

Abstract
Significance: Inflammation is a complex biological process that represents the body's response to infection and/or injury. Endogenous molecules that induce inflammation are called death- or damage-associated molecular patterns (DAMPs). Among cellular constituents with DAMP activity, nuclear molecules can stimulate pattern recognition receptors, including toll-like receptors (TLRs). Current research is elucidating the translocation of nuclear molecules during cell death and identifying novel anti-inflammatory approaches to block their DAMP activity. Recent Advances: High mobility group box protein 1 (HMGB1), a non-histone nuclear protein, can translocate from cells during immune cell activation and cell death. Depending on redox state, HMGB1 can interact with TLR4 although it can bind to molecules such as cytokines to trigger other receptors. DNA and histones, which are bound together in the nucleus, also have important immunological activity. For DNA, DAMP activity may vary depending upon the binding to molecules that affect cell entry and intracellular location. The role of nuclear molecules in disease has been established in animal models using antibodies as inhibitors. Critical Issues: Key issues about the DAMP activity of nuclear molecules relate to (i) the impact on function of biochemical modifications such as redox state and post-translational modification, and (ii) the composition and properties of complexes that nuclear molecules may form with other blood components to affect immunological activity. Future Directions: With the recognition of the immunological activity of the products of dead cells, future studies will define the diversity and properties of nuclear molecules in the extracellular space and develop strategies to block their activity during inflammation. Antioxid. Redox Signal. 20, 1117–1125.
Introduction
Inflammation is a complex biological process that represents the body's response to immunological danger whether arising from infection or injury. This process can occur locally or systemically and result from exogenous or endogenous events that ultimately can culminate in cell death. As shown in in vitro and in vivo experiments, inflammation involves diverse mechanisms to eradicate the proximate threat, restore homeostasis, and promote tissue repair. Repair is frequently compromised, however, with organ dysfunction from scarring and fibrosis, the consequence of a process otherwise successful in eradicating infection or curtailing tissue destruction from trauma.
In a multitude of diseases throughout medicine, improvement in clinical outcomes depends on better understanding of the mechanisms of inflammation and the development of new strategies to modulate this process. While initial studies on inflammation focused on exogenous triggers, recent research has demonstrated that endogenous molecules produced by damaged or dying cells themselves are important mediators. Importantly, as these studies show, endogenous molecules can translocate from the inside to the outside of cells where they can drive the inflammation. This review will provide a perspective on a group of endogenous mediators of inflammation that emanate from the cell nucleus and have dual function depending on their location.
The Immune Response to Infection
As a group, exogenous molecules that trigger inflammation have been termed pathogen-associated molecular patterns (PAMPs). Despite the use of pathogen in the name, these molecules occur generally on bacterial, viral, and fungal organisms and not just pathogenic species. PAMPs differ in chemical structure although, as foreign patterns, they are structurally distinct from endogenous molecules. PAMPs stimulate receptors called pattern recognition receptors (PRRs) of which the toll-like receptors (TLRs) have the most prominent role (13, 31). TLRs can occur in the immune cells on the cell membrane or internal sites such as the endosomes. The cellular location of any TLR likely reflects a balance of two factors: the place where binding to a PAMP occurs and the chance for inadvertent triggering by an endogenous molecule mimicking a PAMP in its receptor binding (5, 6).
Whereas most TLRs, including TLR4, the prototype TLR and the receptor for lipopolysaccharide (LPS), occur on the membrane, TLRs recognizing nucleic acids occupy sites in the endosome; in this locale, TLR3 recognizes double-stranded RNA; TLR7, single-stranded RNA; and TLR9, double-stranded DNA. While this location could suggest that these TLRs respond to intracellular infection, an intracellular location may also minimize exposure to extracellular nucleic acids acting as ligands (5, 6). The interaction of a PAMP with its PRR leads to rapid stimulation of cells such as macrophages, dendritic cells, and B cells and induction of pro-inflammatory molecules such as interleukin (IL)-β, tumor necrosis factor (TNF)-α, and interferon (IFN)-γ.
The Immune Response to Cell Damage and Death
While inflammation occurs abundantly during infection, nevertheless, this process can occur in a host of settings that are otherwise sterile. These settings include physical and chemical injury (e.g., burns or drug toxicity) as well as autoinflammatory diseases (e.g., gout, familial Mediterranean fever). In many instances, the intensity of sterile inflammation rivals that of infection, suggesting the existence of molecules as potent as PAMPs in their immune activity. Furthermore, once inflammation from infection starts, subsequent events in pathogenesis may reflect other processes (e.g., ischemia, circulatory collapse) that are in essence sterile (11, 18, 35).
Endogenous molecules that mediate inflammation are of two kinds. Cytokines are specific products of immune cells that act at low concentrations and trigger specific receptors. The other endogenous mediators of inflammation are cellular molecules that can be released from essentially any cell during cell injury or death. These molecules act at higher concentrations and may involve receptors of other specificities. Endogenous molecules that can trigger inflammation go by a variety of different names, including danger molecules, alarmins, or death- or damage-associated molecular patterns (DAMPs). The term DAMP is particularly popular because of the parallelism with PAMP (9, 43, 48).
While DAMPs are ordinary host components present ubiquitously, they can, nevertheless, acquire immune activity. This activity results from a change in concentration, location, conformation, or biochemical properties and results from various mechanisms. These mechanisms include translocation from the inside to the outside of the cell; cleavage, post-translational modification; redox reactions; or reassembly into a complex with new partners. DAMPs encompass both small (e.g., uric acid and ATP) and large molecules (e.g., DNA). The properties of a series of nuclear DAMPs will next be considered, emphasizing that their immune activity, in addition to their capacity for translocation, may result inherent tendency to associate with other molecules and form both intra- and extracellular complexes (43).
High Mobility Group Box Protein 1
High mobility group box protein 1 (HMGB1) is a non-nuclear histone that consists of a 215 amino acid protein that is comprised of two DNA-binding domains called the A box and B box as well as mobile C terminal tailed (3, 17). This protein was originally characterized as an architectural element that regulates chromatin structure and transcription. HMGB1 has preference for unusual DNA structures such as bends, suggesting that it acts to alter the conformation of DNA to facilitate other interactions (56). While HMB1 is a nuclear component, it also exists in the cytoplasm of nucleated cells and occurs prominently in platelets where it was originally identified as amphoterin (45).
As shown in seminal experiments by Wang et al., HMGB1 has as a dual function and, once outside the cell, can exert powerful immune activity such as cytokine stimulation and serve as the prototype alarmin (60). This translocation can be induced by LPS and, indeed, HMGB1 serves an important mediator of the late effects of LPS. Following immune cell stimulation, HMGB1 can transit from the nucleus, through the cytoplasm and into vesicles for release by a non-classical secretory mechanism.
HMGB1 translocation occurs in macrophages and dendritic cells among other cell types and involves post-translational modification, including acetylation, which alters the intracellular location of HMGB1 (10, 24, 65). Levels of HMGB1 rise in settings such as septic shock in both humans and animal models, with agents that bind to HMGB1 or inhibit its action (i.e., anti-HMGB1 antibodies, A box constructs) attenuating disease manifestations in experimental models. The delayed expression of HMGB1 compared to cytokines provides an extended time frame for therapeutic intervention (3, 17).
In addition to immune activation, HMGB1 can translocate during cell death, with original studies suggesting an important difference between apoptotic and necrotic cell death in this process (47, 49). While cell death is varied and complicated, it can be simplified for experimental models into apoptosis and necrosis. Apoptosis is a form of programmed cell death mediated by enzymes that modify and rearrange cell contents to promote safe removal by phagocytosis and to reduce the chance of inflammation by release of internal constituents. Apoptosis is considered to be immunologically neutral or immunosuppressive. In contrast, necrosis, a sudden and explosive form of cell death induced by physical or chemical trauma, is considered to be pro-inflammatory because of the robust release of intracellular contents (i.e., danger molecules).
As shown in in vitro cell systems, the translocation of HMGB1 can vary depending on the mechanism of cell death. With necrosis of cells induced by treatments like freeze-thaw, HMGB1 rapidly exits cell (47, 49). This translocation occurs readily since HMGB1 is not tightly bound to chromatin and therefore can diffuse away from the nucleus once permeability barriers break down. In contrast, these studies indicated that HMGB1 remains intracellular during apoptosis, reflecting post-translational modifications of HMGB1 (or a chromatin structure with which it interacts) that increases nuclear retention. These findings have suggested that the location of HMGB1 can determine the immune activity of a dead and dying cell, with the “in-out” distinction acting as an “on-off” switch.
While this model has been very appealing, subsequent studies have indicated the need for modification. Thus, as shown in in vitro systems with cell lines, HMGB1 can translocate to the extracellular space during apoptosis as well as necrosis (7). In these experiments, the expression of HMGB1 in the medium increased with the extent of cell death, with significant levels present during a stage termed late apoptosis or secondary necrosis; at this point in time, permeability barriers of the cell have been breached (Fig. 1). These findings are notable since prior increases in HMGB1 interaction with chromatin would have been expected to retain HMGB1 in an inter-nuclear location even during late apoptosis or secondary necrosis. These findings could suggest that events in late apoptosis can modify the chromatin binding of HMGB1 to allow its dissociation and diffusion away from the nucleus.
FIG. 1.

High mobility group box protein 1 (HMGB1) translocation during cell death. As illustrated in the figure, HMGB1 can leave dead and dying cells during both apoptosis and necrosis. During necrosis, HMGB1 exits cells by a passive process once permeability barriers of the nuclear and cytoplasmic membranes break down. Depending on the induction of necrosis, HMGB1 translocation can be very rapid. In contrast, during apoptosis, HMGB1 translocation is more gradual and occurs primarily during a stage of cell death that has been called late apoptosis or secondary necrosis; during early apoptosis, HMGB1 can show increased interaction with chromatin causing cellular retention during this phase. In addition to kinetics, HMGB1 translocation during apoptosis and necrosis differs in the redox state of the molecule.
Studies on HMGB1 release during necrosis in vitro also have indicated nuances that could affect the “in-out, on-off” model. These studies compared the extracellular release of HMGB1 from Jurkat cells subjected to various treatments commonly used to induce experimental necrosis: freeze-thaw, heat, ethanol, and hydrogen peroxide (8). Assessing the presence of extracellular HMGB1 by western blotting, these studies showed major differences in the kinetics of HMGB1 release from necrotic cells and its extent depending on inducing treatment. Whereas freeze–thaw caused a rapid and abundant release of HMGB1, levels were much less and slower to accumulate with heat and hydrogen peroxide. Furthermore, levels of HMGB1 did not appear to rise significantly with necrosis by ethanol (Fig. 2). Together with results on the behavior of HMGB1 during apoptosis, these results suggest that translocation of HMGB1 may be variable and that the immune activity of dead or dying cells may not simply depend on the location of HMGB1.
FIG. 2.
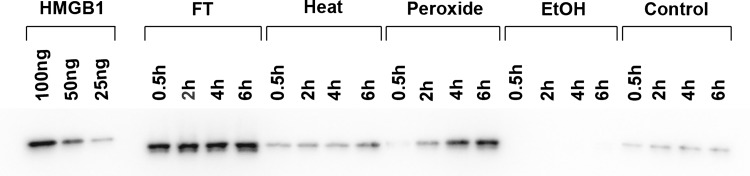
The kinetics of HMGB1 release by necrotic cells. In these experiments, Jurkat T cells were cultured in serum-free medium and induced to undergo necrosis by the following treatments: freeze–thaw, heat, hydrogen peroxide (H2O2), ethanol, or no treatment (controls). After treatment, cells were incubated at 37°C for the times indicated. Culture media were concentrated and then examined by western blotting using a mouse monoclonal anti-HMGB1 antibody followed by a horseradish peroxidase-conjugated goat anti-mouse reagent and detection by enhanced chemiluminescence. Controls for HMGB1 detection (100, 50, and 25 ng of recombinant hHMGB1-histidine tagged protein) were also analyzed. Figure is a digital image of a representative blot. As these data indicate, the release of HMGB1 differs significantly depending on the treatment to cause necrosis. Reproduced with permission from Beyer et al. (8).
Further complicating the story of HMGB1's role as an endogenous mediator of inflammation have been observations on its intrinsic immune activity. While initial studies suggested that HMGB1 could act to stimulate inflammation, subsequent experiments using more highly purified material showed meager activity (2, 46, 66). These observations were confusing and required extensive biochemical investigation to explain this discrepancy. Ultimately, an explanation for the varying activity of HMGB1 was found. Thus, subsequent studies demonstrated that the redox state of HMGB1 is a strong determinant of its immune activity, with the use of reducing agents during isolation and purification affecting HMGB1's activity because of effects on key cytsteine residues. Thus, active HMGB1 requires a C106 thiol and a C23-C45 disulfide. The status of the sulfydryl groups may also explain the differences between the immune activity of HMGB1 during apoptosis and necrosis, with HMGB1 from apoptosis undergoing oxidation by reactive oxygen species (ROS) (33, 59, 63, 64). Table 1 summarizes mechanisms of HMGB1 translocation, while Figure 3 illustrates the differences in HMGB1 structure during apoptosis and necrosis.
Table 1.
Extracellular Translocation of HMGB1
| Induction during activation by TLRs, cytokines, and nitric oxide |
| Secretion by activated cells following post-translational modification |
| Passive release during primary and secondary necrosis |
| Attachment to other nuclear molecules depending on mechanism of cell release |
| Redox changes depending on mechanism of cell release |
TLR, toll-like receptor; HMGB1, high mobility group box protein 1.
FIG. 3.

The relationship of HMGB1 structure to immune activity. The figure indicates the different forms of HMGB1 based on the redox status of sulfhydryl groups at positions 23, 45, and 106. Depending on the formation of a disulfide bond between residues 23 and 45 and a sulfhydryl at position 106, HMGB1 can induce cytokine production. Either an all-thiol structure or terminal oxidation of these residues prevents activity. Reproduced with permission in a modified form from Venereau et al. (59).
With recognition of the importance of redox status, subsequent studies demonstrated that HMGB1 can directly interact with TLR4, in a process dependent on the state of the key sulfydryl groups. While HMGB1 can stimulate responses via TLR4, other studies have demonstrated HMGB1 activation via TLR2 and RAGE (3, 17, 63). It is not yet known whether redox status as well as other post-translational modifications can affect the interaction of the HMGB1 with these other receptors. Since multiple forms of HMGB1 may exist depending on post-translational modification and redox status, a wide array of immunostimulatory interactions are possible. Whether any of the modified forms of HMGB1 are inhibitory is not known although worth exploring.
While HMGB1 can act alone, it can also act in concert with other molecules, including other DAMPs, PAMPs, as well as cytokines, triggering the receptors of the partner molecule (20, 21, 51). Thus, HMGB1 can partner with DNA, LPS, and IL-1β- and IFN-γ among other molecules to create novel structures that can drive innate immunity. The capacity for intermolecular interaction likely reflects the structure of HMGB1 and its content of amino acid sequences, which facilitate binding to structures (protein or nucleic acid) that differ in charge or conformation. This capacity may follow directly from the intranuclear role of HMGB1 whose interactions with chromatin and DNA are varied. Thus, it is possible that HMGB1 may frequently (or always) act in concert with another molecule to induce inflammation. This mechanism is especially difficult to assess in the whole animal since, in experiments in which HMGB1 is administered to promote or accelerate disease, HMGB1 may bind to another molecule present (e.g., a cytokine) in the blood. As such, a complex rather than isolated HMGB1 may be the relevant immunostimulatory moiety (43). Table 2 lists immune properties of HMGB1.
Table 2.
Role of HMGB1 in Inflammation and Repair
| Induce cytokine production |
| Enhance chemotaxis |
| Upregulate adhesion molecule expression |
| Induce dendritic cell maturation and migration |
| Promote angiogenesis |
DNA and Histones
Two other classes of nuclear molecules can serve as endogenous mediators of inflammation following translocation. Thus, both DNA and histones can display immunological activity that may contribute to the pathogenesis of immune-mediated disease. Considering the activity of these molecules separately, however, may be misleading since, in mammalian cells, DNA and histones are intimately and tightly associated in the form of the nucleosome. In the nucleosome, a length of DNA of approximately 147 nucleotides is wrapped around a core octamer comprised of two molecules each of H2A, H2B, H3, and H4. A stretch of linker DNA spans the distance between nucleosomes. Like HMGB1, H1 is not strongly adherent to chromatin although it can interact with this material (34, 58).
As shown with immunochemical assays of nucleosomes, DNA, or histones, levels of these molecules are elevated in the blood in many conditions associated with cell death such as sepsis, autoimmunity, and malignancy (19, 36, 41, 44, 50). Furthermore, levels of these molecules are elevated in animal models of sepsis or liver cell injury, whether apoptosis induced by treatment with anti-Fas or necrosis induced by treatment with carbon tetrachloride or acetaminophen (55). The measurement of other markers such as caspase 3, however, can help identify the form of death since caspase activation is a feature of apoptosis but not necrosis. Like DNA, levels of lactate dehydrogenase (LDH), a commonly used marker of cell death, are increased irrespective of the mechanism of hepatic injury although, interestingly, with apoptosis, the release of LDH into the blood differs from that of DNA or caspase 3, which are similar (Fig. 4). Together, these findings suggest that, in in vivo settings, nuclear molecule release from apoptotic and necrotic cells occurs similarly and that both forms of cell death are associated with abundant translocation of nuclear as well as cytoplasmic contents into the blood.
FIG. 4.

The expression of extracellular DNA during apoptosis induced by anti-Fas. In this experiment, normal BALB/c mice were treated with various doses of a monoclonal anti-Fas antibody (1, 3 or 5 μgs per animal as indicated in the box in panel A). At various times afterward, blood was obtained for analysis of DNA (by PicoGreen) or lactate dehydrogenase (LDH) and caspase 3 by enzymatic assays. (A) Shows values for DNA; (B) for LDH, and (C) for caspase. As these data indicate, DNA, along with LDH, appears in the blood within hours of treatment, with elevated levels of caspase 3 indicative of apoptosis. Reproduced with permission from Tran et al. (55).
The similarity in release of DNA into blood following cell death is also evident in experiments in which cells induced to undergo apoptosis or necrosis in vitro were administered to normal mice (25). In this system, DNA appeared in the blood within about 5–6 h, with similar kinetics irrespective of whether the administered cells were necrotic or apoptotic. As subsequent studies showed, the expression of DNA in the blood is not the simple consequence of a burden of dead and dying cells but rather requires the presence of macrophages and can be modulated by corticosteroids (25, 27). In vitro studies support the role of macrophages in modulating the release of nucleosomal molecules from dead and dying cells (12). In this regard, the release of DNA and histones from cells may differ from that of HMGB1 and occur only with dead and dying cells. While treatment of animals with a TLR agonist-like LPS can lead to the presence of extracellular DNA and nucleosomes in the blood, this stimulation can also produce apoptosis, which is the more likely source of translocated nuclear molecules (26).
Because of the role of DNA as a target antigen in systemic lupus erythematosus, the immune properties of mammalian DNA have been extensively explored in vivo and in vitro (4). These studies, which were conducted over several decades, showed that mammalian DNA alone lacks significant immune activity and may have even inhibitory activity. This situation contrasts sharply with that of bacterial DNA, which is a potent immune activator, triggering responses via TLR9. As now well recognized, mammalian DNA and bacterial DNA differ in structure, with bacterial DNA containing unmethylated CpG motifs. As such, bacterial DNA presents a structural pattern called the CpG motif, which allows bacterial DNA to act as PAMP by triggering TLR9; other DNA sequences and structures, including backbone modifications, may also influence immunostimulatory activity. Mitochondrial DNA, which is structurally related to bacterial DNA, may activity similarly, serving as both a PAMP and a DAMP (65).
While mammalian DNA alone is unable to induce responses in vitro when cultured with cells by itself, it can, nevertheless, induce responses when introduced into cells by agents alter entry into cells and intracellular location. These agents include anti-DNA antibodies; transfection agents such as lipofectin; and proteins as such as LL37 and HMGB1 (14, 28, 37, 38, 54, 57). As a group, these agents alter the pattern of DNA uptake into cells and therefore the access to internal nucleic acid sensors such as DAI (32, 52, 53). Figure 5 illustrates this mechanism and how protein binding can promote an outside to inside translocation and access to receptors that can be either cytoplasmic or endosomal. In a sense, mammalian nuclear DNA is an incomplete DAMP although it can acquire activity by its interaction with another molecule to facilitate and channel cellular uptake. Importantly, in some of these combinations (e.g., antibody and DNA), neither partner alone has activity, with complexation essential for generating a pro-inflammatory structure.
FIG. 5.

The role of proteins in the stimulation of immune cell activation by extracellular DNA. This figure illustrates the role of proteins in the formation of complexes that can promote translocation of DNA from the outside to the inside of immune cells. Once inside an immune cell (e.g., macrophage, plasmacytoid dendritic cell), DNA can stimulate endosomal DNA sensors such as toll-like receptor 9 (TLR9) or cytoplasmic non-TLR DNA sensors such as DAI. In this illustration, DNA encounters TLR9 inside the endosome in a process that can be inhibited by anti-malarial agents. Among cytokines induced, type 1 interferon is an important product of plasmacytoid dendritic cells.
The role of DNA in immune responses in vivo in settings of cell injury is further established by the effects of agents such as TLR9 inhibitors and DNA binding polymers (23, 39). Furthermore, effects of TLR9 knockouts on experimental models support the role of DNA in pathogenesis although these experiments do not allow interpretation of whether DNA acts alone or as part of a complex. Table 3 summarizes immune properties of DNA.
Table 3.
Determinants of DNA's Immune Activity
| Sequence |
| Base methylation |
| Backbone structure |
| Binding of proteins |
| Uptake into cells |
| Access to TLR and non-TLR nucleic acid sensors |
In contrast to studies with DNA, studies indicate that histones have immune activity that may not require intentional addition of another molecule to form an immunoactive complex (1, 22, 61, 62). In animal models, histones can contribute to conditions such as liver and kidney injury as well as sepsis. Since DNA may be present in these settings to form complexes, nevertheless, in several experimental models, histones alone and outside the confine of a nucleosome have activity, whereas DNA does not. Interestingly, antibodies to histones can block disease progression in animal models such as sepsis. These findings suggest that, like HMGB1, histones can be a target of immunotherapy. This type of experiment has not been performed with anti-DNA antibodies, making it unclear whether DNA and histone components of nucleosomes differ in their potential utility as targets of biological therapy. In these models, TLR2, TLR4, and TLR9 all play a role as in the situation with HMGB1. This pattern of receptor utilization could reflect either a promiscuous interaction of histones with TLRs or the role of any attached molecules (e.g., DNA or HMGB1).
An important issue that emerges from these studies concerns the physical–chemical form of nuclear molecules in the blood and the extent to which they exist as biochemically distinct species or complexes. Few studies have explored the state of circulating nuclear molecules although their close relationship is likely for the following reasons: (i) DNA, histones, and HMGB1 are all present in chromatin and have strong interactions; (ii) in human diseases, levels of DNA, histones, nucleosomes, and HMGB1 are elevated in the same conditions; (iii) in in vitro and in vivo disease models, levels of DNA, histones, nucleosomes, and HMGB1 also show similar elevations and kinetics of expression; (iv) the immune activity of DNA and HMGB1 are enhanced by the formation of complexes; (v) antibodies to histones and HMGB1 are effective immunomodulators in animal models of immune-mediated disease involving cell death; and (vi) animals models involving cell death can be attenuated by either TLR9 blockers or the presence of a TLR9 knockout. The latter finding is notable in view of data that DNA alone lacks immunological activity.
Perspective on the Role of Nuclear Molecules in Inflammation
Together, these considerations suggest that further elucidation of the role of nuclear molecules in inflammation would be enhanced by further biochemistry to determine their presence as free versus complexed molecules. Since the immune activity of nuclear molecules may be mutable, determining the partners should provide a better perspective of the ultimate biological activity. These partners could be other DAMPs, PAMPs, or serum proteins, including cytokines, each of which could influence the activity of the molecule, alone or in complexes (Fig. 6). In this regard, in experiments involving the administration of a nuclear molecule (e.g., HMGB1, histones) to enhance disease, the possible formation of a complex with another molecule should be considered, especially nuclear molecules that circulate in the blood as components of the blood nucleome; in this context, the nucleome represents the ensemble of nuclear molecules, both protein and nucleic acid, that are present in the blood. Given the avidity of these interactions and the concentrations of the reactants, complex formation is not unlikely (40, 42).
FIG. 6.

The generation of extracellular complexes with immune activity. As illustrated in this figure, following release of nuclear molecules during cell death, extracellular complexes with immunologically active components can form. These complexes can involve nuclear molecules in concert with each of other as well as with cytokines and pathogen-associated molecular patterns. The existence of multi-component complexes as shown in the circle is hypothetical but consistent with current data. Reproduced with permission from Pisetsky (43). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Another area of investigation concerns the mechanism for nuclear molecule release during different forms of cell death. While the division of cell death into apoptosis and necrosis is a convenient starting point, it is an oversimplification. Cell death, as it occurs in vivo, is far from simple and depends on the metabolic state of the cells, ambient conditions such as hypoxia and acidosis, and the poise of the immune system. The classification of death as apoptosis, necrosis, pyroptosis, and NETosis among others highlights the diversity of death forms that can be described on the basis of in vitro studies (15, 16, 30). In the most simplified form, apoptosis is programmed cell death; necrosis, accidental cell death; pyroptosis, pro-inflammatory programmed cell death; and NETosis, neutrophil cell death with extrusion of extracellular DNA or NETs.
Finding and categorizing the counterparts of these death forms in vivo represents a great challenge, with the visualization of apoptosis, for example, very uncommon in pathological specimens. While difficultly in visualizing apoptotic cells in the tissue may reflect their transient presence because of rapid clearance, another explanation is that apoptosis as defined in vitro does not occur to any great extent in vivo. The ambient conditions may push a cell doomed to die from apoptosis into necrosis, for example. Certainly, during infection, pyroptosis may be a prominent death since it leads to inflammation; the study of nuclear molecule release during this form is just beginning.
Finally, studies on the expression of danger molecules have used terms such as cell injury, cell stress, or cell damage as if these were discrete states of the cell that are different from cell death. While an injured, stressed, or damaged cell can retreat from the brink of death and recover, precise morphological or biochemical definitions of these cell states are often lacking. It is unknown whether there are systems in which cells are poised sufficiently between a vulnerable state (e.g., stress) and death in a way that can be studied experimentally. In this regard, intense stimulation by TLR ligands can drive cells to apoptosis and other forms of death, blurring distinction as well as between activation and death (29).
Conclusions
As these considerations indicate, nuclear molecules may serve as important mediators of inflammation in settings of immunological danger resulting from infection, cell injury, and death as well as their intersection. Future studies will illuminate further the role of these molecules in various diseases as well as the dynamic cell processes that determine their location and structure. Importantly, these studies should determine whether nuclear molecules can be targets of novel biological therapy to improve the course of a wide range of immune-mediated diseases that affect patients throughout the spectrum of medicine.
Abbreviations Used
- DAMP
death- or damage-associated molecular pattern
- HMGB1
high mobility group box protein 1
- IFN
interferon
- IL
interleukin
- LDH
lactate dehydrogenase
- PAMPs
pathogen-associated molecular patterns
- PRRs
pattern recognition receptors
- TLRs
toll-like receptors
Acknowledgments
These studies were supported by a VA Merit review grant, NIH Grant AI056363, and an Alliance for Lupus Research Grant. The expert assistance of Dr. Anirudh Ullal in the preparation of figures is gratefully acknowledged.
References
- 1.Allam R, Scherbaum CR, Darispudi MN, Mulay SR, Hagele H, Lichtnekert J, Hagemann JH, Rupanagudi KV, Ryu M, Schwarzwenberger C, Hohenstein B, Hugo C, Uhl B, Reichel CA, Krombach F, Monestier M, Liapis H, Moreth K, Schaefer L, and Anders H-J. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol 23: 1–14, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersson U, Wang H, Palmblad K, Aveberger A-C, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, and Tracey KJ. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 192: 565–570, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersson U. and Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 29: 139–162, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ardoin SP. and Pisetsky DS. Developments in the scientific understanding of lupus. Arthritis Res Ther 10: 218, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barton GM, Kagan JC, and Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol 7: 49–56, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Barton GM. and Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol 9: 535–542, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell CW, Jiang W, Reich CF, 3rd, and Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol 291: C1318–C1325, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Beyer C, Stearns NA, Giessl A, Distler JH, Schett G, and Pisetsky D. The extracellular release of DNA and HMGB1 from Jurkat T cells during in vitro necrotic cell death. Innate Immun 18: 727–737, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81: 1–5, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, and Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 22: 5551–5560, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen GY. and Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10: 826–837, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi JJ, Reich CF, 3rd, and Pisetsky DS. The role of macrophages in the in vitro generation of extracellular DNA from apoptotic and necrotic cells. Immunology 115: 55–62, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coll RC. and O'Neill LA. New insights into the regulation of signaling by toll-like receptors and nod-like receptors. J Innate Immun 2: 406–421, 2010 [DOI] [PubMed] [Google Scholar]
- 14.Dombrowski Y, Peric M, Koglin S, Kammerbauer C, Gob C, Anz D, Simanski M, Glaser R, Harder J, Hornung V, Gallo RL, Ruzicka T, Besch R, and Schauber J. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med 3: 82ra38, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fink SL. and Cookson BT. Apoptosis, pyroptosis and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73: 1907–1916, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galluzzi L. and Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell 135: 1161–1163, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Harris HE, Andersson U, and Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol 8: 195–202, 2012 [DOI] [PubMed] [Google Scholar]
- 18.Hirsiger S, Simmens H-P, Werner CML, Wanner GA, and Rittirsch D. Danger signals activating the immune response after trauma. Mediators Inflamm 2012: 315941, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holdenrieder S. and Stieber P. Clinical use of circulating nucleosomes. Crit Rev Clin Lab Sci 46: 1–24, 2009 [DOI] [PubMed] [Google Scholar]
- 20.Hreggvidsdottir HS, Ostberg T, Wähämaa H, Schierbeck H, Aveberger AC, Klevenvall L, Palmblad K, Ottosson L, Andersson U, and Harris HE. The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals. J Leukoc Biol 86: 655–662, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Hreggvidsdottir HS, Lundberg AM, Aveberger AC, Klevenvall L, Andersson U, and Harris HE. HMGB1-partner molecule complexes enhance cytokine production by signaling through the partner molecule receptor. Mol Med 18: 224–230, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, Liao X, Billiar T, Xu J, Esmon CT, and Tsung A. Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll-like receptor 9 in mice. Hepatology 54: 999–1008, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, and Mehal WZ. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest 119: 305–314, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ito I, Fukazawa J. and Yoshia M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J Biol Chem 282: 16336–16344, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Jiang N, Reich CF, 3rd, and Pisetsky DS. Role of macrophages in the generation of circulating blood nucleosomes from dead and dying cells. Blood 102: 2243–2250, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Jiang N, Reich CF, 3rd, Monestier M, and Pisetsky DS. The expression of plasma nucleosomes in mice undergoing in vivo apoptosis. Clin Immunol 106: 139–147, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Jiang N. and Pisetsky DS. The effect of dexamethasone on the generation of plasma DNA from dead and dying cells. Am J Pathol 164: 1751–1759, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang W, Reich CF, 3rd, and Pisetsky DS. Mechanisms of activation of the RAW 264.7 macrophage cell line by transfected mammalian DNA. Cell Immunol 229: 31–40, 2004 [DOI] [PubMed] [Google Scholar]
- 29.Jiang W, Bell CW, and Pisetsky DS. The relationship between apoptosis and high-mobility group protein 1 release from murine macrophages stimulated with lipopolysaccharide or polyinosinic-polycytidylic acid. J Immunol 178: 6495–6503, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Kabbe K. and Saleh M. Cell death in the host response to infection. Cell Death Differ 15: 1339–1349, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Kawal T. and Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34: 637–650, 2011 [DOI] [PubMed] [Google Scholar]
- 32.Kawasaki T, Kawai T, and Akira S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev 243: 61–73, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kazama H, Ricci JE, Herndon HM, Hoppe G, Green DR, and Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29: 21–32, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khorasanizadeh S. The nucleosome: from genomic organization to genomic regulation. Cell 116: 259–272, 2004 [DOI] [PubMed] [Google Scholar]
- 35.Kono H. and Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol 8: 279–289, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lam NYL, Rainer TH, Chan LYS, Joynt GM, and Lo YMD. Time course of early and late changes in plasma DNA in trauma patients. Clin Chem 49: 1286–1291, 2003 [DOI] [PubMed] [Google Scholar]
- 37.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang Y-H, Homey B, Cao W, Wang Y-H, Su B, Nestle FO, Zal T, Mellman I, Schröder J-M, Liu Y-J, and Gilliet M. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449: 564–571, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, Bassett R, Amuro H, Fukuhara S, Ito T, Liu Y-J, and Gilliet M. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med 3: 1–11, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee J, Sohn JW, Zhang Y, Leong KW, Pisetsky D, and Sullenger BA. Nucleic acid-binding polymers as anti-inflammatory agents. Proc Natl Acad Sci U S A 108: 14055–14060, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin T, Sammy F, Yang H, Thundivalappil S, Hellman J, Trace KJ, and Warren HS. Identification of hemopexin as an anti-inflammatory factor that inhibits synergy of hemoglobin with HMGB1 in sterile and infectious inflammation. J Immunol 189: 2017–2022, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lui YYN. and Lo YMD. Circulating DNA in plasma and serum: biology, preanalytical issues and diagnostic applications. Clin Chem Lab Med 40: 962–968, 2002 [DOI] [PubMed] [Google Scholar]
- 42.Pemberton AD, Brown JK, and Inglis NF. Proteomic identification of interactions between histones and plasma proteins: implications for cytoprotection. Proteomics 10: 1484–1493, 2010 [DOI] [PubMed] [Google Scholar]
- 43.Pisetsky D. Cell death in the pathogenesis of immune-mediated diseases: the role of HMGB1 and DAMP-PAMP complexes. Swiss Med Wkly 141: w13256, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pisetsky DS. and Ullal AJ. The blood nucleome in the pathogenesis of SLE. Autoimmun Rev 10: 35–37, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rouhiainen A, Imal S, Rauvala H, and Parkkinen J. Occurrence of amphoterin (HMG1) as an endogenous protein of human platelets that is exported to the cell surface upon platelet activation. Thromb Haemost 84: 1087–1094, 2000 [PubMed] [Google Scholar]
- 46.Rouhiainen A, Tumova S, Valmu L, Kalkkinen N, and Rauvala H. Analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (Amphoterin). J Leukoc Biol 81: 1–10, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, Dumitriu IE, Müller S, Iannacone M, Traversari C, Binachi ME, and Manfredi AA. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep 5: 825–830, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rubartelli A. and Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol 28: 429–436, 2007 [DOI] [PubMed] [Google Scholar]
- 49.Scaffidi P, Mistell T, and Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418: 191–195, 2002 [DOI] [PubMed] [Google Scholar]
- 50.Schwarzenbach H, Hoon DSB, and Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 11: 426–437, 2011 [DOI] [PubMed] [Google Scholar]
- 51.Sha Y, Zmijewski J, Xu Z, and Abraham E. HMGB1 developes enhanced proinflammatory activity by binding to cytokines. J Immunol 180: 2531–2537, 2008 [DOI] [PubMed] [Google Scholar]
- 52.Takaoka A, Wang ZC, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, and Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448: 501–506, 2007 [DOI] [PubMed] [Google Scholar]
- 53.Theofilopoulos AN, Kono DH, Beutler B, and Baccala R. Intracellular nucleic acid sensors and autoimmunity. J Interferon Cytokine Res 31: 867–886, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, and Coyle AJ. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 8: 474–477, 2007 [DOI] [PubMed] [Google Scholar]
- 55.Tran TT, Groben P, and Pisetsky DS. The release of DNA into the plasma of mice following hepatic cell death by apoptosis and necrosis. Biomarkers 13: 184–200, 2008 [DOI] [PubMed] [Google Scholar]
- 56.Ueda T. and Yoshida M. HMGB proteins and transcriptional regulation. Biochim Biophys Acta 1799: 114–118, 2010 [DOI] [PubMed] [Google Scholar]
- 57.Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, Kalden JR, Schett G, Rovere-Querini P, Herrmann M, and Voll RE. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med 205: 3007–3018, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, and Sidow A. Determinants of nucleosome organization in primary human cells. Nature 474: 516–522, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Venereau E, Casalgrandi M, Schiradldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, Shams SS, Yang H, Varani L, Andersson U, Tracey KJ, Bachi A, Uguccioni M, and Bianchi ME. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine disease. J Exp Med 209: 1519–1528, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manoque KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, and Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science 285: 248–251, 1999 [DOI] [PubMed] [Google Scholar]
- 61.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, and Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med 15: 1318–1321, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu J, Zhang X, Monestier M, Esmon NL, and Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol 187: 2626–2631, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang H, Lündback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, Al-Abed Y, Andersson U, Tracey KJ, and Antoine DJ. Redox modification of cysteine residues regulates the cytokine activity of HMGB1. Mol Med 18: 250–259, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Youn JH. and Shin J-S. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol 177: 7889–7897, 2006 [DOI] [PubMed] [Google Scholar]
- 65.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, and Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zimmerman K, Völkel D, Pable S, Lindner T, Kramberger F, Bahrami S, and Scheiflinger F. Native versus recombinant high-mobility group B1 proteins: functional activity in vitro. Inflammation 28: 221–229, 2004 [DOI] [PubMed] [Google Scholar]