

Abstract
Background
Excess fibroblast growth factor 23 (FGF23) causes hypophosphatemia in autosomal dominant hypophosphatemic rickets (ADHR) and X-linked hypophosphatemia (XLH). Iron status influences C-terminal FGF23 (incorporating fragments plus intact FGF23) in ADHR and healthy subjects, and intact FGF23 in ADHR. We hypothesized that in XLH serum iron would inversely correlate to C-terminal FGF23, but not to intact FGF23, mirroring the relationships in normal controls.
Methods
Subjects included 25 untreated outpatients with XLH at a tertiary medical center and 158 healthy adult controls. Serum iron and plasma intact FGF23 and C-terminal FGF23 were measured in stored samples.
Results
Intact FGF23 was greater than the control mean in 100% of XLH patients, and >2SD above the control mean in 88%, compared to 71% and 21% respectively for C-terminal FGF23. In XLH, iron correlated negatively to log-C-terminal FGF23 (r= −0.523, p<0.01), with a steeper slope than in controls (p<0.001). Iron was not related to log-intact FGF23 in either group. The log-ratio of intact FGF23 to C-terminal FGF23 was higher in XLH (0.00 ± 0.44) than controls (−0.28 ± 0.21, p<0.01), and correlated positively to serum iron (controls r= 0.276, p<0.001; XLH r= 0.428, p<0.05), with a steeper slope in XLH (p<0.01).
Conclusion
Like controls, serum iron in XLH is inversely related to C-terminal FGF23 but not intact FGF23. XLH patients are more likely to have elevated intact FGF23 than C-terminal FGF23. The relationships of iron to FGF23 in XLH suggest altered regulation of FGF23 cleaving may contribute to maintaining hypophosphatemia around an abnormal set-point.
Keywords: Fibroblast growth factor 23, FGF23, X-linked hypophosphatemia, iron, phosphate
1. Background/Introduction
Under normal physiologic conditions, fibroblast growth factor 23 (FGF23) is produced by osteocytes and osteoblasts, circulates as a hormone, and binds to fibroblast growth factor receptors and the co-receptor klotho in the kidney. In the kidney, FGF23 down-regulates surface expression of sodium phosphate cotransporters (NPT2a and NPT2c), and down-regulates renal 1α-hydroxylase activity[1]. FGF23 excess is the cause of renal phosphate wasting, hypophosphatemia, and inappropriately low or normal 1,25(OH)2D concentrations in both autosomal dominant hypophosphatemic rickets (ADHR) and the more common disorder, X-linked hypophosphatemia (XLH)[2–4].
Part of the FGF23 produced is normally cleaved into inactive fragments before secretion, but mutations in FGF23 impair cleavage of intact FGF23 at an 176RXXR179/S180 cleavage site, causing ADHR[2]. However ADHR has a variable age of onset of clinical features and hypophosphatemia, and some patients resolve their clinical phenotype spontaneously with normalized FGF23 concentrations. We previously demonstrated that the presence of hypophosphatemia versus normophosphatemia in ADHR patients occurred due to varying FGF23 concentrations[4]. In fact, high intact FGF23 concentrations in ADHR occurred in the setting of low serum iron concentration, corresponding to periods of hypophosphatemia[4, 5]. Farrow et al confirmed increased expression of FGF23 in an iron-restricted mouse model of ADHR[6]. Furthermore, iron restriction of wild-type mice also raised FGF23 expression and concentrations of FGF23 fragments, but maintained normal intact FGF23 concentration and normophosphatemia. Likewise healthy adult humans also demonstrated an inverse relationship between serum iron and C-terminal FGF23 measurements (which includes intact protein plus fragments), but no relationship of iron with intact FGF23[5].
XLH is caused by PHEX mutations resulting in deficiency of the protein. PHEX mutations result in increased FGF23 expression and both intact FGF23 and C-terminal FGF23 plasma concentrations [3, 7, 8]. In contrast to ADHR, the phenotypic features of XLH do not vary in timing or resolve spontaneously within an individual, though there is wide clinical variability in biochemical and skeletal features between individuals with XLH. Instead, in XLH the hypophosphatemia phenotype persists through the life of the individual. XLH patients are not known to have any iron related phenotypes.
Despite inappropriate baseline elevations in FGF23 concentrations in XLH, standard treatment with calcitriol and phosphate further increases FGF23 concentrations[9, 10]. In addition, mouse models data suggests that an abnormality in phosphate sensing mechanisms could be involved in the pathogenesis of elevated FGF23 concentrations in XLH[11], with serum phosphate concentrations maintained at an abnormal set point. If this is true, FGF23 production may also increase with low iron status in XLH similar to healthy controls, but the bioactive intact FGF23 would still not be influenced by serum iron concentrations. Thus, we hypothesized that in XLH, serum iron concentrations would be inversely correlated to C-terminal FGF23 measurement, but would not be correlated to intact FGF23 concentrations.
2. Methods
2.1 Study design
This was a cross-sectional analysis of samples obtained during an ongoing observational study of patients with XLH. Control subjects were included from a previously published cross-sectional sample of healthy adult subjects[5]. The purpose of this analysis was to evaluate the relationship between iron status and plasma FGF23 in XLH patients. The study was conducted in accordance with the Declaration of Helsinki and was approved by Indiana University Institutional Review Board. Written informed consent was provided by all subjects or their parents, with assent by minors 7 years of age or older.
2.2 Subjects
All pediatric and adult XLH patients presenting to the practices of the investigators (at the Indiana University Health Hospital and Riley Hospital for Children) are invited to participate in an ongoing observational study of XLH. The diagnosis of XLH is based on usual clinical criteria: hypophosphatemia, renal phosphate wasting, and physical or radiographic evidence of current or past rickets. Family history of hypophosphatemic rickets and personal or family history of PHEX mutation was supportive, but not required. Most subjects had a family history of XLH and/or a confirmed PHEX mutation. Subjects were excluded if there was clinical or genetic evidence of a separate acquired or inherited disorder of phosphate metabolism (such as ADHR or Fanconi syndrome).
Since treating XLH with calcitriol and phosphate increases serum FGF23 concentrations [9], which would confound the analysis of iron effects, only samples from subjects recruited prior to initiating treatment with calcitriol or phosphate were included. All 25 subjects with XLH (15 adults and 10 children) (Table 1) in this analysis were either treatment naïve or had stopped calcitriol and phosphate at least one year before the included sample. For controls, samples from 158 healthy adult controls were pulled from a study on genetics of peak bone mass as controls for the previous study in iron and ADHR and are again used for controls in this analysis[5].
Table 1.
Biochemistries
| Controls | XLH | p=value | |
|---|---|---|---|
| N | 158 | 25 | |
| Male/Female | 79/79 | 6/19 | |
| Age (years) | 27.7 ± 7.4 | 20.3 ± 14.2 | <0.05 |
| Creatinine (mg/dl) | 0.9 ± 0.2 | 0.6 ± 0.2 | <0.001 |
| Calcium (mg/dl) | 9.4 ± 0.4 | 9.3 ± 0.4 | NS |
| Phosphate (mg/dl) | 3.4 ± 0.5 | 2.3 ± 0.7 | <0.001 |
| Phosphate less than age normal (%)* | 4/158 (3%) | 22/25 (88%) | <0.001 |
| Alkaline Phosphatase (U/L) | 58 ± 16 | 258 ± 192 | <0.001 |
| Parathyroid hormone (pg/ml) | 28.8 ± 10.6 | 86.3 ± 58.3 | <0.01 |
| 1,25-dihydroxyvitamin D (pg/ml) | 44.6 ± 17.0 | 45.8 ± 11.6 | NS |
| Iron (mcg/dl) | 93.6 ± 40.5 | 71.2 ± 26.6 | <0.01 |
| Iron <50mcg/dl | 18/158 (11%) | 6/25 (24%) | NS |
| TIBC (mcg/dl) | 329 ± 63 | 341 ± 61 | NS |
| Iron % saturation | 29.1 ± 12.7 | 21.8 ± 9.0 | <0.01 |
| Iron saturation <20% | 37/158 (23%) | 9/25 (36%) | NS |
| Intact FGF23 (pg/ml) Median (25thpercentile–75thpercentile) |
40 ± 13 Median 39 (31–48) |
183 ± 251 Median 111 (86–191) |
<0.001** |
| Log-Intact-FGF23 (log of pg/ml) | 1.58 ± 0.14 | 2.13 ± 0.29 | <0.001 |
| C-terminal FGF23 (RU/ml) Median (25thpercentile–75thpercentile) |
83 ± 52 Median 67 (53–88) |
229 ± 302 Median 106 (78–158) |
<0.001** |
| Log-C-terminal-FGF23 (log of RU/ml) | 1.86 ± 0.20 | 2.12 ± 0.45 | <0.05 |
| Log-ratio of intact to C-terminal FGF23 | −0.28 ± 0.21 | 0.00 ± 0.44 | <0.01 |
Values indicate mean ± standard deviation except where indicated.
Phosphate normal ranges for age: age 1–5 years (3.6–6.5 mg/dl); age 5–10 years (3.4–5.5 mg/dl); age 10–20 years (2.6–5.2 mg/dl); and adults (2.5–4.9 mg/dl). P-values are provided for T-tests, except where marked with ** for Mann-Whitney test. PTH was measured in 129 controls and 18 XLH patients, while 152 controls and 8 XLH patients had 1,25(OH)2D measured. To convert the values for phosphate to millimoles per liter, multiply by 0.32. To convert calcium to millimoles per liter, multiply by 0.25. To convert creatinine to micromoles per liter, multiply by 76.26. To convert 1,25D to picomoles per liter, multiply by 2.6. To convert PTH to picomoles per liter, multiply by 0.1053. To convert iron to micromoles per liter, multiply by 0.179. To convert TIBC to micromoles per liter, multiply by 0.179.
2.3 Measurements
At the time of their routine clinical care (medical history, examination, diagnostic clinical tests, and medication management), enrolled XLH patients are asked to provide fasting serum and plasma samples for research purposes. Blood samples were stored at −80 °C prior to research study measurements.
FGF23 was measured in EDTA-plasma, using two different ELISAs. The intact FGF23 ELISA (Kainos Laboratories, Tokyo, Japan) uses two monoclonal antibodies binding to opposite sides of the FGF23 176RXXR179/S180 cleavage site with a CV of 4.4%. A separate C-terminal FGF23 ELISA (Immutopics, San Clemente, CA) uses polyclonal antibodies against two epitopes on the C-terminal side of the cleavage site, and thus measures the combination of intact FGF23 and C-terminal fragments (CV of 4%). In order to compare the results from the FGF23 assay kits, the standards from each manufacturer’s assay were also measured on the other FGF23 assay. Thus, from the C-terminal FGF23 assay kit purified FGF23 standards at six different concentrations, plus two supplied purified FGF23 controls, were measured on the intact FGF23 ELISA. Likewise, purified FGF23 standards at seven concentrations from the intact FGF23 kit were measured on the C-terminal FGF23 ELISA.
Serum biochemistries were measured with the Roche Cobas Mira S (Roche, Indianapolis, IN) for phosphorus, creatinine and alkaline phosphatase, iron, and total iron binding capacity (TIBC). Reagents for phosphorus, creatinine and alkaline phosphatase were from Thermo Scientific (Waltham, MA). Iron and total iron binding capacity (TIBC) were measured from serum using the Roche Cobas Mira S (Roche, Indianapolis, IN). Reagents for iron and TIBC were purchased from Pointe Scientific, Inc. (Canton, Mi). Percent iron saturation was calculated by serum iron concentration ÷ TIBC. Iron deficiency was defined as iron concentration < 50 mcg/dl, or by percent iron saturation < 20%. Parathyroid hormone (PTH) and 1,25(OH)2D were measured using radioimmunoassays from Diasorin (Stillwater, MN) in controls. Due to sample limitations PTH and 1,25(OH)2D results are reported for XLH from assays run in the hospital clinical lab (not reanalyzed). 129 controls and 18 XLH patients had measures of PTH, while 152 controls and 8 XLH patients had 1,25(OH)2D measured.
2.4 Statistical analysis
The distribution of variables was examined and log-transformations were performed for C-terminal FGF23 and intact FGF23. Equality of variances between groups for continuous variables was tested with Levene test and differences between XLH and control groups were tested with appropriate two-tailed T-tests for means and chi-square tests for proportions. For intact FGF23 and C-terminal FGF23, median values are also provided and the differences in distributions tested with Mann-Whitney test. Pair-wise relationships between variables were examined within each group using scatter plots and Pearson correlation with two-tailed tests for significance. To test whether a pair-wise relationship differed significantly between XLH and control subjects, multiple regression analysis with an appropriate interaction term was used. P-values less than 0.05 were considered statistically significant. Statistical analysis was made using SPSS®, version 20.
3. Results
3.1 FGF23 assay standard comparisons
Assay standards from the intact FGF23 assay were measured on the C-terminal FGF23 assay, and vice versa. The measurements of assay standards from each kit on the other assay provided similar numerical values for the standards in pg/ml and RU/ml (Figure 1). For most assay standards, the ratio of intact FGF23/C-terminal FGF23 was relatively consistent between 0.8 and 1.2 across most values of FGF23 (Figure 1C). Even using assay standards, C-terminal FGF23 (intact plus fragments) was sometimes lower than intact FGF23, indicating differences in measurement ability of the assays.
Figure 1. Comparison of FGF23 assay kit standards.
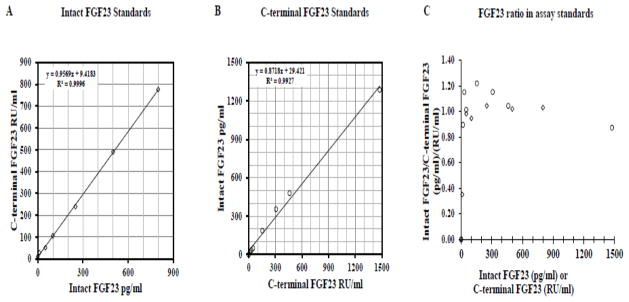
Intact FGF23 assay kit standards were measured on the C-terminal FGF23 ELISA (1A) and the C-terminal FGF23 kit standards were measured on the Intact FGF23 ELISA (1B). The ratio of intact FGF23/C-terminal FGF23 for both sets of assay standards is plotted according to the concentration of the assay standard (1C). Diamonds indicate intact FGF23 assay kit standards and circles indicate C-terminal FGF23 assay kit standards.
3.2 Biochemistries and FGF23 in XLH patients and controls
Biochemistries are listed in Table 1. We have previously reported the adult control results[5]. The 158 adults controls were from a Caucasian population, with age range from 20 to 50 years. For adult controls, males had higher creatinine (1.0 ± 0.2 vs 0.9 ± 0.1, p<0.001), calcium (9.5 ±0.3 vs 9.3 ± 0.4, p<0.001), alkaline phosphatase (62 ± 17 vs 55 ±16, p<0.01), and iron saturation (32 ± 14 vs 26 ±11, p<0.01), and lower TIBC (307 ±50 vs 351 ±66, p<0.01) than adult female controls.
A total of 25 XLH subjects ranged from 1 to 53 years of age. Ten were children and 15 were adults. The children were not on treatment prior to participation. The adults had a past history of treatment with phosphate and vitamin D analogs with discontinuation at least 1 year prior to the analyzed sample. Two subjects were sporadic cases of hypophosphatemia, but all others had family history consistent with X-linked dominant inheritance, including no male-to-male transmission. PHEX mutations were confirmed in 13 subjects, and were not detectable in four (3 of these 4 had strong family history for XLH). For 6 additional subjects from 2 families, there was a strong family history consistent with X-linked dominant inheritance and these were not gene tested. For two additional subjects who did not undergo gene testing there was no family history, but diagnosis was based on clinical grounds consistent with XLH, which is statistically the most common inherited rickets. Notably the detection rate for mutations in PHEX in XLH kindreds has been reported to be about 65%[14]. Three children and 1 adult with XLH were African-American. Their FGF23 concentrations and clinical features were similar to the rest of the XLH population. For XLH children, males did not differ from females in any variable, while for adult XLH subjects, the males had higher alkaline phosphatase than females (197 ±47 vs 94 ±33, p<0.001). However there were only 2 male children and 4 male adults with XLH. The numbers of male subjects and of African American subjects with XLH were too few to determine whether there were race or sex differences in variables or correlations in XLH.
Mean serum phosphorus concentrations were lower in the XLH patients compared with controls (p<0.001). Serum phosphorus was below age-normal reference ranges in 22/25 (88%) of XLH patients and the remaining 3 XLH patients had serum phosphorus near the lower end of normal. Alkaline phosphatase concentrations were also higher in XLH patients than in controls (p<0.001). PTH and 1,25(OH)2D concentration were similar between children and adult subjects with XLH. PTH was also higher in XLH patients than controls (p<0.01), but there was not a difference in 1,25(OH)2D concentration.
FGF23 was not normally distributed in XLH subjects, who had higher median intact and C-terminal FGF23 concentrations than controls (p<0.001). However, intact and C-terminal FGF23 concentrations were similar for adult and pediatric XLH subjects. For regression analysis FGF23 concentrations were log-transformed, and XLH patients had higher mean log-transformed intact FGF23 (p <0.001) compared to controls. Likewise XLH patients had higher log-transformed C-terminal FGF23 concentrations compared to controls (p<0.05). There was not a relationship of age to intact FGF23 or C-terminal FGF23 in the control group, composed of healthy adults, nor in the group of adults and children with XLH.
XLH patients had wide variability in FGF23 concentrations as shown in Table 1 and Figure 2. However, patients with XLH were more likely to have intact FGF23 concentrations elevated above the normal range than to have elevated C-terminal FGF23 concentrations. Log-intact FGF23 concentrations were more than 2 SD above the normal control mean in 88% of patients with XLH (9/10 children and 13/15 adults). Log-C-terminal FGF23 concentrations were higher than 2SD above the normal control mean in only 21% of patients with XLH (2/9 children and 3/15 adults). In addition, despite being hypophosphatemic, 100% of patients with XLH had intact FGF23 concentrations above the control mean of 40 pg/ml, while only 71% had C-terminal FGF23 concentrations above the control mean of 83 RU/ml.
Figure 2. Relationship of iron to FGF23 concentrations in XLH compared to healthy controls.

Iron concentration is inversely related to C-terminal FGF23 in XLH (A) and control (B) subjects, with a greater magnitude of effect in XLH subjects (p<0.001). Iron was not related to intact FGF23 in either XLH (C) or control (D) subjects. The log-ratio of intact to C-terminal FGF23 concentration was positively correlated to iron concentration with a greater magnitude of effect in XLH (E) than in control (F) subjects (p<0.01). Squares represent adults and triangles represent children with XLH. The same vertical axis scales were used for XLH and controls in order to more easily visualize the relative distributions of variables.
3.3 Iron and FGF23
Mean serum iron concentration and percent iron saturation were lower in XLH patients than controls (p<0.01). However the proportion of subjects with iron deficiency, defined by serum iron <50 mcg/dl or percent iron saturation <20%, were not significantly different (Table 1).
For healthy control adults, we previously reported the negative correlation between serum iron concentration and log-C-terminal FGF23 (r= −0.276, p<0.001)[5]. There were no differences in correlation for iron and log-C-terminal FGF23 between female and male controls (r= −0.278 and −0.272 respectively). After adjustment for age, controls retained a significant inverse relationship between iron and log-C-terminal FGF23 (p<0.01).
Similarly in XLH patients there was also a negative correlation between serum iron concentration and log-C-terminal FGF23 (r= −0.523, p<0.01). This relationship did not differ significantly between the adult and pediatric XLH subjects (adult XLH r= −0.689, p<0.01, child XLH r= −0.410, p=0.27; p>0.5 for interaction), and these correlations were similar after age adjustment. However, the C-terminal FGF23 concentrations were higher in XLH, and the regression slope was steeper (p<0.001) compared to controls, suggesting that iron had a greater magnitude of influence on C-terminal FGF23 concentrations in XLH than in controls. Percent iron saturation was similarly related to log-C-terminal FGF23 in controls with r= −0.296 (p<0.001) and in the XLH patients (combined r= −0.476, p <0.05; adults r=−0.690, p<0.01; children r = −0.352, p=0.35). The regression slope of log-C-terminal FGF23 for percent iron saturation was also steeper in XLH subjects (p<0.001). There were no differences in relationships between male and female XLH subjects. However, the small number of pediatric XLH subjects and also of male XLH subjects limited significance of correlations when restricted to these groups. In addition, analysis was run excluding the 4 subjects for whom mutation detection was negative. This group similarly had an inverse relationship between serum iron and log-C-terminal FGF23 concentration (r=−0.455, p<0.05). Limiting analysis to the 13 XLH patients with confirmed mutations the negative relationship was similar (r=−0.497, p<0.01).
We previously reported no relationship between serum iron concentration and log-intact FGF23 in the healthy adult controls (r= 0.023, p>0.5) [5]. Correlations were similar for control males and control females. Though XLH patients had higher log-intact FGF23 than controls, there was no significant relationship between serum iron and log-intact FGF23 whether analyzed separately for adults (r=0.039) and children (r=−0.177) or as a combined XLH group (r = −0.147, all with p>0.5). The regression slopes did not differ between XLH and controls. Likewise, neither serum iron nor percent iron saturation were correlated to serum phosphorus concentration in XLH or controls even when controlling for age.
The mean log-ratio of intact FGF23 to C-terminal FGF23 was higher in XLH compared to controls (log ratio 0.00 ±0.44 versus −0.28 ± 0.21, p<0.01). Furthermore, XLH subjects were more likely to have a log-ratio > 0, (62.5% versus 5.7%, p <0.001) indicating that XLH subjects were more likely to have higher intact, biologically active FGF23 concentration than C-terminal FGF23 concentration, despite their prevailing hypophosphatemia.
The log-ratio of intact FGF23 to C-terminal FGF23 was positively correlated to serum iron concentrations in both controls (r= 0.276, p<0.001) and XLH subjects (r= 0.428, p<0.05), indicating that at lower iron concentrations, more inactive FGF23 fragments are present in both groups. The slope of the regression line between iron and the log-ratio of FGF23 was steeper in XLH than in controls (p<0.01), consistent with the observed steeper relationship between iron and C-terminal FGF23 in XLH. Percent iron saturation was similarly related to the log-ratio of FGF23 in XLH (r= 0.418, p < 0.05) and controls (r= 0.276, p < 0.001), also with a slope of greater magnitude in XLH subjects (p<0.01).
Neither iron nor percent iron saturation were correlated with 1,25(OH)2D concentration in controls or XLH subjects. Furthermore, in controls, 1,25(OH)2D did not correlate with log-C-terminal FGF23 or log-intact FGF23. However in 8 XLH subjects, 1,25(OH)2D was positively correlated to log-C-terminal FGF23 (r= 0.725, p<0.05) but not to log-intact FGF23.
4. Discussion
Recently, we demonstrated that low iron concentrations were inversely related to both intact and C-terminal FGF23 concentrations in ADHR patients (with an FGF23 mutation that impaired cleavage). This provided a potential explanation for the variable age of developing the ADHR phenotype, as well as the waxing and waning of FGF23 concentrations, hypophosphatemia, and clinical symptoms observed in patients with ADHR[5]. In an ADHR mouse model FGF23 expression only became elevated during iron deprivation, consistent with the cross-sectional findings in humans with ADHR[6]. Recently in vitro studies implicated tissue hypoxia in the setting of iron deficiency as a possible mechanism leading to increased FGF23 expression[15].
As opposed to ADHR, the biochemical phenotype of XLH does not fluctuate outside of the effects of calcitriol and phosphate on serum phosphorus, calcium and FGF23 concentrations. Neither does XLH spontaneously remit like ADHR. Here we demonstrate for the first time that FGF23 concentration in XLH is related to iron concentrations in a pattern that is more similar to that of controls than that of ADHR. Iron is negatively correlated to serum C-terminal FGF23 concentration in controls and in XLH, but with a greater magnitude of effect in XLH, as demonstrated by a steeper regression slope.
Furthermore, we demonstrated that like controls, iron does not correlate to intact FGF23 concentrations in XLH. However, the log-ratio of intact to C-terminal FGF23 was positively related to serum iron concentration in both groups, indicating that at lower iron concentrations, a greater proportion of the FGF23 circulates as C-terminal fragments, in both XLH and controls.
Limitations of the current study include small numbers of total subjects with XLH. The sample size was limited by the decision to only include subjects who were not currently receiving treatment with calcitriol and phosphate. This was done to avoid the confounding effect of this treatment, which has been demonstrated to raise FGF23 concentrations in XLH[9, 10]. Furthermore there were small numbers of XLH children and although the direction of relationships was similar, these were sometimes nonsignificant when evaluating children with XLH by themselves. There were too few males with XLH in the study to fully assess whether there might be differences between the males and females in XLH. It is possible that with small numbers of subjects with XLH that a significant relationship between iron and intact FGF23 could have been missed. However, given the lack of correlation, relatively flat slope of the associated regression line and p>0.5), it is more likely that there is no relationship, though future studies in XLH should test to confirm these findings.
Another limitation is that the available assays do not clearly indicate the molar amounts of intact and C-terminal FGF23 in samples and the actual molar relationships of intact and C-terminal FGF23 in circulation are uncertain. In fact, previous publications indicate the relative proportion varies in certain disease states, or even in healthy controls[5, 12, 13]. In addition, western blotting of the C-terminal FGF23 assay standards indicates both intact and C-terminal fragments of FGF23 are present [17]. In a perfectly performing set of such assays, the C-terminal FGF23 level (intact plus fragments) would always be higher than the intact FGF23 in molar terms. Since measuring assay standards from one kit on the other kit resulted in numerically similar values (i.e. 400 units on one assay approximated 400 units on the other), the possible presence of C-terminal fragments in the assay standard did not appear to influence the comparison of assay standards. However, the intact FGF23 concentration is frequently higher than the C-terminal FGF23 concentration in both controls and XLH subjects. Patients with XLH were more likely to have elevated intact FGF23 levels (defined as higher than either the normal mean or than 2SD above the mean) than to have elevated C-terminal FGF23 levels and the ratio of intact to C-terminal FGF23 was higher in XLH than controls.
The data relating iron to FGF23 concentrations in ADHR, XLH and healthy controls would indicate another layer of FGF23 regulation through controlling cleavage of FGF23: that in the setting of inappropriate (i.e., not driven by phosphate, 1,25(OH)2D or CKD) increased FGF23 production, cleavage should normally occur to maintain appropriate intact FGF23 and phosphate concentrations. Unlike ADHR, XLH does not involve an intrinsic resistance to proteolytic cleavage of FGF23, yet in XLH, FGF23 gene expression is generally increased despite ongoing hypophosphatemia[7]. In addition, FGF23 concentration does increase further in XLH [9, 10] and in mouse models of XLH [18, 19] in response to calcitriol and phosphate, which are both usual factors stimulating FGF23 production. Increased FGF23 concentration in XLH during treatment occurs even without normalization of serum phosphate. However, our data suggests that XLH patients also increase production of FGF23 during low iron states similarly to controls, but maintain cleavage of FGF23 such that intact FGF23 is not influenced by iron.
Mouse models expand on the regulation of FGF23 caused by PHEX deficiency. We recently generated an animal model of GALNT3 gene deletion resulting in impaired FGF23 glycosylation, impaired secretion of intact FGF23 and hyperphosphatemia [20]. These mice had a compensatory increased FGF23 gene expression. When crossed to a phex null (XLH) mouse, the resulting double mutant mouse had only partial improvement in hypophosphatemia, and despite impaired secretion of intact FGF23, the intact FGF23 remained quite elevated. While the GALNT3-null mouse increases expression of FGF23 to offset hyperphosphatemia, the double mutant further increases expression in the setting of hypophosphatemia. In fact, the double mutant mouse greatly increased FGF23 gene expression up to 24-fold greater than either the phex-null mouse or the GALNT3-null mouse alone, and 100-fold greater than wild-type mice.
A possible abnormality in phosphate sensing set-point might be responsible for the findings in the double mutant mouse, with an attempt to offset the mild rise in phosphate above that of the phex-null mouse. In support of the concept of altered set-point in phex-deficiency, long term pharmacologic inhibition of fibroblast growth factor receptors (with a receptor antagonist or with MAPK inhibitors) in phex-deficient mice improved serum phosphate, but greatly increased FGF23 expression and serum FGF23 concentrations, even during persistent hypophosphatemia[21, 22]. Consequent to the PHEX mutation, there may also be down-regulation of FGF23 cleavage, due to decreased 7B2-PC2 enzymatic activity [23].
On the converse side, we show in XLH a further increase of C-terminal FGF23 at low iron concentrations, but no relationship of iron to intact FGF23 or serum phosphate. The fact that XLH patients remain hypophosphatemic in the setting of increased FGF23 expression, while still being able to modify intact FGF23 concentrations in response to phosphate or calcitriol[9], and have similar relationships of iron and FGF23 to that of healthy controls would all suggest that some regulation of FGF23 expression and cleavage is still occurring around to some set-point that may be different in XLH than in the normal state.
Since iron concentrations are inversely related to intact FGF23 in ADHR, it is plausible that treatment of iron deficiency ADHR could normalize FGF23 concentrations and resolve hypophosphatemia. In fact in the mouse models of ADHR and wild type mice treated with iron deficient diets during breeding and nursing, increased FGF23 expression was demonstrated, which was improved after iron repletion[15]. There is likely a threshold effect above which FGF23 metabolism is not impacted by iron, as is suggested by the scatterplots of adult control subjects, and by the fact that iron deficient diets were necessary to induce the phenotype in ADHR mice. Similarly in XLH, iron supplementation could lower C-terminal FGF23 concentrations. However, given the absence of relationship of iron and intact FGF23 in XLH patients, which remained elevated at various iron levels, it is unlikely that iron treatment would have any effect on the hypophosphatemia and other clinical features of XLH.
In summary, the relationship of iron to FGF23 continues to be elucidated. In ADHR, the disease state of excess intact FGF23 is related to serum iron concentration. However in XLH, though the absolute levels of FGF23 are higher than in controls, the pattern of relationship of iron and FGF23 is similar to that in healthy controls. In XLH, the persistent elevations in FGF23 result in hypophosphatemia, which is not limited to low iron states. Further studies are needed to determine the mechanisms of the effect of low iron on FGF23, and whether there is any influence of FGF23 fragments on iron metabolism itself.
Highlights.
-
1
Intact FGF3 is more likely to be elevated than C-terminal FGF23 in XLH.
-
2
Despite hypophosphatemia, C-terminal FGF23 is inversely related to iron in XLH.
-
4
Intact FGF23 is not related to serum iron in XLH or controls.
-
5
The presented data suggest FGF23 cleavage is a regulated process even in XLH.
Acknowledgments
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Institute on Aging of the National Institutes of Health under Award Numbers K23AR057096, R01AR42228, P01AG18397, and KL2RR025760. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Disclosures: EAI receives research funding from and is a consultant for Kyowa Hakko Kirin, Pharma Inc. MJE holds a patent on FGF23 and receives royalties from and is a consultant for Kyowa Hakko Kirin, Pharma Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–35. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 2.ADHR_Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345–8. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 3.Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, Fukumoto S. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab. 2002;87:4957–4960. doi: 10.1210/jc.2002-021105. [DOI] [PubMed] [Google Scholar]
- 4.Imel EA, Hui SL, Econs MJ. FGF23 concentrations vary with disease status in autosomal dominant hypophosphatemic rickets. J Bone Miner Res. 2007;22:520–526. doi: 10.1359/jbmr.070107. [DOI] [PubMed] [Google Scholar]
- 5.Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron Modifies Plasma FGF23 Differently in Autosomal Dominant Hypophosphatemic Rickets and Healthy Humans. J Clin Endocrinol Metab. 2011;96:3541–9. doi: 10.1210/jc.2011-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, Robling AG, Stayrook KR, Jideonwo V, Magers MJ, Garringer HJ, Vidal R, Chan RJ, Goodwin CB, Hui SL, Peacock M, White KE. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci U S A. 2011;108:1146–1155. doi: 10.1073/pnas.1110905108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu S, Guo R, Simpson LG, Xiao Z-S, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278:37419–37426. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- 8.Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656–63. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 9.Imel EA, DiMeglio LA, Hui SL, Carpenter TO, Econs MJ. Treatment of X-linked hypophosphatemia with calcitriol and phosphate increases circulating fibroblast growth factor 23 concentrations. J Clin Endocrinol Metab. 2010;95:1846–50. doi: 10.1210/jc.2009-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carpenter TO, Insogna KL, Zhang JH, Ellis B, Nieman S, Simpson C, Olear E, Gundberg CM. Circulating levels of soluble klotho and FGF23 in X-linked hypophosphatemia: circadian variance, effects of treatment, and relationship to parathyroid status. J Clin Endocrinol Metab. 2010;95:E352–7. doi: 10.1210/jc.2010-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ichikawa S, Austin AM, Gray AK, Econs MJ. A Phex mutation in a murine model of X-linked hypophosphatemia alters phosphate responsiveness of bone cells. J Bone Miner Res. 2012;27:453–460. doi: 10.1002/jbmr.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117:2684–91. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ichikawa S, Baujat G, Seyahi A, Garoufali AG, Imel EA, Padgett LR, Austin AM, Sorenson AH, Pejin Z, Topouchian V, Quartier P, Cormier-Daire V, Dechaux M, Malandrinou F, Singhellakis PN, Le Merrer M, Econs MJ. Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations. Am J Med Genet A. 2010;152A:896–903. doi: 10.1002/ajmg.a.33337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho HY, Lee BH, Kang JH, Ha IS, Cheong HI, Choi Y. A clinical and molecular genetic study of hypophosphatemic rickets in children. Pediatr Res. 2005;58:329–33. doi: 10.1203/01.PDR.0000169983.40758.7B. [DOI] [PubMed] [Google Scholar]
- 15.Clinkenbeard EL, Farrow EG, Summers LJ, Cass TA, Roberts JL, Bayt CA, Lahm T, Albrecht M, Allen MR, Peacock M, White KE. Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. J Bone Miner Res. 2013 doi: 10.1002/jbmr.2049. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer DC, Mischek A, Wolf S, Rahn A, Salweski B, Kundt G, Haffner D. Paediatric reference values for the C-terminal fragment of fibroblast-growth factor-23, sclerostin, bone-specific alkaline phosphatase and isoform 5b of tartrate-resistant acid phosphatase. Ann Clin Biochem. 2012;49:546–53. doi: 10.1258/acb.2012.011274. [DOI] [PubMed] [Google Scholar]
- 17.Braithwaite V, Bruggraber SF, Prentice A. Intact fibroblast growth factor 23 and fragments in plasma from Gambian children. Osteoporos Int. 2013;24:1121–4. doi: 10.1007/s00198-012-2029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146:5358–64. doi: 10.1210/en.2005-0777. [DOI] [PubMed] [Google Scholar]
- 19.Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, Quarles LD. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17:1305–15. doi: 10.1681/ASN.2005111185. [DOI] [PubMed] [Google Scholar]
- 20.Ichikawa S, Sorenson AH, Austin AM, Mackenzie DS, Fritz TA, Moh A, Hui SL, Econs MJ. Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology. 2009;150:2543–50. doi: 10.1210/en.2008-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wohrle S, Henninger C, Bonny O, Thuery A, Beluch N, Hynes NE, Guagnano V, Sellers WR, Hofmann F, Kneissel M, Graus Porta D. Pharmacological inhibition of FGFR signaling ameliorates FGF23-mediated hypophosphatemic rickets. J Bone Miner Res. 2013;28:899–911. doi: 10.1002/jbmr.1810. [DOI] [PubMed] [Google Scholar]
- 22.Zhang MY, Ranch D, Pereira RC, Armbrecht HJ, Portale AA, Perwad F. Chronic inhibition of ERK1/2 signaling improves disordered bone and mineral metabolism in hypophosphatemic (Hyp) mice. Endocrinology. 2012;153:1806–16. doi: 10.1210/en.2011-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan B, Feng JQ, Bowman S, Liu Y, Blank RD, Lindberg I, Drezner MK. Hexa-D-Arginine treatment increases 7B2*PC2 activity in hyp-mouse osteoblasts and rescues the HYP phenotype. J Bone Miner Res. 2013;28:56–72. doi: 10.1002/jbmr.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]