained between different CD4+ and CD8+ T cell subsets reflected the quantitative alterations in the T- lymphocyte subpopulations and activation of the immune system during HIV-1 infection104.
Our group also observed an overrepresentation of intermediate (CD27+CD28-) differentiated cells in the naïve, effector memory, central memory and TEMRA CD8 subsets104. Our study has pointed out that this skewed maturation is likely a pathogenic strategy utilized by the virus, whereby CD8 T cells are prevented from achieving full effector functions104. Such abnormal CD8 T cell maturation, in turn may lead to a dearth of effective immune responses to clear the virus, thus establishing disease chronicity. Another factor that has been under intense research is the effect of HAART on maturation of T cells. On examining a group of patients receiving HAART, Lécuroux et al105 found a particular subset of CD8+T cells displaying CD27-CD45RO-/RA+ phenotype, originally which were sparsely present during primary HIV infection, increased in numbers after the early initiation of HAART. This newly identified subset was found to be stable, nonactivated, and resting, presenting memory characteristics, and can develop effector capacities after stimulation thus providing direction for future studies to explore their capacity to control HIV infection. While the ideal vaccine against HIV remains elusive to man, intense research over the decades has revealed and such encouraging findings may prove to be an impetus for future scientists to understand the deleterious effects of HIV on the adaptive immune system.
The dark side of HIV/AIDS
Polyfunctionality: Loss or gain?: In recent years, polyfunctionality of CD8+ T cells has gained popularity and many research groups have identified HIV specific CD8+ T cell subsets capable of producing multiple cytokines simultaneously. The quantitation of these immune parameters has been possibly largely due to the advancements in flow-cytometry where multiple markers can be examined at a time. Using these techniques, investigators have characterized cells producing cytokines such as IFN-γ, TNF-α, and macrophage inflammatory protein (MIP)1-b exhibiting degranulation. It was also demonstrated that these cells produced multiple cytokines on a per cell basis as compared to the monofunctional CD8 T cells. Current notion regarding polyfunctional CD8+T cells suggest that “the more the number of cytokines produced, the better is the quality of the immune response”. Such a response was observed in Elite Controllers (ECs) who are a rare subset of treatment naïve HIV infected individuals capable of maintaining stable CD4 T cell counts for a longer period of time with a viral load of <50 copies/ml106 and long term non-progressors (LTNP) who greatly vary in their levels of viraemia but are able to sustain CD4 T cell counts over 7-10 years in the absence of therapy107. Thus, their natural resistance to HIV disease progression has made them ideal models to delineate the involved mechanisms. ECs and LTNPs have been shown to be able to maintain these responses as compared to non progressors. Additionally, Migueles et al108 revealed that the HIV specific CD8 T cells of LTNPs apart from secreting multiple cytokines also demonstrate high proliferative and cytotoxicity capacity against autologous HIV infected CD4 T cells when compared to progressors. This capacity was attributed to efficient release of perforin and granzyme B production which may be helpful in suppressing HIV replication. However, these polyfunctional HIV-specific CD8 T cells are rarely found in the peripheral blood of individuals with progressive HIV infection42,109. Progressors after mounting a strong polyfunctional anti-HIV CD8 T cell response during the early phase of infection are unable to sustain it over a period of time unlike LTNPs110. Similar findings have also been reported from our laboratory wherein we observed that HIV specific CD8 T cells are significantly impaired with regard not only to IFN-γ but also in IL-2 and TNF-α production in chronically infected individuals111. Interestingly, the degranulation capacity was found to be largely conserved. It was also observed that HIV specific responses were mostly mono functional followed by dual responses and responses with three or more functions were almost absent. The finding also showed that the ability of CD8+ T-cells to respond to Gag and Nef stimulation with cytokine secretion and degranulation did not differ substantially and was irrespective of HIV antigen. Also, the loss of functionality was found to be restricted to only HIV-1-specific CD8+ T cells, whereas no significant changes were found in the functionality of CD8+ T-cell responses to polyclonal stimulation in the same participants concluding that it is a quality of CD8 T cells i.e. paramount for virus control111,112.
Although, CD8+T cells have the potential to suppress viral replication, many LTNPs are completely devoid of these subsets thus, questioning the importance of CD8 T cells as an immune correlate of protection. This clearly indicates either a gap in our knowledge of CTL responses or the possible existence of another subset controlling viral replication.
(ii) Immune exhaustion: As the disease progresses in HIV infected individuals, both HIV specific CD4 and CD8 T cells lose their capacity to mount an effective immune response and gradually become exhausted. Immune exhaustion was initially described in mouse model of lymphocytic choriomeningitis (LCMV) infection113. The LCMV specific CD4 and CD8 T cells were found to be deficient in their cytotoxicity and were termed ‘exhausted’. Subsequently this phenomenon was observed and reported in chronic HIV infection. T cell exhaustion is a slow and steady process, with partial loss of effector functions (proliferation, IL-2 secretion)) in the early phase followed by a hierarchical loss of other functions like TNF-α, IFN-γ secretion during the late phase of infection114. However, this phenomenon of immune exhaustion is not only restricted to the cell but can be seen at a systemic level. On a cellular level, numerous changes have been observed including altered gene expression patterns especially in the expression of inhibitory receptors, exhibition of metabolic and bioenergetic insufficiencies114, dysregulation of TCR and cytokine signalling pathways along with maturational and differentiation defects. On the systemic level Tregs, homeostatic growth factors and regulatory cytokines such as IL-10 and TGF-β have been proposed to have a significant role in immune exhaustion. Although the phenomenon is not fully understood, a few underlying mechanisms have been proposed.
Inhibitory receptors
Many inhibitory receptors (like PD-1, LAG-3, Tim-3, CTLA-4, 2B4, and CD160) have garnered attention due to their upregulation during chronic phase of infection and their possible role in immune suppression.
PD-1: A member of the B7:CD28 family of molecules, PD-1 was first studied in T cell hybridoma undergoing apoptosis and hence was named as programmed death-1. However, further studies have highlighted its association with immune exhaustion. In 2008, Kaufmann and Walker115 elucidated the crucial part PD-1 essays in balancing CD4 and CD8 T cell activation and tolerance. Additionally, this molecule is expressed on an array of cells including natural killer (NK) cells, B cells and monocytes and binds to its ligand, PD-L1, expressed on APCs116. Barber et al117 were the first to report the involvement of PD-1 in chronic viral infections and showed marked upregulation of messenger RNA encoding PD-1 using LCMV mouse models. Expression of PD-1 in LCMV specific CD8 T cells was further confirmed by antibody staining thereby establishing it a marker of T cell impairment. The findings of this study triggered interest in understanding the role of PD-1 in HIV infection. Day et al118 studied the expression of PD-1 in 71 antiretroviral therapy (ART) naïve HIV-infected subjects and found heightened levels of PD-1 expression on both CD4 and CD8 molecules which in turn correlated positively with viral load and inversely with CD4 counts. Furthermore, on blocking PD-1, they found a marked rise in the HIV-1 specific CD4 and CD8 T cells.
The molecular expression of PD-1 in HIV specific CD8 T cells was investigated by Quigley et al119 and they reported that the silencing of Basic leucine transcription factor (BATF), a transcriptional factor downstream of PD-1 receptor, was associated with recovery of IFN-γ secretion along with proliferation of HIV specific CD8 T cells. They also observed an augmentation of IL-2 secretion by CD4 T cells and have concluded that the downstream PD-1/PD-L1 signalling pathway is operative in exhaustion of T cell immune response to HIV. However, these studies failed to explain the association of immune exhaustion with immune activation. Sauce et al120 revealed that the upregulation of PD-1 was accompanied by a concomitant increase in the expression of activation markers: CD38 or HLA-DR. They also proposed that such an increase in inhibitory and activation markers was restricted to antigen experienced cells rather than the naive population. On the contrary, not all exhausted cells express PD-1 indicating the presence of other inhibitory players in T cell exhaustion.
T-cell immunoglobulin and mucin domain-containing molecule-3 (TIM 3)
PD-1 is not the only inhibitory receptor expressed on exhausted T cells, a variety of other molecules including TIM-3, LAG-3, CTLA-4, CD160, CD244 and GP49 are also expressed. TIM-3 is one such inhibitory receptor that has recently been highlighted to play an important role in chronic HIV infection. TIM-3 was initially identified to be expressed on Th1 cells and not on Th2 cells in murine models116. Interaction of TIM-3 with its ligand galactin-9 induces CD4 T cell death. The inhibitory role of TIM-3 was confirmed by blockade of TIM-3 with small interfering RNA (siRNA) or antibodies. In HIV infected individuals, high expression of TIM-3 has been shown to correlate with markers of disease progression along with immune activation121. They also observed that the heightened expression of TIM-3 was more prominent in individuals with progressive acute and chronic HIV infection than in individuals who are able to control viraemia or remain uninfected. In another study it was observed that both TIM-3 and PD-1 were expressed synergistically on LCMV specific CD8 T cells. Also, the severity of immune exhaustion was more when both the markers were co-expressed as compared to singular expression of PD-1. Further on blocking both receptors Jin et al122 recorded restoration of antiviral functions indicating possible therapeutic avenues. However, the role of both the receptors and their co-expression remains to be determined in case of HIV infection.
CTLA-4 and lymphocyte activation gene-3 (LAG-3)
Another negative regulatory receptor belonging to the B7-CD28 family is CTLA-4 which has been found to play an important role in autoimmunity7. Studies in human cohorts have also illustrated its role in tumour reduction and cancer treatment123. Like PD-1 and TIM-3, CTLA 4 expression was found to be elevated in HIV specific CD4 T cells124. Kaufmann et al115 also demonstrated that CTLA-4 expression correlated directly with viral load and indirectly with CD4 T cell counts.
Like TIM-3, LAG-3 is preferentially expressed on Th1 cells and belongs to the immunoglobulin superfamily125. Investigations carried out in LCMV mice models have revealed elevated expression of LAG-3 and like other inhibitory markers, inhibition of LAG-3 resulted in proliferation and production of cytokines by Th1 cells in HIV infection121. Further investigations are required to understand and establish the role of all the inhibitory receptors in T cell exhaustion.
Although, these studies have highlighted the importance of inhibitory receptors in chronic HIV infection, yet many alarming questions are to be answered: (i) whether immune exhaustion is a by-product of high antigen load or is it the cause?, (ii) is the concomitant expression of negative regulatory receptors along with high immune activation a possible regulatory mechanism by the host to control hyper-activation or is it a virus survival tactic?, and (iii) will blockade of inhibitory receptors - with or without therapeutic intervention- abet HIV-specific immune responses or place the immune system in an uncontrolled hyper activated state?
Regulatory cytokines
Apart from inhibitory receptors, regulatory cytokines such as IL-10 and TGF-β have been implicated in T cell exhaustion. LCMV studies have shown enhanced virus specific responses in the absence of IL-10 secretion126. In case of HIV infection elevated levels of IL-10 were shown to have considerable effect on CD41 T cell function127. Recent studies have shown the existence of a novel IL-10 producing HIV specific CD8 T cell population122. Jin et al122 showed phenotypic similarities between IL-10 producing CD8 T cells and severely exhausted CD8 T cells co-expressing PD-1 and TIM-3. Said et al128 studied the possible link between immune exhaustion and microbial translocation and reported upregulation of PD-1 along with plasma IL-10. Also on blocking the IL-10 and PD-1 pathways, viral clearance and improved T cell function were observed in animal models of chronic viral infection. Their findings suggest that translocated microbial products such as LPS result in the upregulation of PD-1 expression on monocytes and positively correlated with high levels of plasma IL-10. The data from this study highlight the need to explore the missing links between microbial translocation and exhaustion.
Conclusion
The vaccine trials have been a turning point in the field of HIV immunopathogenesis and have given enough reasons to re-evaluate our strategies to tackle HIV. The data reviewed here provide new insights into the dynamics of T cells and HIV. Additionally, these findings revealed unique strategies that the virus utilizes in the host system to sustain itself. One of the main pathways that HIV explores to deteriorate the immunity is chronic activation of the immune system. Though the review highlights involvement of multiple mechanisms that are likely to cause aberrant activation of T cells, the role of each factor and their relationship with each other are still incompletely understood and are a matter of debate and speculation. Chronic immune activation has been portrayed as the major driver of CD4 T cell depletion especially in gut associated lymphoid tissues; however, its association with this massive depletion is still ambiguous. Recently the focus has been shifted to investigate the effects of HIV on the intestinal epithelial barrier and studies have shown a link between immune activation and microbial translocation. The details of how infection and loss of intestinal CD4 T cells lead to this leaky gut are unclear, but multiple avenues of investigation have begun to be explored, including the role of Th17 cells and Tregs. Unlocking the role of Th17 and Tregs can provide important clues in unravelling the mystery of HIV pathogenesis.
The last few years have witnessed the exploration of newer avenues of research to understand the impact of HIV on the immune system, with one of these being innate immune system. Emerging data suggest activation of this compartment at an early stage but to what extent this innate activation affects the adaptive immune response and helps in establishment of sustained chronic activation needs to be investigated. Apart from this, immune activation has also been shown to be involved in increased turn over, homeostatic imbalance, exhaustion and apoptosis which represent a massive task for the immune system. To address all these issues, additional studies are required which would help us broaden our view on the dynamics of T cells and HIV.
References
- 1.Dalgleish AG, Beverly PCL, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312:763–7. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 2.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1984;387:183–8. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 3.Igarashi T, Brown CR, Endo Y, Buckler-White A, Plishka R, Bischofberger N, et al. Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4þ T cells by a highly pathogenic simian immunodeficiency virus/HIV type 1 chimera (SHIV): Implications for HIV-1 infections of humans. Proc Natl Acad Sci USA. 2001;98:658–63. doi: 10.1073/pnas.021551798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–71. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 5.Makedonas G, Betts MR. Living in a house of cards: re-evaluating CD8+ T cell immune correlates against HIV. Immunol Rev. 2011;239:109–24. doi: 10.1111/j.1600-065X.2010.00968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staprans SI, Feinberg MB, Shiver JW, Casimiro DR. Role of nonhuman primates in the evaluation of candidate AIDS vaccines: an industry perspective. Curr Opin HIV AIDS. 2010;5:377–85. doi: 10.1097/COH.0b013e32833d2e19. [DOI] [PubMed] [Google Scholar]
- 7.Kindt TJ, Goldsby RA, Osborne BA, Kuby J. Overview of the Immune System. In: Goldsby RA, Kuby J, editors. Kuby immunology. New York: W.H. Freeman & Company; 2006. pp. 10–11. [Google Scholar]
- 8.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–57. [PubMed] [Google Scholar]
- 9.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190:624–31. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 10.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno, et al. Impaired TH17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–6. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–45. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 12.Costantino CM, Baecher-Allan CM, Hafler DA. Human regulatory T cells and autoimmunity. Eur J Immunol. 2008;38:921–4. doi: 10.1002/eji.200738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. 1970;18:723–37. [PMC free article] [PubMed] [Google Scholar]
- 14.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64. [PubMed] [Google Scholar]
- 15.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–9. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 16.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 17.Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol. 2009;10:864–71. doi: 10.1038/ni.1770. [DOI] [PubMed] [Google Scholar]
- 18.Akbar AN, Borthwick N, Salmon M, Gombert W, Bofill M, Shamsadeen N, et al. The significance of low bcl-2 expression by CD45RO T cells in normal individuals and patients with acute viral infections. The role of apoptosis in T cell memory. J Exp Med. 1993;178:427–38. doi: 10.1084/jem.178.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tenca C, Merlo A, Zarcone D, Saverino D, Bruno S, De Santanna A, et al. Death of T cell precursors in the human thymus: a role for CD38. Int Immunol. 2003;15:1105–16. doi: 10.1093/intimm/dxg111. [DOI] [PubMed] [Google Scholar]
- 20.Abbas AK, Lichtman AH, Pillai S. Cellular and molecular immunology. Philadelphia: Elsevier Incorporation; 2007. Properties and overview of immune responses; pp. 15–16. [Google Scholar]
- 21.Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, et al. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med. 1997;186:1407–18. doi: 10.1084/jem.186.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–51. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 23.Mazzucchelli R, Durum SK. Interleukin-7 receptor expression: intelligent design. Nat Rev Immunol. 2007;7:144–54. doi: 10.1038/nri2023. [DOI] [PubMed] [Google Scholar]
- 24.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–45. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romero P, Zippelius A, Kurth I, Pittet MJ, Touvrey C, Iancu EM, et al. Four functionally distinct populations of human effector memory CD8+ T lymphocyte. J Immunol. 2007;178:4112–9. doi: 10.4049/jimmunol.178.7.4112. [DOI] [PubMed] [Google Scholar]
- 26.Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434:1093–7. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- 27.Streeck H, Jolin JS, Qi Y, Yassine-Diab B, Johnson RC, Kwon DS, et al. Human immunodeficiency virus type 1-specific CD [8]+ T-cell responses during primary infection are major determinants of the viral set point and loss of CD [4]+ T cells. J Virol. 2009;83:7641–8. doi: 10.1128/JVI.00182-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaul R, Plummer FA, Kimani J, Dong T, Kiama P, Rostron T, et al. HIV-1-specific mucosal CD8+ lymphocyte responses in the cervix of HIV-1-resistant prostitutes in Nairobi. J Immunol. 2000;164:1602–11. doi: 10.4049/jimmunol.164.3.1602. [DOI] [PubMed] [Google Scholar]
- 29.Paranjape RS. Immunopathogenesis of HIV infection. Indian J Med Res. 2005;121:240–55. [PubMed] [Google Scholar]
- 30.Finkel TH, Tudor-Williams G, Banda NK, Cotton MF, Curiel T, Monks C, et al. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat Med. 1995;1:129–34. doi: 10.1038/nm0295-129. [DOI] [PubMed] [Google Scholar]
- 31.Giorgi JV, Hultin LE, McKeating JA, Johnson TD, Owens B, Jacobson LP, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis. 1999;179:859–70. doi: 10.1086/314660. [DOI] [PubMed] [Google Scholar]
- 32.Silvestri G, Sodora DL, Koup RA, Paiardini M, O’Neil SP, McClure HM, et al. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity. 2003;18:441–52. doi: 10.1016/s1074-7613(03)00060-8. [DOI] [PubMed] [Google Scholar]
- 33.Moanna A, Dunham R, Paiardini M, Silvestri G. CD4+ T-cell depletion in HIV infection: killed by friendly fire? Curr HIV/AIDS Rep. 2005;2:16–23. doi: 10.1007/s11904-996-0004-3. [DOI] [PubMed] [Google Scholar]
- 34.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–26. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 35.Herbeuval JP, Nilsson J, Boasso A, Hardy AW, Kruhlak MJ, Anderson SA, et al. Differential expression of IFN-alpha and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc Natl Acad Sci USA. 2006;103:7000–5. doi: 10.1073/pnas.0600363103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boasso A, Shearer GM. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin Immunol. 2008;126:235–42. doi: 10.1016/j.clim.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–74. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 38.Potula R, Poluektova L, Knipe B, Chrastil J, Heilman D, Dou H, et al. Inhibition of indoleamine 2,3-dioxygenase (IDO) enhances elimination of virus infected macrophages in an animal model of HIV-1 encephalitis. Blood. 2005;106:2382–90. doi: 10.1182/blood-2005-04-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boasso A, Herbeuval JP, Hardy AW, Anderson SA, Dolan MJ, Fuchs D, et al. HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood. 2007;109:3351–9. doi: 10.1182/blood-2006-07-034785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ostrowski SR. Immune activation in chronic HIV infection. Dan Med Bull. 2010;57:B4122. [PubMed] [Google Scholar]
- 41.Lawn SD, Butera ST, Folks TM. Contribution of immune activation to the pathogenesis and transmission of human immunodeficiency virus type 1 infection. Clin Microbiol Rev. 2001;14:753–77. doi: 10.1128/CMR.14.4.753-777.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herbein G, Khan KA. Is HIV infection a TNF receptor signalling-driven disease? Trends Immunol. 2008;29:61–7. doi: 10.1016/j.it.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 43.Muthumani K, Desai BM, Hwang DS, Choo AY, Laddy DJ, Thieu KP, et al. HIV-1 Vpr and anti-inflammatory activity. DNA Cell Biol. 2004;23:239–47. doi: 10.1089/104454904773819824. [DOI] [PubMed] [Google Scholar]
- 44.Park IW, Wang JF, Groopman JE. HIV-1 Tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood. 2001;97:352–8. doi: 10.1182/blood.v97.2.352. [DOI] [PubMed] [Google Scholar]
- 45.Kotler DP, Gaetz HP, Lange M, Klein EB, Holt PR. Enteropathy associated with the acquired immunodeficiency syndrome. Ann Intern Med. 1984;101:421–8. doi: 10.7326/0003-4819-101-4-421. [DOI] [PubMed] [Google Scholar]
- 46.Heise C, Dandekar S, Kumar P, Duplantier R, Donovan RM, Halsted CH, et al. Human immunodeficiency virus infection of enterocytes and mononuclear cells in human jejunal mucosa. Gastroenterology. 1991;100:1521–7. doi: 10.1016/0016-5085(91)90648-5. [DOI] [PubMed] [Google Scholar]
- 47.Maresca M, Mahfoud R, Garmy N, Kotler DP, Fantini J, Clayton F. The virotoxin model of HIV-1 enteropathy: involvement of GPR15/Bob and galactosylceramide in the cytopathic effects induced by HIV-1 gp120 in the HT-29-D4 intestinal cell line. J Biomed Sci. 2003;10:156–66. doi: 10.1007/BF02256007. [DOI] [PubMed] [Google Scholar]
- 48.Brenchley JM, Douek DC. HIV infection and the gastrointestinal immune system. Mucosal Immunol. 2008;1:23–30. doi: 10.1038/mi.2007.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Douek D. Perspective HIV disease progression: Immune activation, microbes and a leaky gut. Top HIV Med. 2007;15:114–7. [PubMed] [Google Scholar]
- 50.Heikenwalder M, Polymenidou M, Junt T, Sigurdson C, Wagner H, Akira S, et al. Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat Med. 2004;10:187–92. doi: 10.1038/nm987. [DOI] [PubMed] [Google Scholar]
- 51.Brenchley JM, Paiardini M, Knox KS, Asher AI, Cervasi B, Asher TE, et al. Differential Th17 CD4 T cell depletion in pathogenic and non-pathogenic lentiviral infections. Blood. 2008;112:2826–35. doi: 10.1182/blood-2008-05-159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Favre D, Lederer S, Kanwar B, Ma ZM, Proll S, Kasakow Z, et al. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. 2009;5:e1000295. doi: 10.1371/journal.ppat.1000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh A, Vajpayee M, Ali SA, Mojumdar K, Chauhan NK, Singh R. HIV-1 diseases progression associated with loss of Th17 cells in subtype ‘C’ infection. Cytokine. 2012;60:55–63. doi: 10.1016/j.cyto.2012.06.288. [DOI] [PubMed] [Google Scholar]
- 54.Terness P, Bauer TM, Röse L, Dufter C, Watzlik A, Simon H, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196:447–57. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Romani L, Fallarino F, De Luca A, Montagnoli C, D’Angelo C, Zelante T, et al. Romani L, Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451:211–5. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]
- 56.Baeuerle PA. The inducible transcription factor NF-Kb: Regulation by distinct protein subunit. Biochim Biophys Acta. 1991;1072:63–80. doi: 10.1016/0304-419x(91)90007-8. [DOI] [PubMed] [Google Scholar]
- 57.Gaynor R. Cellular transcription factor involved in the regulation of HIV-1 gene expression. AIDS. 1992;6:347–63. doi: 10.1097/00002030-199204000-00001. [DOI] [PubMed] [Google Scholar]
- 58.Lawn SD, Pisell TL, Hirsch CS, Wu M, Butera ST, Toossi Z. Anatomically compartmentalized human immunodeficiency HIV replication in HLA-DR+ and CD14 positive macrophages at the site of pleural tuberculosis co-infection. J Infect Dis. 2001;184:1127–33. doi: 10.1086/323649. [DOI] [PubMed] [Google Scholar]
- 59.Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200:749–59. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vajpayee M, Kaushik S, Sreenivas V, Mojumdar K, Mendiratta S, Chauhan NK. Role of immune activation in CD4+ T-cell depletion in HIV-1 infected Indian patients. Eur J Clin Microbiol Infect Dis. 2009;28:69–73. doi: 10.1007/s10096-008-0582-7. [DOI] [PubMed] [Google Scholar]
- 61.Alimonti JB, Ball TB, Fowke KR. Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. J Gen Virol. 2003;84:1649–61. doi: 10.1099/vir.0.19110-0. [DOI] [PubMed] [Google Scholar]
- 62.Fantuzzi L, Belardelli F, Gessani S. Monocyte/macrophage-derived CC chemokines and their modulation by HIV-1 and cytokines: a complex network of interactions influencing viral replication and AIDS pathogenesis. J Leukoc Biol. 2003;74:719–25. doi: 10.1189/jlb.0403175. [DOI] [PubMed] [Google Scholar]
- 63.Wig N, Anupama P, Singh S, Handa R, Aggarwal P, Dwivedi SN, et al. Tumor necrosis factor-alpha levels in patients with HIV with wasting in South Asia. AIDS Patient Care STDS. 2005;19:212–5. doi: 10.1089/apc.2005.19.212. [DOI] [PubMed] [Google Scholar]
- 64.Peter ME, Ehret A, Berndt C, Krammer PH. AIDS and the death receptors. Br Med Bull. 1997;53:604–16. doi: 10.1093/oxfordjournals.bmb.a011633. [DOI] [PubMed] [Google Scholar]
- 65.Herbeuval JP, Nilsson J, Boasso A, Hardy AW, Vaccari M, Cecchinato V, et al. HAART reduces death ligand but not death receptors in lymphoid tissue of HIV-infected patients and simian immunodeficiency virusinfected macaques. AIDS. 2009;23:35–40. doi: 10.1097/QAD.0b013e32831cb907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silvestris F, Cafforio P, Frassanito MA, Tucci M, Romito A, Nagata S, et al. Overexpression of Fas antigen on T cells in advanced HIV-1 infection: differential ligation constantly induces apoptosis. AIDS. 1996;10:131–41. doi: 10.1097/00002030-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 67.Dabrowska A, Kim N, Aldovini A. Tat-induced FOXO3a is a key mediator of apoptosis in HIV-1-infected human CD4+ T lymphocytes. J Immunol. 2008;181:8460–77. doi: 10.4049/jimmunol.181.12.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Algeciras A, Dockrell DH, Lynch DH, Paya CV. CD4 regulates susceptibility to Fas ligand- and tumor necrosis factor mediated apoptosis. J Exp Med. 1998;187:711–20. doi: 10.1084/jem.187.5.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alimonti JB, Ball TB, Fowke KR. Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. Virology. 2003;84:1649–61. doi: 10.1099/vir.0.19110-0. [DOI] [PubMed] [Google Scholar]
- 70.Katsikis PD, Wunderlich ES, Smith CA, Herzenberg LA. Fas antigen stimulation induces marked apoptosis of T lymphocytes in human immunodeficiency virus-infected individuals. J Exp Med. 1995;181:2029–36. doi: 10.1084/jem.181.6.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Poon B, Grovit-Ferbas K, Stewart SA, Chen IS. Cell cycle arrest by Vpr in HIV-1 virions and insensitivity to antiretroviral agents. Science. 1998;281:266–9. doi: 10.1126/science.281.5374.266. [DOI] [PubMed] [Google Scholar]
- 72.Re F, Braaten D, Franke EK, Luban J. Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J Virol. 1995;69:6859–64. doi: 10.1128/jvi.69.11.6859-6864.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nishizawa M, Kamata M, Katsumata R, Aida Y. A carboxy-terminally truncated form of the human immunodeficiency virus type 1Vpr protein induces apoptosis via G (1) cell cycle arrest. J Virol. 2000;74:6058–67. doi: 10.1128/jvi.74.13.6058-6067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Selliah N, Finkel TH. Review biochemical mechanisms of HIV induced T cell apoptosis. Cell Death Differ. 2001;8:127–36. doi: 10.1038/sj.cdd.4400822. [DOI] [PubMed] [Google Scholar]
- 75.Zauli G, Gibellini D, Secchiero P, Dutartre H, Olive D, Capitani S, et al. Human immunodeficiency virus type 1 Nef protein sensitizes CD4(+) T lymphoid cells to apoptosis via functional upregulation of the CD95/CD95 ligand pathway. Blood. 1999;93:1000–10. [PubMed] [Google Scholar]
- 76.Xu XN, Laffert B, Screaton GR, Kraft M, Wolf D, Kolanus W, et al. Induction of Fas ligand expression by HIV involves the interaction of Nef with the T cell receptor zeta chain. J Exp Med. 1999;189:1489–96. doi: 10.1084/jem.189.9.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shedlock DJ, Hwang D, Choo AY, Chung CW, Muthumani K, Weiner DB. HIV-1 viral genes and mitochondrial apoptosis. Apoptosis. 2008;13:1088–99. doi: 10.1007/s10495-008-0239-0. [DOI] [PubMed] [Google Scholar]
- 78.Valdez H, Connick E, Smith KY, Lederman MM, Bosch RJ, Kim RS, et al. Limited immune restoration after 3 years’ suppression of HIV-1 replication in patients with moderately advanced disease. AIDS. 2002;16:1859–66. doi: 10.1097/00002030-200209270-00002. [DOI] [PubMed] [Google Scholar]
- 79.Hazenberg MD, Stuart JW, Otto SA, Borleffs JC, Boucher CA, de Boer RJ, et al. T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART) Blood. 2000;95:249–55. [PubMed] [Google Scholar]
- 80.Hazenberg MD, Hamann D, Schuitemaker H, Miedema F. T cell depletion in HIV-1 infection: how CD4 T cells go out of stock. Nat Immunol. 2001;410:974–89. doi: 10.1038/79724. [DOI] [PubMed] [Google Scholar]
- 81.Jamieson BD, Douek DC, Killian S, Hultin LE, Scripture-Adams DD, Giorgi JV, et al. Generation of functional thymocytes in the human adult. Immunity. 1999;10:569–75. doi: 10.1016/s1074-7613(00)80056-4. [DOI] [PubMed] [Google Scholar]
- 82.Koesters SA, Alimonti JB, Wachihi C, Matu L, Anzala O, Kimani J, et al. IL-7Ralpha expression on CD4+ T lymphocytes decreases with HIV disease progression and inversely correlates with immune activation. Eur J Immunol. 2006;36:336–44. doi: 10.1002/eji.200535111. [DOI] [PubMed] [Google Scholar]
- 83.Mojumdar K, Vajpayee M, Chauhan NK, Singh A, Singh R, Kurapati S. Loss of CD127 & increased immunosenescence of T cell subsets in HIV infected individuals. Indian J Med Res. 2011;134:972–81. doi: 10.4103/0971-5916.92645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Napolitano LA, Grant RM, Deeks SG, Schmidt D, De Rosa SC, Herzenberg LA, et al. Increased production of IL-7 accompanies HIV-1-mediated T-cell depletion: implications for T-cell homeostasis. Nat Med. 2001;7:73–9. doi: 10.1038/83381. [DOI] [PubMed] [Google Scholar]
- 85.Colle JH, Moreau JL, Fontanet A, Lambotte O, Joussemet M, Jacod S, et al. Regulatory dysfunction of the interleukin-7 receptor in CD4 and CD8 lymphocytes from HIV-infected patients-effects of antiretroviral therapy. J Acquir Immune Defic Syndr. 2006;42:277–85. doi: 10.1097/01.qai.0000214823.11034.4e. [DOI] [PubMed] [Google Scholar]
- 86.Potter SJ, Lacabaratz C, Lambotte O, Perez-Patrigeon S, Vingert B, Sinet M, et al. Preserved central memory and activated effector memory CD4+ T-cell subsets in human immunodeficiency virus controllers: an ANRS EP36 study. J Virol. 2007;81:13904–15. doi: 10.1128/JVI.01401-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sodora DL, Silvestri G. Immune activation and AIDS pathogenesis. AIDS. 2008;22:439–46. doi: 10.1097/QAD.0b013e3282f2dbe7. [DOI] [PubMed] [Google Scholar]
- 88.Albuquerque AS, Cortesão CS, Foxall RB, Soares RS, Victorino RM, Sousa AE. Rate of increase in circulating IL-7 and Loss of IL-7R {alpha} expression differ in HIV-1 and HIV-2 infections: two lymphopenic diseases with similar hyperimmune activation but distinct outcomes. J Immunol. 2007;178:3252–9. doi: 10.4049/jimmunol.178.5.3252. [DOI] [PubMed] [Google Scholar]
- 89.Marziali M, De Santis W, Carello R, Leti W, Esposito A, Isgrò A, et al. T-cell homeostasis alteration in HIV-1 infected subjects with low CD4 T-cell count despite undetectable virus load during HAART. AIDS. 2006;20:2033–41. doi: 10.1097/01.aids.0000247588.69438.fd. [DOI] [PubMed] [Google Scholar]
- 90.McCune JM. The dynamics of CD4+ T-cell depletion in HIV disease. Nature. 2001;410:974–9. doi: 10.1038/35073648. [DOI] [PubMed] [Google Scholar]
- 91.Rosenzweig M, Clark DP, Gaulton GN. Selective thymocyte depletion in neonatal HIV-1 infection. AIDS. 1993;7:1601–5. doi: 10.1097/00002030-199312000-00009. [DOI] [PubMed] [Google Scholar]
- 92.Haynes BF, Hale LP, Weinhold KJ, Patel DD, Liao H-X, Bressler PB, et al. Analysis of the adult thymus in reconstitution of T lymphocytes in HIV-1 infection. J Clin Invest. 1999;103:453–60. doi: 10.1172/JCI5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baskin GB, Murphey-Corb M, Martin LN, Davison-Fairburn B, Hu FS, Kuebler D. Thymus in simian immunodeficiency virus-infected rhesus monkeys. Lab Invest. 1991;65:400–7. [PubMed] [Google Scholar]
- 94.Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–5. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- 95.Hazenberg MD, Borghans JA, de Boer RJ, Miedema F. Thymic output: a bad TREC record. Nat Immunol. 2003;4:97–9. doi: 10.1038/ni0203-97. [DOI] [PubMed] [Google Scholar]
- 96.Arron ST, Ribeiro RM, Gettie A, Bohm R, Blanchard J, Yu J, et al. Impact of thymectomy on the peripheral T cell pool in rhesus macaques before and after infection with simian immunodeficiency virus. Eur J Immunol. 2005;35:46–55. doi: 10.1002/eji.200424996. [DOI] [PubMed] [Google Scholar]
- 97.Clerici M, Saresella M, Colombo F, Fossati S, Sala N, Bricalli D, Villa ML, et al. T-lymphocyte maturation abnormalities in uninfected newborns and children with vertical exposure to HIV. Blood. 2000;96:3866–71. [PubMed] [Google Scholar]
- 98.McFarland EJ, Harding PA, Striebich CC, MaWhinney S, Kuritzkes DR, Kotzin BL. Clonal CD8+ T cell expansions in peripheral blood from human immunodeficiency virus type 1-infected children. J Infect Dis. 2002;186:477–85. doi: 10.1086/341939. [DOI] [PubMed] [Google Scholar]
- 99.Appay V, Papagno L, Spina CA, Hansasuta P, King A, Jones L, et al. Dynamics of T cell responses in HIV infection. J Immunol. 2002;168:3660–6. doi: 10.4049/jimmunol.168.7.3660. [DOI] [PubMed] [Google Scholar]
- 100.Tussey L, Speller S, Gallimore A, Vessey R. Functionally distinct CD8+ memory T cell subsets in persistent EBV infection are differentiated by migratory receptor expression. Eur J Immunol. 2000;30:1823–9. doi: 10.1002/1521-4141(200007)30:7<1823::AID-IMMU1823>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 101.Tussey LG, Nair US, Bachinsky M, Edwards BH, Bakari J, Grimm K, et al. Antigen burden is a major determinant of human immunodeficiency virus-specific CD8+ T cell maturation state: Potential implications for therapeutic immunization. J Infect Dis. 2003;187:364–74. doi: 10.1086/367707. [DOI] [PubMed] [Google Scholar]
- 102.Younes SA, Yassine-Diab B, Dumont AR, Boulassel MR, Grossman Z, Routy JP, et al. HIV-1 viremia prevents the establishment of interleukin 2-producing HIV-specific memory CD4+ T cells endowed with proliferative capacity. J Exp Med. 2003;198:1909–22. doi: 10.1084/jem.20031598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Champagne P, Ogg GS, King AS, Knabenhans C, Ellefsen K, Nobile M, et al. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature. 2001;410:106–11. doi: 10.1038/35065118. [DOI] [PubMed] [Google Scholar]
- 104.Mojumdar K, Vajpayee M, Chauhan NK, Singh A, Singh R, Kurapati S. Altered T cell differentiation associated with loss of CD27 and CD28 in HIV infected Indian individuals. Cytometry B Clin Cytom. 2012;82:43–53. doi: 10.1002/cyto.b.20610. [DOI] [PubMed] [Google Scholar]
- 105.Lécuroux C, Girault I, Urrutia A, Doisne JM, Deveau C, Goujard C, et al. Identification of a particular HIV-specific CD8+ T-cell subset with a CD27+ CD45RO-/RA+ phenotype and memory characteristics after initiation of HAART during acute primary HIV infection. Blood. 2009;113:3209–17. doi: 10.1182/blood-2008-07-167601. [DOI] [PubMed] [Google Scholar]
- 106.O’Connell KA, Bailey JR, Blankson JN. Elucidating the elite: mechanisms of control in HIV-1 infection. Trends Pharmacol Sci. 2009;30:631–7. doi: 10.1016/j.tips.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 107.Lambotte O, Boufassa F, Madec Y, Nguyen A, Goujard C, Meyer L, et al. HIV controllers: a homogeneous group of HIV-1-infected patients with spontaneous control of viral replication. Clin Infect Dis. 2005;41:1053–6. doi: 10.1086/433188. [DOI] [PubMed] [Google Scholar]
- 108.Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, et al. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity. 2008;29:1009–21. doi: 10.1016/j.immuni.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T-cells. Blood. 2006;107:4781–9. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ford ES, Puronen CE, Sereti I. Immunopathogenesis of asymptomatic chronic HIV infection: The calm before the storm. Curr Opin HIV AIDS. 2009;4:206–14. doi: 10.1097/COH.0b013e328329c68c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mendiratta S, Vajpayee M, Mojumdar K, Chauhan NK, Sreenivas V. Polyfunctional analysis of Gag and Nef specific CD8+ T-cell responses in HIV-1 infected Indian individuals. Vaccine. 2011;29:1150–8. doi: 10.1016/j.vaccine.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 112.Lopez M, Soriano V, Rallon N, Cascajero A, Gonzalez-Lahoz J, Benito JM. Suppression of viral replication with highly active antiretroviral therapy has no impact on the functional profile of HIV-specific CD8(+) T cells. Eur J Immunol. 2008;38:1548–58. doi: 10.1002/eji.200738054. [DOI] [PubMed] [Google Scholar]
- 113.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–13. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Frebel H, Richter K, Oxenius A. How chronic viral infections impact on antigen-specific T-cell responses. Eur J Immunol. 2010;40:654–63. doi: 10.1002/eji.200940102. [DOI] [PubMed] [Google Scholar]
- 115.Kaufmann DE, Walker BD. Programmed death-1 as a factor in immune exhaustion and activation in HIV infection. Curr Opin HIV AIDS. 2008;3:362–7. doi: 10.1097/COH.0b013e3282f9ae8b. [DOI] [PubMed] [Google Scholar]
- 116.Khaitan A, Unutmaz D. Revisiting immune exhaustion during HIV infection. Curr HIV/AIDS Rep. 2011;8:4–11. doi: 10.1007/s11904-010-0066-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 118.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–4. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 119.Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med. 2010;16:1147–51. doi: 10.1038/nm.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sauce D, Almeida JR, Larsen M, Haro L, Autran B, Freeman GJ, et al. PD-1 expression on human CD8 T cells depends on both state of differentiation and activation status. AIDS. 2007;21:2005–13. doi: 10.1097/QAD.0b013e3282eee548. [DOI] [PubMed] [Google Scholar]
- 121.Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008;205:2763–79. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA. 2010;107:14733–8. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci USA. 2003;100:4712–7. doi: 10.1073/pnas.0830997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zaunders JJ, Ip S, Munier ML, Kaufmann DE, Suzuki K, Brereton C, et al. Infection of CD127_ (interleukin-7 receptor_) CD4_ cells and over expression of CTLA-4 are linked to loss of antigen-specific CD4 T cells during primary human immunodeficiency virus type 1 infection. J Virol. 2006;80:10162–72. doi: 10.1128/JVI.00249-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Richter K, Agnellini P, Oxenius A. On the role of the inhibitory receptor LAG-3 in acute and chronic LCMV infection. Int Immunol. 2010;22:13–23. doi: 10.1093/intimm/dxp107. [DOI] [PubMed] [Google Scholar]
- 126.Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–9. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83:3719–33. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y, El-Far M, et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat Med. 2010;16:452–9. doi: 10.1038/nm.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]