

Abstract
1-(3-Oxocyclobutyl) carboxylic acid (4a) was converted into N-Boc-protected 1-(3-oxocyclobutyl) urea (5a), a key intermediates for the preparation of agonists of metabotropic glutamate receptor 5, in one-step when treated with diphenyl phosphoryl azide and triethylamine in tert-butanol. The mechanism of the reaction involves a nucleophilic addition of the in situ generated tert-butyl carbamate to the isocyanate intermediate. This reaction is applicable to other 1-(3-oxocycloalkyl) carboxylic acids but not to linear γ-keto carboxylic acids.
Keywords: 1-(3-Oxo)ureas, Curtius rearrangement, Carbamoylcarbamate, γ-Keto carboxylic acid, 1-(3-Oxocyclobutyl) carboxylic acid
In the course of our research in developing novel agonists for metabotropic glutamate receptor subtype 5 (mGluR5), we were interested in the synthesis of 1-(3-oxocyclobutyl)urea (1a), a key intermediate to various small molecule agonists of mGluR5. To synthesize 1a, we planned to form the urea beginning with 3-aminocyclobutanone (2a, Scheme 1).1 Although several synthetic routes are known for the generation of 2a, they usually require multiple steps and/or often give poor overall yields.2 To obtain amine 2a rapidly and efficiently, we attempted to use the Curtius rearrangement to directly convert carboxylic acid 4a into carbamate 3a,3 which in turn, can easily lead to 2a after removing the N-protecting group.
Scheme 1.
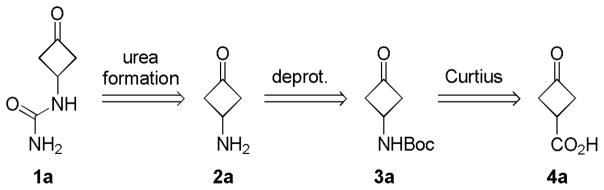
Synthetic plan to 1a.
Our initial effort was to synthesize tert-butyl 3-oxocyclobutylcarbamate (3a). To generate 3a, a solution of 4a in tert-butanol (t-BuOH) was treated with diphenyl phosphoryl azide (DPPA)4 and triethylamine (Et3N). After heating the reaction mixture at 50 °C for 16 h, a major product was isolated in 38% yield. To our surprise, mass spectrum and 1H NMR data of the product did not match those of the anticipated product (3a). This major product only has an (M+H+) peak at 229, which was 43 daltons more than the calculated molecular weight of 3a (M+H+ = 186), implying a possible insertion of a –CONH– fragment into the desired product. This speculation was confirmed by the fact that there was an extra singlet at 8.10 ppm (integrating to one proton) in the 1H NMR spectrum of the product in CDCl3. Further 13C NMR data showed a peak at 204.7 ppm, indicating the retaining of the ketone functionality. More interestingly, after treating this compound with trifluoroacetic acid (TFA), urea 1a was isolated and characterized. On the basis of these results, we assigned the product of the original reaction as tert-butyl N-((3-oxocyclobutyl) carbamoyl)carbamate (5a, Scheme 2). The chemical structure of 5a was confirmed by single crystal X-ray analysis (Figure 1 and Table 1). The crystallographic analysis showed that the N4 participated in intramolecular hydrogen bonding with the carbonyl group, the N4–H···O7 hydrogen bond length was 2.08 Å (Table S7), which explained the downfield shift of the corresponding signal (8.10 ppm) in the 1H NMR. In addition to 5a, the desired product (3a) was isolated as a relatively non-polar compound with only 5% yields. It is also noted that upon heating at 85 °C for 2 h, the cyclobutanone ring of 3a broke to form a significant amount of the α,β -unsaturated methylketone. The thermal instability of 3a might also account for the low yield of previously reported methods to this compound.2c
Scheme 2.
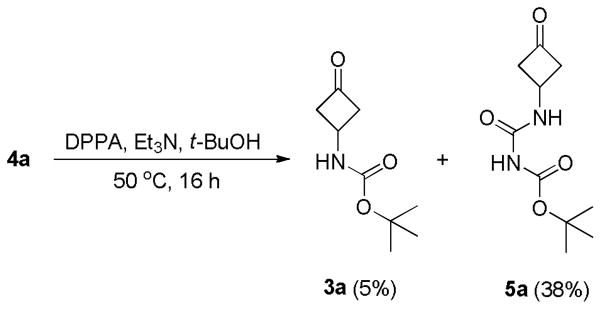
The Curtius rearrangement of 4a in t-BuOH.
Figure 1.
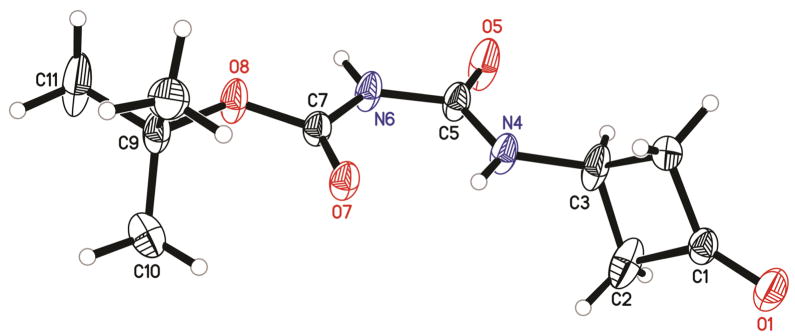
Single crystal structure of compound 5a.
Table 1.
Selected crystallographic data of 5a
| Empirical formula | C10H16N2O4 |
| Formula weight | 228.25 |
| Temperature | 150(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Orthorhombic |
| Space group | P n m a |
| Unit cell dimensions | a = 24.798(2) Å, ϒ= 90° b = 9.0377(7) Å, ϒ= 90° c = 5.2093(4) Å, ϒ= 90° |
| Volume | 1167.49(16) Å3 |
| Z | 4 |
| Density (calculated) | 1.299 Mg/m3 |
| Absorption coefficient | 0.101 mm−1 |
| F(000) | 488 |
| Crystal size | 0.617 × 0.431 × 0.157 mm3 |
To elucidate the origin of 5a, the reaction was repeated in which carboxylic acid 4a was treated with DPPA and Et3N in t-BuOH and monitored closely by thin layer chromatography (TLC). Time course studies clearly showed the disappearance of the starting material (4a) and the emergence of carbamate 3a within the first hour of the reaction. However, the amount of 3a generated did not change significantly during the course of reaction. After stirring the reaction for 2 h, compound 5a, with significantly higher polarity to that of 3a, was formed and built up. Accordingly, we speculated that 3a might be an intermediate, which was transformed into 5a by the insertion of the –CONH– fragment. Specifically, we proposed that at the beginning of the reaction, Curtius rearrangement of 4a formed isocyanate 6a, which was then attacked by t-BuOH to generate carbamate 3a (Scheme 3). Since compound 3a was not stable under basic environment, it lost a molecule of tert-butyl carbamate via elimination to form cyclobut-2-enone.5 The resulting tert-butyl carbamate attacked the isocyanate group of 6a in the reaction mixture to generate compound 5a. It is noted that the theoretical yield of the reaction is 50%, which explains the relatively modest yields of compound 5a.
Scheme 3.
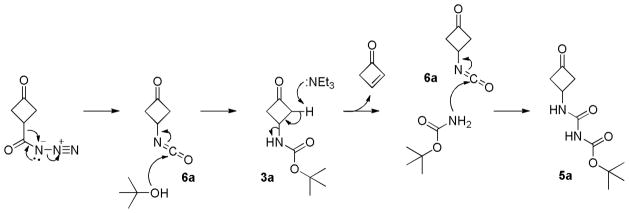
Proposed reaction mechanism for the formation of 5a in t-BuOH.
To test this hypothesis, we carried out reactions under the same conditions starting with cyclobutanecarboxylic acid (7) and 4-oxocyclohexanecarboxylic acid (8). For both reactions, no carbamoylcarbamates were detected (Scheme 4). When 7 was used, the normal Curtius product tert-butyl cyclobutylcarbamate was isolated in good yields. This result indicated that without the presence of the γ-carbonyl group, the Curtius product was stable and no elimination of tert-butyl carbamate happened. On the other hand, treatment of 8 with the same conditions generated bicyclic 6-azabicyclo[3.2.1]octane-2,7-dione.6 These results confirmed that the presence of the γ-keto acid group to the carboxylic acid functionality was essential for the generation of carbamoylcarbamates, which accelerated the elimination of tert-butyl carbamate from carbamate 3a.
Scheme 4.
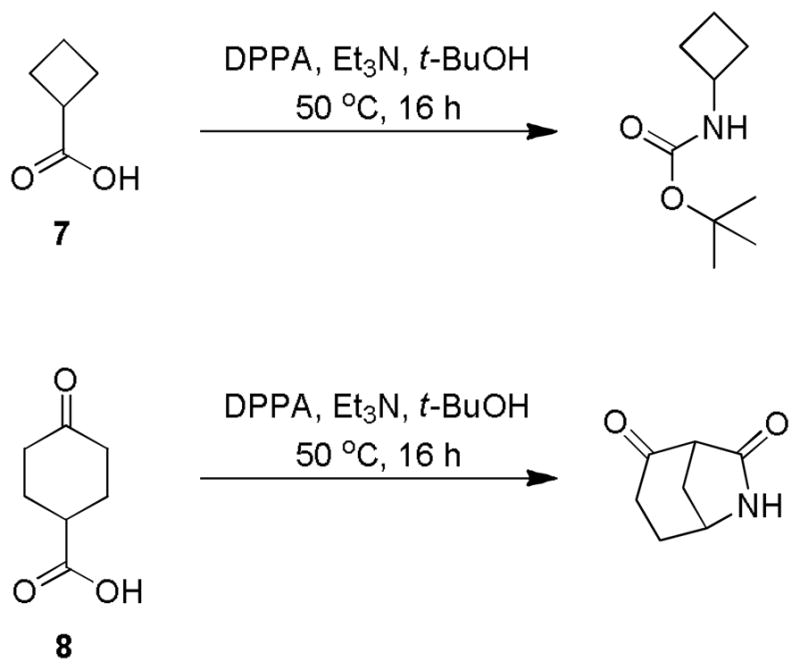
Reactions starting with compounds 7 and 8.
We also repeated the reaction of 4a with DPPA and Et3N in other alcohols (Scheme 5). When sterically less hindered primary alcohols (e.g., MeOH, EtOH, and p-methoxybenzyl) were used, the corresponding esters were isolated as the only products (9a-c) in good yields. On the other hand, the reaction performed in i-PrOH gave carbamate 3b as the major product. Although the iso-propyl N-((3-oxocyclobutyl)carbamoyl)carbamate (5b) was also isolated, the yield was significantly less than that of 5a from the previous reaction in t-BuOH. These results indicated that the outcome of the reaction was largely controlled by the nucleophilicity of the alcohols involved in the reactions.
Scheme 5.
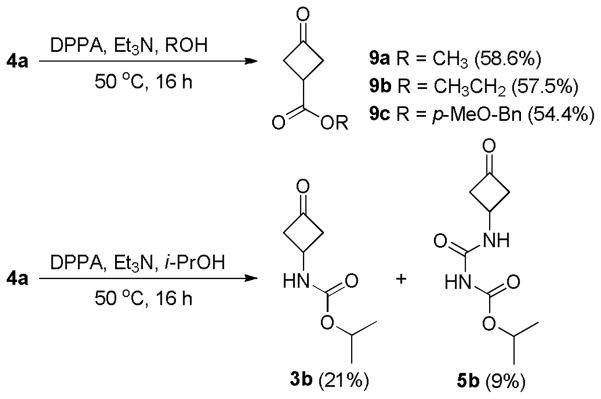
The Curtius rearrangement of 4a in other alcohols.
To study the scope of the reaction, similar reactions were conducted under similar conditions using other γ-keto acids (Scheme 6). When 3-oxocyclopentanecarboxylic acid (4c) and 3-oxocyclo-hexanecarboxylic acid (4d) were used, the corresponding Boc-protected 1-(3-oxo)ureas (5c and 5d) were obtained as the major products. However, the reactions starting with non-cyclic γ–keto acids 4e and 4f generated compounds 5e and 5f in minimum yields, instead, 3,4-dihydro-2H-1,3-oxazin-2-ones 10e and 10f were isolated as the major products. The chemical structure of compound 10e was confirmed by single crystal X-ray analysis (Figure 2). These results showed that the formation of carbamoyl carbamates was only applicable to 1-(3-oxocycloalkyl)carboxylic acids, as for the starting materials employing a non-cyclic γ–keto carboxylic acid functionality, the reaction will favor the formation of 3,4-dihydro-2H-1,3-oxazin-2-ones via an intramolecular cyclization mechanism.
Scheme 6.
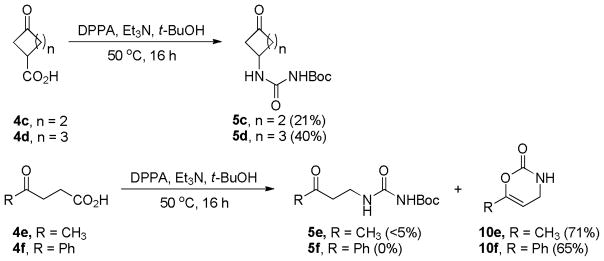
Reaction results using γ–keto acids 4c-f.
Figure 2.

Single crystal structure of compound 10e.
In summary, we report an unexpected one-step formation of Boc-protected 1-(3-oxo)ureas starting with 1-(3-oxo)acids. In the reaction mechanism, the initially generated carbamate product from the Curtius rearrangement was not stable. It eliminated the tert-butyl carbamate that attacks the isocyanate intermediate in the reaction mixture to generate the final product. The application of this method has been highlighted by a rapid preparation of urea 1a, a key fragment for the development of agonists for mGluR5. The conditions described herein is applicable to the preparation of other 1-(3-oxocycloalkyl)ureas.
Supplementary Material
Acknowledgments
We gratefully acknowledge funding from the Department of Pharmaceutical Sciences, University of Maryland School of Pharmacy. The X-ray crystallographic work was supported by NIDA through Interagency Agreement #Y1-DA1101 with the Naval Research Laboratory (NRL).
Footnotes
Supplementary data associated with this article can be found, in the online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Fouladdel S, Khalaj A, Adibpour N, Azizi E. Bioorg Med Chem Lett. 2010;20:5772–5775. doi: 10.1016/j.bmcl.2010.07.137. [DOI] [PubMed] [Google Scholar]; (b) Harriman GC, Brewer M, Bennett R, Kuhn C, Bazin M, Larosa G, Skerker P, Cochran N, Gallant D, Baxter D, Picarella D, Jaffee B, Luly JR, Briskin MJ. Bioorg Med Chem Lett. 2008;18:2509–2512. doi: 10.1016/j.bmcl.2007.07.068. [DOI] [PubMed] [Google Scholar]
- 2.(a) Radchenko DS, Pavlenko SO, Grygorenko OO, Volochnyuk DM, Shishkina SV, Shishkin OV, Komarov IV. J Org Chem. 2010;75:5941–5952. doi: 10.1021/jo101271h. [DOI] [PubMed] [Google Scholar]; (b) Kumar RJ, Chebib M, Hibbs DE, Kim HL, Johnston GAR, Salam NK, Hanrahan JR. J Med Chem. 2008;51:3825–3840. doi: 10.1021/jm7015842. [DOI] [PubMed] [Google Scholar]; (c) Allen JRF, MJ, Harrington PE, Hu E, Pickrell AJ, Rzasa RM, Sham KKC. 0 306 591. US Patent. 2011
- 3.(a) Saunders JH, Slocombe RJ. Chem Rev. 1948;43:203–218. doi: 10.1021/cr60135a001. [DOI] [PubMed] [Google Scholar]; (b) Weinstock J. J Org Chem. 1961;26:3511–3511. [Google Scholar]; (c) Lebel H, Leogane O. Org Lett. 2006;8:5717–5720. doi: 10.1021/ol0622920. [DOI] [PubMed] [Google Scholar]; (d) Lebel H, Leogane O. Org Lett. 2005;7:4107–4110. doi: 10.1021/ol051428b. [DOI] [PubMed] [Google Scholar]
- 4.Shioiri T, Ninomiya K, Yamada S. J Am Chem Soc. 1972;94:6203–6205. doi: 10.1021/ja00772a052. [DOI] [PubMed] [Google Scholar]
- 5.(a) Sieja JB. J Am Chem Soc. 1971;93:2481–2483. [Google Scholar]; (b) Li XH, Danishefsky SJ. J Am Chem Soc. 2010;132:11004–11005. doi: 10.1021/ja1056888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herold P, Herzig JW, Wenk P, Leutert T, Zbinden P, Fuhrer W, Stutz S, Schenker K, Meier M, Rihs G. J Med Chem. 1995;38:2946–2954. doi: 10.1021/jm00015a017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.