

Abstract
Mechanism-based inhibition of cytochrome P450 involves the bioactivation of the drug to a reactive metabolite, which leads to cytochrome inhibition via various mechanisms. This is generally seen in the Phase I of drug metabolism. However, gemfibrozil (hypolipidemic drug) leads to mechanism-based inhibition after generating glucuronide conjugate (gemfibrozil acyl-β-glucuronide) in the Phase II metabolism reaction. The mechanism involves the covalent binding of the benzyl radical (generated from the oxidation of aromatic methyl group in conjugate) to the heme of CYP2C8. This article deals with the development of a 2D QSAR model based on the inhibitory potential of gemfibrozil, its analogues and corresponding glucuronide conjugates in inhibiting the CYP2C8-catalysed amodiaquine N-deethylation. The 2D QSAR model was developed using multiple linear regression analysis in Accelrys Discovery Studio 2.5 and helps in identifying the descriptors, which are actually contributing to the inhibitory potency of the molecules studied. The built model was further validated using leave one out method. The best quantitative structure activity relationship model was selected having a correlation coefficient (r) of 0.814 and cross-validated correlation coefficient (q2) of 0.799. 2D QSAR revealed the importance of volume descriptor (Mor15v), shape descriptor (SP09) and 3D matrix-based descriptor (SpMax_RG) in defining the activity for this series of molecules. It was observed that volume and 3D matrix-based descriptors were crucial in imparting higher potency to gemfibrozil glucuronide conjugate, as compared with other molecules. The results obtained from the present study may be useful in predicting the inhibitory potential (IC50 for CYP2C8 inhibition) of the glucuronide conjugates of new molecules and compare with the standard gemfibrozil acyl-β-glucuronide (in terms of pIC50 values) in early stages of drug discovery and development.
Keywords: Gemfibrozil, glucuronide conjugation, QSAR, 2D descriptors, mechanism-based inhibition, CYP2C8
Cytochrome P450 (CYP450) is a large family of heme-containing enzymes, which catalyse a variety of metabolic reactions, such as oxidation, reduction, hydroxylation and dealkylation[1,2]. The substrates for these reactions include both the exogenous compounds such as drugs, environmental chemicals, natural products as well as endogenous compounds such as hormones. The importance of CYPs in the biotransformation of lipophilic compounds to the more polar metabolites is widely known. The understanding of inhibition of these enzymes also becomes essential, since it leads to several toxicological complications and drug–drug interactions (DDIs)[3,4,5,6,7,8]. The inhibition of CYPs can be reversible or irreversible; Mechanism-based inhibition (MBI) is a type of irreversible inhibition, where the substrate is bioactivated to a more reactive metabolite, which leads to CYP inhibition by different mechanisms[5,6,7,8]. Various mechanisms by which MBI occurs include are (i) coordination with the heme-iron, (ii) alkylation or arylation of heme-porphyrin and (iii) covalent interaction with active site nucleophilic residues. Several examples of drugs leading to MBI include nortriptyline, stiripentol, paroxetine, fluoxetine, ritonavir and verapamil[5,6,7,8].
Gemfibrozil, a hypolipidemic drug, constitutes an interesting example in relation to MBI and DDIs. Gemfibrozil reduces the triglyceride levels and increases high-density lipoprotein (HDL) levels in the patients[9]. Its pharmacological actions are mediated mainly through peroxisome proliferator activated receptor alpha (PPARα) stimulation. It has been reported that the concomitant use of gemfibrozil with other drugs, such as cerivastatin, results in the increased concentration of other drugs and sometimes, fatal cases of rhabdomyolysis may occur. Beckman et al. reported that the plasma concentrations of cerivastatin increased by more than 5 fold in the presence of gemfibrozil, along with a large reduction in the concentrations of the CYP2C8-dependent major metabolite of cerivastatin[9]. Due to certain toxicological complications, the use of gemfibrozil declined. However, this DDI potential of gemfibrozil is currently explored in both in vitro and in vivo studies during drug development.
The glucuronide conjugate of gemfibrozil is believed to be responsible for the toxicological consequences via MBI of cytochrome enzyme (CYP2C8)[10,11,12,13,14]. Mano et al. reported the glucoronidation of gemfibrozil in the hepatocytes by uridine-5′-diphosphoglucuronosyltransferase (UGT) 2B7 enzyme[10]. Ogilvie et al. discussed the inhibitory potential of gemfibrozil glucuronide as the mechanism-based inhibitor of CYP2C8[11]. The mechanism as described by Baer et al. and Jenkins et al. is shown in fig. 1[12,14]. It involves the CYP2C8 catalysed benzylic oxidation of the glucuronide, leading to heme-alkylation and inactivation of CYP2C8. It is interesting to note that gemfibrozil in the parent form does not act as the mechanism-based inhibitor; rather, it acts as a reversible inhibitor of CYP2C9. Also, there were differences reported in the in vitro and in vivo potencies of CYP2C8 and CYP2C9 inhibition by gemfibrozil, which suspects the role of the MBI of CYP2C8 by gemfibrozil 1-O-β-glucuronide (gemfibrozil acyl-β-glucuronide)[10,11,12,13,14].
Fig. 1.
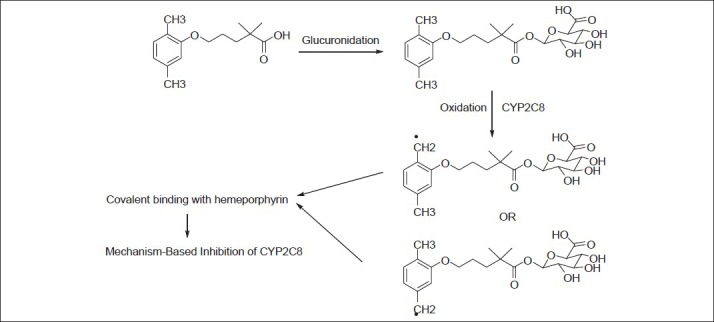
Reaction pathway for conjugation and mechanism-based inhibition.
Reaction pathway for the formation of gemfibrozil glucuronide conjugate, and mechanism-based inhibition of CYP2C8.
Ogilvie et al. and Jenkins et al. recently performed the in vitro studies on conjugates of gemfibrozil and its analogues in causing the inhibition of CYP2C8 catalysed amodiaquine N-deethylation[11,12]. It was observed that only acyl-β-glucuronide of gemfibrozil led to MBI of CYP2C8, whereas few other analogues showed reversible inhibitory properties. The inhibitory potential of all the conjugates and some well-known CYP2C8 inhibitors was reported in terms of IC50 values in micromolar concentration. A higher potency of gemfibrozil glucuronide towards CYP2C8 inhibition was observed in comparison to other analogues and aglycones.
Understanding the specificity of gemfibrozil glucuronide in CYP2C8 inhibition becomes essential, as it would provide information, which may be utilised for the design and development of other CYP inhibitors. Computational methods such as molecular docking and quantitative structure activity studies (QSAR) have been extensively employed to obtain such characteristics features[15,16,17,18]. The importance of in silico methods becomes more indispensable, since the in vitro–in vivo extrapolation (IVIVE) of DDIs caused by MBI is challenging. Because of that, various fatal interactions have remained unrecognised for many years.
In this study, a 2D QSAR model, using the IC50 values of a series of molecules studied by Jenkins et al.[12] was built to account for the important descriptors and parameters, which might play a role in determining the higher potency for gemfibrozil glucuronide conjugate towards inhibition of CYP2C8 (as mechanism-based inhibitor), as compared with other analogues. This also helped in understanding the reported biological data and developing predictive correlations. To the best of our knowledge, this is the first QSAR model study for glucuronide conjugates in relation to their inhibitory potential. This type of study would help in predicting the inhibitory potential of glucuronide conjugates of new molecules, in terms of pIC50 values (CYP2C8 inhibition), and CYP2C8 substrates against the standard acyl-β-glucuronide of gemfibrozil, during the early stages of drug discovery and development.
MATERIALS AND METHODS
QSAR indicates the relationship or correlation between the experimental activity (including inhibitory activity) of a set of compounds and their physicochemical properties using various statistical methods such as multiple linear regression (MLR), partial least squares and others[15,16,17,18]. The biological activity, expressed in IC50 or pIC50 values, is used as the dependent variable, while the descriptor values are used as the independent variables in building a QSAR model. In this study, 2D QSAR model was generated using Accelrys Discovery Studio 2.5[19] on a personal computer with dual core processor and window XP operating system.
Dataset for 2D QSAR model:
The IC50 values for the inhibition of CYP2C8 catalysed amodiaquine N-deethylation by molecules were taken from the published work of Jenkins et al., where the experimental IC50 values were evaluated following incubation of the molecules with NADPH-fortified human liver microsomes. The IC50 values were converted to pIC50 values and were used as the dependent variables for this study. The 3D structures of compounds were sketched using Sybyl 7.1[20]. The built molecules were minimised using Powell method for 10 000 iterations with 0.05 kcal/mol Ε as gradient. Tripos force field and Gasteiger–Huckel charges were implemented during energy minimisation and geometry optimisations. The energy minimised structures were utilised for calculation of 491 descriptors, including steric, topological, surface, thermodynamic, electronic, charge, spatial, such as molecular weight, molecular refractivity, logP, topological index, partial charge, element count, atom count (all atoms), atom count (carbon), atom count (hydrogen), atom count (oxygen), bond count (all bonds), dipole moment (debye), group count (amine), group count (carboxyl), group count (ether), group count (hydroxyl), group count (methyl), ionisation potential (eV), LogP, polarisability, ring count (all rings), solvent accessibility surface area (Å), using E-Dragon[21]. The 3D descriptors calculated take into account the difference between enantiomers in the dataset of molecules.
Selection of training and test set:
The dataset of 29 molecules (fig. 2) was randomly divided into training set (20 compounds) and test set (9 compounds) for MLR models, using pIC50 value as the dependent variable and various 2D descriptors as the independent variables. The structures of training set and test set are tabulated in Tables 1 and 2, respectively, along with the experimental and predicted pIC50 values. The definition of the descriptors utilised in deriving the QSAR equation is given in Table 3. The statistical method utilised in deriving the QSAR equation was MLR method[15,16,17,18,22,23,24,25].
Fig. 2.
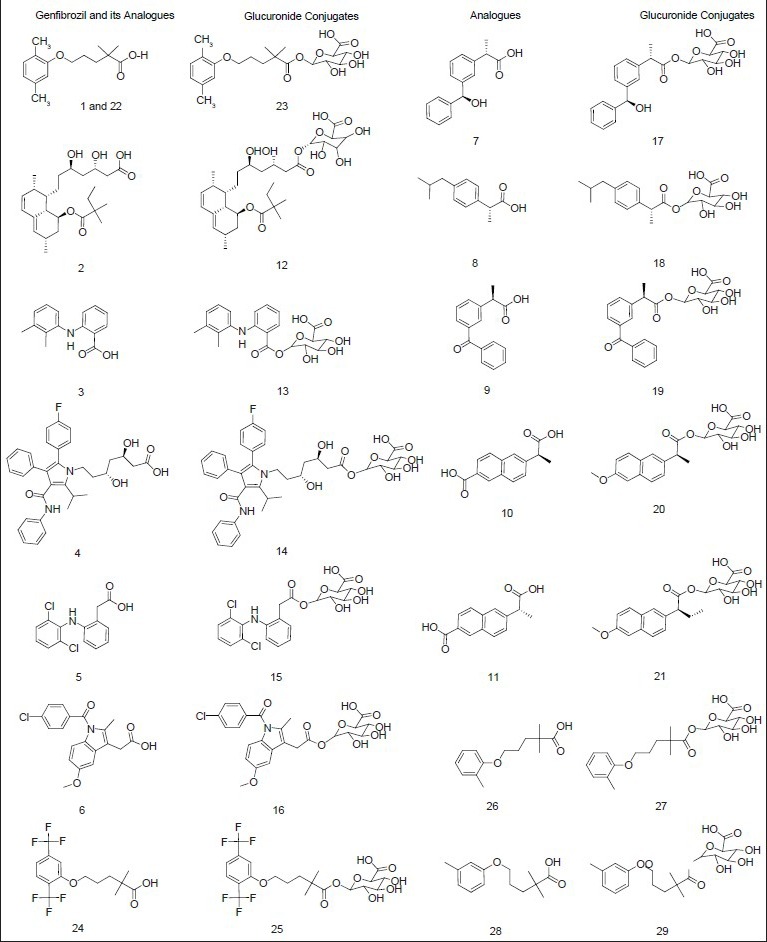
Structures of gemfibrozil, analogues and conjugates.
Structures of gemfibrozil, gemfibrozil analogues and their corresponding glucuronide conjugates and other known CYP2C8 inhibitors. Gemfibrozil (1) at 200 micromolar concentration and (22) at 100 micromolar concentration (Jenkins et al., 2011).
TABLE 1.
STRUCTURES, EXPERIMENTAL AND PREDICTED IC50 VALUES FOR TRAINING SET OF COMPOUNDS
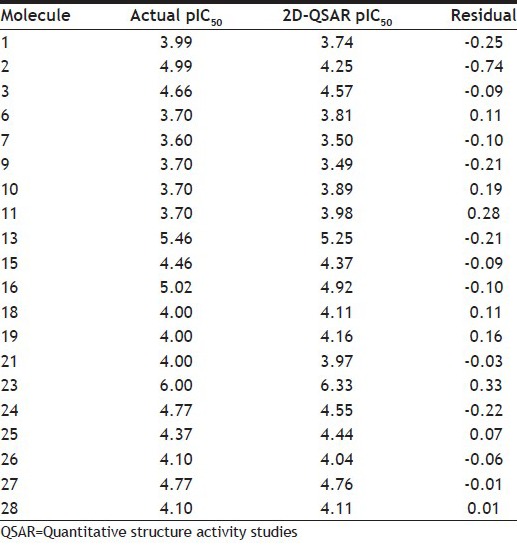
TABLE 2.
STRUCTURES, EXPERIMENTAL AND PREDICTED IC50 VALUES FOR TEST SET OF COMPOUNDS
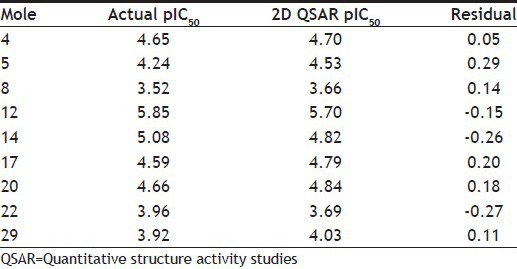
TABLE 3.
DEFINITIONS OF MOLECULAR DESCRIPTORS USED IN THE QSAR MODEL

Descriptor selection:
The selection of potential descriptors from a set of multiple descriptors is a critical step in QSAR analysis. Out of the various 1D, 2D and 3D descriptors calculated by E-Dragon[21], highly correlated descriptors were removed. Pearson's correlation coefficient was calculated for each pair of descriptors as well as between a descriptor and the activity of a molecule. The correlation matrix was, thereafter, built using the above analysis (Table 4). Simultaneously, the contribution of descriptors for the activity is also calculated in the correlation matrix. This strategy afforded a total of 145 descriptors. The resulting sets of 2D properties in terms of descriptors were given as an input for Genetic Function Approximation (GFA)[25] model protocol under QSAR cluster of protocols in DS 2.5. GFA algorithm is considered to be a stochastic optimisation method, which is inspired by evolutionary principles. It investigates many solutions of a given problem simultaneously to explore various regions of the parameter space[22,23,24,25]. In GFA, population size was kept as 100 and the run was set up to 10 000 generations. As the number of compounds in the study is less than 30, maximum equation length was set to 3 with R2 as scoring function. Ten models were generated to select the best set of descriptors, which was tested during MLR analysis.
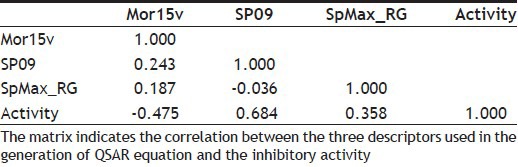
TABLE 4.
THE CORRELATION MATRIX OF THE THREE DESCRIPTORS USED FOR QSAR MODEL
Regression analysis:
The dataset of 29 molecules was subjected to regression analysis using MLR as the method for model development. As mentioned earlier, QSAR model was generated using pIC50 value as the dependent variable and various descriptors selected during GFA analysis (Table 3) as the independent variables. The number of variables utilised in the final equation was three for this model. The selection criteria for the best model covered a range of statistical parameters such as correlation coefficient r, cross-validated correlation coefficient r2cv/q2, predictive squared correlation coefficients r2pred, number of compounds in regression (n) and others.
Multiple linear regression analysis:
This method is widely used for 2D QSAR model development and is utilised in modelling a linear relationship between the dependent variable (pIC50) and independent variables (descriptors). In the fitting of the MLR model (based on least squares), the sum of squares of differences of observed and predicted pIC50 values is minimised. The values of regression coefficient r2 in MLR are estimated using least squares curve fitting method. The relationship is depicted in the form of a straight line from the model that shows best approximation for all the individual data points. In MLR, the conditional mean of pIC50 (dependent variable) is dependent on many descriptors (more than one independent variable) used in the study. The regression Eqn 1 is generally represented as, Y=B1×x1+b2×x2+c....(1), where, Y refers to the dependent variable (pIC50 in this case), b refers to the regression coefficients for the corresponding independent variables (x), c refers to the regression constant.
Validation of QSAR model:
The QSAR model can be validated using leave one out (LOO) method, which is mostly used. Herein, one value from the dependent variable (biological activity in pIC50) is eliminated from the training set, followed by the division of the training dataset into subsets of equal size. The values of dependent variable are determined using the same subsets (utilised in building QSAR model), and this gives the predictive value for the model. The same procedure is thereafter repeated, until each value from the dependent variable has been eliminated once. The q2 is calculated using Eqn 2, q2 = 1−∑(Ypred−Yact)2/∑(Yact−Ymean).....(2), where Ypred, Yact, and Ymean refer to the predicted, actual and mean values of the pIC50, respectively. Σ(Ypred-Yact)2 is the predictive residual error sum of squares (PRESS).
The predicted ability of the final QSAR equation obtained was checked using cross-validated squared correlation coefficient (q2) and predictive squared correlation coefficients (r2pred). The fitness plot for the QSAR model supports the statistical significance of the model.
RESULTS AND DISCUSSION
Several equations (20) were obtained when the data set was subjected to MLR analysis in order to develop a 2D QSAR model. Finally, the best model was derived after a number of permutations and trials using three descriptors as shown in Table 3. The best model was selected on the basis of statistically significant parameters, such as correlation coefficient (r), its square (r2), cross-validation (q2) and others as discussed before. The best 2D QSAR equation obtained using MLR as, pIC50 =−4.2548+3.9824*Mor15v+0.7154*SP09 −2.6936 SpMax_RG, where, n=20training and 9test, r=0.814, r2=0.847, r2adj=0.838, q2=0.799, r2pred=0.718.
In this QSAR study, r2 refers to variance in the biological activity (including inhibitory activity), obtained by the multiplication of the correlation coefficient with 100. Predictive ability of the built QSAR model was evaluated by q2 (LOO method for internal validation), as discussed earlier. Predictive ability was also confirmed for test compounds by external validation (r2pred).
The role of the major descriptors in the QSAR model can be visualised in the correlation matrix, shown in Table 4. It indicates a significant correlation between all the three descriptors. As per the definition of the descriptors, it can be ascertained that the information conveyed by these descriptors is different and distinct. Randic recommends that the descriptors, which are different and distinct in their information content, can be retained for building the QSAR model, even if they show a high correlation. The QSAR equation derived showed a good value of correlation coefficient, r of 0.814, with 84.7% of the variation observed in the inhibitory activity. The validation of the model showed a good internal predictive value (q2) of 0.799.
2D QSAR model generated indicate the positive contribution of Mor15v and SP09 descriptors, and negative contribution of SpMax_RG descriptor. Gemfibrozil glucuronide conjugate contains one hydrophobic phenyl ring and one polar glucuronide moiety, whereas other analogues contain more than two hydrophobic aromatic rings. This tends to increase their lipophilicity, which is indicated by 3D matrix-based descriptor such as SpMax_RG. Therefore, more the value of SpMax_RG, greater negative contribution and lesser would be the inhibitory potential. The descriptors SpMax_RG and Mor15v were found to contribute more significantly to the activity, owing to their higher coefficients in the equation as shown before. This indicates that the volume, shape and 3D matrix-based descriptor of the molecule play a significant role in determining the inhibitory potential of the molecules. Hence, out of the descriptors studied, few play a crucial determining role in imparting higher potency to gemfibrozil glucuronide conjugate, as compared to other molecules.
Fig. 3a shows the plot of experimental v/s predicted pIC50 values for the training set using the obtained QSAR equation (mentioned earlier). The external validation of the test set showed a good predictive ability of 0.718. Table 2 shows the predicted pIC50 values for the test set. Fig. 3b shows the plot of experimental versus predicted pIC50 values for the test set using the obtained QSAR equation. It shall be noted that the different set of descriptors may give different correlation with the inhibitory activity, and this should be taken into consideration while interpreting the derived equation.
Fig. 3.
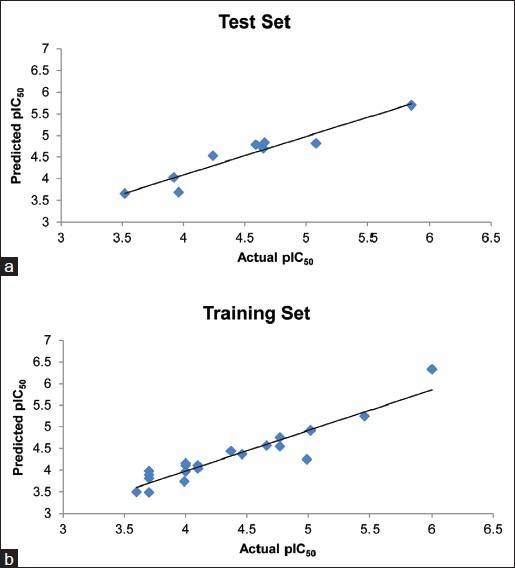
The plots of experimental v/s predicted pIC50.
(a) The plot of experimental v/s predicted pIC50 values for the training set, and (b) test set using the obtained QSAR equation
Glucuronidation plays a pivotal role in the MBI of CYP2C8 by gemfibrozil. 2D QSAR model predicted the contribution of volume, shape and 3D matrix-based descriptors in the inhibitory potential of conjugates of gemfibrozil and its analogues. 2D QSAR studies revealed that Mor15v, SP09 and SpMax_RG were the major contributing descriptors. Mor15v and SP09 contributed positively, while, SpMax_RG contributed negatively to the inhibitory activity. The descriptors Mor15v and SpMax_RG were found to contribute more significantly to the activity, owing to their higher coefficients in the equation. This indicates that the van der Waals volume along with the 3D matrix-based properties of the molecule play a significant role in determining the inhibitory interaction with CYP2C8. A good correlation coefficient (r) of 0.814 was obtained from the QSAR model. The model when validated using LOO method showed a cross-validated correlation coefficient (q2) value of 0.799, and a good predictive value (r2pred, external validation) of 0.718. In this QSAR model, 84.7% of the variance in biological activity was predicted, as indicated by r2 value of 0.847 multiplied by 100.
This QSAR study thus, shows that the inhibitory potential of glucuronide conjugates of gemfibrozil and its analogues is governed by the contribution of volume, shape and 3D matrix-based descriptors. This model would be useful in the prediction of inhibitory potential in terms of pIC50 values (CYP2C8 inhibition) of glucuronide conjugates of new molecules and CYP2C8 substrates against the standard acyl-β-glucuronide of gemfibrozil (potent mechanism-based inhibitor of CYP2C8), during the early stages of drug discovery and development.
ACKNOWLEDGEMENTS
NT is thankful to Department of Science and Technology (DST) for DST Inspire Fellowship. The authors thank Mr. Anup Shah for his help and the reviewers for their valuable suggestions.
Footnotes
Taxak and Bharatam: QSAR Study on Gemfibrozil Glucuronide Conjugates
REFERENCES
- 1.de Montellano PR, editor. New York: Kluwer Academic/Plenum Publishers; 2005. Cytochrome P450: Structure, mechanism, and biochemistry. [Google Scholar]
- 2.Lee J, Obach RS, Fisher MB, editors. New York: Marcel Dekker Inc; 2005. Drug metabolizing enzymes: Cytochrome P450 and other enzymes in drug discovery and development. [Google Scholar]
- 3.Kalgutkar AS, Didiuk MT. Structural alerts, reactive metabolites, and protein covalent binding: How reliable are these attributes as predictors of drug toxicity? Chem Biodivers. 2009;6:2115–37. doi: 10.1002/cbdv.200900055. [DOI] [PubMed] [Google Scholar]
- 4.Kalgutkar AS, Dalvie D, Obach RS, Smith DA, editors. New York: John Wiley and Sons; 2012. Reactive Drug Metabolites. [Google Scholar]
- 5.Taxak N, Bharatam PV. An insight into the concept and details of mechanism-based inhibition of CYP450. Curr Res Inform Pharm Sci. 2010;11:62–7. [Google Scholar]
- 6.Taxak N, Patel B, Bharatam PV. Carbene generation by cytochromes and electronic structure of heme-iron-porphyrin-carbene complex: A quantum chemical study. Inorg Chem. 2013;52:5097–109. doi: 10.1021/ic400010d. [DOI] [PubMed] [Google Scholar]
- 7.Taxak N, Desai PV, Patel B, Mohutsky M, Klimkowski VJ, Gombar V, et al. Metabolic-intermediate complex formation with Cytochrome P450: Theoretical studies in elucidating the reaction pathway for the generation of reactive nitroso intermediate. J Comput Chem. 2012;33:1740–7. doi: 10.1002/jcc.23008. [DOI] [PubMed] [Google Scholar]
- 8.Orr ST, Ripp SL, Ballard TE, Henderson JL, Scott DO, Obach RS, et al. Mechanism-Based Inactivation (MBI) of Cytochrome P450 Enzymes: Structure-Activity relationships and discovery strategies to mitigate drug-Drug interaction risks. J Med Chem. 2012;55:4896–933. doi: 10.1021/jm300065h. [DOI] [PubMed] [Google Scholar]
- 9.Wang JS, Neuvonen M, Wen X, Backman JT, Neuvonen PJ. Gemfibrozil inhibits CYP2C8-mediated cerivastatin metabolism in human liver microsomes. Drug Metab Dispos. 2002;30:1352–6. doi: 10.1124/dmd.30.12.1352. [DOI] [PubMed] [Google Scholar]
- 10.Mano Y, Usui T, Kamimura H. The UDP-Glucuronosyltransferase 2B7 Isozyme Is Responsible for Gemfibrozil Glucuronidation in the Human Liver. Drug Metab Dispos. 2007;35:2040–4. doi: 10.1124/dmd.107.017269. [DOI] [PubMed] [Google Scholar]
- 11.Ogilvie BW, Zhang D, Li W, Rodrigues AD, Gipson AE, Holsapple J, et al. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: Implications for drug-drug interactions. Drug Metab Dispos. 2006;34:191–7. doi: 10.1124/dmd.105.007633. [DOI] [PubMed] [Google Scholar]
- 12.Jenkins SM, Zvyaga T, Johnson SR, Hurley J, Wagner A, Burrell R, et al. Studies to further investigate the inhibition of human liver microsomal CYP2C8 by the acyl-â-glucuronide of gemfibrozil. Drug Metab Dispos. 2011;39:2421–30. doi: 10.1124/dmd.111.041947. [DOI] [PubMed] [Google Scholar]
- 13.Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Mechanism-based inactivation of CYP2C8 by Gemfibrozil occurs rapidly in humans. Clin Pharmacol Ther. 2011;89:579–86. doi: 10.1038/clpt.2010.358. [DOI] [PubMed] [Google Scholar]
- 14.Baer BR, DeLisle RK, Allen A. Benzylic oxidation of gemfibrozil-1-O-beta-glucuronide by P450 2C8 leads to heme alkylation and irreversible inhibition. Chem Res Toxicol. 2009;22:1298–309. doi: 10.1021/tx900105n. [DOI] [PubMed] [Google Scholar]
- 15.Dearden JC. In silico prediction of drug toxicity. J Comput Aided Mol Des. 2003;17:119–27. doi: 10.1023/a:1025361621494. [DOI] [PubMed] [Google Scholar]
- 16.Gramatica P. Principles of QSAR models validation: Internal and external. QSAR Comb Sci. 2007;26:694–701. [Google Scholar]
- 17.Perkins R, Fang H, Tong W, Welsh WJ. Quantitative structure-activity relationship methods: Perspectives on drug discovery and toxicology. Environ Toxicol Chem. 2003;22:1666–79. doi: 10.1897/01-171. [DOI] [PubMed] [Google Scholar]
- 18.Deeb O. Recent applications of quantitative structure-Activity relationships in drug design. In: Ekinci D, editor. Biochemistry, genetics and molecular biology, medicinal chemistry and drug design. Rijeka, Croatia: InTech; 2012. [Last accesed on 2013 Oct 02]. Available from: http://www.intechopen.com/books/medicinal-chemistry-and-drug-design/recent-applications-of-quantitative-structure-activity-relationships-in-drug-design . [Google Scholar]
- 19.San Diego: Accelrys Software Inc; 2012. Accelrys Software Inc., Discovery Studio Modeling Environment, Release 3.5. [Google Scholar]
- 20.St. Louis, MO 63144, USA: SYBYL7.1, Tripos Inc., 1699 South Hanley Rd. [Google Scholar]
- 21.Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, et al. Virtual computational chemistry laboratory - design and description. J Comput Aided Mol Des. 2005;19:453–63. doi: 10.1007/s10822-005-8694-y. Sybyl7.1. [DOI] [PubMed] [Google Scholar]
- 22.Joshi UJ, Tikhele SH, Shah FH. 2D QSAR of arylpiperazines as 5-HT 1A receptor agonists. Indian J Pharm Sci. 2007;69:800–4. [Google Scholar]
- 23.Khan N, Soni LK, Gupta AK, Wakode SR, Wagh RD, Kashedikar SG. QSAR analysis of N-Alkyl imidazole analogues as antibacterial agents. Indian J Pharm Sci. 2006;68:341–6. [Google Scholar]
- 24.Kirubakaran P, Muthusamy K, Singh KH, Nagamani S. Ligand-based pharmacophore modeling; atom-based 3D-QSAR analysis and molecular docking studies of phosphoinositide-dependent kinase-1 inhibitors. Indian J Pharm Sci. 2012;74:141–51. doi: 10.4103/0250-474X.103846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogers D, Hopfinger AJ. Application of genetic function approximation to quantitative structure-activity relationships and quantitative structure property relationships. J Chem Inf Comput Sci. 1994;34:854–66. [Google Scholar]