

Summary
The Rho family of GTP binding proteins – also commonly referred to as the Rho GTPases – are master regulators of the platelet cytoskeleton and platelet function. These low molecular weight or “small” GTPases act as signaling switches in the spatial and temporal transduction and amplification of signals from platelet cell surface receptors to the intracellular signaling pathways that drive platelet function. The Rho GTPase family members RhoA, Cdc42 and Rac1 have emerged as key regulators in the dynamics of the actin cytoskeleton in platelets and play key roles in platelet aggregation, secretion, spreading and thrombus formation. Rho GTPase regulators, including GEFs and GAPs and downstream effectors, such as the WASPs, formins and PAKs, may also regulate platelet activation and function. In this review, we provide an overview of Rho GTPase signaling in platelet physiology. Studies of Rho GTPases and platelets have had a shared history, as platelets have served as an ideal, non-transformed cellular model to characterize Rho function. Likewise, recent studies of the cell biology of Rho GTPase family members have helped to build an understanding of the molecular regulation of platelet function and will continue to do so through the further characterization of Rho GTPases as well as Rho GAPs, GEFs, RhoGDIs and Rho effectors in actin reorganization and other Rho-driven cellular processes.
Keywords: actin, cytoskeleton, platelet, Rac, Rho, Cdc42
Introduction
Platelets function as the primary cellular mediators of hemostasis and thrombosis [1]. As the guardians of vascular integrity, platelets patrol the circulation for vessel leakage in a quiescent, bidiscoid shape. Upon detecting molecular cues of vessel damage, platelets undergo a dramatic change in shape, bind to adhesive protein substrates and aggregate to form vascular plugs to ultimately halt bleeding. As platelets encounter exposed extracellular matrix proteins such as collagen and laminin, receptors on platelets trigger intracellular signaling events that rapidly result in platelet activation and a complex rearrangement of platelet morphology to form filopodia and lamellipodia (Fig. 1). Finger-like platelet filopodial extensions and actin-rich sheets of lamellipodia dramatically increase platelet surface area and stabilize platelet aggregates to form thrombotic plugs (Fig. 1). The molecular events that drive platelet surface adhesion, aggregation and thrombus formation are critical to platelet function in physiology as well as pathology, as platelet activation is a contributing factor to atherothrombosis, sepsis, and cancer as well as other disease states [2–4]. Cellular biological paradigms of signal transduction have refined the understanding of the molecular basis of platelet regulation as a crosstalk between phosphorylation-, cyclic nucleotide-, calcium-, lipid-based signaling pathways that commit platelets to an activated state through rapid changes in the platelet cytoskeleton [5]. These systems that control platelet function are interdependently coordinated by a number of molecular mediators that integrate, amplify and regulate intracellular signals, including GTP-binding proteins, or small GTPases.
Figure 1. Platelet filopodia and lamellipodia formation.
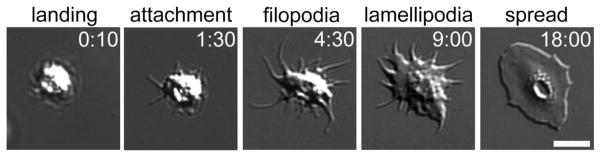
As platelets land upon and attach to surfaces of adhesive proteins, such as fibrinogen, they spread upon surfaces by first forming actin-rich, finger-like filopodia (examples indicated with white arrow) which are then filled in by actin-rich sheets of lamellipodia (example indicated by white arc). Rho GTPase proteins RhoA, Cdc42 and Rac1 have specific roles in these actin-driven processes. Visualized by DIC microscopy [170]. Times shown in [min:sec]. Scale bar = 5 μm.
Small GTPase proteins serve as signaling mediators in the control of diverse cellular processes [6, 7]. These intracellular mediators of signal transduction are homologous to the Gα subunits of heterotrimeric G proteins that couple to G protein coupled receptors (GPCRs) and similarly bind GTP to promote an interaction with specific effectors, and in turn activate downstream signaling events (Fig. 2). Like Gαs, the intrinsic GTPase activity of small GTPases leads to the hydrolysis of GTP to GDP, serving as a molecular and temporal “on-off” switch in the control of effector activation and function. Small GTPases differ in part from Gαs in that they lack intrinsic GTPase activating domains, which accounts for their “small” molecular weights in the range of 21 kD, and their slower GTP hydrolysis rates. A fortunate convergence of findings in the cell biology, virology and oncology fields described the rat sarcoma virus gene product, Ras, as the first small GTPase protein, linking retrovirus gene expression to aberrant GTPase signaling, cellular proliferation and oncogenesis [8, 9]. A quest to identify other Ras-like genes led to the discovery of a set of Ras homolog, or Rho, GTPase proteins first in marine snails [10, 11] and subsequently in yeast [12], where the Rho homologous Cdc42 GTPase was found to be required for budding and cell polarity [13–15]. Unlike the Ras GTPase, the Rho proteins were found to be targets of the Clostridium botulinum C3 transferase enzyme, which modifies and inactivates Rho GTPases by ADP ribosylation at a specific, conserved asparagine residue to promote microfilament disassembly [16, 17]. The roles of the Rho proteins in actin cytoskeletal dynamics became further evident in early experiments showing that botulinum C3 enzyme induced a loss of actin stress fibers in epithelial cells through Rho modifications [16]. Similar experiments led to the discovery of Rac, a Ras-related C3 botulinum toxin substrate and small GTP binding protein that was noted to be abundant in platelets [18, 19]. While over 20 Rho family members have been identified in mammalian cells, RhoA, Cdc42 and Rac have held the spotlight as central players in cellular functions including cell division, exocytosis and actin cytoskeletal dynamics [20, 21].
Figure 2. Rho GTPase activation and regulation.

In their GTP-bound states, the Rho GTPases, including RhoA, Cdc42 and Rac1, associate with specific downstream effectors to regulate cytoskeletal remodeling events and platelet function. RhoA, Cdc42 and Rac1 are cyclically regulated by specific GEF and GAP proteins. GEFs promote GDP to GTP exchange to activate Rho GTPases. GAP proteins accelerate the hydrolysis of Rho-GTP to GDP and effectively inhibit Rho GTPase activation. Rho proteins may also be regulated by RhoGDI proteins, which “grab” and “release” Rho GTPases to sequester their activities [153].
Initial studies of Rho GTPases in platelet function alluded to their important functional roles in platelet cytoskeletal rearrangements [22, 23]. Accordingly, platelets have served as an important model for studies of Rho GTPase function owing to their conserved actin pathways [24, 25]. Studies of the Rho GTPases have fueled discoveries central to the functional biology of platelets and hemostasis (Table 1). In platelets, RhoA plays a role in actin contractility and contributes to platelet shape change upon activation as well as thrombus stability [26–29]. Rac1 controls the formation of platelet lamellipodia [30–33]. The specific roles of Cdc42 in platelet function are controversial [34, 35]; however, Cdc42 activation is associated with platelet filopodia formation and granule secretion [25, 36]. Despite these generalized functions of the Rho GTPases in specific steps of platelet activation, an interdependent model of Rho function in platelet physiology is emerging that integrates a crosstalk amongst integrins, GPCRs and Rho proteins in platelet cytoskeletal regulation, aggregation, secretion, thrombus formation and other cellular processes.
Table 1.
Summary of physiological functions reported for Rho GTPases in platelets from selected mouse model and inhibitor studies.
| Rho GTPase | Function | Reference |
|---|---|---|
| RhoA | Platelet production | [28] |
| Platelet shape change (U46619 stimulation) | [27, 28] | |
| Platelet aggregation (U46619 or PAR-4 stimulation) | [28] | |
| Platelet integrin αIIbβ3 activation (U46619+ADP or thrombin stimulation) | [28] | |
| Platelet granule secretion (U46619+ADP or thrombin stimulation) | [28] | |
| Regulates platelet rolling velocity on vWF | [28] | |
| Maintains strength of αIIbβ3 – vWF contacts under shear | [29] | |
| Clot retraction | [28] | |
| Thrombus formation in vivo | [28] | |
| Hemostasis in vivo | [28] | |
| Pathology of cerebral ischemia in vivo | [28] | |
| Cdc42 | Platelet production | [34, 35] |
| Regulates platelet life span | [35] | |
| Platelet filopodia formation (on vWF surface) | [35] | |
| Platelet spreading (on collagen or fibrinogen) | [34] | |
| Platelet granule secretion | [34] | |
| Platelet ATP secretion | [34] | |
| Regulates secretion output | [35] | |
| Platelet aggregation (collagen or thrombin stimulation) | [34] | |
| Controls platelet aggregate size on collagen | [35] | |
| Hemostasis in vivo | [34] | |
| Regulates rate of thrombus formation | [35] | |
| Rac1 | Platelet aggregation (collagen or collagen peptide stimulation) | [30, 32, 71, 84] |
| Platelet aggregation (thrombin stimulation) | [30, 116] | |
| Platelet aggregation (ADP or U46619 stimulation) | [30] | |
| Platelet aggregation (vWF stimulation) | [86] | |
| Platelet spreading on FG, collagen or laminin | [32] | |
| Platelet αIIbβ3 activation (collagen stimulation) | [84] | |
| Platelet αIIbβ3 activation (vWF stimulation) | [86] | |
| Platelet ATP secretion (thrombin stimulation) | [30] | |
| Platelet granule secretion (collagen peptide, convulxin or ristocetin stimulation) | [84] | |
| Platelet granule secretion (thrombin stimulation) | [30, 116] | |
| Calcium mobilization (collagen peptide stimulation) | [84] | |
| PLCγ2 activation (convulxin stimulation) | [84] | |
| Thromboxane synthesis (vWF stimulation) | [86] | |
| Aggregation on collagen surface under flow | [32, 84] | |
| Aggregation on vWF surface under flow | [32, 86] | |
| Clot Retraction | [31] | |
| Thrombus formation in vivo | [32, 84] | |
| Hemostasis in vivo | [30] |
RhoA, platelet contractility and thrombus stability
The highly homologous Rho GTPase proteins RhoA, RhoB and RhoC are best known for their roles in focal adhesion formation and actomyosin contractions, as microinjection or overexpression of these Rho isoforms in fibroblasts promotes actin stress fiber formation [11, 37]. Experiments using the clostridial enzyme C3 transferase, which inhibits these three Rho isoforms through ADP ribosylation, has helped to identify Rho protein family members and define their functions in a number of cell types, including platelets [22, 23, 38]. RhoA is likely the most highly expressed Rho isoform in human platelets [23, 39, 40] and is most noted for its general role in actomyosin contractility to drive changes in shape from a quiescent discoid shape to small spheres upon activation [5, 27, 28]. In platelets, RhoA works downstream of a number of G-protein coupled receptor pathways to control platelet shape, as RhoA maintains platelet sphericity and promotes contractility, contributing to platelet shape change upon activation (Fig. 3).
Figure 3. RhoA in platelet shape change, spreading and clot retraction.
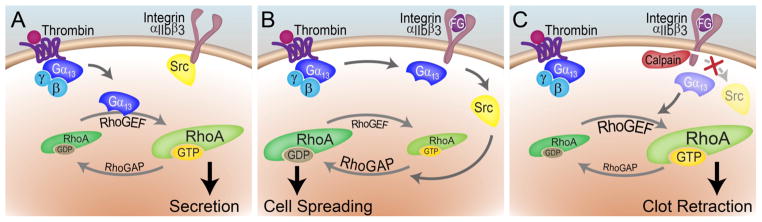
RhoA activation and inactivation drives separate steps of platelet activation. (A) Upon activation with thrombin, Gαq activates p115RhoGEF to promote RhoA-GTP formation, ROCK activation and MLC phosphorylation to drive platelet shape change and secretion events. (B) Gαq later supports the activation of c-Src through an association with the cytosolic domain of integrin αIIbβ3. c-Src activates p190RhoGAP to stimulate RhoA-GTP to GDP hydrolysis to effectively shut down RhoA contractile activity and facilitate platelet spreading. (C) Later, calcium signaling activates the calpain protease which cleaves integrin β3, resulting in the loss of c-Src activity, driving platelet contraction and clot retraction [5, 29, 51, 54].
Over the past decade, multidisciplinary studies in the GPCR, integrin and platelet biology fields have synergistically established a model of how RhoA mediates platelet function from the levels of protein structure to thrombus formation in vivo [5, 41]. The Rho proteins were first suspected as regulators of platelet function after studies demonstrated that C3 enzyme blocked platelet function and that RhoA was a major C3 target in platelets [22, 23]. An investigation of GPCR signaling pathways downstream of thromboxane A2 (TxA2) receptors first found that upon stimulation with the prostaglandin PGH2 analog and thromboxane mimetic, U46619, platelet Gαq activates PLC to drive lipid and calcium signaling. While Gαq-deficient platelets fail to aggregate or release granules upon agonist stimulation, they still undergo a characteristic shape change upon stimulation [42]. However, Rho inhibition with C3 enzyme or the ROCK inhibitor, Y-27632, blocks this shape change and actin polymerization in Gαq-deficient systems in which Gα12/Gα13 are active [27]. These classic studies were the first to show that stimulation of platelets with U46619 in Gαq-deficient systems leads to the activation of Gα12/Gα13 and MLC phosphorylation and shape change in a Rho and ROCK dependent manner. In its active form, RhoA-GTP binds to and activates ROCK to drive actin remodeling events [43]. Once active, ROCK phosphorylates and inactivates the myosin light chain (MLC) phosphatase to result in a net increase in MLC phosphorylation [44, 45] and the subsequent actomyosin contractions that promote a change in platelet shape from discoid to spheres [27, 46]. In addition to the Gα13-Rho-ROCK pathway, MLC phosphorylation is also promoted through Gαq signaling, which stimulates MLC kinase activation and MLC phosphorylation directly [47].
Platelets express both Gα12 and Gα13; however, Gα12-null platelets have limited phenotypic alterations and thus Gα13 has emerged as a dominant player in platelet RhoA activation downstream of GPCR stimulation [48, 49]. In its GTP-bound form, Gα13 directly activates a specific guanine nucleotide exchange factor (GEF), p115RhoGEF, which promotes the activation of RhoA through its Gα13 switch region 1 (SRI). This switch region of Gα13 interacts with p115RhoGEF to mediate Rho activation [50] and is critical for protease-activated receptor (PAR) mediated platelet shape changes and aggregation [50]. The RhoA-ROCK-MLC axis is cyclically targeted through a number of steps in the platelet activation pathway to temporally mediate cell spreading, thrombus stability and clot retraction (Fig. 3). In the initial stages of platelet activation, the coordinated inhibition of the MLC phosphatase and activation of MLC kinase is thought to promote platelet secretion and rounding (Fig. 3A). After this first wave of platelet RhoA activation, Gα13-GPCR-integrin crosstalk, in which Gα13 binds to the cytosolic domain of integrin β3, mediates Src family kinase (SFK) activation and platelet spreading through RhoA inhibition (Fig. 3B) [5, 51]. In this step, Gα13 associates with the cytosolic domain of integrin β3 to promote the activation of c-Src. c-Src then phosphorylates and activates p190RhoGAP, a GTPase activating protein that stimulates Rho-GTP to GDP hydrolysis and RhoA inactivation as platelets spread upon a surface [52]. The cytosolic domain of integrin β3 that activates c-Src is targeted by the calcium-dependent calpain protease [26, 31]. After platelets mobilize calcium from intracellular stores during the early stages of platelet activation, calpains eventually cleave integrin β3 to effectively inhibit c-Src activation to again promote contractility and clot retraction (Fig. 3C) [26]. In accordance with this model, compared to wild type platelets, platelets from calpain−/− mice show temporal alterations in RhoA and Rac1 activation as they spread on extracellular matrix proteins [53]. In addition to shape changes, spreading and clot retraction, RhoA-mediated platelet contraction may also have a role in thrombus stability under shear flow conditions, as RhoA and ROCK stabilize and maintain platelet-matrix and platelet-platelet interactions [29, 54].
Studies of megakaryocyte/platelet-specific RhoA-deficient mice have confirmed that RhoA has roles in the platelet shape change, Gα13-mediated GPCR crosstalk activation of integrin αIIbβ3, granule secretion, clot retraction and thrombus formation and stability [28]. Platelets from RhoA-null mice have impaired shape changes and mild aggregation deficits downstream of Gα13 as well as Gαq activation. While RhoA is not required for platelets to spread upon a surface of the integrin αIIbβ3 substrate fibrinogen, RhoA is required for full integrin activation and granule secretion in response to thrombin, or combined ADP and U46619 stimulation, as well as for clot retraction [28]. These in vitro observations translate into in vivo functions as RhoA-deficient mice show reduced thrombus formation, prolonged tail-bleeding times and partial protection from ischemic stroke [28].
Cdc42 in platelet filopodia formation and granule secretion
Since its discovery as a Rho-related GTP-binding protein and mediator of budding and cell polarity in yeast [14, 15, 55], Cdc42 has become established as a generalized regulator of filopodia formation [21, 56, 57] as well as exocytosis and secretion [58, 59]. While Cdc42 activation is commonly associated with microspike and filopodia formation at the periphery of a number of cell types [56, 57, 60], including platelets [34, 36], the requirement of Cdc42 in filopodial formation is not clear. Some studies have reported filopodia formation in Cdc42-free or Cdc42-inhibited conditions [61–63], including studies of platelets from Cdc42 megakaryocyte/platelet deficient mice [35]. In contrast, other studies have shown that Cdc42 has very specific roles in platelet filopodia formation and plays a critical part in granule secretion [34].
While Cdc42 activation is associated with platelet spreading and secretion, the specific roles of Cdc42 in platelet biology remain vague. Early experimental observations supported the hypothesis that Cdc42 would play critical roles in platelet filopodia formation, as Cdc42 was found to be abundant in platelets [15]. Upon stimulation with thrombin receptor activating peptide (TRAP) or ADP, Cdc42 translocates to the platelet cytoskeleton in an integrin-dependent manner, also requiring actin polymerization and tyrosine kinase activation [25]. Cdc42 is activated by PAR stimulation in solution [64, 65] and also as platelets spread on collagen [66]. Treatment of platelets with the Cdc42-specific inhibitor, secramine A, blocks platelet adhesion on collagen, and collagen-induced aggregation [36].
To date, two separate approaches to characterizing the roles of platelet Cdc42 in knockout systems of mouse megakaryocyte/platelet or hematopoietic cells have provided varied results regarding the roles of Cdc42 in platelet function [34, 35]. The reasons for these inconsistent conclusions are not clear but may be due to the different knockout strategies employed to eliminate Cdc42 expression by gene excision in hematopoietic cells after the administration of multiple doses of polyinosinic acid-polycytidylic acid [34, 67] versus a Cre-recombinase based model that targets Cdc42 in megakaryocytes and platelets directly through the platelet factor 4 (Pf4) promoter during megakaryocyte maturation [35, 68].
A study of mice with conditional excision of the Cdc42 gene under the Pf4 promoter in megakaryocyte lineage specific cells suggests that the role of Cdc42 in platelet biology is complex [35]. Consistent with a role for GPIb upstream of Cdc42 activation in the formation of filopodia, Cdc42-null platelets from these mice show a reduced ability to form filopodia on a surface of the GPIb ligand, von Willebrand Factor (vWF), but not on surfaces of collagen-related peptide (CRP) or fibrinogen, suggesting that Cdc42 has a specific role in platelet filopodia formation downstream of GPIb but not GPVI or integrin αIIbβ3. Unlike the requisite role of Cdc42 in secretory processes in nucleated cells, Cdc42 deletion was found to increase granule secretion in response to low doses of U46619, CRP or collagen, but showed no differences in aggregation in response to thrombin. Consistent with this increased secretion, Cdc42−/− platelets formed larger aggregates when flowed over collagen and increased thrombus formation in vivo. However, although erratic, tail bleeding times were generally increased in these Cdc42−/− mice.
Studies of conditional knockout mice lacking Cdc42 in hematopoietic cells, including platelets, have shown results more consistent with the hypothesized functions of Cdc42 in filopodia formation [34]. Platelets from these mice showed reduced phosphorylation of the Cdc42 effector PAK in response to stimulation with thrombin. Such Cdc42-deficient platelets demonstrated reduced filopodia formation and spreading on surfaces of fibrinogen and CRP as well as reduced secretion, Akt phosphorylation, and aggregation. These Cdc42-null mice also displayed prolonged tail-bleeding times. Despite the phenotypic disparities between the two described knockout models of platelet Cdc42 function, both studies demonstrate that Cdc42 deletion leads to reduced platelet counts, suggesting that Cdc42 plays a key role in platelet production.
Rac regulates platelet lamellipodia formation
Since their initial discovery as substrates of botulinum C3 enzyme and regulators of actin dynamics [18], the Rac proteins have been an active topic of investigation in platelet biology. In nucleated cells, the Rac proteins are known for roles in lamellipodia formation, as dominant-negative Rac mutants inhibit lamellipodia extension, membrane ruffling and cell migration in a variety of cell types [21]. Of the Rac1, Rac2 and Rac3 isoforms, only Rac1 is expressed in platelets [32]. Early studies of Rac in platelet function found that Rac is activated in platelets upon stimulation with a number of agonists, including collagen and thrombin [69], and surfaces such as fibrinogen [66]. Similar to Cdc42, Rac translocates to the platelet cytoskeleton upon activation [25, 64]. Inhibition of Rho GTPase signaling with clostridial toxin B blocks the activation of the Rac/Cdc42 effector PAK and platelet lamellipodia formation [65], while inhibition of RhoA signaling with the ROCK inhibitor Y-27632 reduced platelet stress fiber formation but not the degree of platelet spreading [54], providing the first suggestion that Rac or Cdc42 would play critical roles in platelet lamellipodia formation [66].
A specific role for Rac in platelet lamellipodia formation and function was established through studies of mice with hematopoietic cell deletion of Rac1. Rac1 is required for lamellipodia formation as platelet spread upon surfaces of fibrinogen, vWF, laminin and collagen [32, 70]. Rac is also required to maintain the stability of platelet aggregates formed under physiological conditions of shear as well as for thrombus stability at sites of injury in vivo. Experiments taking advantage of the Rac-specific inhibitor, NSC23766, as well as genetic models of Rac activation show that Rac has a role in granule secretion and PAK activation downstream of thrombin or U46619, as well as in aggregation in response to ADP, collagen, thrombin and U46619 [30]. Pharmacological inhibition of Rac with a separate Rac-specific inhibitor, EHT 1864, also provides evidence for Rac function downstream of platelet integrin αIIbβ3 activation as well as platelet aggregation in response to collagen and platelet aggregate stability under flow in vitro [71]. Rac, but not ROCK, activation is also required for platelet integrin-mediated p38 and ERK phosphorylation, suggesting that in addition to roles in cell spreading, Rac may also have a role in clot retraction via MLC phosphorylation through p38 and ERK, independent of the Rho and ROCK pathway [31].
The mechanism by which platelet agonists activate Rac is not well defined, but may be similar to Cdc42 activation in that it is similarly regulated by the release of 14-3-3ζ from the glycoprotein GPIb-IX subunit of the adhesion receptor GPIb (Fig. 4) [72]. The exact manner by which 14-3-3ζ mediates Rac activation is not yet understood, but may involve recruitment of the Rac GEF and T-cell Lymphoma Invasion and Metastasis protein TIAM1 to membranes [73] or 14-3-3 dimerization [74]. Like RhoA, Rac1 activation by thromboxane A2 (TxA2) occurs in an αIIbβ3-independent manner [75]. Unlike RhoA, which is activated by Gα13, Rac is activated by Gαq and is not required for platelet shape change [75]. In nucleated cells, Rac can also be activated through Gi as well as through Gβγ activation of PI3K [76, 77]. In platelets, however, activation of Gi alone is not sufficient for Rac1 activation, as thrombin, ADP, or combined ADP and U46619 treatments do not activate Rac in Gαq-deficient platelets. However, full activation of Rac requires direct Gαq signaling as well as a Gαq-mediated release of Gi coupled receptor agonists (Fig. 4) [75, 78].
Figure 4. Regulation of platelet Rac1 activation and lamellipodia formation.
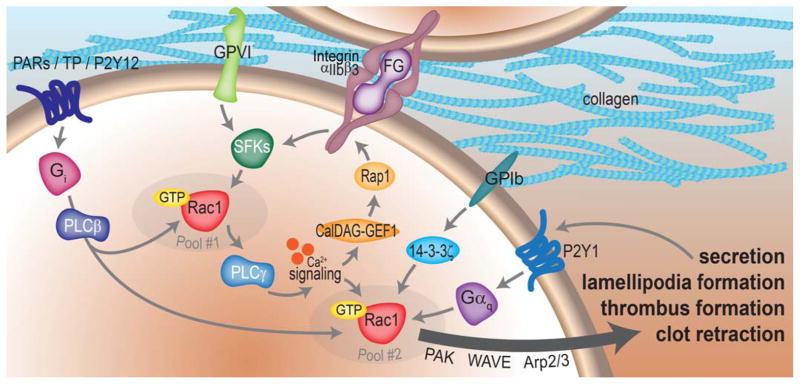
Upon stimulation of platelet GPCRs such as the PARs, TPs or P2Y12, Gαq mediates the activation of Rac1 through PLCβ [42]. Integrin- and GPVI-coupled Src family kinase (SFK) activation also supports Rac1 activation [30, 32, 71]. GPIb release of 14-3-3ζ also has a role in platelet Rac1 activation [72]. Gi coupled to P2Y1 receptors is also required for full platelet Rac1 activation [78]. Separate pools of Rac1 may work in distinct steps of the platelet activation process. A first pool proximal to SFK-coupled receptor activation drives PLCγ activation and calcium signaling to activate CalDEG-GEF1 and the Rap1 GTPase to support integrin αIIbβ3 activation [33, 84]. A second pool of Rac1 downstream of PLCγ and calcium signaling then supports secretion, aggregation and spreading events through the activation of Rac effectors, such as the WAVE and Arp2/3 system or the PAK kinases.
While Rac has a role in platelet PI3K activation, PI3K activation is also required for Rac to become completely active, supporting a model of platelet activation in which Rho GTPases regulate multidirectional, feed-forward and feed-back signaling between receptors, phospholipase enzymes, calcium signaling systems and PI3Ks to control platelet lamellipodia formation, secretion and thrombus stability (Fig. 4) [79]. In agreement with such a multi-modal model of platelet Rac1 function, platelet Rac1 activation is calcium dependent [69, 75]. Intracellular calcium signaling plays a key role in platelet activation, adhesion, secretion, and thrombus formation [5, 80, 81]. Platelets use two major pathways to elevate intracellular calcium. Soluble agonists (ADP, thrombin, TxA2) stimulate GPCRs coupled to Gαq to stimulate phospholipase Cβ (PLCβ) [42]. Alternatively, PLCγ2 is activated downstream of immunoreceptor tyrosine activation motif (ITAM) receptors GPVI or CLEC-2, which are receptors for collagen or rhodocytin, respectively [82]. PLCs generate inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 triggers calcium mobilization from internal stores; DAG activates PKC which may also contribute to calcium entry from non-internal stores. Rac may play a role in the activation of PLCγ2 through a mechanism independent of tyrosine phosphorylation [83]. In platelets, Rac1 is required for PLCγ2 activation downstream of GPVI/ITAM, but not for PLCβ activation downstream of PARs (Fig. 4) [84, 85]. Accordingly, Rac1 is required for GPVI-mediated platelet aggregation, αIIbβ3 activation, granule secretion, and ADP secretion downstream of calcium mobilization but plays only a minor role in thrombin responses. Rac1 is also required for vWF-induced activation of αIIbβ3 upstream of PI3K/Akt activation to drive platelet responses to GPIb-IX engagement [86]. Many of the observed defects in thrombus formation in Rac-deficient platelets can be rescued with exogenous ADP and U46619 [84], supporting a role for Rac1 in exocytosis and secretion [87, 88]. Consistent with platelet Rac signaling upstream and downstream of both calcium and PI3K signaling, a separate pool of Rac1 has recently been proposed to work immediately downstream of GPVI receptor activation to regulate CalDAG-GEF1- and P2Y12-dependent activation of the non-Rho, Ras-related small GTPase, Rap1, leading to integrin αIIbβ3 activation [33]. A second pool of Rac1 downstream of PLCγ and calcium signaling regulates platelet secretion, aggregation and spreading events through the activation of Rac effectors such as the WAVE and Arp2/3 system or the PAK kinases (Fig. 4) [33].
Actin reorganization downstream of Rho GTPase activation
The current paradigms of Rho GTPase action in the mediation of actin remodeling, filopodia extension and lamellipodia formation rely on the activation of the actin related protein Arp2/3 complex [21] and the formin proteins. Through this model, Cdc42 mediates filopodia formation through the Wiskott–Aldrich syndrome proteins WASP and neuronal N-WASP to induce actin branching via Arp2/3 [89–91]; Rac activates Arp2/3 to form lamellipodial structures through the WASP-family verprolin-homologous WAVE complex proteins [92, 93]. Rho GTPases also activate formin proteins, including mDia and Daam, to nucleate, elongate and bundle actin fibers in the control of cell spreading, motility and division [94]. In this regard, RhoA mediates actin assembly through the formins mDia1, Daam [95] and FHOD1 [96] while Rac works through mDia2 [11].
Early studies of platelet Rac1 implicated Rho GTPases in processes of platelet actin elongation, as exogenous Rac protein stimulated phosphoinositide synthesis and actin filament uncapping in permeabilized platelets [97]. In platelets, Rac1 interacts with the phosphatidylinositol kinase, PIP5K, which synthesizes phosphatidylinositol-4,5-bisphosphate, or PI(4,5)P(2), a lipid that dissociates capping proteins such as gelsolin from actin filament barbed ends [98, 99]. This uncapping process has a role in platelet Arp2/3 complex activity at the barbed ends of actin filaments [100, 101]. Platelets express WAVE-1, WAVE-2 and low levels of WAVE-3 [102]. Like Rac1-deficient platelets, lamellipodia formation and aggregation in response to GPVI agonists including CRP and laminin are disrupted in Scar/WAVE-1-null platelets [103]. These findings suggest that platelet spreading may be driven by a Rac-WAVE-Arp2/3 axis rather than Cdc42-WASP, as platelets deficient in WASP activate Arp2/3 normally and have no deficits in actin cytoskeletal function, spreading, lamellipodia formation and aggregation [102, 104].
A serial analysis of gene expression (SAGE) library study of formin RNA expression in megakaryocytes found that six of the 15 mammalian formins (mDia1, mDia2, Daam1, Fmnl1, Fmnl3 and FHOD1) are present in mouse megakaryocytes, as well as human megakaryocytes, which also contain mDia3 [96]. Notably, FHOD1 was found to be the most highly expressed formin in megakaryocytes. FHOD1 is activated by ROCK phosphorylation in platelets in a RhoA-dependent and Rac1-independent manner by thrombin, CRP, or a combined ADP and U49919 treatment, but not in platelets on a surface of fibrinogen [96]. FHOD1 has a role in actin stress fiber formation following thrombin treatment of endothelial cells [105]; however, a similar function of FHOD1 in platelets remains to be confirmed.
A number of downstream effectors of Cdc42 have been identified in non-platelet systems with roles in filopodia formation. For instance, Cdc42 activates IRSp53, an insulin receptor substrate protein known to mediate actin bundling and filopodia formation [106, 107]. The role of IRSp53 in platelet physiology has not yet been determined. Given the lack of a requirement for WASP in platelet Arp2/3 activation [108] and the minimal role of N-WASP in platelet physiology due to its low levels of expression [108, 109], platelet filopodia formation may be regulated by other mechanisms, perhaps involving Rif [110], a newly characterized Rho GTPase in filopodia that works independently of Rac, Cdc42 and Arp2/3 to drive filopodia formation through mDia1 [62] and mDia2 [63]. In cultured cells, Cdc42 and Rac can also mediate actin microspike formation downstream of thrombospondin stimulation through the actin bundling protein fascin [111]. Interestingly, Rac plays a role in controlling the interaction of PKC with fascin in cell migration [112]. The role of Rho GTPases in fascin-mediated actin processes in platelets has not yet been defined.
The PAKs as Rho GTPase effectors
The ~21 kD Rho GTPases Cdc42 and Rac support the autocatalytic activation of the p21 activated kinases, or PAKs, to mediate actin reorganization processes in focal adhesion formation and cell migration [113]. The PAKs represent perhaps the best characterized effectors of Rac and Cdc42 [114]. Upon activation by GTP-bound Rac or Cdc42, the PAKs phosphorylate a number of substrates to coordinate adhesion complex formation and actin dynamics [114]. Interestingly, PAK2, the most abundant and ubiquitously expressed PAK isoform [114], was originally described as a thrombin-activated kinase in platelets [115]. Platelets express a number of PAK isoforms [116], and like Rac, PAK is activated as platelets spread on collagen in a Src- and PI3K-dependent manner [65, 66]. In platelets, PAK acts downstream of Rho GTPases to regulate the intracellular distribution of cortactin [65], an established regulator of actin polymerization that associates with the Arp2/3 complex and has role in actin polymerization in lamellipodial structures [117, 118]. The adaptor protein SLP-76 has also been proposed to potentiate PAK activity downstream of Rac activation to mediate platelet lamellipodia formation [119]. However, the exact roles of PAKs in platelet function have not yet been characterized.
A notable PAK effector linked to platelet function is LIM domain kinase, LIMK1, which phosphorylates and inactivates the actin binding protein cofilin to regulate actin assembly and disassembly [120]. Stimulation of platelets with thrombin causes a rapid dephosphorylation of cofilin [121] that is associated with platelet secretion [116] but not with the platelet shape change [122]. However, platelet cofilin regulation is complex, as cofilin is rephosphorylated minutes following its dephosphorylation [121]. LIMK1 activity is regulated by PAK phosphorylation of LIMK1 in a Rac- and calcium-dependent manner that does not require PKC or PI3K activation [116]. Platelet LIMK1 activity is also regulated by RhoA [122]. It has been proposed that RhoA activation of LIMK has a role in the platelet shape change independent of cofilin phosphorylation, while calcium-dependent LIMK signaling regulates platelet secretion and aggregation [123]. Hence, the LIMK-cofilin system may represent an important coordinator of RhoA as well as Cdc42/Rac1-PAK signaling in multiple stages of the platelet activation process.
Other PAK substrates of potential significance to platelet function include the G protein-coupled receptor kinase interactor GIT and the PAK interacting exchange factor PIX proteins, which lie both upstream and downstream of Rac and Cdc42 activation in the regulation of actin processes [114, 124]. Upon integrin engagement, platelet GIT1, is phosphorylated in a Src-dependent manner and may play a role at recruiting βPIX to areas of active actin remodeling [125]. The functions of GIT and PIX in platelet actin dynamics have not yet been defined; however, a computational analysis of proteomics data suggests a role for GIT proteins in platelet function [126]. Moreover, in endothelial cells, GIT1 regulates focal adhesion assembly and cell contractility in response to thrombin stimulation [127]. PIX and GIT proteins are PAK effectors downstream of Rac activation but also serve as GAPs, respectively, in the regulation of Rac activation. Accordingly, GIT and PIX may cyclically regulate feed-back and feed-forward waves of Rac and PAK activation at sites of active actin remodeling in platelets and other cells [128].
Rho GEFs, GAPs and GDIs in platelet function
A number of intracellular regulatory mechanisms control Rho GTPase function. Guanine nucleotide exchange factors, or GEFs, interact with Rho GTPases to catalyze the exchange of GDP for GTP to activate GTP binding proteins (Fig. 2 and reviewed in [129]). While over 60 different GEFs for Rho family and other GTPases have been identified in mammalian cells, few have been examined for expression or function in platelets. Notably, as described above, upon platelet activation, the RhoA GEF p115RhoGEF is activated by Gα13 directly to mediate RhoA and subsequently ROCK activation to promote platelet shape changes (Fig. 3).
To date, no Cdc42-specific GEFs have been described with roles in platelet function. The Vav proteins represent perhaps the best-characterized Rac GEFs in platelet function. Vav was originally identified as a protooncogene from hematopoietic cells [130]. The three Vav isoforms (Vav1, Vav2 and Vav3) have GEF activities towards Rac, Cdc42 and Rho proteins, with preference for Rac. Vav proteins have been shown to mediate several cytoskeletal-associated cellular processes downstream of receptor activation and tyrosine kinase signal transduction. Thrombin-, collagen- and integrin-mediated platelet activation, but not ADP or thromboxanes, activate Vav in platelets [131]. Vav1, unlike Vav2, has a role in Rac activation in response to GPVI agonists but not thrombin [132]. Double knockout mouse studies have shown that Vav1 and Vav3 together have redundant roles in platelet activation, as platelets from Vav1−/−/Vav3−/− knockout mice show defects in platelet spreading on fibrinogen [133] and aggregation in response to GPVI agonists such as collagen [134].
In addition to Vav, platelets also express the Rac GEF TIAM1, which localizes to the leading edge of platelets spread on fibrinogen surfaces [71]. Platelets from mice deficient for expression of the phosphatidylinositol 3,4,5-trisphosphate-dependent Rac exchanger 1 (P-Rex1) show that the Rac GEF P-Rex1 is not required for platelet spreading on surfaces of fibrinogen, collagen or thrombin [135]. However, P-Rex1 has a role in secretion and aggregation, suggesting that the roles of Rho GEFs in platelet physiology are complex [136]. Notably, P-Rex1 links the mammalian target of rapamycin, or mTOR, signaling network to Rac regulation and the control of cell motility [137]. Traditionally, mTOR serves as a master orchestrator of a number of cellular signaling systems, linking cellular homeostasis to protein translation and cellular growth [138]. Platelet express mTOR, which has a role in platelet function upstream of Rac [71, 139] and protein translation events associated with clot retraction [140]. A number of studies show that mTOR activation is under the control of small GTPases, including Rac [141, 142]. Other studies show a role for mTOR in the control of cell motility, specifically through the regulation of Rho GTPase activity [143, 144]. Given the emerging co-dependent functions of mTOR and small GTPases in the regulation of diverse cellular activities, the mTOR system may emerge as centralized nexus of Rho GTPase regulation in platelets [139]. In addition, the mTOR-Rac axis has roles in platelet function, as inhibition of mTOR prevents platelet Rac activation downstream of αIIbβ3, blocks platelet aggregation in response to collagen, and has a role in platelet aggregate stability [71]. These effects may be specific to integrin and glycoprotein receptor signaling, as mTOR inhibitors have no effect on platelet aggregation in response to PAR agonists [145] but inhibit αIIbβ3-mediated clot retraction [140]. In addition to mTOR, other proteins with more traditional roles in gene expression and disease are also emerging as Rho GTPase regulators in platelets. For instance, Wnt-3a was recently shown to play a role in Rho GEF activation through Dvl and Daam [146, 147].
While Rho GEFs promote Rho-GTP binding and activation, Rho GTPases are inactivated by specific GTPase activating proteins, or GAPs, that accelerate the hydrolysis of GTP to GDP to inactivate GTPase signaling (Fig. 2) [148]. Platelets have served important roles in the history of GAP studies, as the first RhoGAP for Cdc42 was originally discovered using platelet as a model cellular system [149]. While over 70 RhoGAPs have been identified, the roles of RhoGAPs in platelet function remain mostly uncharacterized [148]. The Rho GAP p190RhoGAP has a role in contractility in the control of clot retraction (Fig. 3). Emerging evidence suggests that other RhoGAPs may have roles in platelet function. Thrombin, but not collagen treatment, leads to the translocation of the Rac/Cdc42 IQ-domain containing GAP IQGAP2 to the cytoskeleton of platelet filopodia [150]. Oligophrenin1 (OPHN1) as well as Nadrin - Rho GAPs that activate Rac, Cdc42 and Rho GTPase activity - have also been proposed to regulate platelet function [151, 152].
In addition to GEFs and GAPs, Rho GTPases are also held in check by specific Rho- guanine nucleotide dissociation inhibitor RhoGDI proteins that act as an “invisible hand” in the regulation of total cellular Rho GTPase activity (Fig. 2) [153]. As Rho GTPase binding proteins, the RhoGDIs regulate Rho GTPase function by sequestering Rho GTPases to specific intracellular locations, regulating GTPase enzymatic activity and also controlling Rho GTPase expression. Like Rho GAPs, the first RhoGDI discovered for Cdc42 was also first found using platelets [154, 155]. The roles of RhoGDIs in platelet function remain unexplored.
Future perspectives
Platelets have served as an ideal non-transformed system in the studies of signaling processes independent of nuclear events, and will continue to do so as novel paradigms of Rho GTPase regulation develop. For instance, recent studies have shown that Rho GTPases and many Rho regulators are targets of micro RNAs (miRNAs) [156]. miRNAs have been increasingly recognized as important regulators of platelet function and hemostasis [157]. Given the abundance of Rho-related RNA transcripts in platelets [40] and the roles of RNA translation in platelet function [158], real-time miRNA regulation of Rho GTPase expression and activity may emerge as an important mechanism of platelet physiological function, and platelets may serve as a model cell for the study of miRNA regulation of cytoskeletal dynamics.
The roles of the Rho GTPases in regulating non-actin based platelet cytoskeletal structures is less known. For instance, as platelets activate, a ring-like marginal band of tubulin at the platelet periphery that holds platelet shape disappears to allow for the platelet shape change [24]. Studies from nucleated cells suggest a close relationship between Rho GTPases and tubulin dynamics in cell motility [159, 160], but the relation of Rho signaling to microtubule dynamics in platelets remains unclear. Interestingly, RhoA−/− platelets show altered tubulin structures [28]. Actin and tubulin dynamics play a critical role in platelet formation by megakaryocytes [161, 162]. Accordingly, Rho GTPases have been and will continue to be a topic of investigation in platelet biogenesis and may reveal insights into roles of cytoskeletal dynamics in hematopoesis and hematological disease [163, 164].
Conclusions
Since their discovery as mediators of intracellular signaling and cytoskeletal dynamics, the Rho GTPases have continually been demonstrated to play critical roles in actin mediated processes in a number of cellular systems, including platelets [11, 21, 82, 165]. Cell biological studies of the Rho GTPases have linked specific Rho GTPase signaling axes to the dynamics of platelet function over the course of platelet activation, thrombus formation, thrombus stability and clot retraction. Newly emerging methods for studies of Rho GTPase function that take advantage of single-molecule and single-cell systems combined with computational and “omics” tools will serve to define the link between platelet Rho signaling and platelet cellular mechanics [166–168]. These studies may lead to the development of Rho GTPases-based pharmacologic therapies for platelet-centric disease states [169].
Acknowledgments
We thank J. Gertz for technical assistance, T. Howard for illustration services, and K. Haley, S. Baker and C. Loren (OHSU) for critical reading of the manuscript. This work was supported in part by the NIH (T32-HL007781 to J.E.A. and R01-HL101972 to O.J.T.M.). J.E.A is a 2012–2013 Fulbright Scholar. The authors have no competing or financial interests to declare.
References
- 1.Furie B, Furie BC. Thrombus formation in vivo. J Clin Invest. 2005;115:3355–62. doi: 10.1172/JCI26987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson SP, Schoenwaelder SM. Antiplatelet therapy: in search of the ‘magic bullet’. Nat Rev Drug Discov. 2003;2:775–89. doi: 10.1038/nrd1198. [DOI] [PubMed] [Google Scholar]
- 3.Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002;8:1227–34. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- 4.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11:264–74. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 5.Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30:2341–9. doi: 10.1161/ATVBAHA.110.207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature. 1991;349:117–27. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 7.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 8.Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395–413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 9.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 10.Madaule P, Axel R. A novel ras-related gene family. Cell. 1985;41:31–40. doi: 10.1016/0092-8674(85)90058-3. [DOI] [PubMed] [Google Scholar]
- 11.Ridley AJ. Historical overview of Rho GTPases. Methods Mol Biol. 2012;827:3–12. doi: 10.1007/978-1-61779-442-1_1. [DOI] [PubMed] [Google Scholar]
- 12.Madaule P, Axel R, Myers AM. Characterization of two members of the rho gene family from the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1987;84:779–83. doi: 10.1073/pnas.84.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munemitsu S, Innis MA, Clark R, McCormick F, Ullrich A, Polakis P. Molecular cloning and expression of a G25K cDNA, the human homolog of the yeast cell cycle gene CDC42. Mol Cell Biol. 1990;10:5977–82. doi: 10.1128/mcb.10.11.5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson DI, Pringle JR. Molecular characterization of CDC42, a Saccharomyces cerevisiae gene involved in the development of cell polarity. J Cell Biol. 1990;111:143–52. doi: 10.1083/jcb.111.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polakis PG, Snyderman R, Evans T. Characterization of G25K, a GTP-binding protein containing a novel putative nucleotide binding domain. Biochem Biophys Res Commun. 1989;160:25–32. doi: 10.1016/0006-291x(89)91615-x. [DOI] [PubMed] [Google Scholar]
- 16.Chardin P, Boquet P, Madaule P, Popoff MR, Rubin EJ, Gill DM. The mammalian G protein rhoC is ADP-ribosylated by Clostridium botulinum exoenzyme C3 and affects actin microfilaments in Vero cells. EMBO J. 1989;8:1087–92. doi: 10.1002/j.1460-2075.1989.tb03477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sekine A, Fujiwara M, Narumiya S. Asparagine residue in the rho gene product is the modification site for botulinum ADP-ribosyltransferase. J Biol Chem. 1989;264:8602–5. [PubMed] [Google Scholar]
- 18.Didsbury J, Weber RF, Bokoch GM, Evans T, Snyderman R. rac, a novel ras-related family of proteins that are botulinum toxin substrates. J Biol Chem. 1989;264:16378–82. [PubMed] [Google Scholar]
- 19.Polakis PG, Weber RF, Nevins B, Didsbury JR, Evans T, Snyderman R. Identification of the ral and rac1 gene products, low molecular mass GTP-binding proteins from human platelets. J Biol Chem. 1989;264:16383–9. [PubMed] [Google Scholar]
- 20.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–35. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 21.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 22.Morii N, Teru-uchi T, Tominaga T, Kumagai N, Kozaki S, Ushikubi F, Narumiya S. A rho gene product in human blood platelets. II. Effects of the ADP-ribosylation by botulinum C3 ADP-ribosyltransferase on platelet aggregation. J Biol Chem. 1992;267:20921–6. [PubMed] [Google Scholar]
- 23.Nemoto Y, Namba T, Teru-uchi T, Ushikubi F, Morii N, Narumiya S. A rho gene product in human blood platelets. I. Identification of the platelet substrate for botulinum C3 ADP-ribosyltransferase as rhoA protein. J Biol Chem. 1992;267:20916–20. [PubMed] [Google Scholar]
- 24.Fox JE. The platelet cytoskeleton. Thromb Haemost. 1993;70:884–93. [PubMed] [Google Scholar]
- 25.Dash D, Aepfelbacher M, Siess W. Integrin alpha IIb beta 3-mediated translocation of CDC42Hs to the cytoskeleton in stimulated human platelets. J Biol Chem. 1995;270:17321–6. doi: 10.1074/jbc.270.29.17321. [DOI] [PubMed] [Google Scholar]
- 26.Flevaris P, Stojanovic A, Gong H, Chishti A, Welch E, Du X. A molecular switch that controls cell spreading and retraction. J Cell Biol. 2007;179:553–65. doi: 10.1083/jcb.200703185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klages B, Brandt U, Simon MI, Schultz G, Offermanns S. Activation of G12/G13 results in shape change and Rho/Rho-kinase-mediated myosin light chain phosphorylation in mouse platelets. J Cell Biol. 1999;144:745–54. doi: 10.1083/jcb.144.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pleines I, Hagedorn I, Gupta S, May F, Chakarova L, van Hengel J, Offermanns S, Krohne G, Kleinschnitz C, Brakebusch C, Nieswandt B. Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood. 2012;119:1054–63. doi: 10.1182/blood-2011-08-372193. [DOI] [PubMed] [Google Scholar]
- 29.Schoenwaelder SM, Hughan SC, Boniface K, Fernando S, Holdsworth M, Thompson PE, Salem HH, Jackson SP. RhoA sustains integrin alpha IIbbeta 3 adhesion contacts under high shear. J Biol Chem. 2002;277:14738–46. doi: 10.1074/jbc.M200661200. [DOI] [PubMed] [Google Scholar]
- 30.Akbar H, Kim J, Funk K, Cancelas JA, Shang X, Chen L, Johnson JF, Williams DA, Zheng Y. Genetic and pharmacologic evidence that Rac1 GTPase is involved in regulation of platelet secretion and aggregation. J Thromb Haemost. 2007;5:1747–55. doi: 10.1111/j.1538-7836.2007.02646.x. [DOI] [PubMed] [Google Scholar]
- 31.Flevaris P, Li Z, Zhang G, Zheng Y, Liu J, Du X. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood. 2009;113:893–901. doi: 10.1182/blood-2008-05-155978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCarty OJ, Larson MK, Auger JM, Kalia N, Atkinson BT, Pearce AC, Ruf S, Henderson RB, Tybulewicz VL, Machesky LM, Watson SP. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem. 2005;280:39474–84. doi: 10.1074/jbc.M504672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stefanini L, Boulaftali Y, Ouellette TD, Holinstat M, Desire L, Leblond B, Andre P, Conley PB, Bergmeier W. Rap1-Rac1 circuits potentiate platelet activation. Arterioscler Thromb Vasc Biol. 2012;32:434–41. doi: 10.1161/ATVBAHA.111.239194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akbar H, Shang X, Perveen R, Berryman M, Funk K, Johnson JF, Tandon NN, Zheng Y. Gene targeting implicates Cdc42 GTPase in GPVI and non-GPVI mediated platelet filopodia formation, secretion and aggregation. PLoS One. 2011;6:e22117. doi: 10.1371/journal.pone.0022117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pleines I, Eckly A, Elvers M, Hagedorn I, Eliautou S, Bender M, Wu X, Lanza F, Gachet C, Brakebusch C, Nieswandt B. Multiple alterations of platelet functions dominated by increased secretion in mice lacking Cdc42 in platelets. Blood. 2010;115:3364–73. doi: 10.1182/blood-2009-09-242271. [DOI] [PubMed] [Google Scholar]
- 36.Pula G, Poole AW. Critical roles for the actin cytoskeleton and cdc42 in regulating platelet integrin alpha2beta1. Platelets. 2008;19:199–210. doi: 10.1080/09537100701777303. [DOI] [PubMed] [Google Scholar]
- 37.Wheeler AP, Ridley AJ. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res. 2004;301:43–9. doi: 10.1016/j.yexcr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 38.Nishioka H, Horiuchi H, Tabuchi A, Yoshioka A, Shirakawa R, Kita T. Small GTPase Rho regulates thrombin-induced platelet aggregation. Biochem Biophys Res Commun. 2001;280:970–5. doi: 10.1006/bbrc.2001.4237. [DOI] [PubMed] [Google Scholar]
- 39.Martens L, Van Damme P, Van Damme J, Staes A, Timmerman E, Ghesquiere B, Thomas GR, Vandekerckhove J, Gevaert K. The human platelet proteome mapped by peptide-centric proteomics: a functional protein profile. Proteomics. 2005;5:3193–204. doi: 10.1002/pmic.200401142. [DOI] [PubMed] [Google Scholar]
- 40.Rowley JW, Oler AJ, Tolley ND, Hunter BN, Low EN, Nix DA, Yost CC, Zimmerman GA, Weyrich AS. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118:e101–11. doi: 10.1182/blood-2011-03-339705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwartz MA, Shattil SJ. Signaling networks linking integrins and rho family GTPases. Trends Biochem Sci. 2000;25:388–91. doi: 10.1016/s0968-0004(00)01605-4. [DOI] [PubMed] [Google Scholar]
- 42.Offermanns S, Toombs CF, Hu YH, Simon MI. Defective platelet activation in G alpha(q)-deficient mice. Nature. 1997;389:183–6. doi: 10.1038/38284. [DOI] [PubMed] [Google Scholar]
- 43.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–56. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 44.Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M. Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem. 1998;273:21867–74. doi: 10.1074/jbc.273.34.21867. [DOI] [PubMed] [Google Scholar]
- 45.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–8. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki Y, Yamamoto M, Wada H, Ito M, Nakano T, Sasaki Y, Narumiya S, Shiku H, Nishikawa M. Agonist-induced regulation of myosin phosphatase activity in human platelets through activation of Rho-kinase. Blood. 1999;93:3408–17. [PubMed] [Google Scholar]
- 47.Bauer M, Retzer M, Wilde JI, Maschberger P, Essler M, Aepfelbacher M, Watson SP, Siess W. Dichotomous regulation of myosin phosphorylation and shape change by Rho-kinase and calcium in intact human platelets. Blood. 1999;94:1665–72. [PubMed] [Google Scholar]
- 48.Offermanns S, Laugwitz KL, Spicher K, Schultz G. G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc Natl Acad Sci U S A. 1994;91:504–8. doi: 10.1073/pnas.91.2.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moers A, Nieswandt B, Massberg S, Wettschureck N, Gruner S, Konrad I, Schulte V, Aktas B, Gratacap MP, Simon MI, Gawaz M, Offermanns S. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat Med. 2003;9:1418–22. doi: 10.1038/nm943. [DOI] [PubMed] [Google Scholar]
- 50.Huang JS, Dong L, Kozasa T, Le Breton GC. Signaling through G(alpha)13 switch region I is essential for protease-activated receptor 1-mediated human platelet shape change, aggregation, and secretion. J Biol Chem. 2007;282:10210–22. doi: 10.1074/jbc.M605678200. [DOI] [PubMed] [Google Scholar]
- 51.Gong H, Shen B, Flevaris P, Chow C, Lam SC, Voyno-Yasenetskaya TA, Kozasa T, Du X. G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin “outside-in” signaling. Science. 2010;327:340–3. doi: 10.1126/science.1174779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arthur WT, Petch LA, Burridge K. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr Biol. 2000;10:719–22. doi: 10.1016/s0960-9822(00)00537-6. [DOI] [PubMed] [Google Scholar]
- 53.Kuchay SM, Wieschhaus AJ, Marinkovic M, Herman IM, Chishti AH. Targeted gene inactivation reveals a functional role of calpain-1 in platelet spreading. J Thromb Haemost. 2012;10:1120–32. doi: 10.1111/j.1538-7836.2012.04715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calaminus SD, Auger JM, McCarty OJ, Wakelam MJ, Machesky LM, Watson SP. MyosinIIa contractility is required for maintenance of platelet structure during spreading on collagen and contributes to thrombus stability. J Thromb Haemost. 2007;5:2136–45. doi: 10.1111/j.1538-7836.2007.02696.x. [DOI] [PubMed] [Google Scholar]
- 55.Bender A, Pringle JR. Multicopy suppression of the cdc24 budding defect in yeast by CDC42 and three newly identified genes including the ras-related gene RSR1. Proc Natl Acad Sci U S A. 1989;86:9976–80. doi: 10.1073/pnas.86.24.9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kozma R, Ahmed S, Best A, Lim L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol. 1995;15:1942–52. doi: 10.1128/mcb.15.4.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 58.Zhang X, Bi E, Novick P, Du L, Kozminski KG, Lipschutz JH, Guo W. Cdc42 interacts with the exocyst and regulates polarized secretion. J Biol Chem. 2001;276:46745–50. doi: 10.1074/jbc.M107464200. [DOI] [PubMed] [Google Scholar]
- 59.Hong-Geller E, Cerione RA. Cdc42 and Rac stimulate exocytosis of secretory granules by activating the IP(3)/calcium pathway in RBL-2H3 mast cells. J Cell Biol. 2000;148:481–94. doi: 10.1083/jcb.148.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang L, Wang L, Zheng Y. Gene targeting of Cdc42 and Cdc42GAP affirms the critical involvement of Cdc42 in filopodia induction, directed migration, and proliferation in primary mouse embryonic fibroblasts. Mol Biol Cell. 2006;17:4675–85. doi: 10.1091/mbc.E06-05-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Czuchra A, Wu X, Meyer H, van Hengel J, Schroeder T, Geffers R, Rottner K, Brakebusch C. Cdc42 is not essential for filopodium formation, directed migration, cell polarization, and mitosis in fibroblastoid cells. Mol Biol Cell. 2005;16:4473–84. doi: 10.1091/mbc.E05-01-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goh WI, Sudhaharan T, Lim KB, Sem KP, Lau CL, Ahmed S. Rif-mDia1 interaction is involved in filopodium formation independent of Cdc42 and Rac effectors. J Biol Chem. 2011;286:13681–94. doi: 10.1074/jbc.M110.182683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pellegrin S, Mellor H. The Rho family GTPase Rif induces filopodia through mDia2. Curr Biol. 2005;15:129–33. doi: 10.1016/j.cub.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 64.Azim AC, Barkalow K, Chou J, Hartwig JH. Activation of the small GTPases, rac and cdc42, after ligation of the platelet PAR-1 receptor. Blood. 2000;95:959–64. [PubMed] [Google Scholar]
- 65.Vidal C, Geny B, Melle J, Jandrot-Perrus M, Fontenay-Roupie M. Cdc42/Rac1-dependent activation of the p21-activated kinase (PAK) regulates human platelet lamellipodia spreading: implication of the cortical-actin binding protein cortactin. Blood. 2002;100:4462–9. doi: 10.1182/blood.V100.13.4462. [DOI] [PubMed] [Google Scholar]
- 66.Suzuki-Inoue K, Yatomi Y, Asazuma N, Kainoh M, Tanaka T, Satoh K, Ozaki Y. Rac, a small guanosine triphosphate-binding protein, and p21-activated kinase are activated during platelet spreading on collagen-coated surfaces: roles of integrin alpha(2)beta(1) Blood. 2001;98:3708–16. doi: 10.1182/blood.v98.13.3708. [DOI] [PubMed] [Google Scholar]
- 67.Yang L, Wang L, Geiger H, Cancelas JA, Mo J, Zheng Y. Rho GTPase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc Natl Acad Sci U S A. 2007;104:5091–6. doi: 10.1073/pnas.0610819104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tiedt R, Schomber T, Hao-Shen H, Skoda RC. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood. 2007;109:1503–6. doi: 10.1182/blood-2006-04-020362. [DOI] [PubMed] [Google Scholar]
- 69.Soulet C, Gendreau S, Missy K, Benard V, Plantavid M, Payrastre B. Characterisation of Rac activation in thrombin- and collagen-stimulated human blood platelets. FEBS Lett. 2001;507:253–8. doi: 10.1016/s0014-5793(01)02984-2. [DOI] [PubMed] [Google Scholar]
- 70.McCarty OJ, Calaminus SD, Berndt MC, Machesky LM, Watson SP. von Willebrand factor mediates platelet spreading through glycoprotein Ib and alpha(IIb)beta3 in the presence of botrocetin and ristocetin, respectively. J Thromb Haemost. 2006;4:1367–78. doi: 10.1111/j.1538-7836.2006.01966.x. [DOI] [PubMed] [Google Scholar]
- 71.Aslan JE, Tormoen GW, Loren CP, Pang J, McCarty OJ. S6K1 and mTOR regulate Rac1-driven platelet activation and aggregation. Blood. 2011;118:3129–36. doi: 10.1182/blood-2011-02-331579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bialkowska K, Zaffran Y, Meyer SC, Fox JE. 14-3-3 zeta mediates integrin-induced activation of Cdc42 and Rac. Platelet glycoprotein Ib-IX regulates integrin-induced signaling by sequestering 14-3-3 zeta. J Biol Chem. 2003;278:33342–50. doi: 10.1074/jbc.M301217200. [DOI] [PubMed] [Google Scholar]
- 73.O’Toole TE, Bialkowska K, Li X, Fox JE. Tiam1 is recruited to beta1-integrin complexes by 14-3-3zeta where it mediates integrin-induced Rac1 activation and motility. J Cell Physiol. 2011;226:2965–78. doi: 10.1002/jcp.22644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goc A, Abdalla M, Al-Azayzih A, Somanath PR. Rac1 activation driven by 14-3-3zeta dimerization promotes prostate cancer cell-matrix interactions, motility and transendothelial migration. PLoS One. 2012;7:e40594. doi: 10.1371/journal.pone.0040594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gratacap MP, Payrastre B, Nieswandt B, Offermanns S. Differential regulation of Rho and Rac through heterotrimeric G-proteins and cyclic nucleotides. J Biol Chem. 2001;276:47906–13. doi: 10.1074/jbc.M104442200. [DOI] [PubMed] [Google Scholar]
- 76.Belisle B, Abo A. N-Formyl peptide receptor ligation induces rac-dependent actin reorganization through Gbeta gamma subunits and class Ia phosphoinositide 3-kinases. J Biol Chem. 2000;275:26225–32. doi: 10.1074/jbc.M002743200. [DOI] [PubMed] [Google Scholar]
- 77.Ueda H, Morishita R, Yamauchi J, Itoh H, Kato K, Asano T. Regulation of Rac and Cdc42 pathways by G(i) during lysophosphatidic acid-induced cell spreading. J Biol Chem. 2001;276:6846–52. doi: 10.1074/jbc.M007541200. [DOI] [PubMed] [Google Scholar]
- 78.Soulet C, Hechler B, Gratacap MP, Plantavid M, Offermanns S, Gachet C, Payrastre B. A differential role of the platelet ADP receptors P2Y1 and P2Y12 in Rac activation. J Thromb Haemost. 2005;3:2296–306. doi: 10.1111/j.1538-7836.2005.01588.x. [DOI] [PubMed] [Google Scholar]
- 79.Welch HC, Coadwell WJ, Stephens LR, Hawkins PT. Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett. 2003;546:93–7. doi: 10.1016/s0014-5793(03)00454-x. [DOI] [PubMed] [Google Scholar]
- 80.McCarty OJ, Zhao Y, Andrew N, Machesky LM, Staunton D, Frampton J, Watson SP. Evaluation of the role of platelet integrins in fibronectin-dependent spreading and adhesion. J Thromb Haemost. 2004;2:1823–33. doi: 10.1111/j.1538-7836.2004.00925.x. [DOI] [PubMed] [Google Scholar]
- 81.Varga-Szabo D, Braun A, Nieswandt B. Calcium signaling in platelets. J Thromb Haemost. 2009;7:1057–66. doi: 10.1111/j.1538-7836.2009.03455.x. [DOI] [PubMed] [Google Scholar]
- 82.Watson SP, Auger JM, McCarty OJ, Pearce AC. GPVI and integrin alphaIIb beta3 signaling in platelets. J Thromb Haemost. 2005;3:1752–62. doi: 10.1111/j.1538-7836.2005.01429.x. [DOI] [PubMed] [Google Scholar]
- 83.Piechulek T, Rehlen T, Walliser C, Vatter P, Moepps B, Gierschik P. Isozyme-specific stimulation of phospholipase C-gamma2 by Rac GTPases. J Biol Chem. 2005;280:38923–31. doi: 10.1074/jbc.M509396200. [DOI] [PubMed] [Google Scholar]
- 84.Pleines I, Elvers M, Strehl A, Pozgajova M, Varga-Szabo D, May F, Chrostek-Grashoff A, Brakebusch C, Nieswandt B. Rac1 is essential for phospholipase C-gamma2 activation in platelets. Pflugers Arch. 2009;457:1173–85. doi: 10.1007/s00424-008-0573-7. [DOI] [PubMed] [Google Scholar]
- 85.Pollitt AY, Grygielska B, Leblond B, Desire L, Eble JA, Watson SP. Phosphorylation of CLEC-2 is dependent on lipid rafts, actin polymerization, secondary mediators, and Rac. Blood. 2010;115:2938–46. doi: 10.1182/blood-2009-12-257212. [DOI] [PubMed] [Google Scholar]
- 86.Delaney MK, Liu J, Zheng Y, Berndt MC, Du X. Role of Rac1 in Glycoprotein Ib-IX Mediated Signal Transduction and Integrin Activation. Arterioscler Thromb Vasc Biol. 2012 doi: 10.1161/ATVBAHA.112.254920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li J, Luo R, Kowluru A, Li G. Novel regulation by Rac1 of glucose- and forskolin-induced insulin secretion in INS-1 beta-cells. Am J Physiol Endocrinol Metab. 2004;286:E818–27. doi: 10.1152/ajpendo.00307.2003. [DOI] [PubMed] [Google Scholar]
- 88.Li Q, Ho CS, Marinescu V, Bhatti H, Bokoch GM, Ernst SA, Holz RW, Stuenkel EL. Facilitation of Ca(2+)-dependent exocytosis by Rac1-GTPase in bovine chromaffin cells. J Physiol. 2003;550:431–45. doi: 10.1113/jphysiol.2003.039073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Higgs HN, Pollard TD. Activation by Cdc42 and PIP(2) of Wiskott-Aldrich syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. J Cell Biol. 2000;150:1311–20. doi: 10.1083/jcb.150.6.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner MW. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–31. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- 91.Tomasevic N, Jia Z, Russell A, Fujii T, Hartman JJ, Clancy S, Wang M, Beraud C, Wood KW, Sakowicz R. Differential regulation of WASP and N-WASP by Cdc42, Rac1, Nck, and PI(4,5)P2. Biochemistry. 2007;46:3494–502. doi: 10.1021/bi062152y. [DOI] [PubMed] [Google Scholar]
- 92.Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998;17:6932–41. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Soderling SH, Scott JD. WAVE signalling: from biochemistry to biology. Biochem Soc Trans. 2006;34:73–6. doi: 10.1042/BST0340073. [DOI] [PubMed] [Google Scholar]
- 94.Campellone KG, Welch MD. A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol. 2010;11:237–51. doi: 10.1038/nrm2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Higashi T, Ikeda T, Shirakawa R, Kondo H, Kawato M, Horiguchi M, Okuda T, Okawa K, Fukai S, Nureki O, Kita T, Horiuchi H. Biochemical characterization of the Rho GTPase-regulated actin assembly by diaphanous-related formins, mDia1 and Daam1, in platelets. J Biol Chem. 2008;283:8746–55. doi: 10.1074/jbc.M707839200. [DOI] [PubMed] [Google Scholar]
- 96.Thomas SG, Calaminus SD, Machesky LM, Alberts AS, Watson SP. G-protein coupled and ITAM receptor regulation of the formin FHOD1 through Rho kinase in platelets. J Thromb Haemost. 2011;9:1648–51. doi: 10.1111/j.1538-7836.2011.04357.x. [DOI] [PubMed] [Google Scholar]
- 97.Hartwig JH, Bokoch GM, Carpenter CL, Janmey PA, Taylor LA, Toker A, Stossel TP. Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell. 1995;82:643–53. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- 98.Barkalow K, Witke W, Kwiatkowski DJ, Hartwig JH. Coordinated regulation of platelet actin filament barbed ends by gelsolin and capping protein. J Cell Biol. 1996;134:389–99. doi: 10.1083/jcb.134.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tolias KF, Hartwig JH, Ishihara H, Shibasaki Y, Cantley LC, Carpenter CL. Type Ialpha phosphatidylinositol-4-phosphate 5-kinase mediates Rac-dependent actin assembly. Curr Biol. 2000;10:153–6. doi: 10.1016/s0960-9822(00)00315-8. [DOI] [PubMed] [Google Scholar]
- 100.Barkalow KL, Falet H, Italiano JE, Jr, van Vugt A, Carpenter CL, Schreiber AD, Hartwig JH. Role for phosphoinositide 3-kinase in Fc gamma RIIA-induced platelet shape change. Am J Physiol Cell Physiol. 2003;285:C797–805. doi: 10.1152/ajpcell.00165.2003. [DOI] [PubMed] [Google Scholar]
- 101.Falet H, Hoffmeister KM, Neujahr R, Italiano JE, Jr, Stossel TP, Southwick FS, Hartwig JH. Importance of free actin filament barbed ends for Arp2/3 complex function in platelets and fibroblasts. Proc Natl Acad Sci U S A. 2002;99:16782–7. doi: 10.1073/pnas.222652499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kashiwagi H, Shiraga M, Kato H, Honda S, Sako M, Kurata Y, Kanakura Y, Tomiyama Y. Expression and subcellular localization of WAVE isoforms in the megakaryocyte/platelet lineage. J Thromb Haemost. 2005;3:361–8. doi: 10.1111/j.1538-7836.2004.01082.x. [DOI] [PubMed] [Google Scholar]
- 103.Calaminus SD, McCarty OJ, Auger JM, Pearce AC, Insall RH, Watson SP, Machesky LM. A major role for Scar/WAVE-1 downstream of GPVI in platelets. J Thromb Haemost. 2007;5:535–41. doi: 10.1111/j.1538-7836.2007.02377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oda A, Miki H, Wada I, Yamaguchi H, Yamazaki D, Suetsugu S, Nakajima M, Nakayama A, Okawa K, Miyazaki H, Matsuno K, Ochs HD, Machesky LM, Fujita H, Takenawa T. WAVE/Scars in platelets. Blood. 2005;105:3141–8. doi: 10.1182/blood-2003-04-1319. [DOI] [PubMed] [Google Scholar]
- 105.Takeya R, Taniguchi K, Narumiya S, Sumimoto H. The mammalian formin FHOD1 is activated through phosphorylation by ROCK and mediates thrombin-induced stress fibre formation in endothelial cells. EMBO J. 2008;27:618–28. doi: 10.1038/emboj.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lim KB, Bu W, Goh WI, Koh E, Ong SH, Pawson T, Sudhaharan T, Ahmed S. The Cdc42 effector IRSp53 generates filopodia by coupling membrane protrusion with actin dynamics. J Biol Chem. 2008;283:20454–72. doi: 10.1074/jbc.M710185200. [DOI] [PubMed] [Google Scholar]
- 107.Goh WI, Lim KB, Sudhaharan T, Sem KP, Bu W, Chou AM, Ahmed S. mDia1 and WAVE2 proteins interact directly with IRSp53 in filopodia and are involved in filopodium formation. J Biol Chem. 2012;287:4702–14. doi: 10.1074/jbc.M111.305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Falet H, Hoffmeister KM, Neujahr R, Hartwig JH. Normal Arp2/3 complex activation in platelets lacking WASp. Blood. 2002;100:2113–22. [PubMed] [Google Scholar]
- 109.Egile C, Loisel TP, Laurent V, Li R, Pantaloni D, Sansonetti PJ, Carlier MF. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J Cell Biol. 1999;146:1319–32. doi: 10.1083/jcb.146.6.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ellis S, Mellor H. The novel Rho-family GTPase rif regulates coordinated actin-based membrane rearrangements. Curr Biol. 2000;10:1387–90. doi: 10.1016/s0960-9822(00)00777-6. [DOI] [PubMed] [Google Scholar]
- 111.Adams JC, Schwartz MA. Stimulation of fascin spikes by thrombospondin-1 is mediated by the GTPases Rac and Cdc42. J Cell Biol. 2000;150:807–22. doi: 10.1083/jcb.150.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Parsons M, Adams JC. Rac regulates the interaction of fascin with protein kinase C in cell migration. J Cell Sci. 2008;121:2805–13. doi: 10.1242/jcs.022509. [DOI] [PubMed] [Google Scholar]
- 113.Szczepanowska J. Involvement of Rac/Cdc42/PAK pathway in cytoskeletal rearrangements. Acta Biochim Pol. 2009;56:225–34. [PubMed] [Google Scholar]
- 114.Arias-Romero LE, Chernoff J. A tale of two Paks. Biol Cell. 2008;100:97–108. doi: 10.1042/BC20070109. [DOI] [PubMed] [Google Scholar]
- 115.Teo M, Manser E, Lim L. Identification and molecular cloning of a p21cdc42/rac1-activated serine/threonine kinase that is rapidly activated by thrombin in platelets. J Biol Chem. 1995;270:26690–7. doi: 10.1074/jbc.270.44.26690. [DOI] [PubMed] [Google Scholar]
- 116.Pandey D, Goyal P, Dwivedi S, Siess W. Unraveling a novel Rac1-mediated signaling pathway that regulates cofilin dephosphorylation and secretion in thrombin-stimulated platelets. Blood. 2009;114:415–24. doi: 10.1182/blood-2008-10-183582. [DOI] [PubMed] [Google Scholar]
- 117.Kaksonen M, Peng HB, Rauvala H. Association of cortactin with dynamic actin in lamellipodia and on endosomal vesicles. J Cell Sci. 2000;113(Pt 24):4421–6. doi: 10.1242/jcs.113.24.4421. [DOI] [PubMed] [Google Scholar]
- 118.Uruno T, Liu J, Zhang P, Fan Y, Egile C, Li R, Mueller SC, Zhan X. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat Cell Biol. 2001;3:259–66. doi: 10.1038/35060051. [DOI] [PubMed] [Google Scholar]
- 119.Obergfell A, Judd BA, del Pozo MA, Schwartz MA, Koretzky GA, Shattil SJ. The molecular adapter SLP-76 relays signals from platelet integrin alphaIIbbeta3 to the actin cytoskeleton. J Biol Chem. 2001;276:5916–23. doi: 10.1074/jbc.M010639200. [DOI] [PubMed] [Google Scholar]
- 120.Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol. 2007;39:1071–6. doi: 10.1016/j.biocel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 121.Falet H, Chang G, Brohard-Bohn B, Rendu F, Hartwig JH. Integrin alpha(IIb)beta3 signals lead cofilin to accelerate platelet actin dynamics. Am J Physiol Cell Physiol. 2005;289:C819–25. doi: 10.1152/ajpcell.00587.2004. [DOI] [PubMed] [Google Scholar]
- 122.Pandey D, Goyal P, Bamburg JR, Siess W. Regulation of LIM-kinase 1 and cofilin in thrombin-stimulated platelets. Blood. 2006;107:575–83. doi: 10.1182/blood-2004-11-4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pandey D, Goyal P, Siess W. Lysophosphatidic acid stimulation of platelets rapidly induces Ca2+-dependent dephosphorylation of cofilin that is independent of dense granule secretion and aggregation. Blood Cells Mol Dis. 2007;38:269–79. doi: 10.1016/j.bcmd.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 124.Frank SR, Hansen SH. The PIX-GIT complex: a G protein signaling cassette in control of cell shape. Semin Cell Dev Biol. 2008;19:234–44. doi: 10.1016/j.semcdb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sato H, Suzuki-Inoue K, Inoue O, Ozaki Y. Regulation of adaptor protein GIT1 in platelets, leading to the interaction between GIT1 and integrin alpha(IIb)beta3. Biochem Biophys Res Commun. 2008;368:157–61. doi: 10.1016/j.bbrc.2008.01.064. [DOI] [PubMed] [Google Scholar]
- 126.Wright B, Stanley RG, Kaiser WJ, Mills DJ, Gibbins JM. Analysis of protein networks in resting and collagen receptor (GPVI)-stimulated platelet sub-proteomes. Proteomics. 2011;11:4588–92. doi: 10.1002/pmic.201100410. [DOI] [PubMed] [Google Scholar]
- 127.van Nieuw Amerongen GP, Natarajan K, Yin G, Hoefen RJ, Osawa M, Haendeler J, Ridley AJ, Fujiwara K, van Hinsbergh VW, Berk BC. GIT1 mediates thrombin signaling in endothelial cells: role in turnover of RhoA-type focal adhesions. Circ Res. 2004;94:1041–9. doi: 10.1161/01.RES.0000125627.77235.0C. [DOI] [PubMed] [Google Scholar]
- 128.Kuo JC, Han X, Hsiao CT, Yates JR, 3rd, Waterman CM. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for beta-Pix in negative regulation of focal adhesion maturation. Nat Cell Biol. 2011;13:383–93. doi: 10.1038/ncb2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–80. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 130.Bustelo XR, Ledbetter JA, Barbacid M. Product of vav proto-oncogene defines a new class of tyrosine protein kinase substrates. Nature. 1992;356:68–71. doi: 10.1038/356068a0. [DOI] [PubMed] [Google Scholar]
- 131.Cichowski K, Brugge JS, Brass LF. Thrombin receptor activation and integrin engagement stimulate tyrosine phosphorylation of the proto-oncogene product, p95vav, in platelets. J Biol Chem. 1996;271:7544–50. doi: 10.1074/jbc.271.13.7544. [DOI] [PubMed] [Google Scholar]
- 132.Pearce AC, Wilde JI, Doody GM, Best D, Inoue O, Vigorito E, Tybulewicz VL, Turner M, Watson SP. Vav1, but not Vav2, contributes to platelet aggregation by CRP and thrombin, but neither is required for regulation of phospholipase C. Blood. 2002;100:3561–9. doi: 10.1182/blood.V100.10.3561. [DOI] [PubMed] [Google Scholar]
- 133.Pearce AC, McCarty OJ, Calaminus SD, Vigorito E, Turner M, Watson SP. Vav family proteins are required for optimal regulation of PLCgamma2 by integrin alphaIIbbeta3. Biochem J. 2007;401:753–61. doi: 10.1042/BJ20061508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pearce AC, Senis YA, Billadeau DD, Turner M, Watson SP, Vigorito E. Vav1 and vav3 have critical but redundant roles in mediating platelet activation by collagen. J Biol Chem. 2004;279:53955–62. doi: 10.1074/jbc.M410355200. [DOI] [PubMed] [Google Scholar]
- 135.Aslan JE, Spencer AM, Loren CP, Pang J, Welch HC, Greenberg DL, McCarty OJ. Characterization of the Rac guanine nucleotide exchange factor P-Rex1 in platelets. J Mol Signal. 2011;6:11. doi: 10.1186/1750-2187-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Qian F, Le Breton GC, Chen J, Deng J, Christman JW, Wu D, Ye RD. Role for the Guanine nucleotide exchange factor phosphatidylinositol-3,4,5-trisphosphate-dependent rac exchanger 1 in platelet secretion and aggregation. Arterioscler Thromb Vasc Biol. 2012;32:768–77. doi: 10.1161/ATVBAHA.111.243675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hernandez-Negrete I, Carretero-Ortega J, Rosenfeldt H, Hernandez-Garcia R, Calderon-Salinas JV, Reyes-Cruz G, Gutkind JS, Vazquez-Prado J. P-Rex1 links mammalian target of rapamycin signaling to Rac activation and cell migration. J Biol Chem. 2007;282:23708–15. doi: 10.1074/jbc.M703771200. [DOI] [PubMed] [Google Scholar]
- 138.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Aslan JE, McCarty OJT. Regulation of the mTOR-Rac1 axis in platelet function. Small GTPases. 2012;3:16–9. doi: 10.4161/sgtp.19137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Weyrich AS, Denis MM, Schwertz H, Tolley ND, Foulks J, Spencer E, Kraiss LW, Albertine KH, McIntyre TM, Zimmerman GA. mTOR-dependent synthesis of Bcl-3 controls the retraction of fibrin clots by activated human platelets. Blood. 2007;109:1975–83. doi: 10.1182/blood-2006-08-042192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Duran RV, Hall MN. Regulation of TOR by small GTPases. EMBO Rep. 2012;13:121–8. doi: 10.1038/embor.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Saci A, Cantley LC, Carpenter CL. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol Cell. 2011;42:50–61. doi: 10.1016/j.molcel.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Liu L, Parent CA. Review series: TOR kinase complexes and cell migration. J Cell Biol. 2011;194:815–24. doi: 10.1083/jcb.201102090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhou H, Huang S. Role of mTOR signaling in tumor cell motility, invasion and metastasis. Curr Protein Pept Sci. 2011;12:30–42. doi: 10.2174/138920311795659407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Moore SF, Hunter RW, Hers I. mTORC2 protein complex-mediated Akt (Protein Kinase B) Serine 473 Phosphorylation is not required for Akt1 activity in human platelets [corrected] J Biol Chem. 2011;286:24553–60. doi: 10.1074/jbc.M110.202341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Steele BM, Harper MT, Macaulay IC, Morrell CN, Perez-Tamayo A, Foy M, Habas R, Poole AW, Fitzgerald DJ, Maguire PB. Canonical Wnt signaling negatively regulates platelet function. Proc Natl Acad Sci U S A. 2009;106:19836–41. doi: 10.1073/pnas.0906268106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Steele BM, Harper MT, Smolenski AP, Alkazemi N, Poole AW, Fitzgerald DJ, Maguire PB. WNT-3a modulates platelet function by regulating small GTPase activity. FEBS Lett. 2012;586:2267–72. doi: 10.1016/j.febslet.2012.05.060. [DOI] [PubMed] [Google Scholar]
- 148.Tcherkezian J, Lamarche-Vane N. Current knowledge of the large RhoGAP family of proteins. Biol Cell. 2007;99:67–86. doi: 10.1042/BC20060086. [DOI] [PubMed] [Google Scholar]
- 149.Hart MJ, Shinjo K, Hall A, Evans T, Cerione RA. Identification of the human platelet GTPase activating protein for the CDC42Hs protein. J Biol Chem. 1991;266:20840–8. [PubMed] [Google Scholar]
- 150.Bahou WF, Scudder L, Rubenstein D, Jesty J. A shear-restricted pathway of platelet procoagulant activity is regulated by IQGAP1. J Biol Chem. 2004;279:22571–7. doi: 10.1074/jbc.M402561200. [DOI] [PubMed] [Google Scholar]
- 151.Beck S, Fotinos A, Lang F, Gawaz M, Elvers M. Isoform-specific roles of the GTPase activating protein Nadrin in cytoskeletal reorganization of platelets. Cell Signal. 2012 doi: 10.1016/j.cellsig.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 152.Elvers M, Beck S, Fotinos A, Ziegler M, Gawaz M. The GRAF family member oligophrenin1 is a RhoGAP with BAR domain and regulates Rho GTPases in platelets. Cardiovasc Res. 2012;94:526–36. doi: 10.1093/cvr/cvs079. [DOI] [PubMed] [Google Scholar]
- 153.Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12:493–504. doi: 10.1038/nrm3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Leonard D, Hart MJ, Platko JV, Eva A, Henzel W, Evans T, Cerione RA. The identification and characterization of a GDP-dissociation inhibitor (GDI) for the CDC42Hs protein. J Biol Chem. 1992;267:22860–8. [PubMed] [Google Scholar]
- 155.Hart MJ, Maru Y, Leonard D, Witte ON, Evans T, Cerione RA. A GDP dissociation inhibitor that serves as a GTPase inhibitor for the Ras-like protein CDC42Hs. Science. 1992;258:812–5. doi: 10.1126/science.1439791. [DOI] [PubMed] [Google Scholar]
- 156.Liu M, Bi F, Zhou X, Zheng Y. Rho GTPase regulation by miRNAs and covalent modifications. Trends Cell Biol. 2012;22:365–73. doi: 10.1016/j.tcb.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dangwal S, Thum T. MicroRNAs in platelet biogenesis and function. Thromb Haemost. 2012:108. doi: 10.1160/TH12-03-0211. [DOI] [PubMed] [Google Scholar]
- 158.Rowley JW, Schwertz H, Weyrich AS. Platelet mRNA: the meaning behind the message. Curr Opin Hematol. 2012 doi: 10.1097/MOH.0b013e328357010e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wittmann T, Waterman-Storer CM. Cell motility: can Rho GTPases and microtubules point the way? J Cell Sci. 2001;114:3795–803. doi: 10.1242/jcs.114.21.3795. [DOI] [PubMed] [Google Scholar]
- 160.Ridley AJ. Life at the leading edge. Cell. 2011;145:1012–22. doi: 10.1016/j.cell.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 161.Patel SR, Richardson JL, Schulze H, Kahle E, Galjart N, Drabek K, Shivdasani RA, Hartwig JH, Italiano JE., Jr Differential roles of microtubule assembly and sliding in proplatelet formation by megakaryocytes. Blood. 2005;106:4076–85. doi: 10.1182/blood-2005-06-2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tablin F, Castro M, Leven RM. Blood platelet formation in vitro. The role of the cytoskeleton in megakaryocyte fragmentation. J Cell Sci. 1990;97 (Pt 1):59–70. doi: 10.1242/jcs.97.1.59. [DOI] [PubMed] [Google Scholar]
- 163.Chang Y, Aurade F, Larbret F, Zhang Y, Le Couedic JP, Momeux L, Larghero J, Bertoglio J, Louache F, Cramer E, Vainchenker W, Debili N. Proplatelet formation is regulated by the Rho/ROCK pathway. Blood. 2007;109:4229–36. doi: 10.1182/blood-2006-04-020024. [DOI] [PubMed] [Google Scholar]
- 164.Mulloy JC, Cancelas JA, Filippi MD, Kalfa TA, Guo F, Zheng Y. Rho GTPases in hematopoiesis and hemopathies. Blood. 2010;115:936–47. doi: 10.1182/blood-2009-09-198127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Watson SP. Platelet activation by extracellular matrix proteins in haemostasis and thrombosis. Curr Pharm Des. 2009;15:1358–72. doi: 10.2174/138161209787846702. [DOI] [PubMed] [Google Scholar]
- 166.Lam WA, Chaudhuri O, Crow A, Webster KD, Li TD, Kita A, Huang J, Fletcher DA. Mechanics and contraction dynamics of single platelets and implications for clot stiffening. Nat Mater. 2011;10:61–6. doi: 10.1038/nmat2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kita A, Sakurai Y, Myers DR, Rounsevell R, Huang JN, Seok TJ, Yu K, Wu MC, Fletcher DA, Lam WA. Microenvironmental geometry guides platelet adhesion and spreading: a quantitative analysis at the single cell level. PLoS One. 2011;6:e26437. doi: 10.1371/journal.pone.0026437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Coller BS. Historical perspective and future directions in platelet research. J Thromb Haemost. 2011;9 (Suppl 1):374–95. doi: 10.1111/j.1538-7836.2011.04356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Dwivedi S, Pandey D, Khandoga AL, Brandl R, Siess W. Rac1-mediated signaling plays a central role in secretion-dependent platelet aggregation in human blood stimulated by atherosclerotic plaque. J Transl Med. 2010;8:128. doi: 10.1186/1479-5876-8-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Aslan JE, Itakura A, Gertz JM, McCarty OJ. Platelet shape change and spreading. Methods Mol Biol. 2012;788:91–100. doi: 10.1007/978-1-61779-307-3_7. [DOI] [PubMed] [Google Scholar]