

Abstract
Renal hypodysplasia (RHD) is a heterogeneous condition encompassing a spectrum of kidney development defects including renal agenesis, hypoplasia, and (cystic) dysplasia. Heterozygous mutations of several genes have been identified as genetic causes of RHD with various severity. However, these genes and mutations are not associated with bilateral renal agenesis, except for RET mutations, which could be involved in a few cases. The pathophysiological mechanisms leading to total absence of kidney development thus remain largely elusive. By using a whole-exome sequencing approach in families with several fetuses with bilateral renal agenesis, we identified recessive mutations in the integrin α8-encoding gene ITGA8 in two families. Itga8 homozygous knockout in mice is known to result in absence of kidney development. We provide evidence of a damaging effect of the human ITGA8 mutations. These results demonstrate that mutations of ITGA8 are a genetic cause of bilateral renal agenesis and that, at least in some cases, bilateral renal agenesis is an autosomal-recessive disease.
Main Text
Congenital abnormalities of the kidney and urinary tract (CAKUT) are frequently observed in children and represent a significant cause of morbidity, accounting for more than 40% of pediatric end-stage renal failure.1 The most severe forms lead to perinatal death or justify termination of pregnancy. Among CAKUT defects, renal hypodysplasia (RHD [MIM 191830]) is a heterogeneous condition encompassing a spectrum of kidney development defects including renal agenesis, hypoplasia, and cystic and noncystic dysplasia. Although most RHD cases are simplex, the occurrence of familial and syndromic cases indicates the existence of a genetic component.
Kidney development is initiated by a reciprocal induction between the ureteric bud (UB) and the metanephric mesenchyme (MM). Reiteration of this process induces branching of the UB to form the collecting ducts and mesenchyme-to-epithelial differentiation of the MM to generate the nephrons. Kidney development is orchestrated by a large number of genes encoding transcription factors, signaling and adhesion molecules, and extracellular matrix proteins. Mutations in ∼15 of these genes have been reported in individuals with RHD, demonstrating the genetic heterogeneity of these pathologies. The most frequent mutations, observed in individuals with syndromic RHD, affect the transcription factor encoding PAX2 (renal-coloboma syndrome [MIM 120330]), EYA1 and SIX2 (branchio-oto-renal syndrome [MIM 113650]), and HNF1B (renal cysts and diabetes association [MIM 137920]).2–6 Other rarer forms of syndromic RHD are associated with mutations in SALL1 (Townes-Brocks syndrome [MIM 107480]), KAL1 (Kallman syndrome [MIM 308700]), FRAS1 and FREM2 (Fraser syndrome [MIM 219000]), FREM1 (bifid nose and anorectal and renal anomalies [MIM 608980]), GATA3 (RHD, hypoparathyroidism, and sensorineural deafness [MIM 146255]), GLI3 (Pallister-Hall syndrome [MIM 146510]), LRP4 (Cenani-Lenz syndrome [MIM 212780]), or RET (Hirschsprung disease [MIM 142623] and CAKUT).7–15 Mutations in most of these genes and a few others (e.g., BMP4) have also been described in individuals with isolated RHD.16,17 Almost all the mutations reported to date are heterozygous, suggesting that haploinsufficiency is a common mechanism leading to RHD.
Genotype-phenotype correlation analyses show that the severity of the renal defects associated with mutations in a given gene can be extremely variable. This is notably well documented for PAX2 and HNF1B mutations.2,3 However, it is striking that these two genes, whose mutations can lead to very severe phenotypes, diagnosed in utero and justifying termination of pregnancy, have not been reported to date as mutated in fetuses with bilateral renal agenesis (BRA).18 It was proposed that heterozygous loss-of-function mutations in RET, encoding the GDNF receptor (which plays a crucial role in branching of the UB), could be a major mechanism leading to isolated BRA.19 However, this was questioned by another study based on a larger series of cases.20 The genetic mechanisms leading to BRA thus remain largely unresolved.
Absence of kidney development is reported in a large number of homozygous knockout mouse models (>45 knockout genes associated with the phenotype “absent kidney” in the Mouse Genome Informatics [MGI] database), suggesting that BRA in humans could be due to recessive mutations in some of these genes. Accordingly, by whole-exome sequencing, we have recently identified a homozygous mutation in FGF20 in fetuses with BRA from a consanguineous family.21 By the same approach, we analyzed ten other families with BRA cases. In two of these families, we identified recessive mutations in another gene with a major role during kidney development, the integrin α8-encoding gene ITGA8.22
Family F1 was a consanguineous Roma (Gypsy) family originating from Serbia and Spain. In this large family, three couples experienced one to four terminations of pregnancy for BRA associated with the Potter sequence due to anhydramnios (facial dysmorphism, pulmonary hypoplasia, and clubbed feet). DNA from three fetuses and the mother of one of them was available for analysis (Figure 1A and Figure S1 available online). Family F2 was a nonconsanguineous family originating from Guinea-Bissau (Figure 1B). F2-1 (II-2 in Figure 1B) was a fetus, spontaneous aborted in the 24th gestational week for BRA with anhydramnios. F2-2 (II-3 in Figure 1B) died perinatally and presented with BRA and bilateral cryptorchidism. DNA from F2-1, F2-2, and their parents was available. The study was approved by the Comité de Protection des Personnes pour la Recherche Biomédicale Ile de France 2 and informed consent was obtained from the parents. Whole-exome sequencing was performed for individuals F1-1 (II-3 in Figure 1A), F2-1, and F2-2 via the 50 Mb Agilent SureSelect all exon V3 and a SOLiD4 (Life Technologies) sequencer (50 base fragment reads) for F1-1 and the 51 Mb Agilent SureSelect all exon V5 and the HiSeq2500 (Illumina) sequencer (paired-end 76-76 base reads) for F2-1 and F2-2. Sequence data were aligned to the human genome reference sequence (UCSC Genome Browser, hg19 build) by the Lifescope suite from Lifetech (SOLID) or BWA aligner (Hiseq).23 Downstream processing was carried out with the Genome Analysis Toolkit (GATK), SAMtools, and Picard.24 Variants were annotated with a software system developed by the Paris Descartes University Bioinformatics Platform, based on the Ensembl (release 72), dbSNP (135), EVS (ESP6500SI-V2), and 1000 Genomes (May 2011) databases.
Figure 1.
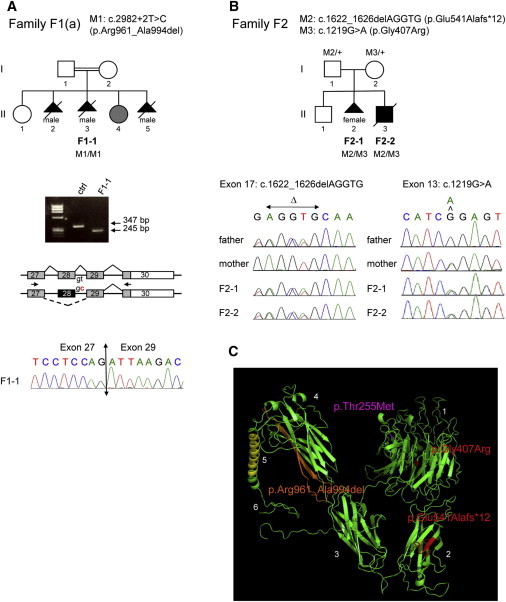
Identification of Recessive ITGA8 Mutations in Two Families with Bilateral Renal Agenesis
(A) Characterization of a homozygous 5′ splice site mutation, c.2982+2T>C, in consanguineous family F1. Pedigree of branch F1(a) of the family, with three fetuses with bilateral renal agenesis (in black) and one child with unilateral renal agenesis (in gray). Exome sequencing was performed for fetus F1-1 (II-3 in the pedigree), leading to the identification of the c.2982+2T>C homozygous mutation (RefSeq NM_003638.1). For validation of the effect of this mutation on splicing, RT-PCR analysis of the ITGA8 transcript was performed on fetal lung cDNAs of F1-1 and a control, using primers located in exons 27 and 30. Whereas a 347 bp band corresponding to normal splicing was obtained for the control, a 245 bp band was amplified for fetus F1-1. Sanger sequencing of the 245 bp fragment confirmed that the c.2982+2T>C mutation leads to aberrant splicing and skipping of exon 28, resulting in in-frame deletion of 34 amino acids (p.Arg961_Ala994del).
(B) Characterization of compound heterozygous mutations in family F2. Exome sequencing was performed for F2-1 and F2-2 with bilateral renal agenesis (II-2 and II-3 in the pedigree), leading to the identification of two heterozygous mutations, a 5 bp deletion in exon 17 (c.1622_1626delAGGTG) resulting in a frameshift and a truncated protein (p.Glu541Alafs∗12), and a point mutation in exon 13 (c.1219G>A) resulting in a missense change (p.Gly407Arg). Sanger sequencing chromatograms showing paternal origin of the deletion and maternal origin of the missense are presented.
(C) Structure of the ITGA8 protein in bent (inactive) configuration, designed with the Phyre2 program and visualized with PyMOL. The position of the part of the protein that is deleted as a consequence of the homozygous splice mutation identified in family F1 is shown in orange (p.Arg961_Ala994del), the position of the amino acids that are changed as a consequence of the compound heterozygous mutations identified in family F2 are in red (p.Gly407Arg and p.Glu541Alafs∗12), and the position of the amino acid that is changed as a result of the heterozygous mutation identified by Sanger sequencing in a simplex BRA case is in magenta (p.Thr255Met). The protein domains are numbered from N-ter to C-ter: 1, seven-bladed β propeller head domain; 2, thigh domain; 3, calf1 domain; 4, calf2 domain (domains 1–4 constituting the extracellular part of the protein); 5, transmembrane domain; 6, intracellular domain.
Mean coverage was 45, 53, and 52, with 75% of the targeted bases covered at least 5 times for individual F1-1 and 99% for F2-1 and F2-2. Prioritization of the variants was performed by the following strategy: (1) filtration of the variants present in public databases or in our in-house exome database; (2) selection of genetic variants located in the coding regions or splice sites and with a predicted deleterious effect on the protein (Polyphen2, Sift); and (3) expression of the selected candidate genes in early kidney development (GUDMAP database). For F1-1 belonging to a consanguineous family, we looked for homozygous variants and identified three candidate genes, whereas for F2-1 and F2-2 we focused on compound heterozygous variants common to both cases, which were detected in four genes (Table S1). Interestingly, ITGA8 was one of the candidate genes in both families. Because of its crucial role for renal development, this gene was a very good candidate whose mutations could explain the lack of kidneys in the fetuses. Indeed, integrin α8 is expressed in the MM, notably in the cap mesenchyme surrounding the UB, and, in association with integrin β1, signals though binding with nephronectin, an extracellular ligand produced by the UB, for activation of the expression of GDNF.25 Itga8 knockout mice display renal agenesis resulting from an altered invasion of the MM by the UB.22
A homozygous 5′-splice variant (c.2982+2T>C [RefSeq accession number NM_003638.1]) was detected in fetus F1-1 from family F1 (Figure 1A, Table S1). The mutation was predicted to affect splicing. Sequencing of a fetal lung cDNA fragment spanning exons 27 to 30 allowed us to demonstrate that the mutation led to total skipping of exon 28, resulting in in-frame deletion of 34 amino acids (p.Arg961_Ala994del) (Figure 1A). By Sanger sequencing, we showed that the two other fetuses of the family with DNA available were also homozygous for the mutation, and the mother of fetus F1-3 was heterozygous (Figure S1). Although the c.2982+2T>C variant was absent from public as well as our in-house databases, we sought to determine its frequency in the Gypsy population. We analyzed a panel of 541 unrelated anonymized Gypsy controls from three different migratory populations, comprising multiple endogamous groups (Vlax, 190 samples; Balkan, 238 samples; Western European, 113 samples including 57 Spanish Roma).26 The mutation screening was performed by high-resolution melting (LightCycler 480 Real-Time PCR System, Roche) with primers CAGCAAAACTCCCAGAAGGA and TTGGTGAGCTGAAGAGGAAA producing a 160 bp fragment spanning the variation. Two heterozygous individuals were identified in the Balkanic population (confirmed by Sanger sequencing), resulting in an overall allelic frequency of 0.04% in Balkan Gypsy groups. This suggests that the ITGA8 c.2982+2T>C change is a rare variant in the Gypsy population family F1 originates from.
In family F2, we identified compound heterozygosity in F2-1 and F2-2: a 5 base pair deletion in exon 17 (c.1622_1626delAGGTG) resulting in a frameshift (p.Glu541Alafs∗12) and a point mutation (c.1219G>A) in exon 13 leading to a missense (p.Gly407Arg) with a predicted damaging effect (Polyphen2 = 1/Sift = 0.01) (Figure 1B, Table S1). The deletion was absent from variant databases and the missense is reported once in EVS, with a very low frequency (1/13,005). By analysis of the parents’ DNA, we demonstrated that the two mutations were located on different alleles.
Integrins are noncovalently bound αβ heterodimeric transmembrane receptors that mediate various biological functions, notably cell adhesion, cytoskeleton rearrangement, and activation of cell signaling pathways, through conformational changes and interaction with extracellular ligands.27 These processes play a major role during organogenesis and epithelial organization, and lack of integrin α8, integrin β1, or the α8β1 ligand nephronectin dramatically affects kidney development in mice.22,25,28 The α chain consists of four extracellular domains, a transmembrane helicoidal domain, and a small intracellular tail.27 With the Phyre2 and PyMOL softwares, we analyzed the position of the variations relative to these domains (Figure 1C). The splice mutation identified in family F1 leads to the deletion of two blade-shaped β sheets in calf 2 domain, which probably destabilizes the structure of this whole domain. It is expected that this should affect the conformation of the α8β1 heterodimer and the transition from bent and inactive to extended and active conformation necessary for interaction with nephronectin.27 In family F2, the p.Gly407Arg missense change, occurring in combination with a frameshift mutation that results in a truncated protein in calf 1 domain, is located in one of the seven blade-shaped β sheets of the β propeller, suggesting that it also affects the function of the protein (Figure 1C).
In order to validate the effect of this amino acid change, the c.1219G>A mutation was introduced by site-directed mutagenesis in a human WT ITGA8 cDNA cloned in pcDNA3.1.29 The WT and mutated ITGA8 constructs were transiently transfected in HEK293 cells, which do not endogenously express ITGA8.30 We characterized the subcellular localization of ITGA8 by immunofluorescence in nonpermeabilized and permeabilized cells. Although both ITGA8 WT and ITGA8 p.Gly407Arg proteins could be detected in the cytoplasm of permeabilized cells where they colocalize with a marker of the endoplasmic reticulum (Figure 2A, left), only the WT protein was detected at the cell surface in nonpermeabilized cells (Figure 2A, right). These results suggested that the p.Gly407Arg variant protein was unable to reach the plasma membrane and was retained in the reticulum. In order to go further in the functional characterization of this protein variant, we generated stably transfected cell lines. We evaluated the ITGA8 mRNA and protein levels in the two cell lines by quantitative RT-PCR (not shown) and immunoblot (Figure 2B). Absence of the ITGA8 p.Gly407Arg protein at the cell surface was confirmed by flow cytometry analysis (Figure 2C).
Figure 2.
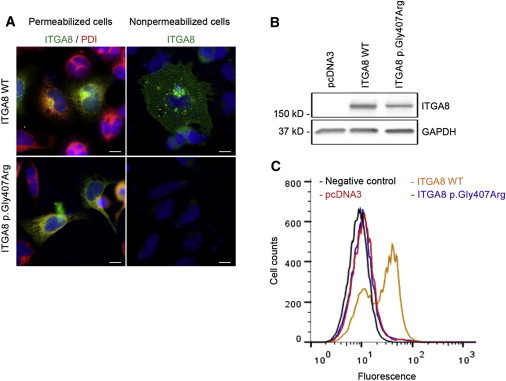
Analysis of the Subcellular Localization of the ITGA8 p.Gly407Arg Protein
(A) Analysis of the subcellular localization of WT and variant ITGA8 proteins by immunofluorescence: 2 × 105 HEK293 cells transiently transfected with plasmid constructs containing the WT or the mutated ITGA8 cDNA cloned in pcDNA3 were plated on slides previously coated for 1 hr at 37°C with 10 μg/ml of nephronectin (R&D Systems) and rinsed for an additional 1 hr with 1% BSA/PBS. After a 24 hr incubation at 37°C, cells were fixed with PFA 4%. For cell permeabilization, a 10 min treatment with Triton 0.2% in PBS was applied before incubation with the antibodies. The following primary antibodies were used in 1% BSA/PBS: rabbit anti-integrin α8 1/100 (Sigma-Aldrich) and mouse anti-PDI 1/200 (Assay designs).
(B and C) Analysis of α8 integrin at the cell surface by flow cytometry.
(B) HEK293 cells were stably transfected with WT or mutated ITGA8 cDNA constructs or empty pcDNA3 vector and ITGA8 protein level was controlled by immunoblot (rabbit anti-integrin α8 1/1,000, mouse anti-GAPDH [Millipore] 1/3,000).
(C) For flow cytometry analysis, cells were detached from culture plates with a Costar cell lifter. Cell pellets were incubated with rabbit anti-integrin α8 antibody (1/50) for 30 min at room temperature in 3% BSA/PBS. After washing with PBS, cells were labeled with secondary antibody (polyclonal donkey anti rabbit, Alexa Fluor 488, 1/1,000). Cells were fixed with 1% PFA/PBS and the amount of α8 integrin at the surface was measured with FACS Calibur (Becton Dickinson). Data were analyzed with the CELLQuest program (Becton Dickinson). Negative control was cell line ITGA8 WT incubated with secondary antibody alone (black curve).
Then, we analyzed the effect of the mutation on cell adhesion and spreading, two mechanisms that are regulated by integrins. Cell adhesion (Figure 3B) and spreading (Figure 3C) were observed by immunofluorescence after staining for the actin cytoskeleton and focal adhesions (phalloidin and paxillin, respectively; Figure 3A). In parallel, we used the xCELLigence technology (ACEA Biosciences, Roche), which allows real-time analysis of cell index, an arbitrary value representing the number of adherent cells and the intensity of their interaction with the substrate (Figure 3D). ITGA8 WT cells showed both increased adhesion and spreading on nephronectin compared to ITGA8 p.Gly407Arg cells and to control cells, as visualized by the number of adherent cells and the proportion of extensively spread cells (Figures 3A–3C). This was confirmed by analysis of cell index by xCELLigence, showing that presence of ITGA8 WT led to a two times increase in cell adhesion velocity, represented by the slope of the curves over 45 min from seeding, compared to ITGA8 p.Gly407Arg (Figures 3D and 3E). Altogether, these results demonstrate that the p.Gly407Arg change affects localization of ITGA8 at the plasma membrane and consequently integrin α8β1-dependent cell adhesion and spreading.
Figure 3.
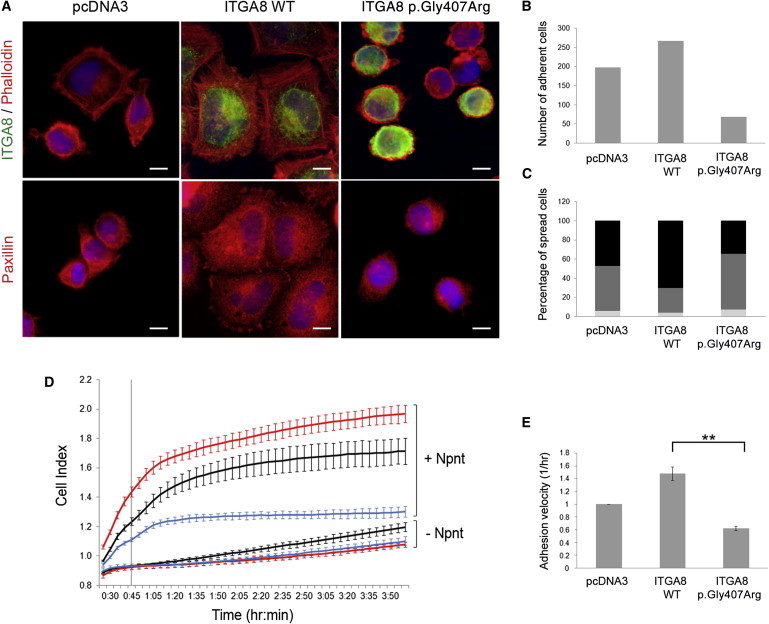
Characterization of the Effect of the ITGA8 p.Gly407Arg Variation on Cell Adhesion and Spreading
(A–C) Analysis of adhesion and spreading by immunofluorescence.
(A) For each cell line, 2 × 105 cells were plated on glass slides previously coated with nephronectin. After a 45 min incubation at 37°C, cells were fixed with PFA 4%. Staining of ITGA8 (rabbit anti-integrin α8 1/100), filamentous actin (rhodamine coupled phalloidin 1/400, Sigma-Aldrich), and paxillin (mouse anti-paxillin 1/50, BD Biosciences) showed enhanced spreading of ITGA8 WT cells compared to ITGA8 p.Gly407Arg cells.
(B) Cell adhesion was measured by counting the number of adhered cells in 12 fields (at magnification ×63) for each cell line.
(C) The percentage of spread and unspread cells was estimated by counting of 100 cells for each cell line (light gray, unspread; medium gray, weakly spread; black, strongly spread).
(D and E) Lifetime cell adhesion and spreading was evaluated with the impedance-based system xCELLigence (ACEA Biosciences, Roche). E-type plates were coated for 1 hr at 37°C with 10 μg/ml of nephronectin (Npnt) and rinsed for an additional 1 hr with 1% BSA/PBS. 8 × 104 stably transfected HEK293 cells (ITGA8 WT, ITGA8 p.Gly407Arg, or empty pcDNA3) were seeded in each well.
(D) Cell index (CI) was measured from 25 min after seeding. Each CI value corresponds to the average of triplicates ± standard deviation (red curves, ITGA8 WT; blue curves, ITGA8 p.Gly407Arg; black curves, empty pcDNA3).
(E) Adhesion velocity is represented as the slope of the curves, calculated on the first 45 min after seeding (indicated by the vertical line in D). The graph represents the mean ± SEM of two independent experiments, each one in triplicate. ∗∗p < 0.01 calculated by t test.
Finally, in order to identify other ITGA8 mutations, we performed Sanger sequencing of the 30 exons of the gene in a cohort of 57 fetuses with bilateral RHD (47 with BRA, 10 with unilateral renal agenesis and hypodysplasia). Seven cases were familial cases. No other recessive mutation was identified. However, one fetus with BRA carried a heterozygous missense variant (c.764C>T [p.Thr255Met]). This variant is not a reported polymorphism and is predicted to be damaging (Polyphen2 = 0.999/Sift = 0). As for the p.Gly407Arg missense change, the p.Thr255Met change is located in one of the blade-shaped β sheets of the β propeller domain, suggesting that is could also affect the function of the protein (Figure 1C). Although the second mutation remains to be identified, alteration of ITGA8 could also be the causative mechanism of BRA in this fetus.
In conclusion, our results demonstrate that recessive ITGA8 mutations can cause BRA in humans. Accurate evaluation of the frequency of ITGA8 mutations associated with this phenotype, as well as of the potential variability of the kidney development defects resulting from these mutations, will require analysis of a larger series of families. However, our identification of ITGA8 mutations in two families out of ten analyzed by whole-exome sequencing suggests that ITGA8 mutations could be a frequent cause of familial BRA. These results also clearly confirm the hypothesis that the most severe forms of human RHD, notably BRA, can be autosomal-recessive disorders, as observed in mouse models with severe kidney development defects.
Acknowledgments
We thank Yuya Sato for providing us with the ITGA8 cDNA construct and William Baron for his help for FACS analysis. We also thank Christine Bole-Feysot and Patrick Nitschké for their effective support in exome sequencing and bioinformatic analyses. We are grateful to the families for their participation. We also thank the fetopathologists of the SOFFOET for providing us with fetal tissue samples. The research leading to these results has received funding from the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 305608 (EURenOmics), GIS-Maladies Rares (AAE11010KSA), and the Programme Hospitalier de la Recherche Clinique Assistance Publique (AOM07129). C.H. was funded by EUrenOmics.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Ensembl Genome Browser, human genome, http://useast.ensembl.org/Homo_sapiens/Info/Index
GenitoUrinary Molecular Anatomy Project (GUDMAP), http://www.gudmap.org/
Mouse Genome Informatics, http://www.informatics.jax.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Phyre2, http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
PyMOL, http://www.pymol.org
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Sanna-Cherchi S., Ravani P., Corbani V., Parodi S., Haupt R., Piaggio G., Innocenti M.L., Somenzi D., Trivelli A., Caridi G. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney Int. 2009;76:528–533. doi: 10.1038/ki.2009.220. [DOI] [PubMed] [Google Scholar]
- 2.Heidet L., Decramer S., Pawtowski A., Morinière V., Bandin F., Knebelmann B., Lebre A.S., Faguer S., Guigonis V., Antignac C., Salomon R. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin. J. Am. Soc. Nephrol. 2010;5:1079–1090. doi: 10.2215/CJN.06810909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krug P., Morinière V., Marlin S., Koubi V., Gabriel H.D., Colin E., Bonneau D., Salomon R., Antignac C., Heidet L. Mutation screening of the EYA1, SIX1, and SIX5 genes in a large cohort of patients harboring branchio-oto-renal syndrome calls into question the pathogenic role of SIX5 mutations. Hum. Mutat. 2011;32:183–190. doi: 10.1002/humu.21402. [DOI] [PubMed] [Google Scholar]
- 4.Orten D.J., Fischer S.M., Sorensen J.L., Radhakrishna U., Cremers C.W., Marres H.A., Van Camp G., Welch K.O., Smith R.J., Kimberling W.J. Branchio-oto-renal syndrome (BOR): novel mutations in the EYA1 gene, and a review of the mutational genetics of BOR. Hum. Mutat. 2008;29:537–544. doi: 10.1002/humu.20691. [DOI] [PubMed] [Google Scholar]
- 5.Ulinski T., Lescure S., Beaufils S., Guigonis V., Decramer S., Morin D., Clauin S., Deschênes G., Bouissou F., Bensman A., Bellanné-Chantelot C. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J. Am. Soc. Nephrol. 2006;17:497–503. doi: 10.1681/ASN.2005101040. [DOI] [PubMed] [Google Scholar]
- 6.Weber S., Moriniere V., Knüppel T., Charbit M., Dusek J., Ghiggeri G.M., Jankauskiené A., Mir S., Montini G., Peco-Antic A. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J. Am. Soc. Nephrol. 2006;17:2864–2870. doi: 10.1681/ASN.2006030277. [DOI] [PubMed] [Google Scholar]
- 7.Alazami A.M., Shaheen R., Alzahrani F., Snape K., Saggar A., Brinkmann B., Bavi P., Al-Gazali L.I., Alkuraya F.S. FREM1 mutations cause bifid nose, renal agenesis, and anorectal malformations syndrome. Am. J. Hum. Genet. 2009;85:414–418. doi: 10.1016/j.ajhg.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albuisson J., Pêcheux C., Carel J.C., Lacombe D., Leheup B., Lapuzina P., Bouchard P., Legius E., Matthijs G., Wasniewska M. Kallmann syndrome: 14 novel mutations in KAL1 and FGFR1 (KAL2) Hum. Mutat. 2005;25:98–99. doi: 10.1002/humu.9298. [DOI] [PubMed] [Google Scholar]
- 9.Cain J.E., Islam E., Haxho F., Chen L., Bridgewater D., Nieuwenhuis E., Hui C.C., Rosenblum N.D. GLI3 repressor controls nephron number via regulation of Wnt11 and Ret in ureteric tip cells. PLoS ONE. 2009;4:e7313. doi: 10.1371/journal.pone.0007313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller E.M., Hopkin R., Bao L., Ware S.M. Implications for genotype-phenotype predictions in Townes-Brocks syndrome: case report of a novel SALL1 deletion and review of the literature. Am. J. Med. Genet. A. 2012;158A:533–540. doi: 10.1002/ajmg.a.34426. [DOI] [PubMed] [Google Scholar]
- 11.Pitera J.E., Scambler P.J., Woolf A.S. Fras1, a basement membrane-associated protein mutated in Fraser syndrome, mediates both the initiation of the mammalian kidney and the integrity of renal glomeruli. Hum. Mol. Genet. 2008;17:3953–3964. doi: 10.1093/hmg/ddn297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Esch H., Groenen P., Nesbit M.A., Schuffenhauer S., Lichtner P., Vanderlinden G., Harding B., Beetz R., Bilous R.W., Holdaway I. GATA3 haplo-insufficiency causes human HDR syndrome. Nature. 2000;406:419–422. doi: 10.1038/35019088. [DOI] [PubMed] [Google Scholar]
- 13.van Haelst M.M., Maiburg M., Baujat G., Jadeja S., Monti E., Bland E., Pearce K., Hennekam R.C., Scambler P.J., Fraser Syndrome Collaboration Group Molecular study of 33 families with Fraser syndrome new data and mutation review. Am. J. Med. Genet. A. 2008;146A:2252–2257. doi: 10.1002/ajmg.a.32440. [DOI] [PubMed] [Google Scholar]
- 14.Pini Prato A., Musso M., Ceccherini I., Mattioli G., Giunta C., Ghiggeri G.M., Jasonni V. Hirschsprung disease and congenital anomalies of the kidney and urinary tract (CAKUT): a novel syndromic association. Medicine (Baltimore) 2009;88:83–90. doi: 10.1097/MD.0b013e31819cf5da. [DOI] [PubMed] [Google Scholar]
- 15.Li Y., Pawlik B., Elcioglu N., Aglan M., Kayserili H., Yigit G., Percin F., Goodman F., Nürnberg G., Cenani A. LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am. J. Hum. Genet. 2010;86:696–706. doi: 10.1016/j.ajhg.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renkema K.Y., Winyard P.J., Skovorodkin I.N., Levtchenko E., Hindryckx A., Jeanpierre C., Weber S., Salomon R., Antignac C., Vainio S., EUCAKUT consortium Novel perspectives for investigating congenital anomalies of the kidney and urinary tract (CAKUT) Nephrol. Dial. Transplant. 2011;26:3843–3851. doi: 10.1093/ndt/gfr655. [DOI] [PubMed] [Google Scholar]
- 17.Yosypiv I.V. Congenital anomalies of the kidney and urinary tract: a genetic disorder? Int. J. Nephrol. 2012;2012:909083. doi: 10.1155/2012/909083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madariaga L., Morinière V., Jeanpierre C., Bouvier R., Loget P., Martinovic J., Dechelotte P., Leporrier N., Thauvin-Robinet C., Jensen U.B. Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin. J. Am. Soc. Nephrol. 2013;8:1179–1187. doi: 10.2215/CJN.10221012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skinner M.A., Safford S.D., Reeves J.G., Jackson M.E., Freemerman A.J. Renal aplasia in humans is associated with RET mutations. Am. J. Hum. Genet. 2008;82:344–351. doi: 10.1016/j.ajhg.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeanpierre C., Macé G., Parisot M., Morinière V., Pawtowsky A., Benabou M., Martinovic J., Amiel J., Attié-Bitach T., Delezoide A.L., Société Française de Foetopathologie RET and GDNF mutations are rare in fetuses with renal agenesis or other severe kidney development defects. J. Med. Genet. 2011;48:497–504. doi: 10.1136/jmg.2010.088526. [DOI] [PubMed] [Google Scholar]
- 21.Barak H., Huh S.H., Chen S., Jeanpierre C., Martinovic J., Parisot M., Bole-Feysot C., Nitschké P., Salomon R., Antignac C. FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Dev. Cell. 2012;22:1191–1207. doi: 10.1016/j.devcel.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Müller U., Wang D., Denda S., Meneses J.J., Pedersen R.A., Reichardt L.F. Integrin alpha8beta1 is critically important for epithelial-mesenchymal interactions during kidney morphogenesis. Cell. 1997;88:603–613. doi: 10.1016/s0092-8674(00)81903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linton J.M., Martin G.R., Reichardt L.F. The ECM protein nephronectin promotes kidney development via integrin alpha8beta1-mediated stimulation of Gdnf expression. Development. 2007;134:2501–2509. doi: 10.1242/dev.005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gresham D., Morar B., Underhill P.A., Passarino G., Lin A.A., Wise C., Angelicheva D., Calafell F., Oefner P.J., Shen P. Origins and divergence of the Roma (gypsies) Am. J. Hum. Genet. 2001;69:1314–1331. doi: 10.1086/324681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hynes R.O. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X., Mernaugh G., Yang D.H., Gewin L., Srichai M.B., Harris R.C., Iturregui J.M., Nelson R.D., Kohan D.E., Abrahamson D. beta1 integrin is necessary for ureteric bud branching morphogenesis and maintenance of collecting duct structural integrity. Development. 2009;136:3357–3366. doi: 10.1242/dev.036269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato Y., Uemura T., Morimitsu K., Sato-Nishiuchi R., Manabe R., Takagi J., Yamada M., Sekiguchi K. Molecular basis of the recognition of nephronectin by integrin alpha8beta1. J. Biol. Chem. 2009;284:14524–14536. doi: 10.1074/jbc.M900200200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schnapp L.M., Hatch N., Ramos D.M., Klimanskaya I.V., Sheppard D., Pytela R. The human integrin alpha 8 beta 1 functions as a receptor for tenascin, fibronectin, and vitronectin. J. Biol. Chem. 1995;270:23196–23202. doi: 10.1074/jbc.270.39.23196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.