

Abstract
The current paradigm of unidirectional migration of neutrophils from circulation to sites of injury in tissues has been recently challenged by observations in zebrafish showing that neutrophils can return from tissues back into the circulation. However, the relevance of these observations to human neutrophils remains unclear, the forward and reversed migration of neutrophils is difficult to quantify, and the precise conditions modulating the reverse migration cannot be isolated. Here, we designed a microfluidic platform inside which we observed human neutrophil migration in response to chemoattractant sources inside channels, simulating the biochemical and mechanical confinement conditions at sites of injury in tissues. We observed that, after initially following the direction of chemoattractant gradients, more than 90% of human neutrophils can reverse their direction and migrate persistently and for distances longer than one thousand micrometers micrometers away from chemoattractant sources (retrotaxis). Retrotaxis is enhanced in the presence of lipoxin A4 (LXA4), a well-established mediator of inflammation resolution, or Tempol, a standard antioxidant. Retrotaxis stops after neutrophils encounter targets which they phagocytise or on surfaces presenting high concentrations of fibronectin. Our microfluidic model suggests a new paradigm for neutrophil accumulation at sites of inflammation, which depends on the balance of three simultaneous processes: chemotaxis along diffusion gradients, retrotaxis following mechanical guides, and stopping triggered by phagocytosis.
Introduction
Neutrophils, the first and most abundant of the white blood cells to respond against bacterial and fungal pathogens invading tissues, play an essential physiological role during innate immune responses.1 They can be recruited from the circulation to inflamed tissues and guided to the site of injury by chemical and mechanical cues.2 Once they reach their targets in the tissues, neutrophils perform their sterilizing functions to neutralize the invading microorganisms.3 This process eventually triggers neutrophil apoptosis and subsequent macrophage-mediated clearance, which restores tissue homeostasis.4 However, this neutrophil unidirectional migration paradigm has recently been challenged by observations in zebrafish showing that neutrophils can return to circulation after migrating long distances away from inflammation sites.5–9 Careful analysis of the neutrophil trajectories inside the tissues suggested that the reverse migration phenotype is best described by random diffusion rather than directional drift.10 Yet, before a new paradigm of bi-directional neutrophil migration could be established, several issues remain to be addressed. The frequency of neutrophil reversed migration at sites of injury is difficult to evaluate in vivo. The complexity of conditions during the in vivo experiments limits our understanding of the precise stimuli under which reversed migration can occur. More importantly, the question whether or not human neutrophils are capable of reversing their migration for long distances in tissues has not yet been answered.
Over the past decade, soft lithography in transparent biocompatible materials, such as polydimethylsiloxane (PDMS), has emerged as a remarkable technology for biological studies. Its application to the study of neutrophil migration under controlled conditions11 has revealed several surprising neutrophil behaviours. These include neutrophil fugetaxis in response to steep gradients,12 U-turns and reversal of polarity in response to temporal changes of chemical gradients,13,14 directional decision making in response to opposing chemoattractant gradients15 or during encounters with mechanical obstacles.16 Studies using microfluidic devices to analyze neutrophil migration in clinical context are also emerging.17, 18
Here, we employ soft lithography to build and validate a microfluidic platform for studying neutrophil reversed migration. Using the new tools, we can trigger reverse migration over long distances in nine out of ten migrating human neutrophils. This migration pattern, which we name retrotaxis (retrograde, contrary to the direction of others; taxis, movement) is independent of the nature of the chemoattractant triggering the migration and requires mechanical guiding cues. Retrotaxis is enhanced in the presence of lipid mediators of inflammation resolution, such as lipoxin A4 (LXA4), and antioxidants, such as 4-hydroxy-2,2,6,6- tetramethylpiperidine-1-oxyl (Tempol). Retrotaxis is inhibited when neutrophils encounter targets they phagocytise and on surfaces presenting fibronectin at high concentrations. Our platform for inducing neutrophil retrotaxis complements current in vivo models for neutrophil reverse migration and could enable systematic, higher throughput studies of neutrophils roles in inflammation.
Materials and Methods
Microfluidic device fabrication
The microfluidic devices to study the effect of gradients and mechanical confinement on neutrophils were designed to mimic some of the biomechanical features encountered by neutrophils in tissues. These devices consist of a main loading channel and symmetric side migration channels shaped like an inverted letter U (Figure 1a). A gradient of the chemoattractant is established by diffusion between the side channels and the main channel with the highest concentration region located at the tip of the U. All migration channels are 8 μm wide, a 1000 μm long, and of varied height.
Figure 1. Human neutrophils migrate persistently against chemical gradients in U-shaped channels.
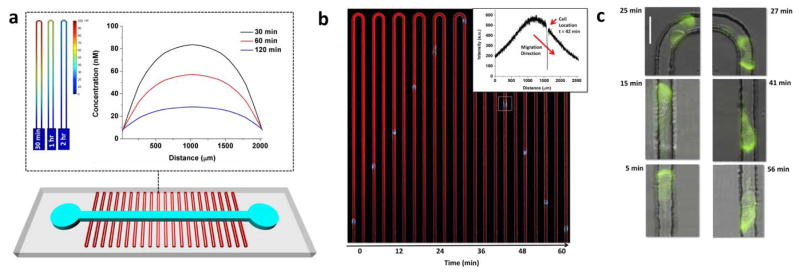
(a) 3D sketch of the PDMS-based microfluidic device. Two arrays of U-shaped migration channels (w = 8 μm, length of one arm = 1000 μm, and varied heights) are orthogonal to a main loading channel (w = 500 μm and h = 50 μm). The main channel has an inlet and an outlet to load chemoattractant and cells. Top inset presents a COMSOL simulation image of the gradient profile along a single U-shaped channel at times 30 min, 1 hour, and 2 hours after cell loading. The highest concentration is at the tip of the U and decreasing concentrations are observed along the two arms of the U. Concentration profile along the U-shaped channel over time (0 and 2000 μm indicate the entrance and exit location to and from the U-shaped Channel). Slope of the profile after 2 hours provides at least a 1% concentration difference along any 20 μm length of the channel. (b) Single-channel montage showing a differentiated HL-60 cell with fluorescently labelled nucleus during chemotaxis and retrotaxis in a U-shaped channel. The gradient is represented by a red gradient of Dextran-conjugated Texas Red dye. Inset demonstrates the gradient profile of the Dextran dye along the U-channel 42 minutes after the entrance of a cell through the left arm of the channel. The location of the cell corresponds to the region with no Texas-red fluorescent signal. Length of the channel is 1000 μm. (c) Successive images of one transfected HL-60 cell with PH-Akt-GFP during migration through the U-shaped channel. The fluorescent signal corresponding to PH-Akt-GFP is localized at the leading edge of the cell throughout its journey toward and away from the high concentration. Scale bar is 20 μm.
Microfluidic devices were fabricated by soft lithography. Briefly, polydimethylsiloxane (PDMS, Dow Corning, Midland, MI) was moulded on a master wafer, fabricated using standard photolithographic technologies. A silicon wafer (Desert Silicon, Grandale, AZ) was baked in an oven set to 200 °C for 30 minutes, cleaned with oxygen plasma (March, Concord, CA), and then spun coated with either SU-8 2, 5, or 10 (SU8, Microchem, Newton, MA), depending on the desired thickness of the first layer of the design (representing the migration channels). A second layer of SU-8 50 photoresist was then spun and baked following the standard protocol as recommended by the manufacturer to define the cell-loading channel of the device. PDMS and its curing agent were mixed (10:1, respectively) and poured on the master and left to de-gas for 1 hour in a vacuum chamber. After baking for 8 hours at 65°C, the PDMS layer covering the master was peeled off, punched with a 0.75 mm puncher (Harris Uni-Core, Ted Pella Inc., Reading, Ca) to define the inlets and outlets, and then exposed to 35 s of oxygen plasma along with a glass slide (Fisherbrand 1×3″, Fisher Scientific, Pittsburgh, PA). The PDMS device was then bonded to the glass slide and baked at 75 C for 10 minutes. Afterwards, two pieces of flexible plastic tubing were cut to a length of ~ 3 cm (Tygon, Greene Rubber Co., Woburn, MA). One was connected to blunt 30G needle from one side, to serve as an inlet port of the device, while the other was inserted into the outlet of the device.
Device Priming
Prior to cell loading, the device was primed with Iscove’s Modified Dulbecco’s Medium (IMDM, ATCC, Manassas, VA) containing human-fibronectin, diluted to either 100 nM or 1 μM (Sigma-Aldrich, St. Louis, MO), and the desired concentration of the chemoattractant. To establish the gradient along the side channels, a 1 mL syringe containing the chemoattractant-fibronectin solution was connected to one port of the device while blocking the other port. Gentle pressure was then applied to replace the air in the side channels with the solution. Afterwards, a new 1 mL syringe containing only IMDM medium was used to push a fresh solution through the main channel. Neutrophils or HL-60s were then slowly loaded into the cell-loading channel of the device and allowed to settle.
For experiments involving 5(S),6(R)-Lipoxin A4 (LXA4) (Cayman Chemical, Ann Arbor, MI), stock concentrations were vigorously diluted to 10 nM, 100 nM and 1 μM and loaded into the side channels along with the chemoattractant and/or fibronectin. Therefore, a spatial gradient of LXA4 and 100 nM fMLP was established along the arms of the U-shaped channels. Similarly, device priming for experiments involving Alexa-Fluor-488- or Texas-Red-labelled zymosan A (S. cerevisiae) (Life Technologies, Grand Island, NY) was done by first reconstituting the bioparticles in a glass vial at 20 mg/mL in IMDM. The vial was then vigorously vortexed according to the manufacturer’s protocol and diluted 50 times into the chemoattractant mixture, to produce a homogenous population of zymosan particles along the U-shaped channels upon loading (maximum of 5 particles per channel).
Cell Preparation
De-identified, fresh human blood samples from healthy volunteers, aged 18 years and older, were purchased from Research Blood Components (Alston, MA). Peripheral blood was drawn in heparinized-tubes (Vacutainer; Becton Dickinson) and processed within 2 hours after blood collection. Human neutrophils were separated from the blood samples as previously described.19 Briefly, neutrophils were isolated from whole blood using HetaSep followed by the EasySep Human Neutrophil Enrichment Kits (STEMCELL Technologies) following the manufacturer’s protocol. The final aliquots of neutrophils were re-suspended in IMDM containing 20% FBS (Sigma-Aldrich, St. Louis, MO) at a concentration of 5×106 cells/mL and kept at 37°C. Cells were then immediately introduced into the microfluidic device. For experiments involving 4-hydroxy-2,2,6,6- tetramethylpiperidine-1-oxyl (Tempol, Tocris Bioscience, Bristol, UK), isolated neutrophils were incubated for 30 minutes in diluted concentrations (10 mM, 1 mM, and 0.2 mM) of Tempol in IMDM at 37°C and 5% CO2.
Human leukemia cell line (HL-60, CCL-240, ATCC, Manassas, VA), was used as a model to test the different designs prior to their use with human neutrophils. After DMSO-induced differentiation, HL-60 cells prime and polarize in response to chemoattractants and migrate in a gradient at rates comparable to those of human neutrophils.20, 21 Stable HL-60 cells expressing PH-Akt-GFP were generously provided by the laboratory of Dr. Jagesh Shah (Harvard Medical School). Both wild and transfected types, were differentiated to neutrophil-like cells by culturing the cells for a period of at least 5 days in IMDM with 1.3% DMSO and 20% FBS at 37°C and 5% CO2. Upon loading, cells were spun and re-suspended in IMDM containing 20% FBS at a density of 5×106 cells/mL.
Chemotaxis Imaging and Measurements
Time-lapse imaging was performed on a Nikon Eclipse Ti microscope equipped with a biochamber, heated to 37°C, and with 5% CO2. Separate experiments to characterize the formation and dissipation of gradients inside the device in the absence of cells were performed under similar temperature and gas conditions but by replacing the chemoattractant with fluorescein (Sigma-Aldrich, St. Louis, MO) of comparable molecular weight.
Cell velocities were measured manually using NIS Elements Viewer, only for the first cell to enter each channel within the first two hours, and from the time required to travel a distance of 600–800 μm. Distance travelled after phagocytosis was measured between the initial position of a zymosan particle in the U channel before phagocytosis and the final position of the neutrophils that stops after phagocytosis.
Statistical Analysis
Chemotaxis and retrotaxis velocities were compared using independent Student t-test or two-way analysis of variance (ANOVA) followed by Bonferroni post-tests. For the analysis of migration speed and patterns in different conditions, at least 50 cells were measured, from three independent experiments. For the analysis of phagocytosis, at least 30 cells were measured, from three independent experiments. The statistical significance for the changes in size of various neutrophil subpopulations, defined by distinct migration patterns in the U-shaped channels, was calculated using Chi-square analysis. All differences were considered significant above a 95% confidence level. Details of statistical analysis for each condition are included in the figure legends. All values are expressed as mean ± standard deviation of the mean (SD) unless otherwise indicated.
Results
In order to determine if human neutrophils are capable of reversed migration and identify the conditions that favour this migration pattern, we designed a microfluidic platform combining chemical gradients and mechanical constraints. The critical design feature of these devices is the U shape of the channels through which neutrophils can migrate. Inside the channels, chemical gradients are established by diffusion, with the highest concentration at the tip of the U and symmetrically decreasing concentrations along the two arms of the U (Figure 1a). Biophysical modelling of chemoattractant diffusion between the U-shaped channel and the main loading channel predicts that chemoattractant gradients of a small molecule are formed in less than 5 min after introducing the neutrophils in the main channel (Figure 1a). At 2 hours, the gradient decreases by 70%, however it remains greater than 1% concentration difference along any 20 μm-length (the average length of moving neutrophils), the threshold for neutrophil chemotaxis.
Human neutrophils, as well as differentiated HL-60 cells, follow the chemoattractant gradients, enter the U-shaped channels, migrate towards the highest concentrations at the tip of the U, and, surprisingly, continue their movement through the second arm and against the chemoattractant gradient, until they reach the main loading channel again (retrotaxis - Figure 1b and Movie S1, ESI†). To verify the continuous presence of the gradients during chemotaxis and retrotaxis, we mixed the chemoattractant with fluorescent dyes of comparable molecular weight. We found that strong gradients are still present in both arms of the U during neutrophil retrotaxis (Figure 1b-inset). To verify that the characteristic, asymmetric distribution of signalling molecules is preserved during retrotaxis, we employed differentiated HL-60s expressing a GFP-labeled PH-Akt (Akt pleckstrin homology domain). We found that the asymmetric distribution of the PH-Akt reporter is maintained throughout chemotaxis and retrotaxis (Figure 1c; Movie S2, ESI†), suggesting that common mechanisms of cell migration are active during both chemotaxis and retrotaxis.
Over 90% of human neutrophils and over 80% of differentiated HL-60 cells undergo retrotaxis in the presence of fMLP gradients (Figure 2). The fraction of human neutrophils and differentiated HL-60 cells undergoing retrotaxis was significant regardless of the chemoattractant types (fMLP, C5a, or LTB4, P > 0.05, Figure 2a) or concentrations (fMLP, from 1 to 100nM, P > 0.05, Figure 2b). Remarkably, average velocities of human neutrophils during retrotaxis were 22% higher than during chemotaxis for all three chemoattractants tested (37.3 ± 4.4 vs. 30.5 ± 5.9 μm/min, P < 0.001, Figure 2c). The 5 μm channel height demonstrated persistent velocities across all chemokines and concentrations and therefore it was used throughout the rest of the study (Figure 2d).
Figure 2. Robust retrotaxis of human neutrophils and HL-60 cell line through U-shaped channels.

(a) More than 90% of primed human neutrophils retrotaxed in gradients of 100 nM fMLP, LTB4, or C5a (p > 0.05, Two-way ANOVA). (b) Similar to human neutrophils, retrotaxis was observed in more than 80% of differentiated HL-60 cells. We observed no significant variation in the percentage of retrotaxing cells in channels of different dimensions (8×3, 8×5, and 8×10 μm) and various fMLP concentrations (p > 0.05) (c) Human neutrophils migrated at significantly different speeds during chemotaxis and retrotaxis in response to 100 nM chemoattractants of fMLP, LTB4, and C5a (N=3 experimental repeats, N > 100 cells for each condition, ***p < 0.001, Student T test). (c) Differentiated HL-60 cells migrate at comparable speeds during chemotaxis and retrotaxis in channels of different channel heights (8×3, 8×5, and 8×10 μm) and for various fMLP concentrations (p > 0.05, Two-way ANOVA). Cells migrated faster through taller channels under 1 and 10 nM concentrations of fMLP (***p < 0.001, Two-way ANOVA)
Three distinct patterns of human neutrophil migration in U-shaped channels can be differentiated based on the length of the migration paths: a pattern named “long”, where neutrophils pass the tip of the U and return to the main channel through either arm, a pattern named “short”, when neutrophils travel for at least 200 μm in the channels and then return to the main channel before reaching the tip of the U, and a pattern named “trapped” when neutrophils cease their persistent migrating in the channel and remain alive for at least 2 hours (Figure 3a). In the presence of fMLP gradients, 62 ± 20 and 22 ± 4% of the neutrophils migrated on “long” and “short” patterns, respectively, while only 10 ± 2% of cells were “trapped”. To verify the role of the chemical gradients during retrotaxis, we observed the migration patterns of human neutrophils inside channels with uniform concentrations of fMLP, or culture media with no fMLP. For the same cell density in the main channels, four times fewer neutrophils entered the side channels, and rarely any cell reached the tip of the U in the uniform and no fMLP conditions (Fig 3b, P < 0.001). The fraction of cells migrating on “long” patterns decreased by an order of magnitude to below 5%, while the fraction of neutrophils trapped inside channels increased to more than 50%. The “short” and “trapped” human neutrophil populations also travelled inside the channels for shorter distances in uniform and no fMLP conditions (a decrease from 837 ± 339 μm under gradient conditions to 398 ± 238 μm). Moreover, neutrophils migrated significantly faster in the presence of fMLP gradients compared to uniform or no fMLP conditions (P < 0.001, Figure 3c). These results indicate that retrotaxis phenotype is distinct from chemokinesis induced by uniform concentrations of chemoattractant and from random migration in the absence of chemoattractants. Finally, to test if the migration patterns are related to the time when the neutrophil enters the channel, we compared the frequencies of each migration pattern in 15 minutes time intervals. We found that the frequencies of the three patterns change proportionally for all the patterns during the experiments (Figure 3d). The changes in frequencies with time are expected due to the reduction of the number of neutrophils remaining in the main loading channel while the experiment progresses.
Figure 3. The retrotaxis phenotype is distinct from chemokinesis or random migration in the absence of chemoattractants.

(a) Retrotaxis in U-shaped channels was divided into three distinguishable phenotypes: “long”, “short”, and “trapped”. “Long” indicates that the cell reaches the tip of the U channel and returns back to the main channel through either arm. “Short” indicates a cell that travels for at least 200 μm and returns back to the main channel, before reaching the tip of the U. “Trapped” indicates a cell that stops before, at, or after the tip of the U-shaped channel and does not return to the main channel. Scale bar is 40 μm (b) Results of 3 control experiments demonstrated that in the presence of fMLP gradients, 125 neutrophils migrated on a “long” pattern, compared to 50 and 24 on short and trapped patterns. The “long” pattern for gradient experiments was significantly higher than that of experiments with uniform concentrations of fMLP (1 cell), or culture media with no fMLP (no cells) (***p < 0.001, Two-way ANOVA). The fraction of neutrophils trapped inside channels increased to more than 50% in uniform and no fMLP experiments. (c) Forward and reverse cell velocities in uniform and no fMLP experiments were significantly lower than velocities recorded for cells in fMLP gradient devices (***p < 0.001, Two-way ANOVA). (d) The frequency of the three patterns was not dependent on the gradient in the channel. This is shown by the comparable entry time of the cells across the three migration patterns (p > 0.05 for all 15-minute intervals across the three phenotypes, One-way ANOVA).
The distinction of three patterns of migration during retrotaxis enabled us to probe the activity of various factors on neutrophil retrotaxis. First, we tested the effect of lipid mediator of inflammation resolution, LXA4, on the retrotaxis patterns of human neutrophils (Figure 4a). We measured a significant increase in frequency of “long” patterns (from 62 ± 8 % to 86 ± 6 %, P < 0.001) accompanied by a significant decrease in the number of cells in the “short” and “trapped” categories (from 30 ± 6 % to 9 ± 5 %, and from 8 ± 2 % to 3 ± 5 %, respectively, P < 0.001). Second, we probed the role of fibronectin on cell behaviour. Interestingly, neutrophils migrated equally well in the absence and presence of moderate amounts of fibronectin (surfaces coated in the presence of 100 nM fibronectin for 30 minutes, p > 0.05). However, on surfaces coated with 1 μM fibronectin for 30 minutes prior to cell loading, we observed a 5-fold increase (from 14 ± 8% to 72 ± 4%, P < 0.001) in the number of “trapped” neutrophils (Figure 4b). Neutrophil forward and reverse velocities remained essentially the same and were not affected by the exposure to LXA4 (Figure 4c). However, we measured significantly faster velocity for reverse than forward migration for all LXA4 concentrations tested. We observed similar differences across the various fibronectin concentrations. The forward velocity on channels with 1000 nM fibronectin was significantly lower than that at 100nM fibronectin (a decrease from 27.8 ± 5.3 to 14.2 ± 4.7 μm/min, P < 0.001, Figure 4d). These results suggest that chemoattractants, surface proteins, and lipid mediators could modulate neutrophil retrotaxis during the initiation and resolution phases of inflammation.
Figure 4. The patterns of neutrophil retrotaxis are modulated by inflammation resolution mediator, lipoxin A4, and adhesion molecule, fibronectin.

(a) Diffusing gradients of lipoxin A4 as low as 10 nM along with 100 nM fMLP demonstrated a significant increase in the number of cells that follow the “long” behaviour and a decrease in those that follow the “short” behaviour (***p < 0.001, Two-way ANOVA). This may explain the functionality of resolution-stage mediators in keeping the cell persistently polarized away from the injury site. (b) High fibronectin concentrations in channels of 100 nM fMLP gradients show a drastic increase in the number of cells that become “trapped” in the channel and do not return back to the main channel (***p < 0.001, Two-way ANOVA). This may explain the role of adhesion molecules during acute inflammation in neutrophils fate in tissue. (N = 2 experiments, N > 100 cells for all conditions). (c) Neutrophil forward and reverse velocities were not affected by the exposure to LXA4. Significantly higher reverse velocities than forward velocities were measured for all LXA4 concentrations (**p < 0.01 and *p < 0.05, Two-way ANOVA). (d) Forward velocity in channels with 1000 nM fibronectin was significantly lower than that at 100nM fibronectin (***p < 0.001, two-way ANOVA).
To further understand the role of neutrophil retrotaxis during inflammation and infections, we examined the modulation of neutrophil retrotaxis by phagocytosis events (Figure 5a). We found that the ratio of neutrophils that display “trapped” versus “long” migration patterns is reversed in the presence of zymosan particles (P < 0.001, Figure 5b). The number of “trapped” neutrophils inside the channels increased 2.7 fold after encountering zymosan particles (from 28.5 ± 10.0 % to 77.0 ± 10.8%, Figure 5b). At the same time, the fraction of “long” retrotaxis patterns decreased 5.2 fold (from 55 ± 1% to 10.5 ± 3%). The fraction of “short” migration patterns did not change significantly (16 ± 9 % vs. 12 ± 12 %). With randomly dispersed zymosan particles of maximum size of 3 μm along the 8×5 μm U channel, the stop in migration was observed before and after the tip, during forward as well as reversed migration.
Figure 5. Human neutrophil retrotaxis stops during phagocytosis and is enhanced upon exposure to various compounds.

(a) Montage of a human neutrophil during chemotaxis in one of the U-shaped channel arms encountering a red zymosan particle and stopping after migrating for a short distance. Scale bar is 100 μm. Inset demonstrates a difference in the cell before and after phagocytising a 2-μm zymosan particle. Channel width = 8 μm (b) Comparison between the migration patterns of human neutrophils in U-channels containing 100 nM fMLP, with and without zymosan particles. More than 90% of the cells in the control experiment demonstrated retrotaxis toward the main channel (“long” and “short” combined). Only 15% of the neutrophils returned back to the main channel in zymosan-containing migration channels after phagocytosis. (N=3 experiments, N=163 cells counted, ***p < 0.001, Two-way ANOVA). (c) The fraction of cells retrotaxing after phagocytosis increased while the fraction of cells trapped decreased in the presence of lipoxin A4 and zymosan particles in channels with 100 nM fMLP. (N=3 experiments, N=116 cells counted, ***p < 0.001, Two-way ANOVA) (d) Pre-treatment of cells for 30 minutes with 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (Tempol) demonstrated a similar effect to that of LXA4. (e) The distribution of distances travelled by human neutrophils after phagocytosis showed no significant change in the presence of LXA4. (N= 3 experiments, N = 146 cells counted) (f) Average distance travelled by Tempol-pre-treated neutrophils increased by approximately 100 μm after phagocytosis compared to control, untreated cells. (N=3 experiments, N=196 cells counted).
To validate the microfluidic devices as a platform for screening compounds that could modulate various aspects of retrotaxis after phagocytosis, we examined the effect of various concentrations of LXA4 and 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (Tempol). Human neutrophils migrating in U-shaped channels with 100 nM fMLP gradient and randomly dispersed zymosan particles were either pre-treated with Tempol for 30 minutes before migration, or exposed to LXA4 mixed with the chemoattractant as they migrated through the channels. We observed a significant increase in the frequency of “long” retrotaxis migration patterns (10.3 ± 3 % without LXA4 vs. 48.8 ± 9.5 % in a 100 nM LXA4 gradient, P < 0.001, and 7.1 ± 6.4 % without Tempol pre-treatment vs. 30.4 ± 8.3 % with a 30-minute, 1 mM Tempol pre-treatment, P < 0.01). This was accompanied by a significant decrease in the number of “trapped” cells (from 70.1 ± 1.2% without LXA4 to 40.2 ± 5.4 % in the presence of a 100 nM LXA4 gradient, P < 0.001, and from 76.4 ± 7.9% without Tempol pre-treatment to 45.3 ± 10.5% with a 30-minute pre-treatment with 1 mM Tempol, P < 0.01, Figures 5c, d).
Interestingly, neutrophils did not stop migrating immediately after encountering zymosan particles. Instead, neutrophils continued moving in their original direction for on average 250 μm before stopping completely (Figure 5e,f; Movie S3, ESI†). Exposure to LXA4 effectively reduced the number of trapped neutrophils. Interestingly, this took place with no significant alterations of the average migration distance after phagocytosis. On the other hand, pre-treatment for 30 minutes with Tempol increased the average distance travelled after phagocytosis by at least 100 μm (245 ± 224 μm vs. 379 ± 318 μm in control vs. 1 mM Tempol conditions, respectively).
Discussion
We developed a microfluidic platform that combines chemical gradients and mechanical constraints to induce the reverse migration (retrotaxis) of human neutrophils. Inside channels, neutrophils enter at the lowest chemoattractant concentration and migrate toward higher concentrations. Upon reaching the tip of the U, neutrophils continue their migration and move persistently toward lower chemoattractant concentrations. The critical feature enabling retrotaxis is the array of U-shaped channels, inside which similar chemical gradients form along the two arms. The highest concentration is at the tip of the U and the lowest concentration is at the connection to the main cell-loading channel. The slope of the two gradients depends on the concentration of chemoattractant loaded in the channels and the length of the branches. The gradients decay with time due to the diffusion of chemoattractants into the cell-loading channel. However, for the two hour duration of the experiments, these gradients remain steep enough and compatible with chemotaxis.
The retrotaxis patterns of migration observed in our devices are consistent with in vivo observations in zebrafish, of neutrophils migrating away from sites of tissue injury7 and they are in contrast to previous in vitro studies of neutrophil migration patterns on flat surfaces during changes in the direction of chemical gradients. In micropipette experiments, for instance, after triggering neutrophil migration in one direction, relocating the pipette to the back of the moving cells, turns neutrophils away from the original direction, toward the new location of the pipette, quickly and without hesitation.22 In studies using microfluidic devices to generate “hill shaped” stable gradients, neutrophils reaching the highest concentration rarely overshoot and always return to the highest concentrations, migrating less than 50 μm down the concentration gradients.11 In the presence of fast switching gradients, neutrophils stop immediately after the gradient change and re-polarize in the new direction within 5 minutes.14 By contrast, in our platform, neutrophils continue their persistent migration after passing the point of maximal chemoattractant concentration, for more than 20 minutes, with increased velocity, and for distances longer than 1000 μm.
The contrasting behaviour of human neutrophils inside confining channels compared to neutrophils on flat surfaces emphasizes the critical role that mechanical guidance plays during retrotaxis. Consistent with this role, previous in vitro studies have shown that when squeezed in between agarose and glass, human neutrophils undergo “chimneying migration” independent of the presence of calcium.23 When confronted with mechanical obstacles inside chemotaxis channels, human neutrophils maintain their persistent migration and squeeze around these.16 Additional support for the role of mechanical guidance during retrotaxis is provided by the detailed analysis of neutrophil migration tracks from in vivo studies in zebrafish.6 These studies have suggested the existence of fixed and preferred channels that neutrophils migrate along during the reverse migration from tissue back to the circulation. The physical support for such tracks in tissues has not been elucidated and one suggestion is that these may represent cleavage planes inside tissues.6 Further studies comparing the results from in vitro and in vivo models will reveal if additional elements would have to be explored and implemented in future designs of the microfluidic devices, in addition to chemical gradients and mechanical confinement,.
The retrotaxis migration patterns are distinct from those of fugetaxis12 in that they do not appear to depend on the nature of the chemoattractant or its concentration. They are also distinct from the reversed transmigration (RT) of neutrophils back and forth through layers of endothelial cells24,25 because the neutrophil behaviour we observe occurs without the need for interactions with the endothelial cells. Retrotaxis can be blocked by phagocytosis and requires a pre-existent chemoattractant gradient, mechanical confinement, and takes place along migration paths that are several hundred microns long. In contrast, reversed transmigration is triggered by activation of the endothelial cells by pro-inflammatory cytokines (e.g TNF),24 involves neutrophils migrating back and forth through a thin endothelial layer,24, 25 and could take place independent of phagocytosis in the peritonitis model.25 In vivo, retrotaxis and reversed transmigration could complement each other to assure that returning neutrophils could pass through tissues and endothelial layers to re-enter the circulation. Retrotaxis, is also distinct from haptotaxis,26 because the mechanical guidance alone in the absence of guiding gradients was not sufficient to induce retrotaxis patterns of cell migration, and only cells already “primed” by chemotaxis could reverse their migration direction. Finally, we showed that retrotaxis is distinct from chemokinesis because pre-activated cells in a uniform chemoattractant channel display significantly shorter migration patterns and no significant reversed migration compared to the neutrophils exposed to chemoattractant gradients. Overall, retrotaxis describes a new behaviour for human neutrophils that has to be triggered by chemical gradients and guided by microscale mechanical confinement.
Our results, showing that nine out of ten neutrophils entering the channels are capable of moving persistently against chemical gradients, raise the question of the potential physiological implications of retrotaxis during inflammation. The current dogma for neutrophil infiltration is that most neutrophils entering a tissue are trapped inside the tissue, where they degranulate or undergo apoptosis. These neutrophils are ultimately cleared by macrophages, which restore the tissue environment to the original state. However, the high proportion of neutrophils capable of retrotaxis in our assay suggests that alternative mechanisms for neutrophil clearance may play significant roles. Based on our in vitro experiments, we suggest a new hypothesis for neutrophil accumulation in tissue, which could be a result of three fluxes: neutrophil chemotaxis along diffusion gradients, neutrophil retrotaxis along mechanical guides, and local trapping of neutrophils triggered by phagocytosis. During sterile inflammation, retrotaxis could accelerate neutrophil clearance and contribute to the restoration of homeostasis. During infections, turning off retrotaxis after phagocytosis could act as safety mechanism to prevent the dissemination of infectious agents by the neutrophils if their neutralization inside phagosomes is not successful. The balance between the two processes could be modulated by LXA4, a physiologic inflammation-resolving lipid mediator27 that is known to decrease the migration19 and transmigration28 of resting human neutrophils. Overall, retrotaxis could play a critical role in controlling the accumulation of neutrophils in tissues, depending upon the nature of inflammation and other modulating factors.
Our microfluidic platform could also be useful for testing new approaches for controlling acute and chronic inflammation. For example, our results suggest an interesting hypothesis regarding a potential role for increased levels of reactive oxygen species (ROS)29 induced by phagocytosis during the switch between trapping and retrotaxis. Some support for this hypothesis come from our observations using LXA4 and Tempol, already known for the ability to reduce the levels of ROS in neutrophils30,31. In our experiments both restored retrotaxis in neutrophils after phagocytosis. Future studies probing this hypothesis could lead to practical approaches for controlling inflammation or enhancing the defences against infections in patients.
In conclusion, the microfluidic device for inducing neutrophil retrotaxis provides a robust method to study neutrophil reversed trafficking in vitro, complementing current in vivo models. It enables higher throughput experiments, in precisely controlled conditions. Such experiments are critical for understanding the mechanisms that integrate mechanical, chemical, and phagocytic interactions during neutrophil migration. Together, the new tools and better understanding of neutrophil unique functional abilities could help expand our knowledge of processes limiting inflammation during acute and chronic diseases, while preserving the ability to respond to new infections.
Supplementary Material
Insight, Innovation, Integration.
Using an innovative microfluidic assay, we found that more than 90% of human neutrophils have the ability move persistently against chemoattractant gradients over long distances (retrotaxis). This finding was enabled by the design of microscale channels in which human neutrophils move in conditions of mechanical confinement and encounter successive spatial chemical gradients in opposite directions. Using these new experimental tools, we found that human neutrophil retrotaxis is inhibited by phagocytosis and can be restored by lipid mediators of resolution, suggesting a new, more complex paradigm for neutrophil trafficking in and out of tissues during the course of inflammation and infection processes.
Acknowledgments
We thank Dr. A.J. Aranyosi and Mr. Octavio Hurtado for assistance with device fabrication at the BioMEMS Resource Center, Mr. Harrison Prentice-Mott for helpful discussions and Dr. Jagesh Shah for sharing of various HL-60 cell lines. This work was supported by the National Institutes of Health (GM092804, EB0025003).
Footnotes
Electronic Supplementary Information (ESI) available: Movie S1: Chemotaxis followed by retrotaxis in U-shaped Channels. Movie S2: Ph-Akt-GFP at the leading edge of the cell during retrotaxis. Movie S3: Retrotaxis of neutrophil-like HL-60 cell stops after encountering a zymosan particle See DOI: 10.1039/b000000x/
References
- 1.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Annual review of immunology. 2012;30:459–489. doi: 10.1146/annurev-immunol-020711-074942. [DOI] [PubMed] [Google Scholar]
- 2.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 3.Phillipson M, Kubes P. Nature medicine. 2011;17:1381–1390. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kennedy AD, DeLeo FR. Immunologic research. 2009;43:25–61. doi: 10.1007/s12026-008-8049-6. [DOI] [PubMed] [Google Scholar]
- 5.Starnes TW, Huttenlocher A. Advances in hematology. 2012;2012:398640. doi: 10.1155/2012/398640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmes GR, Anderson SR, Dixon G, Robertson AL, Reyes-Aldasoro CC, Billings SA, Renshaw SA, Kadirkamanathan V. Journal of the Royal Society, Interface/the Royal Society. 2012;9:3229–3239. doi: 10.1098/rsif.2012.0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng Q, Huttenlocher A. Journal of cell science. 2012;125:3949–3956. doi: 10.1242/jcs.093633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elks PM, van Eeden FJ, Dixon G, Wang X, Reyes-Aldasoro CC, Ingham PW, Whyte MK, Walmsley SR, Renshaw SA. Blood. 2011;118:712–722. doi: 10.1182/blood-2010-12-324186. [DOI] [PubMed] [Google Scholar]
- 9.Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT, Huttenlocher A. Journal of leukocyte biology. 2006;80:1281–1288. doi: 10.1189/jlb.0506346. [DOI] [PubMed] [Google Scholar]
- 10.Holmes GR, Dixon G, Anderson SR, Reyes-Aldasoro CC, Elks PM, Billings SA, Whyte MK, Kadirkamanathan V, Renshaw SA. Advances in hematology. 2012;2012:792163. doi: 10.1155/2012/792163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Jeon N, Baskaran H, Dertinger SK, Whitesides GM, Van de Water L, Toner M. Nature biotechnology. 2002;20:826–830. doi: 10.1038/nbt712. [DOI] [PubMed] [Google Scholar]
- 12.Tharp WG, Yadav R, Irimia D, Upadhyaya A, Samadani A, Hurtado O, Liu SY, Munisamy S, Brainard DM, Mahon MJ, Nourshargh S, van Oudenaarden A, Toner MG, Poznansky MC. Journal of leukocyte biology. 2006;79:539–554. doi: 10.1189/jlb.0905516. [DOI] [PubMed] [Google Scholar]
- 13.Herzmark P, Campbell K, Wang F, Wong K, El-Samad H, Groisman A, Bourne HR. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13349–13354. doi: 10.1073/pnas.0705889104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irimia D, Liu SY, Tharp WG, Samadani A, Toner M, Poznansky MC. Lab Chip. 2006;6:191–198. doi: 10.1039/b511877h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin F, Nguyen CM, Wang SJ, Saadi W, Gross SP, Jeon NL. Annals of biomedical engineering. 2005;33:475–482. doi: 10.1007/s10439-005-2503-6. [DOI] [PubMed] [Google Scholar]
- 16.Ambravaneswaran V, Wong IY, Aranyosi AJ, Toner M, Irimia D. Integr Biol (Camb) 2010;2:639–647. doi: 10.1039/c0ib00011f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butler KL, Ambravaneswaran V, Agrawal N, Bilodeau M, Toner M, Tompkins RG, Fagan S, Irimia D. PloS one. 2010;5:e11921. doi: 10.1371/journal.pone.0011921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sackmann EK, Berthier E, Young EW, Shelef MA, Wernimont SA, Huttenlocher A, Beebe DJ. Blood. 2012;120:e45–53. doi: 10.1182/blood-2012-03-416453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones CN, Dalli J, Dimisko L, Wong E, Serhan CN, Irimia D. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:20560–20565. doi: 10.1073/pnas.1210269109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Servant G, Weiner OD, Herzmark P, Balla T, Sedat JW, Bourne HR. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu J, Van Keymeulen A, Wakida NM, Carlton P, Berns MW, Bourne HR. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9296–9300. doi: 10.1073/pnas.0703153104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu J, Wang F, Van Keymeulen A, Herzmark P, Straight A, Kelly K, Takuwa Y, Sugimoto N, Mitchison T, Bourne HR. Cell. 2003;114:201–214. doi: 10.1016/s0092-8674(03)00555-5. [DOI] [PubMed] [Google Scholar]
- 23.Malawista SE, de Boisfleury Chevance A. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:11577–11582. doi: 10.1073/pnas.94.21.11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, Stone PC, Salmon M, Matharu NM, Vohra RK, Nash GB, Rainger GE. J Leukoc Biol. 2006;79:303–311. doi: 10.1189/jlb.0905496. [DOI] [PubMed] [Google Scholar]
- 25.Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE, Meda P, Imhof BA, Nourshargh S. Nat Immunol. 2011;12:761–769. doi: 10.1038/ni.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ganiko L, Martins AR, Freymuller E, Mortara RA, Roque-Barreira MC. Biochim Biophys Acta. 2005;1721:152–163. doi: 10.1016/j.bbagen.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Serhan CN, Maddox JF, Petasis NA, Akritopoulou-Zanze I, Papayianni A, Brady HR, Colgan SP, Madara JL. Biochemistry. 1995;34:14609–14615. doi: 10.1021/bi00044a041. [DOI] [PubMed] [Google Scholar]
- 28.Patcha V, Wigren J, Winberg ME, Rasmusson B, Li J, Sarndahl E. Experimental cell research. 2004;300:308–319. doi: 10.1016/j.yexcr.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 29.Forman HJ, Torres M. American journal of respiratory and critical care medicine. 2002;166:S4–8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- 30.Carlo T, Kalwa H, Levy BD. FASEB J. 2013;27:2733–2741. doi: 10.1096/fj.12-223982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hosseinzadeh A, Messer PK, Urban CF. Front Immunol. 2012;3:391. doi: 10.3389/fimmu.2012.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.