

Abstract
Cellular exposure to reactive oxygen species induces the rapid oxidation of DNA, proteins, lipids, and other biomolecules. At the proteome level, cysteine thiol oxidation is a prominent post-translational process implicated in normal physiology and numerous pathologies. Methods for investigating protein oxidation include direct labeling with selective chemical probes and indirect tag-switch techniques. Common to both approaches is a chemical blocking of free thiols with reactive electrophiles to prevent post-lysis oxidation or other thiol-mediated cross-reactions. These reagents are used in large excess and their reactivity with cysteine sulfenic acid, a critical oxoform in numerous proteins, has not been investigated. Here we report the reactivity of three thiol-blocking electrophiles, iodoacetamide, N-ethylmaleimide, and methyl methanethiosulfonate, with protein sulfenic acid and dimedone, the structural core of many sulfenic acid probes. We demonstrate that covalent cysteine -SOR (product) species are susceptible to reduction by DTT, TCEP, and ascorbate, regenerating protein thiols, or in the case of ascorbate, more highly oxidized species. The implications of this reactivity on detection methods for protein sulfenic acids and S-nitrosothiols are discussed.
Keywords: Protein sulfenic acid, thiol blocking, S-nitrosation, dimedone, mass spectrometry
Introduction
Protein cysteine oxidation results from a multitude of physiological events occurring in both normal cellular processes and disease states. Cysteine oxidation initially generates sulfenic acid (-SOH), a labile species susceptible to attack by thiols and nitrogen nucleophiles to form disulfides and sulfenamides (-SN), respectively. Under reducing conditions, sulfenic acid readily reverts to the reduced thiol; however in the presence of H2O2 or other oxidants, -SOH is hyperoxidized to form the thermodynamically stable sulfinic (-SO2H) and sulfonic (-SO3H) acid products. Sulfenic acids are therefore a key intermediate of both reversible and irreversible processes and detection of this species pinpoints the initial site of oxidation allowing for temporal resolution of oxidative signaling [1–5]. Recent investigations to quantify the oxidized proteome in HEK and HeLa cells found that fewer than 10% of cysteines are involved in disulfide bond formation [6]. Uncovering protein oxidation targets and mechanisms of regulation therefore requires methods for detecting the less abundant thiol oxoforms in the presence of large amounts of reduced cysteine.
Chemically, sulfenic acids have been widely regarded as electrophiles that react with dimedone and other enol nucleophiles to generate thioethers [7–9]. Recent studies have shown that 1,3-cyclopentanediones and linear beta-ketoesters are effective chemical reporters for -SOH and offer elevated rates of reaction, improved compatibility with downstream analysis by mass spectrometry, and simple synthetic schemes [10,11]. With the aim of further enhancing the methods for sulfenic acid detection in cell lysates, we explored the potential cross-reactivity of thiol-quenching reagents with protein sulfenic acids and dimedone, which represents the core structure of the 1,3-dicarbonyl probes. The most widely used blocking reagents (e.g., iodoacetamide (IAM), methyl methanethiosulfonate (MMTS), N-ethylmaleimide (NEM)) are small organic electrophiles intended to stably block cysteine thiols through covalent modification. Techniques to probe for oxidized cysteine in vitro often include the use of thiol blocking strategies to quench reduced cysteine, diminishing non-physiologically relevant thiol oxidation during or subsequent to cell lysis [12]. In the biotin-switch assay and related methods to detect S-nitrosothiols (-SNO), S-glutathionylation, and other oxidative post-translational cysteine modifications, thiol-selective reagents are used both to block free thiols during lysis and to label nascent thiols generated in situ from the reduction of the oxidized species (Figure 1).
Figure 1.

Approaches for detecting oxidized and nitrosated cysteine using thiol blocking and substrate-specific reduction.
Efforts to optimize sulfenic acid labeling procedures when working with complex samples (e.g., cells) have led to the consistent observation that regardless of the -SOH probe employed, labeling efficiency is decreased in the presence of a thiol-blocking electrophile. Aside from lysis-induced oxidation in the absence of a thiol-blocking compound, other possible explanations considered for decreased efficiency were the cross-reactivity between the nucleophilic probe and electrophilic blocking reagent as well as the inadvertent reaction of protein -SOH with blocking electrophiles. Alkylation of cysteine thiols is a long-established and widely used approach with selectivity largely based on the nucleophilicity of thiols surpassing that of all other amino acid residues [13]. Despite our earlier tentative identification of sulfenic acid reactivity toward thiol alkylating reagents [14], this cross-reactivity has remained poorly characterized, and to our knowledge, has not been investigated in terms of its impact on various detection techniques for modified cysteines.
The nucleophilicity of organic sulfenic acids has been noted in synthetically useful routes toward sulfones, sulfoxides, and alkenes, and forms the basis for addition-elimination chemistry with NBD-Cl for spectroscopic protein -SOH detection [15–18]. Lone electron pairs on the -SOH oxygen provide α-effect sulfur nucleophilicity, and the potential for oxygen nucleophilicity has also been documented [19, 20]. The nucleophilic nature of protein sulfenic acids has been much less explored and undoubtedly has profound implications for biochemical methods aimed at both -SOH detection and other cysteine-based chemistry performed in the presence of -SOH. We report here the reaction of a protein sulfenic acid with widely utilized thiol-blocking agents. The effects of this cross-reactivity on approaches for detecting proteome-wide cysteine oxidation, pertaining especially to sulfenic acids and S-nitrosothiols, are discussed.
Results and Discussion
To obtain detailed chemical information regarding the potential reactivity of cellular -SOH with thiol-blocking electrophiles, we focused our studies on a recombinant bacterial peroxiredoxin, AhpC. The C165S mutant of this 2-cysteine enzyme from S. typhimurium was chosen because it allows for accumulation of -SOH at C46 and for its documented stability across various pH and temperature conditions, its efficient MS-induced ionization, and the lack of other cysteine residues that could otherwise complicate the interpretation of results. These characteristics allow the direct observation of chemical reactivity than cannot be achieved in cells or with cell lysates due to sample complexity. Preliminary investigations with C165S AhpC revealed that the treatment of C165S AhpC-SOH with the thiol-blocking reagent IAM led to an unexpected decrease in the effectiveness of subsequent -SOH labeling using cyclic or linear 1,3-dicarbonyl probes (e.g., dimedone). To examine suspected -SOH quenching by IAM, freshly prepared C165S AhpC-SOH (30 μM) was incubated with IAM (2 mM) in ammonium bicarbonate (50 mM, pH 7.5) at ambient temperature for 3 hours (Figure 2A, first step). The reaction was quenched by passing the mixture through a Bio-Gel P6 spin column pre-equilibrated with 0.1% formic acid in water. Analysis by electrospray ionization-time of flight mass spectrometry (ESI-TOF MS) revealed the formation of a protein adduct at 20,673 a.m.u. in 72% yield (Figure 2B). Unreacted AhpC-SOH is observed in the gas phase as the presumed dehydrated sulfenamide (-SN) species at 20,598 a.m.u. [21]. The deconvoluted adduct mass is +16 a.m.u. relative to the adduct formed via the reaction of IAM with reduced cysteine (+57 a.m.u. relative to -SH), suggesting the presence of one oxygen atom and corresponding to the addition of the carboamidomethyl moiety to AhpC-SOH.
Figure 2.

A. General reaction scheme for AhpC-SOH addition to thiol-blocking electrophiles. B. Representative ESI-TOF mass spectra of the reactions of C165S AhpC-SOH (30 μM) with 2 mM IAM (i), NEM (ii), and MMTS (iii) at 3 hours at pH 7.5. The signal at 20,598 amu corresponds to sulfenamide (-SN). Signals labeled with * and ** indicate 20,632 (-SO2H) and 20,648 (-SO3H) a.m.u., respectively. Evidence for nucleophilic -SOH addition is indicated at 20,673 (i), 20,742 (ii), and 20,662 (iii) a.m.u. The signal at 20,566 a.m.u. (iii) corresponds to the conversion of cysteine to dehydroalanine.
To further characterize this reactivity with other electrophilic reagents, AhpC-SOH (30 μM) was similarly screened for reaction with NEM (2 mM) and MMTS (2 mM) under the conditions described above. A 3 hour incubation period with each of these electrophiles resulted in the formation of stable adducts at 20,742 and 20,662 a.m.u., respectively, with higher observed yields (89% for NEM and 100% for MMTS in terms of -SOH consumption) as compared to IAM (Figure 2B). Trypsin digestion of the three samples with subsequent nanoLC-MS/MS peptide analysis confirmed that the modification includes oxygen, presumably from the sulfenic acid, and that the modification occurs at peroxidatic C46 (Figure S1).
Kinetic analysis
Rates for the reaction of AhpC-SOH with saturating amounts of IAM, NEM, and MMTS were obtained by monitoring adduct formation as a function of time using ESI-TOF MS. Product yields at various timepoints were plotted against reaction time using an exponential increase equation to yield pseudo-first order rate constants (kobs) of 1.1 ± 0.3 min−1 (MMTS), 0.07 ± 0.02 min−1 (NEM), and 0.030 ± 0.003 min−1 (IAM) all obtained at pH 7.5 and ambient temperature (Figure 3). Accounting for electrophile concentration, the peroxidatic C46 of AhpC had similar reactivity towards IAM in both the thiol (0.15 M−1 s−1 at pH 7.0) [22] and sulfenic acid (0.10 M−1 s−1 at pH 7.5) states. Higher rate constants have been reported for other redox-sensitive proteins, and have illustrated the same order of observed reactivity (MMTS > NEM > IAM) as demonstrated for AhpC-SOH [23–26]. More importantly for the objective of this study, the rate constants for electrophilic -SOH capture exceed those reported for labeling fully folded C165S AhpC-SOH with nucleophilic 1,3-dicarbonyl probes like dimedone (e.g., 0.10 M−1 s−1 for the slowest electrophile IAM vs. 0.05 M−1 s−1 for the dimedone-based DCP-Bio1 at pH 7, each calculated from pseudo-first order rate constants and reagent concentration) [27]. Such comparable rate constants illustrate that blocking reagents and -SOH-targeted probes will compete for the oxidized thiol site, emphasizing the risk of quenching cellular -SOH with blocking reagents when added prior to or alongside a 1,3-dicarbonyl probe.
Figure 3.

Timecourse of adduct formation from the reactions of MMTS (closed circles), NEM (open circles), and IAM (triangles), each 5 mM, with C165S AhpC-SOH (30 μM). Concentrations of adducts were determined based on adduct abundance among the total ion abundances of the prominent species in the mass spectra as shown in Figure 2. Reactions were carried out at r.t. in ammonium bicarbonate buffer (50 mM, pH 7.5).
In addition to sulfenic acid, thiol-blocking electrophiles were screened for reactivity with protein sulfinic acid (AhpC-SO2H) and S-nitrosothiol (AhpC-SNO). AhpC-SO2H was incubated in the presence of each electrophile for 2 hours at pH 7.5 then analyzed by ESI-TOF MS (Figure S2). A small amount of adduct at the expected mass was observed only in the case of NEM, though subsequent digestion and nanoLC-MS showed that the succinimide modification occurred at histidine sites and not at C46. Indeed, the slow alkylation of non-cysteine amino acid residues by NEM has been documented at longer reaction times [28–31]. AhpC-SNO was generated via transnitrosation by S-nitrosocysteine (CySNO), then incubated at pH 7.5 in the presence of each electrophile following CySNO removal. Product formation corresponding to electrophile addition to AhpC-SNO was not observed by ESI-TOF MS for the three electrophiles surveyed (Figure S3).
Reductive cleavage
Reduction of oxidized species and tagging of the nascent thiols is performed subsequent to thiol blocking in the biotin-switch assay and related methods for detection of oxidized cysteine. This approach is often tailored for the detection of disulfides (including sites of glutathionylation), sulfenic acids, and S-nitrosothiols through the use of chemoselective reducing agents [25]. Additionally, reduction chemistry is an integral component of protein sample processing for in-gel and in-solution digestion followed by mass spectrometric analysis. As a result, we investigated the stability of the -SOH/electrophile (-SOR) adducts toward reducing agents at concentrations relevant to the biotin-switch and other biochemical techniques in an effort to understand the implications for inadvertent -SOH reactivity with thiol-blocking electrophiles on overall outcome of these assays (Figure 2A, second step).
Freshly generated AhpC-SOH was first incubated with IAM, NEM, or MMTS (5–10 mM) at pH 7.5 for 2 hours to maximize the abundance of -SOR. Unreacted electrophiles were removed by gel filtration and the resulting protein species were treated with biochemical reductants (10 mM) at room temperature. Product ratios were measured at time points ranging from 10 min to 3 hours by ESI-TOF MS (Figures 4, S4–5). Cleavage of IAM -SOR adducts by equal concentrations of DTT, TCEP, and sodium ascorbate proceeded similarly, leading to a 40–50% reduction in adduct concentration (Table 1). Succinimide adducts, generated with NEM, were more susceptible to reduction compared with the IAM -SOR adducts. With DTT, there was a ~87% loss of adduct upon a 1 hour incubation, while with TCEP the loss of -SOR adduct was ~60% (Figure S4). Least stable were the MMTS adducts, with full reduction by DTT and TCEP, and 85% reduction by ascorbate (Figure S5). DTT and TCEP reductions of all -SOR adducts proceeded with concomitant generation of C165S AhpC-SH. In contrast, all ascorbate-mediated reductions were instead accompanied by an increase in the amount of AhpC-SO2H. Sulfinic acid formation was also observed upon direct treatment of AhpC-SOH with sodium ascorbate (10 mM) under the same reaction conditions, suggesting an ascorbate-mediated oxidation of thiol and sulfenic acid to AhpC-SO2H catalyzed by trace metals in the reaction medium as previously described [32].
Figure 4.
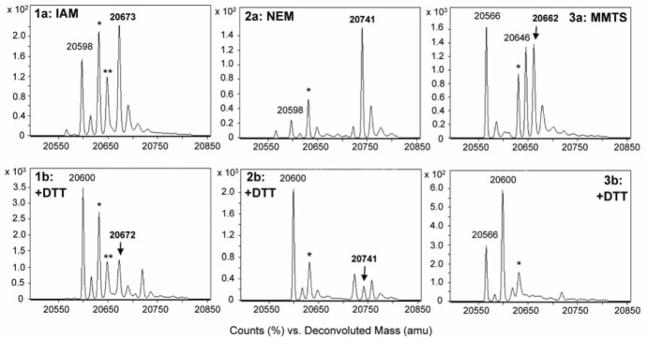
ESI-TOF mass spectra demonstrating (a) the formation of sulfenic acid-electrophile (-SOR) adducts and (b) subsequent reduction by DTT (10 mM, pH 7.5). Adducts in upper panels were generated via incubation of C165S AhpC-SOH (30 μM) with electrophile (2 mM): IAM (1), NEM (2), or MMTS (3) for 3 hours at r.t. Excess electrophile was removed prior to DTT treatment. The signal at 20,598 amu corresponds to sulfenamide (-SN), and the signals at 20,566 and 20,646 amu (3a–b) correspond to cysteine conversion to dehydroalanine (20,566) and AhpC-SH addition to MMTS (20,646). Signals labeled with * and ** indicate 20,632 (-SO2H) and 20,648 (-SO3H) a.m.u., respectively. Evidence for thiol regeneration is indicated by the appearance of a mass peak at 20,600 a.m.u. (-SH) with concomitant loss of signal corresponding to the -SOR adducts.
Table 1.
Percentage of AhpC-SOR adducts remaining following room temperature incubation (1 h) with reducing agents DTT, TCEP, and sodium ascorbate.
| entry | -SOR adduct | +10 mM DTT | +10 mM TCEP | +10 mM Asc |
|---|---|---|---|---|
| a | IAM | 60.3 | 58.0 | 50.4 |
| b | NEM | 13.1 | 40.2 | 27.7 |
| c | MMTS | 0.5 | 0.0a | 15.0 |
no evidence of MMTS adduct as determined by ESI-TOF MS.
The structural identity of the -SOR adducts, whether a sulfoxide or a thioperoxide (also called a sulfenate ester; thiosulfinate when derived from MMTS), is not well-defined. Structures of both types have been proposed for nucleophilic addition of sulfenic acids, and formation of the thermodynamically stable sulfoxide product represents the favored proposed mechanism [18]. However, reduction of the -SOR adducts to thiols by these relatively mild reductants suggest a less stable bond connectivity and/or the possibility for β-elimination. Sulfoxide reduction to thioethers is well-defined enzymatically and has been documented for chemical reductants like TCEP; reduction of these species to thiols typically requires boron or other Lewis acids [33, 34]. With the exception of MMTS adduct reduction, the products observed subsequent to -SOR reduction do not broadly suggest that the β-elimination mechanism dominates, as expected products include NEM-blocked cysteine and the conversion of cysteine to dehydroalanine [35]. Molecular structure and stability of protein -SOR adducts are certainly site-dependent and warrant further investigation.
The accuracy of biochemical methods for cysteine oxoform detection relies profoundly on the chemoselectivity of both the thiol blocking and chemical reduction steps prior to downstream analysis (spectroscopy, Western blotting, mass spectrometry, etc). The rates of -SOH reaction with electrophiles and subsequent adduct reduction are undoubtedly protein-dependent and expected to vary according to the microenvironment of each -SOH moiety, complicating the view of how this chemistry ultimately affects the interpretation of results obtained using thiol blocking and reduction techniques. Clearly, the implications of the sulfenic acid reactivity described herein on the outcomes of oxoform detection will vary according to specific experimental details, most critically sample complexity and cysteine accessibility. A few general scenarios are examined below based on the chemical data obtained with AhpC.
Implications for the global detection of reversibly oxidized cysteine species
In the case of biotin-switch techniques, thiol blocking is immediately followed by a presumably chemoselective reduction of a given oxidation product to generate nascent thiols (Figure 1) [36]. We have demonstrated that the thiol-blocking step also consumes the sulfenic acid of AhpC and anticipate that similar cross-reactivity may exist with other protein sulfenic acids. Reductants such as DTT and TCEP, often utilized to quantify the content of reversibly oxidized cysteines, cleave the majority of -SOR species (the extent determined by the blocking reagent and reduction conditions) to generate a pool of cysteine thiols with varying fractions originating from disulfide (-SS-), -SOH, and -SNO species (Figure 1, path 2a) [37]. In this experimental scenario, and given the low preponderance of the -SOH species relative to disulfides, the -SOR chemistry is not be expected to significantly affect the global qualitative or quantitative analysis of reversibly oxidized cysteines in the cellular proteome. (Note: This is aside from caveats related to indirect methods of detection: e.g., incomplete blocking of free -SH during the first workflow step.)
Implications for the detection of -SOH species in complex samples using the biotin-switch assay
The biotin-switch assay has been tailored for -SOH detection via the use of sodium arsenite as reductant, and although unexplored here, this reagent is expected to similarly reduce -SOR species based on its demonstrated selectivity for S-O bonds [38–40]. Here as well, and considering the caveats described above, the -SOR chemistry will not impact the qualitative identification of -SOH sites, but may affect the quantitative analysis by underestimating the -SOH content. It is however possible that -SOR formation in this assay could enable the identification of -SOH sites otherwise lost to adventitious hyperoxidation during sample workup had they not been alkylated in the initial step.
Implications for the direct detection of -SOH species in complex samples using chemical probes
Uncovering protein sulfenic acids by mass spectrometry or Western blotting involves the use of nucleophilic 1,3-dicarbonyl probes often in conjunction with a thiol-blocking reagent [2, 41–44]. As noted above, the cross-reactivity of the -SOH species and nucleophilic -SOH probes with thiol-capturing electrophiles was suspected of decreasing the overall yield of probe incorporation in our cellular oxidation studies. Indeed, the incubation of equimolar amounts of dimedone with IAM, NEM, and MMTS each resulted in the formation of covalent adducts as observed by LC-MS (Figure S6). With m/z values corresponding to a 1:1 stoichiometric addition, proposed structures involve the new bond connecting the nucleophilic methylene C2 of dimedone to the blocking group.
These observations led to an experiment designed to investigate the cross-reactivity among several probes and electrophiles when one reactive partner is protein-bound. Samples of AhpC-SOH (30 μM) were incubated separately with dimedone and 1,3-cyclopentanedione probe BP1 (2 mM each) for 1 h to generate the protein thioethers [10]. Excess probe was removed, and the proteins were treated separately with either IAM or NEM (10 mM each) for 45 minutes, then digested using trypsin with analysis by nanoLC-MS. The MS2 spectra acquired for the cysteine-containing peptide in each sample provided evidence only for cysteine modified by dimedone or BP1; no evidence for subsequent addition of IAM or NEM was observed with either of the probes once bound to AhpC. Such results corroborate the dimedone reactivity described above, suggesting that the 1,3-dicarbonyl enol attacks both the -SOH and the electrophiles at C2. Once the C2 site formed a thioether at a protein cysteine residue, the dicarbonyl adduct was no longer reactive with electrophiles despite opportunities to form other less reactive enols.
As a result of the reactivity of thiol-blocking electrophiles with both -SOH itself and the 1,3-dicarbonyl probes for its labeling, adjustments to the workflows for -SOH labeling and detection are needed. There is a clear need for more chemoselective thiol-blocking reagents. A recent report details the use of a new thiol-quenching reagent shown to circumvent the cross-reactivity of IAM and NEM with non-cysteine amino acids, but this reagent has not yet been tested with oxidized cysteine species [45]. While new approaches are in development, the addition of thiol-blocking reagents after -SOH tagging with the 1,3-dicarbonyl probes is recommended. More stringent efforts to prevent lysis-associated oxidation (e.g., lowering lysis buffer pH, using degassed lysis buffers supplemented with catalase and superoxide dismutase, and others) are also critical.
Implications for the detection of -SNO species in complex samples using the biotin-switch assay
The replacement of DTT/TCEP with ascorbate as the reductant has been widely utilized as a method for the indirect detection of protein S-nitrosothiols (Fig. 1) [46]. The limitations of this technique have been critically investigated in recent years with particular attention given to the chemoselectivity of ascorbate and its Cu(I) dependency in the reduction of S-nitroso moieties [47–49]. Cumulatively, these studies question the capacity of ascorbate to reduce -SNO species without affecting disulfide content and generating false-positive identifications of -SNO species.
Here also, ascorbate is shown to be a promiscuous reductant with respect to -SOH and the -SOR adduct (Figure S5, Table 1). These results suggest that the use of ascorbate in the presence of protein -SOH may well lead to false-positive identifications of -SNO content by generating cysteine thiol sites derived from -SNO, -SOH, and -SOR functional groups with an inability to distinguish their precursor states. In fact the potential for ascorbate to reduce protein sulfenic acids has been long established although evidence to the contrary has been reported as well [50–52]. Nevertheless, the current results clearly indicate the conversion of -SOH species to -SOR during the thiol-blocking step of the biotin-switch assay. We investigated next the chemical response of -SOR to ascorbate reduction under conditions described in the biotin-switch workflow.
In contrast to earlier biotin-switch reports requiring 10–20 mM ascorbate, more recent investigations have uncovered the role of Cu(I) ions in promoting nitrosothiol reduction, allowing for a decrease to 1 mM ascorbate with catalytic amounts of Cu(I) or Cu(II) [53]. The C165S AhpC-SOR, where R is N-ethylsuccinimide, was treated with 1 mM ascorbate in the presence of 1 μM CuSO4 at pH 7.5 for one hour in the absence of light. In comparison to the 73% decrease of this -SOR species by 10 mM ascorbate (Table 1), ESI-TOF MS analysis of the 1 mM ascorbate reduction showed less -SOR reduction at the lower ascorbate levels (38% remaining, Table 2). Again, AhpC-SO2H was the primary product of -SOR reduction (and oxidation) by ascorbate, and neither the degree of -SOR cleavage nor the amount of -SO2H formed was altered by carrying out the reduction in the presence of catalase and superoxide dismutase (SOD).
Table 2.
Susceptibility of AhpC-SOR (R = succinimide) to reduction by sodium ascorbate (1 and 10 mM). Each reaction contained 1 μM CuSO4 and was incubated 1 h at room temperature.
| Asc (mM) | 1 | 1 | 10 | 10 | 10 | 10 | 10 |
| cat/SOD | − | + | − | + | − | + | − |
| 1% SDS | − | − | − | − | − | − | + |
|
| |||||||
| AhpC-SOR | 38 | 37 | 35 | 39 | 77 | 89 | 77 |
|
|
|
||||||
| NH4 HCO3 pH 7.5 | HEPES pH 7.7 | ||||||
A notable difference in the ascorbate reduction among reported tag-switch procedures and the -SOR investigations described herein is reaction buffer composition. The reduction step in the tag-switch workflows is often performed in HEPES in the presence of 1% SDS, whereas -SOR chemistry has been largely uncovered in the MS-compatible NH4HCO3. To assess buffer effects of the ascorbate reduction, the succinimide-based AhpC-SOR was incubated with 10 mM ascorbate in HEPES (25 mM, pH 7.7) and NH4HCO3 (50 mM, pH 7.5). Interestingly, the reductive cleavage of -SOR by ascorbate is weakened in HEPES buffer as compared to NH4HCO3: 33% cleavage in HEPES compared to 73% cleavage in NH4HCO3. (Note: The extent of AhpC-SOR formation was unchanged when comparing reactions carried out in HEPES vs. NH4HCO3 buffers.) The use of denaturing conditions (1% SDS) did not affect -SOR stability in HEPES as measured by ESI-TOF MS. Furthermore, the principal product of AhpC-SOR reduction by ascorbate in the presence or absence of Cu(I) was sulfinic acid, which is unreactive toward biotin-HPDP and other biotinylated electrophiles (Figure S5). Together, these results imply that -SOR reduction by ascorbate could impact the analysis of S-nitrosation in tag-switch assays. A full biotin switch assay with Western blot detection was performed using MS-identified oxoforms of AhpC to further test these conclusions.
Cysteine oxoforms of interest, -SOH, -SOR (R = N-ethylsuccinimide), and -SNO, were generated using mutant AhpC and the mass of each modified protein species confirmed by ESI-TOF MS (Figure S7). AhpC-SH (40 μM) was also treated with biotin-HPDP or NEM (5 mM) for positive (lane 1) and negative controls (lane 3), respectively. Analysis by ESI-TOF MS confirmed fully blocked thiol in both control samples. Biotin-switch blocking was performed with 20 mM NEM and 2.5% SDS as described previously [53, 54]. Following precipitation, resuspended samples were treated in the dark with ascorbate (1 mM) in the presence of CuSO4 (1 μM) and 1% SDS. Non-reducing SDS-PAGE followed by Western blot illustrates that along with -SNO (lane 6), both -SOH and pre-generated -SOR (lanes 4 and 5, respectively) are reduced by ascorbate under biotin-switch conditions, confirming the extensive cross-reactivity of these species with ascorbate and illustrating the potential for misinterpretation using this assay (Figure 5). These results also reveal a relatively low overall yield of the biotin-switch assay, perhaps a result of nascent AhpC-SH oxidation to AhpC-SO2H out-competing thiol capture by biotin-HPDP.
Figure 5.
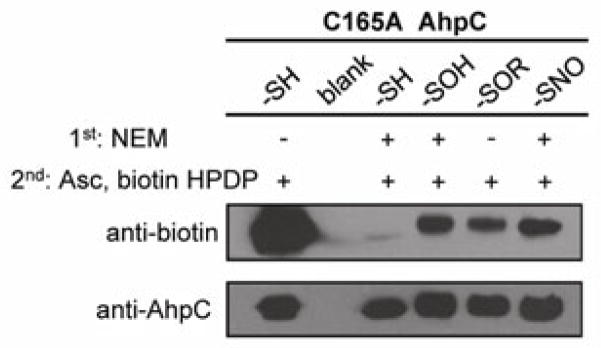
Biotin-switch assay of C165A AhpC reduced and oxidized cysteine species provides evidence of biotinylation akin to levels observed for AhpC-SNO. Where noted, the initial blocking step was carried out using NEM (20 mM); thiols generated by sodium ascorbate (1 mM) reduction were tagged with biotin-HPDP (5 mM). Steps 1 and 2 above reflect order of treatment. ESI-TOF MS spectra confirming the identities of starting AhpC species may be found in Figure S7.
Despite the wide use of biotin and other switch-tagging techniques, the fate of -SOH in such approaches has been largely under-investigated aside from the report of Forrester et al. where biotin-switch specificity was assessed using cysteine oxoforms of human PTP1B [51]. Oxidatively modified PTP1B, presumably a mixture of -SOH/-SN, was confirmed by a sharply decreased phosphatase activity of which < 40% was restored upon DTT treatment, suggesting the presence of hyperoxidized enzyme as well as the reversibly oxidized species [51]. The lack of evidence for biotinylated PTP1B in the described biotin-switch assay with up to 100 mM ascorbate is perhaps a result of the mildly oxidized enzyme’s propensity to reside in the sulfenamide (-SN) state as has been described in great detail [21, 55]. Our results taken with these previous observations illustrate the critical importance of careful control experiments and the challenge of assessing specificity in such indirect detection assays.
In summary, we have demonstrated the promiscuity of several thiol-blocking reagents by detailing their chemical reactivity with the sulfenic acid of peroxiredoxin AhpC. Treatment of AhpC-SOH with IAM, NEM, and MMTS at concentrations relevant to the biotin-switch assay and other biochemical methods produces covalent adducts that are partly or fully susceptible to reduction and thiol generation. This reactivity has profound effects on the approaches for detecting cellular cysteine oxidation, in particular the methods seeking selective detection of labile signaling species like sulfenic acid and S-nitrosothiol.
Experimental procedures
General
Unless otherwise stated, all reagents and enzymes were obtained from Sigma-Aldrich (St. Louis, MO, USA). Bio-Gel P6 Gel was obtained from Bio-Rad (Hercules, CA, USA); HEPES and sodium dodecyl sulfate (SDS) from American Bioanalytical (Natick, MA, USA); formic acid (99+%, LC-MS grade) from Thermo Scientific (Waltham, MA, USA).
Generation of AhpC-SOH and AhpC-SO2H
The C165S and C165A mutants of Salmonella typhimurium AhpC were overexpressed and purified from Escherichia coli as previously described [56, 57]. Mutant AhpC was pre-reduced with DTT (10 mM) for 30 min at ambient temperature, at which time DTT was removed by passing the solution through a Bio-Gel P6 spin column equilibrated with ammonium bicarbonate (50 mM). Protein concentration was determined using the solution absorbance at 280 nm (ε = 24,300 M−1 cm−1) [56]. The sulfenic acid species was generated via treatment with 1 equivalent of hydrogen peroxide for 30–45 seconds at room temperature (pH 7–7.5 buffer) with quenching by passage through a Bio-Gel spin column equilibrated with ammonium bicarbonate (50 mM). The sulfinic acid was generated via treatment of AhpC-SH with 2 equivalents of hydrogen peroxide for 45–75 seconds at room temperature with similar removal of unreacted substrate. Formation of each oxidized species was confirmed by ESI-TOF MS.
Generation of AhpC-SNO
C165S AhpC was pre-reduced as detailed above and then transnitrosated with freshly prepared S-nitrosocysteine (CySNO) for 1 h in the dark as previously described [58]. CySNO was removed using Bio-Gel spin columns equilibrated with HEPES buffer (50 mM) and formation of AhpC-SNO was confirmed by ESI-TOF MS.
Reactivity of AhpC oxoforms with thiol-blocking electrophiles
Freshly prepared AhpC-SOH, AhpC-SO2H, or AhpC-SNO (30–40 μM) was incubated at room temperature in the presence of IAM, NEM, or MMTS (5 mM) in ammonium bicarbonate buffer (50 mM, pH 7.5). Reactions involving IAM and/or AhpC-SNO were protected from light. Aliquots (40–50 μL) were quenched at various time points by passage through Bio-Gel spin columns pre-equilibrated with either 0.1% formic acid in water for analysis by ESI-TOF MS or with ammonium bicarbonate (50 mM, pH 7.5) or HEPES (25 mM, pH 7.7) to assay subsequent reactivity of adducts. Reduction of the adducts was investigated by treatment with DTT, TCEP, or sodium ascorbate (1–10 mM) for lengths of time ranging from 10 min to 2 h. All ascorbate reductions were performed in the absence of light, and when noted, ascorbate reductions were carried out in the presence of bovine liver catalase (100 U/mL), bovine erythrocyte superoxide dismutase (100 U/mL), and/or SDS (1%). When applicable, samples were passed through detergent removal spin columns (Thermo Scientific). All samples were passed through Bio-Gel spin columns pre-equilibrated with 0.1% formic acid prior to ESI-TOF MS analysis.
Biotin-switch assay of protein -SOH, -SOR, and -SNO
Mutant AhpC-SOH and AhpC-SNO were prepared as described above. A representative AhpC-SOR species was generated via the reaction of AhpC-SOH (40 μM) with NEM (5 mM) for one hour as described above. Biotinylated and maleimide-labeled AhpC were formed upon treatment of AhpC-SH (40 μM) with biotin-HPDP (5 mM) and NEM (5 mM), respectively. Resulting protein species were each desalted and their identities confirmed by ESI-TOF MS. All steps of the biotin switch assay were carried out in the absence of light. Blocking was achieved using NEM (20 mM) in 2.5% SDS in 25 mM HEPES, 1 mM DTPA (pH 7.7) for 20 min at 50°C, followed by a cold acetone precipitation. Protein pellets were resuspended in 25 mM HEPES (pH 7.7) containing 1% SDS, 1 μM CuSO4, 1 mM ascorbate, and 1 mM biotin-HPDP and incubated for 2 hrs at r.t. Excess reagents were again removed by cold acetone precipitation. Protein precipitates were suspended in 25 mM HEPES (pH 7.7) containing 1% SDS, normalized based on protein concentration, and separated by non-reducing SDS-PAGE. Biotinylated protein species were identified by Western blot using an anti-biotin-HRP antibody (Cell Signaling Technologies). Total protein was measured using anti-AhpC (Lampire Biological Laboratories) primary and anti-rabbit-HRP (Cell Signaling) secondary antibodies.
Electrospray ionization - mass spectrometry
ESI-TOF MS analyses were performed on an Agilent 6120 MSD-TOF system operating in positive ion mode with the following settings: capillary voltage of 3500 V, nebulizer gas pressure of 30 psig, drying gas flow of 5 L/min, fragmentor voltage of 175 V, skimmer voltage of 65 V, and gas temperature of 325°C. Samples were introduced via direct infusion at a flow rate of 20 μL/min using a syringe pump (KD Scientific). Mass spectra were averaged and deconvoluted using the Agilent MassHunter Workstation software v B.02.00. AhpC digests were analyzed on a Dionex UltiMate3000 nanoLC system coupled to a Thermo Orbitrap Velos Pro high-resolution mass spectrometer and dimedone-electrophile reactions were analyzed on an Accela Open UPLC coupled to a Thermo Orbitrap LTQ XL high-resolution mass spectrometer. Additional experimental details are included in the SI.
Supplementary Material
Acknowledgments
Financial support was provided by funds from NIH R01 CA136810 (C.M.F.), R01 GM050389 (L.B.P.), R33 CA126659/CA126659Z (L.B.P.), R33 CA177461 (C.M.F., L.B.P., and S.B.K.), start-up funds from Wake Forest School of Medicine (C.M.F.), and pilot funds from the Center for Molecular Signaling and Communication at Wake Forest University (C.M.F., S.B.K., and L.B.P.). The Thermo Orbitrap LTQ XL mass spectrometer was purchased with support from NSF-CRIF 0947028. The authors kindly thank Dr. Derek Parsonage (WFSM) for preparation of the C165A AhpC mutant and Drs. Kimberly Nelson and Chananat Klomsiri (WFSM) for helpful discussions of this work.
Abbreviations
- a.m.u
atomic mass units
- biotin-HPDP
N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide
- DTT
dithiothreitol
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IAM
iodoacetamide
- m/z
mass to charge ratio
- NBD-Cl
4-chloro-7-nitrobenzofurazan
- MMTS
methyl methanethiosulfonate
- NEM
N-ethylmaleimide
- TCEP
tris(2-carboxyethyl)phosphine
Footnotes
Supporting Information containing Figures S1–S7 and expanded experimental details is available.
Figure S1: Positive ion MS2 (CID) spectrum of the Cys-containing peptide of C165S AhpC resulting from the reaction of AhpC-SOH (30 μM) with IAM (5 mM) at pH 7.5.
Figure S2: ESI-TOF mass spectra for the incubation of C165S AhpC-SO2H (30 μM) with electrophiles IAM, NEM, or MMTS (5 mM) for 2 hours at r.t. and pH 7.5. The signals at 20,566 a.m.u. and 20,632 a.m.u. correspond to dehydroalanine and sulfinic acid (-SO2H), respectively. The adduct at 20,758 a.m.u., corresponding to the addition of one equivalent of NEM, was not found to contain a NEM-modified cysteine peptide as determined via tryptic digest and nanoLC-MS/MS.
Figure S3: ESI-TOF mass spectra for the incubation of C165S AhpC-SNO (40 μM) with electrophiles IAM, NEM, or MMTS (5 mM) for 2 hours at r.t. and pH 7.5. The signals at 20,629 and 20,719 a.m.u correspond to nitrosothiol (AhpC-SNO) and mixed disulfide (AhpC-S-S-Cys), respectively.
Figure S4: ESI-TOF mass spectra demonstrating the reduction of sulfenic acid-electrophile adducts (a) by TCEP (10 mM) over 1 h at pH 7.5 (b). Adducts shown in upper panels were generated via incubation of C165S AhpC-SOH (30 μM) with electrophile (5 mM): IAM (1), NEM (2), or MMTS (3) for 3 hours at r.t. Excess electrophile was removed prior to TCEP treatment. The signals at 20,566 and 20,598 a.m.u correspond to dehydroalanine and sulfenamide (-SN), respectively. Signals labeled with * and ** indicate 20,632 (-SO2H) and 20,648 (-SO3H) a.m.u, respectively. Ammonium (+NH4) adducts result from the NH4HCO3 reaction buffer. Evidence for thiol regeneration is indicated at 20,600 a.m.u with concomitant loss of signal corresponding to the covalent adducts.
Figure S5: ESI-TOF mass spectra demonstrating the reduction of sulfenic acid-electrophile adducts (a) by sodium ascorbate (10 mM) over 1 h at pH 7.5 (b). Adducts shown in a panels were generated via incubation of C165S AhpC-SOH (30 μM, 4a) with electrophile (5 mM) IAM (1), NEM (2), or MMTS (3) for 2 hours at r.t. Excess electrophile was removed prior to ascorbate treatment. The signals at 20,566, 20,598, 20,616, and 20,632 a.m.u correspond to dehydroalanine, sulfenamide (-SN), sulfenic acid (-SOH) and sulfinic acid (-SO2H), respectively.
Figure S6: a: LC-MS spectrum indicating that the equimolar reaction of dimedone and iodoacetamide at pH 7.5 generates small amounts of a covalent adduct.
b: LC-MS spectrum resulting from the equimolar reaction of dimedone and NEM at pH 7.5.
c: LC-MS spectrum resulting from the equimolar reaction of dimedone and MMTS at pH 7.5.
Figure S7: ESI-TOF mass spectra identifying cysteine oxoforms of C165A AhpC: biotin-HPDP-treated thiol, NEM-treated thiol, -SOH, -SOR (R = succinimide), and -SNO prior to biotin-switch assay. The observed mass of reduced C165A AhpC is 20,584 a.m.u., representing the [M+H]+ ion.
Text: Experimental details for mass spectrometry analyses
References
- 1.Poole LB, Nelson KJ. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr Opin Chem Biol. 2008;12:18–24. doi: 10.1016/j.cbpa.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wani R, Qian J, Yin L, Bechtold E, King SB, Poole LB, Paek E, Tsang AW, Furdui CM. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc Natl Acad Sci USA. 2011;108:10550–10555. doi: 10.1073/pnas.1011665108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wani R, Bharathi NS, Field J, Tsang AW, Furdui CM. Oxidation of Akt2 kinase promotes cell migration and regulates G1-S transition in the cell cycle. Cell Cycle. 2011;10:3263–3268. doi: 10.4161/cc.10.19.17738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saurin AT, Neubert H, Brennan JP, Eaton P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc Natl Acad Sci USA. 2004;101:17982–17987. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol. 2012;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen RE, Roth D, Winther JR. Quantifying the global cellular thiol-disulfide status. Proc Natl Acad Sci USA. 2009;106:422–427. doi: 10.1073/pnas.0812149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benitez LV, Allison WS. The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphate dehydrogenase by dimedone and olefins. J Biol Chem. 1974;249:6234–6243. [PubMed] [Google Scholar]
- 8.Poole LB, Klomsiri C, Knaggs SA, Furdui CM, Nelson KJ, Thomas MJ, Fetrow JS, Daniel LW, King SB. Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjugate Chem. 2007;18:2004–2017. doi: 10.1021/bc700257a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seo YH, Carroll KS. Facile synthesis and biological evaluation of a cell-permeable probe to detect redox-regulated proteins. Bioorg Med Chem Lett. 2009;19:356–359. doi: 10.1016/j.bmcl.2008.11.073. [DOI] [PubMed] [Google Scholar]
- 10.Qian J, Klomsiri C, Wright MW, King SB, Tsang AW, Poole LB, Furdui CM. Simple synthesis of 1,3-cyclopentanedione derived probes for labeling sulfenic acid proteins. Chem Commun (Camb) 2011;47:9203–9205. doi: 10.1039/c1cc12127h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian J, Wani R, Klomsiri C, Poole LB, Tsang AW, Furdui CM. A simple and effective strategy for labeling cysteine sulfenic acid in proteins by utilization of β-ketoesters as cleavable probes. Chem Commun (Camb) 2012;48:4091–4093. doi: 10.1039/c2cc17868k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hansen RE, Winther JR. An introduction to methods for analyzing thiols and disulfides: Reactions, reagents, and practical considerations. Anal Biochem. 2009;394:147–158. doi: 10.1016/j.ab.2009.07.051. [DOI] [PubMed] [Google Scholar]
- 13.Dickens F. Interaction of haloacetates and SH compounds. The reaction of haloacetic acids with glutathione and cysteine. The mechanism of iodoacetate poisoning of glyoxalase. Biochem J. 1933;27:1141–1151. doi: 10.1042/bj0271141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poole LB, Ellis HR. Identification of cysteine sulfenic acid in AhpC of alkyl hydroperoxide reductase. In: Helmut S, Lester P, editors. Methods in Enzymology. Academic Press; 2002. pp. 122–136. [DOI] [PubMed] [Google Scholar]
- 15.Aversa MC, Barattucci A, Bonaccorsi P, Giannetto P, Jones DN. Synthesis and asymmetric Diels-Alder reactions of enantiopure 3-(alkylsulfinyl)-1-methoxy-1,3-butadienes. J Org Chem. 1997;62:4376–4384. doi: 10.1021/jo962286p. [DOI] [PubMed] [Google Scholar]
- 16.Aversa MC, Barattucci A, Bonaccorsi P, Temperini A. Regio- and stereocontrolled synthesis of (Z)-α-(phenylseleno)sulfinyl and -sulfonyl alkenes via sulfenic acids, and a study of their reactivity. Eur J Org Chem. 2011;2011:5668–5673. [Google Scholar]
- 17.Aversa MC, Barattucci A, Bonaccorsi P, Giannetto P. Recent advances and perspectives in the chemistry of sulfenic acids. Curr Org Chem. 2007;11:1034–1052. [Google Scholar]
- 18.Ellis HR, Poole LB. Novel application of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole to identify cysteine sulfenic acid in the AhpC component of alkyl hydroperoxide reductase. Biochemistry. 1997;36:15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 19.Kice JL. Mechanisms and reactivity in reactions of organic oxyacids of sulfur and their anhydrides. In: Gold V, Bethell D, editors. Advances in Physical Organic Chemistry. Academic Press; 1981. pp. 65–181. [Google Scholar]
- 20.Davis FA, Billmers RL. Chemistry of sulfenic acids. 4. The first direct evidence for the involvement of sulfenic acids in the oxidation of thiols. J Am Chem Soc. 1981;103:7016–7018. [Google Scholar]
- 21.Salmeen A, Andersen JN, Myers MP, Meng T-C, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 22.Nelson KJ, Parsonage D, Hall A, Karplus PA, Poole LB. Cysteine pKa values for the bacterial peroxiredoxin AhpC. Biochemistry. 2008;47:12860–12868. doi: 10.1021/bi801718d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts DD, Lewis SD, Ballou DP, Olson ST, Shafer JA. Reactivity of small thiolate anions and cysteine-25 in papain toward methyl methanethiosulfonate. Biochemistry. 1986;25:5595–5601. doi: 10.1021/bi00367a038. [DOI] [PubMed] [Google Scholar]
- 24.Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J Biol Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 25.Rogers LK, Leinweber BL, Smith CV. Detection of reversible protein thiol modifications in tissues. Anal Biochem. 2006;358:171–184. doi: 10.1016/j.ab.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 26.Guidotti G. The rates of reaction of the sulfhydryl groups of human hemoglobin. J Biol Chem. 1965;240:3924–3927. [PubMed] [Google Scholar]
- 27.Klomsiri C, Nelson KJ, Bechtold E, Soito L, Johnson LC, Lowther WT, Ryu S, King SB, Furdui CM, Poole LB. Use of dimedone-based chemical probes for sulfenic acid detection: Evaluation of conditions affecting probe incorporation into redox-sensitive proteins. In: Enrique C, Lester P, editors. Methods in Enzymology. Academic Press; 2010. pp. 77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papini A, Rudolph S, Siglmueller G, Musiol HJ, Goehring W, Moroder L. Alkylation of histidine with maleimido-compounds. Int J Pept Protein Res. 1992;39:348–355. doi: 10.1111/j.1399-3011.1992.tb01594.x. [DOI] [PubMed] [Google Scholar]
- 29.Aliverti A, Gadda G, Ronchi S, Zanetti G. Identification of Lys116 as the target of N-ethylmaleimide inactivation of ferredoxin: NADP+ oxidoreductase. Eur J Biochem. 1991;198:21–24. doi: 10.1111/j.1432-1033.1991.tb15981.x. [DOI] [PubMed] [Google Scholar]
- 30.Brewer CF, Riehm JP. Evidence for possible nonspecific reactions between N-ethylmaleimide and proteins. Anal Biochem. 1967;18:248–255. [Google Scholar]
- 31.Paulech J, Solis N, Cordwell SJ. Characterization of reaction conditions providing rapid and specific cysteine alkylation for peptide-based mass spectrometry. Biochim Biophys Acta - Proteins and Proteomics. 2013;1834:372–379. doi: 10.1016/j.bbapap.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 32.Schmalhausen EV, Pleten’ AP, Muronetz VI. Ascorbate-induced oxidation of glyceraldehyde-3-phosphate dehydrogenase. Biochem Biophys Res Commun. 2003;308:492–496. doi: 10.1016/s0006-291x(03)01421-9. [DOI] [PubMed] [Google Scholar]
- 33.Faucher A, Grand-Maitre C. tris(2-Carboxyethyl)phosphine (TCEP) for the reduction of sulfoxides, sulfonylchlorides, N-oxides, and azides. Synth Commun. 2003;33:3503–3511. [Google Scholar]
- 34.Friedmann C, Brase S. Synthesis of paracyclophane thiols via an unprecedented reduction-deprotection sequence: direct conversion of tert-butyl sulfoxides into thiols with boron tribromide. Synlett. 2010;5:774–776. [Google Scholar]
- 35.Fishkin N, Maloney EK, Chari RVJ, Singh R. A novel pathway for maytansinoid release from thioether linked antibody-drug conjugates (ADCs) under oxidative conditions. Chem Commun (Cambridge, UK) 2011;47:10752–10754. doi: 10.1039/c1cc14164c. [DOI] [PubMed] [Google Scholar]
- 36.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 37.Leichert LI, Jakob U. Protein thiol modifications visualized in vivo. PLoS Biol. 2004;2:1723–1737. doi: 10.1371/journal.pbio.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gutmann A. Über die einwirkung von laugen auf athylnatriumthiosulfat. Berichte der deutschen chemischen Gesellschaft. 1908;41:1650–1655. [Google Scholar]
- 39.Parker DJ, Allison WS. The mechanism of inactivation of glyceraldehyde 3-phosphate dehydrogenase by tetrathionate, o-iodosobenzoate, and iodine monochloride. J Biol Chem. 1969;244:180–189. [PubMed] [Google Scholar]
- 40.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls: The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 41.Reddie KG, Seo YH, Muse WB, Leonard SE, Carroll KS. A chemical approach for detecting sulfenic acid-modified proteins in living cells. Mol Biosyst. 2008;4:521–531. doi: 10.1039/b719986d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem Biol. 2009;4:783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 43.Kaplan N, Urao N, Furuta E, Kim S, Razvi M, Nakamura Y, McKinney RD, Poole LB, Fukai T, Ushio-Fukai M. Localized cysteine sulfenic acid formation by vascular endothelial growth factor: role in endothelial cell migration and angiogenesis. Free Radical Res. 2011;45:1124–1135. doi: 10.3109/10715762.2011.602073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michalek RD, Nelson KJ, Holbrook BC, Yi JS, Stridiron D, Daniel LW, Fetrow JS, King SB, Poole LB, Grayson JM. The requirement of reversible cysteine sulfenic acid formation for T cell activation and function. J Immunol. 2007;179:6456–6467. doi: 10.4049/jimmunol.179.10.6456. [DOI] [PubMed] [Google Scholar]
- 45.Zhang D, Devarie-Baez NO, Li Q, Lancaster JR, Xian M. Methylsulfonyl benzothiazole (MSBT): A selective protein thiol blocking reagent. Org Lett. 2012;14:3396–3399. doi: 10.1021/ol301370s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:PL1–PL9. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 47.Landino LM, Koumas MT, Mason CE, Alston JA. Ascorbic acid reduction of microtubule protein disulfides and its relevance to protein S-nitrosylation assays. Biochem Biophys Res Commun. 2006;340:347–352. doi: 10.1016/j.bbrc.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 48.Huang B, Chen C. An ascorbate-dependent artifact that interferes with the interpretation of the biotin switch assay. Free Radic Biol Med. 2006;41:562–567. doi: 10.1016/j.freeradbiomed.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 49.Giustarini D, Dalle-Donne I, Colombo R, Milzani A, Rossi R. Is ascorbate able to reduce disulfide bridges? A cautionary note. Nitric Oxide. 2008;19:252–258. doi: 10.1016/j.niox.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 50.You K, Benitez LV, McConachie WA, Allison WS. The conversion of glyceraldehyde-3-phosphate dehydrogenase to an acylphosphatase by trinitroglycerin and inactivation of this activity by azide and ascorbate. Biochim Biophys Acta - Enzymology. 1975;384:317–330. doi: 10.1016/0005-2744(75)90033-9. [DOI] [PubMed] [Google Scholar]
- 51.Forrester MT, Foster MW, Stamler JS. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J Biol Chem. 2007;282:13977–13983. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- 52.Monteiro G, Horta BB, Pimenta DC, Augusto O, Netto LES. Reduction of 1-Cys peroxiredoxins by ascorbate changes the thiol-specific antioxidant paradigm, revealing another function of vitamin C. Proc Natl Acad Sci USA. 2007;104:4886–4891. doi: 10.1073/pnas.0700481104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Kettenhofen NJ, Shiva S, Hogg N, Gladwin MT. Copper dependence of the biotin switch assay: Modified assay for measuring cellular and blood nitrosated proteins. Free Radic Biol Med. 2008;44:1362–1372. doi: 10.1016/j.freeradbiomed.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kettenhofen NJ, Wang X, Gladwin MT, Hogg N. In-gel detection of S-nitrosated proteins using fluorescence methods. Methods Enzymol. 2008;441:53–71. doi: 10.1016/S0076-6879(08)01204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Montfort RLM, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 56.Poole LB, Ellis HR. Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 1. Purification and enzymatic activities of overexpressed AhpF and AhpC proteins. Biochemistry. 1996;35:56–64. doi: 10.1021/bi951887s. [DOI] [PubMed] [Google Scholar]
- 57.Ellis HR, Poole LB. Roles for the two cysteine residues of AhpC in catalysis of peroxide reduction by alkyl hydroperoxide reductase from Salmonella typhimurium. Biochemistry. 1997;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- 58.Bechtold E, Reisz JA, Klomsiri C, Tsang AW, Wright MW, Poole LB, Furdui CM, King SB. Water-soluble triarylphosphines as biomarkers for protein S-nitrosation. ACS Chem Biol. 2010;5:405–414. doi: 10.1021/cb900302u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.