

Abstract
A family of detoxifying enzymes called aldehyde dehydrogenases (ALDHs) has been a subject of recent interest, as its role in detoxifying aldehydes that accumulate through metabolism and to which we are exposed from the environment has been elucidated. Although the human genome has 19 ALDH genes, one ALDH emerges as a particularly important enzyme in a variety of human pathologies. This ALDH, ALDH2, is located in the mitochondrial matrix with much known about its role in ethanol metabolism. Less known is a new body of research to be discussed in this review, suggesting that ALDH2 dysfunction may contribute to a variety of human diseases including cardiovascular diseases, diabetes, neurodegenerative diseases, stroke, and cancer. Recent studies suggest that ALDH2 dysfunction is also associated with Fanconi anemia, pain, osteoporosis, and the process of aging. Furthermore, an ALDH2 inactivating mutation (termed ALDH2*2) is the most common single point mutation in humans, and epidemiological studies suggest a correlation between this inactivating mutation and increased propensity for common human pathologies. These data together with studies in animal models and the use of new pharmacological tools that activate ALDH2 depict a new picture related to ALDH2 as a critical health-promoting enzyme.
I. INTRODUCTION
Throughout their lifespan, every organism is exposed to numerous damaging agents. Some are formed endogenously, while others accumulate following ingestion of food or exposure to environmental pollutants. There are three sets of active responses to these damaging events. The first set provides a shield from these damaging agents by quickly detoxifying them through enzymatic reactions or through sequestration of these agents (e.g., phase I and II detoxifying enzymes). The second set of responses involves a damage repair system (e.g., proteolysis, DNA repair), and the third set activates programmed cell death (e.g., apoptosis), thus preventing propagation of the injury. It is not surprising that many diseases are associated with a failure of one or more of these three sets of protective mechanisms.
Many recent reviews cover literature on the repair mechanisms and on programmed cell death activation (52, 92, 263, 328). In this review, we focus on one detoxifying enzyme that provides a critical shield from damaging agents that arise both endogenously and exogenously from exposures to the environment: aldehyde dehydrogenase-2 (ALDH2). However, many other detoxifying enzymes have also evolved. The best characterized detoxification enzymatic system (also termed phase I system) is encoded by the cytochrome P-450 gene superfamily, consisting of 57 different genes, that encode the enzymes involved in ∼75% of all drug metabolism in humans (103, 104, 340). Aldehyde dehydrogenase (ALDH) is another gene superfamily of phase I oxidizing enzymes that is responsible for the detoxification of biogenic and xenogenic aldehydes. Over the years, the field of ALDH research has been frequently reviewed and updated. Notably, Dr. Vasilis Vasiliou's laboratory at the University of Colorado's Health Sciences Center has maintained a database of the ALDH gene superfamily for public viewing at www.aldh.org. Table 1 lists these ALDHs and some of their characteristics.
Table 1.
19 Human ALDH isozymes
| ALDH | Subcellular Location | Preferred Substrate | Disease, Mutational Phenotypes, Functions | Chromosome | GeneBank Accession No. |
|---|---|---|---|---|---|
| ALDH1A1 | Cytosol | Retinal | Alcohol sensitivity, alcoholism, Parkinsonism with ALDH2 double knockout animal (334) | 9q21.13 | NM_000689 |
| ALDH1A2 | Cytosol | Retinal | Increased risk for neural tube defect | 15q22.1 | NM_003888 |
| ALDH1A3 | Cytosol | Retinal | Embryonic lethal in knockout animal | 15q26.2 | NM_000693 |
| ALDH1B1 | Mitochondria | Aliphatic aldehydes | Unknown | 9q11.1 | NM_000692 |
| ALDH1L1 | Cytosol | 10-Formyl tetrahydrofolate | Low fertility and decreased hepatic folate in knockout animal | 3q21.2 | NM_012190 |
| ALDH1L2 | Unknown | Unknown | Unknown | 12q23.3 | NM_001034173 |
| ALDH2 | Mitochondria | Acetaldehyde, 4-HNE and MDA | See Figure 4 in this review | 12q24.2 | NM_000690 |
| ALDH3A1 | Cytosol, nucleus | Aromatic, aliphatic aldehydes | Cataracts in knockout animal | 17p11.2 | NM_000691 |
| ALDH3A2 | Microsomes, peroxisomes | Fatty aldehydes | Sjögren-Larsson syndrome | 17p11.2 | NM_000382 |
| ALDH3B1 | Cytosol | Unknown | Unknown, locus linked to paranoid schizophrenia | 11q13.2 | NM_000694 |
| ALDH3B2 | Unknown | Unknown | Unknown | 11q13.2 | NM_000695 |
| ALDH4A1 | Mitochondria | Glutamate γ-semi-aldehyde | Type II hyperprolinemia | 1p36.13 | NM_003748 |
| ALDH5A1 | Mitochondria | Succinate semi-aldehyde | γ-Hydroxybutric aciduria | 6p22.2 | NM_001080 |
| ALDH6A1 | Mitochondria | Malonate semi-aldehyde | Development delay, metabolic abnormality, methylmalonic aciduria | 14q24.3 | NM_005589 |
| ALDH7A1 | Cytosol, nucleus, mitochondria | α-Aminoadipic semi-aldehyde | Pyridoxine-dependent seizures, locus linked to osteoporosis (107) | 5q31 | NM_001182 |
| ALDH8A1 | Cytosol | Retinal | Unknown | 6q23.2 | NM_022568 |
| ALDH9A1 | Cytosol | γ-Aminobutyr-aldehyde | Unknown | 1q23.2 | NM_000696 |
| ALDH16A1 | Unknown | Unknown | Unknown | 19q13.33 | NM_153329 |
| ALDH18A1 | Mitochondria | Glutamic γ-semi-aldehyde | Hyperammonemia, hypoprolinemia, neurodegeneration, cataract | 10q24.3 | NM_002860 |
ALDH, aldehyde dehydrogenases. Data from the following sources: Marchitti et al. (194), Marchitti et al. (195), http://www.aldh.org/.
ALDH has an ancient origin and is found in all living organisms from Archaea and Eubacteria to eukaryotes (131, 291). Complete human genome and expression studies revealed that there are 19 functional ALDH genes with a wide range of tissue expression and substrate specificity. The ALDH gene superfamily has been reviewed recently (for comprehensive reviews on ALDH, see Refs. 59, 194, 320). Here, we focus solely on the physiology and pathology associated with one member of this family, the mitochondrial ALDH2. Significant discussion is dedicated to the potential health impact of an inactivating single point mutation in ALDH2 that is found in ∼560 million East Asians (ALDH2*2 mutation), or nearly 8% of the world's population (24).
Human ALDH2 is a 517-amino acid polypeptide encoded by a nuclear gene located at chromosome 12q24 (256). The protein is transported to the mitochondrial matrix in a process that is dependent on its NH2 terminus 17-amino acid mitochondrial targeting sequence, which is cleaved as part of the complete folding and maturation of the enzyme inside the mitochondria (20). Like most members of the ALDH family, ALDH2 is a tetrameric enzyme with ∼56 kDa identical subunits. The tetramer is regarded as a dimer of dimers with only 2 of the catalytic sites on each enzyme complex maintaining the activity. Each subunit consists of three main domains: the catalytic domain, the coenzyme- or NAD+-binding domain, and the oligomerization domain. In addition to its dehydrogenase activity, ALDH2 has also reductase and esterase activities (49, 163, 211). However, no physiologically relevant substrates for these two additional enzymatic activities have been reported, except for the reductase activity-dependent bioconversion of nitroglycerin to 1,2-glyceryl dinitrate (GDN) resulting in the release of nitric oxide (NO) (48–50, 312). ALDH2 is expressed ubiquitously in all tissues but is most abundant in the liver and also found in high amounts in organs that require high mitochondrial oxidative phosphorylation, such as the heart and brain (236, 295). Over the past few decades, the biochemical and molecular characterizations of ALDH2 have been studied extensively (see reviews in Refs. 68, 330, 331).
ALDH2 is better known for its critical role in ethanol metabolism. The ethanol detoxifying pathway in humans occurs mainly in the liver and is carried out by two enzymatic steps. The first step is catalyzed by alcohol dehydrogenase (ADH), and the second step is mainly catalyzed by ALDH2 (Figure 1). The ALDH2 gene was first identified and fully characterized in 1987 (20, 21), and the X-ray crystal structure of ALDH2 was fully elucidated in 1999 (222). Among the 19 human ALDH isozymes, ALDH2 is the most efficient one for the metabolism of ethanol-derived acetaldehyde; it has the lowest Km (∼0.2 μM) towards this substrate (72). This Km is 900-fold lower than that of the abundant cytosolic ALDH, ALDH1, and therefore, in humans, ALDH2 is probably the only ALDH enzyme that contributes significantly to acetaldehyde metabolism (149). Less known is that ALDH2 is also capable of metabolizing numerous other short-chain aliphatic aldehydes, as well as some aromatic and polycyclic aldehydes (148), thus providing an important protective enzymatic function against these toxic agents. In particular, ALDH2 plays a key role in oxidizing endogenous aldehydic products that arise from lipid peroxidation under oxidative stress, such as 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA) as well as environmental aldehydes, such as acrolein (present, for example, in tobacco smoke and in car exhaust; Refs. 41, 368).
FIGURE 1.
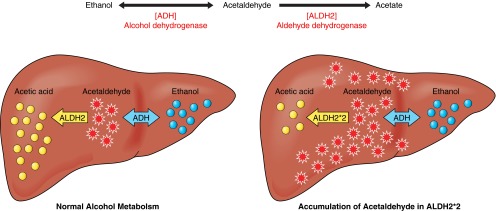
Ethanol metabolism. A: ethanol metabolism occurs in two steps: ethanol is metabolized quickly by alcohol dehydrogenase (ADH) to generate acetaldehyde. Acetaldehyde is then metabolized by the mitochondrial aldehyde dehydrogenase 2 (ALDH2) to acetate. The first step of metabolism, catalyzed by ADH, is a reversible reaction. The second step, catalyzed by ALDH2, is the rate-limiting step in ethanol metabolism, and it takes ∼1 h to metabolize the amount of ethanol that is found in a single alcoholic beverage. B: ethanol metabolism occurs mainly in the liver. In normal ALDH2 individuals, acetaldehyde is quickly oxidized to nontoxic acetate. In ALDH2 enzyme-deficient (ALDH2*2) individuals, a significant amount of acetaldehyde is rapidly accumulated even after a moderate amount of alcohol ingestion. Acetaldehyde, which is very diffusible and crosses biological membranes, can be circulated in the blood and metabolized in all tissues, as ALDH2 is present in all mitochondria.
Genetic polymorphisms of human ALDH2 have been well surveyed among a wide range of ethnic groups (44, 99, 172, 227, 319, 366). The most relevant ALDH2 variant is the ALDH2*2 allele, which is found in as many as ∼35–45% of East Asians (i.e., Chinese, Japanese, Korean, and Taiwanese) (59, 99). The ALDH2*2 carriers have a lower ALDH2 enzymatic activity, and this deficiency is manifested by the characteristic facial flushing, headaches, nausea, dizziness, and cardiac palpitations after consumption of alcoholic beverages. This ethanol-induced flushing syndrome in ALDH2*2 individuals is caused by a single G to A nucleotide change, which leads to a substitution of glutamate to lysine at position 487 (E487K, or E504K in some literature, when the first 17 amino acid mitochondrial targeting sequence, which are removed in the mature enzyme, are included) in the ALDH2 monomer (366). The E487K mutation exerts a dominant effect over the wild-type monomer encoding the ALDH2*1 allele. Therefore, heterozygotic individuals (ALDH2*1/*2) are expected to have dramatically lower than 50% of the wild-type's enzymatic activity, and ALDH2*2/*2 homozygotes have <1–4% of the wild-type activity (77, 78, 378). With ∼560 million (or 8%) of the world population having this mutation, ALDH2*2 is probably the most common human enzyme deficiency, exceeding other well-known human enzymopathies, such as glucose-6-phosphate dehydrogenase (G6PD; affecting ∼400 million; Ref. 249), or sickle cell anemia and thalassemia (hemoglobin disorders; affecting ∼350 million; Ref. 3). Remarkably, it appears that all of the ∼560 million ALDH2*2 East Asians carry the identical single nucleotide mutation, which can be traced back to the Han Chinese in Southeast China ∼2,000–3,000 years ago (184).
As discussed above, although human mitochondrial aldehyde dehydrogenase was first described in the early 1980s, much of the work on the enzyme relates to its role in ethanol metabolism. In particular, these studies investigated the negative health impacts of the ALDH2*2 mutation associated with alcohol consumption and reduced prevalence of alcoholism among ALDH2*2 carriers, due to alcohol avoidance because of the unpleasant effect of acetaldehyde accumulation following alcohol consumption.
However, a significant amount of evidence has recently emerged to indicate a greater role of ALDH2: its role in preventing numerous pathologies. Critical participation of ALDH2 in cytoprotection and its implication in many human diseases have only been recently elucidated. The following review summarizes these new data. As will be discussed, some of the observations on the protective role of ALDH2 in a variety of pathologies were based on epidemiological studies, whereas other data include studies in model systems of human diseases. A number of novel small molecule modulators of ALDH2 have been discovered and developed recently. In addition to the well-characterized inhibitors of ALDH2, new research tools, including more selective inhibitors, transgenic mice with altered ALDH2 activity, and selective ALDH2 activators have shed new light on the role of ALDH2 in human diseases. The description of these tools and how they helped to determine the role of ALDH2 in human pathologies is provided in this review. Importantly, the ability to specifically enhance or repair the enzymatic activity by a newly discovered class of ALDH2 activators has stimulated new interest in this enzyme and may provide new opportunities for the development of pharmaceuticals for the treatment of important human diseases.
II. MOUSE MODELS WITH MODIFIED ALDH2 ACTIVITY
Since a deficiency in ALDH2 activity is common in East Asians, transgenic mice with ALDH2 deficiency were generated to serve as models to recapitulate the phenotypes of the diseases or physiological responses associated with this mitochondrial enzyme deficiency. These animal models can also be used in proof-of-concept studies of potential interventions and therapeutics targeting ALDH2 in humans. Four types of transgenic ALDH2 mice have been developed. Table 2 summarizes the description of these transgenic mouse models.
Table 2.
Transgenic ALDH2 animal models
| Mouse Strain | Nature of the Change in ALDH2 | Phenotype | Reference Nos. |
|---|---|---|---|
| ALDH2 knockout | Endogenous wild-type ALDH2 gene replaced by an inactivated ALDH2 gene | Hypersensitivity to ethanol and acetaldehyde; elevated blood ethanol and acetaldehyde levels; increased ethanol-induced tissue oxidative markers, osteopenia, tissue damage and DNA modification; increased ischemia-reperfusion damage, cardiomyocyte dysfunction, 4-HNE adducts | 79, 126–129, 184, 200–202, 215, 227, 228, 235, 283 |
| ALDH2*2 transgenic | Multiple insertions of overexpressed ALDH2*2 gene, endogenous wild-type ALDH2 gene is intact | Accelerated aging, shortened lifespan, neuronal loss and impaired cognitive function, smaller body size, increased 4-HNE adducts; mitochondria stress, metabolic remodeling, tolerance to ischemia-reperfusion stress; hypersensitivity of osteoblasts to acetaldehyde, kyphosis, osteoporosis | 68, 230, 119 |
| ALDH2 overexpression | Multiple insertions of overexpressed ALDH2 wild-type gene, endogenous wild-type ALDH2 gene is intact | Reduced blood ethanol and acetaldehyde levels, reduced 4-HNE adducts; protection against acute and chronic ethanol administration and oxidative stress in liver, heart, and brain | 65, 95, 95, 106, 172, 185, 258, 259, 372, 374, |
| ALDH2 E487K knock-in | Endogenous wild-type ALDH2 gene replaced by an ALDH2*2 gene | Hypersensitivity to ethanol and acetaldehyde | Unpublished data |
| ALDH2, ALDH1A1 double knockout | Endogenous wild-type ALDH1A1 and ALDH2 genes replaced by inactivated ALDH1A1 and ALDH2 genes | Increased neurodegeneration, age-dependent motor dysfunction, increased DOPAL, 4-HNE adduct formation | 335 |
A. ALDH2 Knockout Mice
Many manifestations of defects in alcohol metabolism, alcohol-induced pathology, and hypersensitivity to acetaldehyde observed in the ALDH2*2 East Asians can be reproduced in the ALDH2 knockout (denoted ALDH2−/−) mouse model. Pronounced susceptibility to ischemia-reperfusion injuries and accumulation of aldehydic adducts are also demonstrated in the ALDH2−/− mice. Two independent ALDH2 knockout alleles, tagged with a neomycin-resistant marker within the ALDH2 gene, were introduced to the C57BL/6 mouse genome to disrupt the gene function (80, 147). Western blot analyses confirmed that no immunoreactive ALDH2 protein was produced in the homozygous ALDH2−/− mice. ALDH2 knockout mice provided useful research tools to explore the physiology, phenotypes, and pathologies derived from a complete lack of ALDH2 function. However, these mice may not completely reflect the phenotype of the human ALDH2*2 population, since the ALDH2*2 allele in human carries residual enzyme activity. It is conceivable that subtle biological differences may exist between the genotypes of ALDH2−/−, ALDH2+/−, and ALDH2*1/*2, which represents the largest affected human population. A comprehensive review on the ALDH2 knockout mouse has been published recently by Yu et al. (371).
B. ALDH2*2 Overexpressing Mice
The E487K ALDH2 mutation exerts a dominant-negative effect on the wild-type gene in humans; heterotetramerization of the mutated and wild-type subunits of ALDH2 results in a dramatically reduced catalytic activity. Therefore, the first mice to carry the ALDH2*2 mutation were transgenic mice that overexpressed mouse E487K ALDH2 subunits (69, 231). These mice showed 2- to 20-fold increased expression of ALDH2*2 gene over the endogenous wild-type ALDH2 transcripts and suppressed catalytic activity relative to the endogenous wild-type ALDH2 enzyme (69, 120, 231). These ALDH2*2 transgenic mice better represent the human phenotype of ALDH2*2 homozygotes, since they have some residual ALDH2 enzyme activity (120), and unlike the ALDH2 knockout mice, they can be used to test the ability of ALDH2 modulators that correct the structural defect of the enzyme to reverse the phenotype.
Two strains of transgenic ALDH2*2 have been constructed to express full-length human ALDH2 cDNA containing the E487K point-mutation under the control of elongation factor, EF1α and chicken β-actin promoters, respectively (69, 231). In the EF1α ALDH2*2 transgenics, expression of exogenous ALDH2*2 transcripts was observed in all tissues of these mice. In particular, high levels of the ALDH2*2 transcripts were detected in olfactory bulb, cortex, hippocampus, and midbrain, where their levels were more than twice as high as the endogenous wild-type ALDH2 transcripts. In the second transgenic strain, overexpressing the mutated mouse E487K cDNA under the control of the chicken-β-actin promoter, tissue specific overexpression of the ALDH2*2 mutant subunits was particularly pronounced in cardiac and skeletal muscles (69). Reduced ALDH enzyme activity correlated well with increased aldehydic load and severity of the phenotypes. It is important to note that many developmental abnormalities and exacerbated pathophysiological conditions described in both strains of the ALDH2*2 overexpressing mice were not observed in the ALDH2−/− mice. Considerations should be therefore made to determine how applicable and relevant the phenotypes and the results derived from ALDH2*2 overexpressing transgenic mice are to humans carrying the ALDH2*2 mutation.
C. ALDH2 Overexpressing Mice
Transgenic mice overexpressing the wild-type ALDH2 gene driven by the chicken-β-actin promoter have also been generated (66). These ALDH2 overexpressing mice were used to evaluate the effect of augmented ALDH2 function on organ protection against acetaldehyde, oxidative stress, and lipid peroxidation-derived 4-HNE. The ALDH2 overexpressing mice have been challenged with both acute and chronic ethanol administration and were subjected to diabetes-induced cardiomyopathy to explore different signal transduction pathways mediated by ALDH2 (66, 96, 97, 107, 138, 173, 185, 186, 260, 372, 375, 376). Overall, this different genetic model demonstrated the critical role of ALDH2 in the protection against toxic aldehydes and oxidative stress in various organs and physiological conditions. As expected, the outcomes were, in general, the opposite of what were observed in the ALDH2 knockout or the ALDH2*2 overexpressing mice, which showed increased susceptibility to aldehydes and pathological conditions. The studies in ALDH2 overexpressing mice demonstrated, in principle, that enhancing the ALDH2 activity can ameliorate many of the deleterious effects of aldehydes and may provide a better protection against both acute and chronic injuries induced by ethanol toxicity or oxidative stress.
D. ALDH2*2 Knock-in Mice
An ALDH2*2 knock-in mouse model was recently developed in our laboratory. In these mice, we replaced the mouse wild-type ALDH2 allele with a mouse E487K substituted ALDH2 allele by homologous recombination. The ALDH2*2 knock-in mice therefore differ only by one single amino acid within the ALDH2 gene compared with the wild-type mice. Western blot analysis indicated that the ALDH2*2 heterozygous animals expressed similar levels of ALDH2 protein to the wild-type mice. However, the enzymatic activity of the knock-in animal is dramatically reduced as expected from the dominant-negative effect of the E487K mutation. The ALDH2 knock-in mice carry a single amino acid substitution that is equivalent to the E487K substitution that defines the human ALDH2*2 mutation. Unlike ALDH2 knockout or ALDH2 overexpression, the overall level of ALDH2 gene expression in the ALDH2*2 knock-in mice is not affected and should mimic the expression level of human ALDH2*2. The enzymology, structural defect, and phenotype of the ALDH2*2 mice are, therefore, a true representation of the human ALDH2*2 carriers. Because of the identical nature of the genetic defect, the ALDH2*2 knock-in mice can serve as an ideal experimental model for the research of human diseases associated with ALDH2 deficiency, and also for efficacy studies using small molecule activators (Aldas, described below in the section of pharmacological modulators of ALDH2 activity) in animal. Indeed, in our in vitro enzyme assays, Alda-1 was capable of activating both human and mouse ALDH2*2 recombinant enzymes and with similar potency profile and activation kinetics. In our view, the ALDH2*2 knock-in mice should be the best model for the studies of human ALDH2*2 mutation phenotype.
III. PHARMACOLOGICAL MODULATORS OF ALDH2 ACTIVITY
A. Inhibitors
Inhibition of ALDH2 as a treatment for alcohol abuse has a long history that predates modern pharmacological interventions using small molecule-based drugs. Kudzu (Pueraria lobata), a traditional Chinese medicine, has been used for more than 1,000 years for its antidipsotropic effect (144). Daidzin and daidzein are two active isoflavones identified in the root and flowers of Kudzu. Daidzin binds to the catalytic site of ALDH2 and is a potent reversible competitive inhibitor of ALDH2 with IC50 of ∼80 nM (181). Daidzin inhibits mitochondrial ALDH2 activity, which results in higher blood acetaldehyde levels, thus mimicking the ALDH2*2 phenotype (143). Disulfiram (Antabuse) is, so far, the only United States Food and Drug Administration (FDA)-approved drug that putatively targets ALDH2. Disulfiram and its metabolites irreversibly inactivate catalytic Cys302 in ALDH2 by carbamylation in the substrate site of the enzyme (178, 283). Similar to daidzin, inhibition of ALDH2 by disulfiram leads to acetaldehyde accumulation, facial flushing, nausea, and vertigo after alcohol drinking, thus mimicking the ALDH2*2 phenotype. As a result of these discomforts, disulfiram has been approved and used as an alcohol aversion drug in the treatment of dependency since 1951 (12). However, poor compliance of drug usage has been an issue that compromises the effectiveness of disulfiram as a treatment for alcoholism (297) (See further discussion below).
Based on the structure of daidzin and its co-crystal structure with ALDH2, other potent ALDH2 inhibitors have been designed (7, 94). Most recently, CVT-10216, which was developed based on structure-activity relationship studies of daidzin, was found to be a potent ALDH2 inhibitor (IC50 of ∼30 nM; ∼2- to 3-fold better than the parent molecule, daidzin) (7, 235). Another new class of irreversible inhibitors has been identified using an unbiased high-throughput screening using purified ALDH2 and measuring the decline in catalytic activity in vitro (40, 145). This class of aldehyde dehydrogenase inhibitors (Aldis) is characterized by a core structure of 3-amino-1-phenylpropan-1-one (145). Due to an enzyme-mediated β-elimination reaction, which generates a vinyl ketone intermediate, these inhibitors covalently modify the active site cysteine residue (Cys 302), thus irreversibly abolishing ALDH enzymatic activity. These first generation Aldis, which are not ALDH isozyme-selective, represent a novel class of broad and generally applicable suicide inhibitors for ALDH. Additional ALDH2 inhibitors have also been discovered by other efforts of high-throughput screening and virtual computational screens, based on crystal structures of ALDH2 and the identification of critical interacting amino acids in the catalytic site of the enzyme (239). A comprehensive review of the pharmacology, mechanism of action, isozyme selectivity, substrate specificity, and clinical applications of an additional 13 known ALDH inhibitors is available (151).
B. Activators
Only a couple of small molecule activators for metabolic enzymes have been identified. One example is a class of novel molecules called Aldas, for aldehyde dehydrogenase activators. We discovered Aldas using a fluorescence-based high-throughput screening. A family of ALDH2 isozyme-specific agonists, represented by Alda-1 [N-(1,3-benzodioxol-5-ylmethyl)-2,6-dichlorobenzamide, MW 324], were found to enhance the catalytic activity of ALDH2 in vitro and in vivo (40). Alda-1 has several useful features. 1) It enhances, specifically, the catalytic activity of ALDH2 by nearly twofold, with EC50 of ∼20 μM. 2) It protects ALDH2 enzymatic activity from 4-HNE-induced inactivation and is able to maintain the catalytic activity of ALDH2 in the presence of a high concentration of 4-HNE. 4-HNE, a substrate of ALDH2, also inhibits the enzyme by forming a Michael adduct or Schiff base with cysteine 302 at the catalytic site (65, 182). 3) Importantly, and of potential clinical relevance, Alda-1 effectively restores the enzymatic activity of the E487K mutant ALDH2. Alda-1 increases the catalytic activity of ALDH2*2 homozygotic enzyme by 11-fold and brings the activity of the heterozygotic enzyme to wild-type levels (40). Cocrystal structures of Alda-1 with wild-type ALDH2 and Alda-1 with the E487K mutant ALDH2 demonstrated that Alda-1 binds at the entrance of the catalytic tunnel in close proximity to Cys302 and Glu286, which are critical to its substrate catalysis (248) (Figure 2). Kinetically, Alda-1 dramatically decreases the Km of the ALDH2 for the cofactor NAD and increases the Vmax of substrate catalysis of aldehydes up to four carbons in length (14, 248). The binding of Alda-1 near the substrate entrance may facilitate productive encounters between the activated water molecule and the thio-acyl intermediate of the substrate, thus resulting in an accelerated hydrolysis and release of the catalytic product. The close proximity between Alda-1 and Cys302 likely also shields this crucial catalytic residue from the nucleophilic attack of reactive aldehydes, such as 4-HNE.
FIGURE 2.

Alda-1, an allosteric agonist of ALDH2, corrects the structural defect in the ALDH2*2 mutant present in 8% of the human population. Top: crystal structure of Alda-1 (stick presentation) in the catalytic tunnel (shown as filled structure) of ALDH2 (Protein Data Bank Accession Code PDB: 3INJ). Highlighted are the critical amino acids in this interaction. Bottom: crystal structure of wild-type ALDH2 (left panel, Protein Data Bank Accession Code PDB: 1O05) and mutant ALDH2*2 (middle panel, Protein Data Bank Accession Code, PDB: 1ZUM). Co-crystal structure of ALDH2*2 with Alda-1 (shown as a ball-filled structure and highlighted in yellow; see arrows) is provided in the right panel (PDB: 3INL). Circles focus on the alpha helix structure (αG) that is missing in the mutant ALDH2*2 enzyme (middle panel vs. left panel) and is restored (right panel) when Alda-1 is bound to ALDH2*2. Images produced using UCSF Chimera package from the Computer Graphics Laboratory, University of California, San Francisco.
Another important feature of Alda-1 is its ability to restore the activity of a defective ALDH2*2 enzyme. E487K substitution in ALDH2 causes a loss of electron density near residues 245–262, which form helix αG, and residues 466–478, which form the active-site loop (162, 163). As revealed by the crystal structure of Alda-1 in complex with ALDH2 E487K mutant, binding of Alda-1 restores the α helix structure and the loop at these regions nearly to the native wild-type state even though Alda-1 has no direct contact with these residues (248) (Figure 2). Alda-1 is therefore an agonist and simultaneously functions as a chemical chaperone, which exerts its allosteric effect to restore the structural defect of a catalytically impaired enzyme.
Another example of generating novel ALDH agonists was guided by the information generated from X-ray cocrystal structure of Alda-1 with ALDH2. With the use of a computer-assisted virtual screening of chemical libraries, three-dimensional molecular docking of 19,943 compounds on ALDH2 generated 21 hits (152).
In summary, the novel small molecule modulators of ALDH represented by Aldas and Aldis are useful tools to determine the role of ALDH2 in a variety of human pathologies. They may also serve as a new class of therapeutics.
IV. DISEASES ASSOCIATED WITH ALDH2
Aldehydes are formed as a part of normal catabolism (e.g., from amino acids, lipids, and nucleotides). Aldehydes and their precursors are present in food: ethanol is a precursor to acetaldehyde, fermented or pickled fruits and vegetables contain aldehydes, many spices and flavorings like vanilla and cinnamon are aldehydes, and aldehydes are also products of frying or smoking meat and fish. Finally, aldehydes are also present in our environment in a variety of aromas and perfumes, in cigarette smoke and car exhaust, and in a variety of chemicals and pollutants.
The potential role of aldehydes in general and aldehydes that are the substrates of ALDH2 in particular, in human pathologies has not been the focus of much research until recently. ALDH2 is well conserved throughout evolution, from bacteria to humans (131, 291, 319). Because much of its biochemical characterization occurred about 30 years ago (132, 177, 281), and because an inactivating point mutation present in 8% of the human population was thought to be benign (24, 183, 285, 358), there was little active research on the role of ALDH2 in human diseases apart from its role in ethanol metabolism and complications associated with excessive ethanol drinking.
Yet, the toxic nature of aldehydes has been known for a long time. Aldehydes can easily diffuse through cell membranes, form adducts with macromolecules including proteins, DNA, and lipids; such aldehydic adduction usually modulates or disrupts the function of these macromolecules (Figure 3; Refs. 73, 223, 250, 266, 307). Reactive aldehydes are readily formed during oxidative stress as products of lipid peroxidation (Figure 3). Therefore, a mechanism for rapid clearance of these highly diffusible and harmful aldehydes is crucial to protect cells and tissues from damage. In diseases where there is excessive aldehydic load, at least some of the pathologies may be alleviated by faster metabolism and inactivation of these toxic aldehydes.
FIGURE 3.

4-HNE, an aldehydic product of lipid peroxidation, causes cytotoxicity. Buildup of aldehydes, such as 4-hydroxynonenal (4-HNE), due to oxidative stress promotes cell death. Listed are steps in aldehydic injury to cells, using 4-HNE (depicted as a zigzag line), as an example. 1) 4-HNE is produced through ROS-mediated lipid peroxidation of mitochondrial and plasma membranes. 4-HNE can in turn 2) reduce membrane integrity, 3) inhibit proteasomal function, 4) trigger unfolded protein accumulation, 5) inhibit electron transport chain activity, 6) reduce Kreb's cycle activity, 7) inhibit ALDH2 activity, 8) increase mitochondrial permeability and dysfunction, or 9) cause DNA damage.
In the following, we argue that ALDH2 is critical to the health of cells and that it serves as an important shield from the damage occurring under oxidative stress. Since oxidative stress is associated with numerous human pathologies, increasing the catalytic activity of ALDH2 may provide a novel and effective means to reduce oxidative stress-induced cell and organ dysfunction and therefore support human health. Figure 4 depicts some of the diseases in which reports from human specimens demonstrated an increase in the aldehydic load and/or in which ALDH2 was associated with or played a positive role in reducing the pathology. These diseases are discussed in more detail in the following.
FIGURE 4.
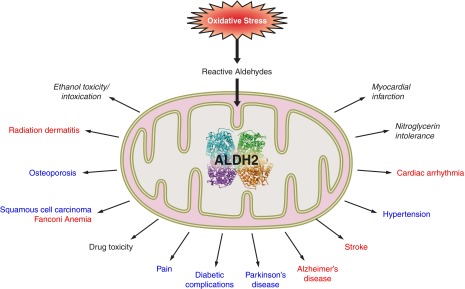
Diseases in which activators of ALDH2 may be beneficial. See text for full discussion. Red highlights diseases where the evidence for ALDH2 role came from preclinical proof-of-concept studies. Blue highlights diseases where ALDH2 role was supported by clinical observations. Black highlights diseases where evidence from both preclinical studies and from human epidemiological or pathohistological studies supports a role for ALDH2.
A. Cardiovascular Diseases
1. Myocardial infarction, heart failure, and stroke
Cardiovascular diseases, including myocardial infarction, cardiac hypertrophy, and heart failure, are a major cause of morbidity and mortality worldwide. Although early research efforts focused on the damaging effects of free radicals in the heart (19, 280), recent findings revealed that accumulation of cardiotoxic reactive aldehydes, derived from reactive oxygen species-induced stress, can also impair myocardial function in rodents and humans (40, 41, 255).
Of the 19 different ALDH isozymes expressed in the human body, the mitochondrial ALDH2 has rapidly emerged as a crucial enzyme involved in protecting the heart from oxidative stress. Although the contribution of ALDH2 in cardioprotection was initially discovered by our group downstream of protein kinase C type ϵ (ϵPKC) activation (40), ϵPKC has multiple protein partners intracellularly including ion channels (45). Additionally proteomic analysis suggests ϵPKC in models of cardioprotection have many subcellular protein partners (323). In this regard, targeting ALDH2, instead of ϵPKC, will provide a more specific therapeutic target. Experimental approaches using either pharmacological activation or genetic overexpression of ALDH2 have shown that improved detoxification of reactive aldehydes, such as 4-HNE, is protective against acute (i.e., ischemia) and chronic (i.e., heart failure) cardiovascular diseases (25, 26, 40, 41, 97, 185, 186, 259, 298, 375–377). The cardioprotective effects of ALDH2 have also been highlighted by epidemiological studies, demonstrating that individuals carrying a point mutation in the ALDH2 gene (ALDH2*2), which results in a dramatic reduction in enzymatic activity, are more susceptible to cardiac diseases and to the morbidity associated with them (16, 38).
Most of the cardiac damage occurring during ischemia and reperfusion injury and in heart failure is due to excessive generation of reactive oxygen species (19), which leads to exacerbated peroxidation of polyunsaturated fatty acids (i.e., linoleic acid, arachidonic acid, and cardiolipin) present in biological membranes (266). These lipid hydroperoxides can subsequently form toxic secondary end products, such as 4-HNE and malondialdehyde (266). As mentioned above, 4-HNE is a highly reactive carbonyl compound that readily interacts with cysteine, histidine, and lysine residues and forms protein adducts via Michael addition or Schiff base reaction (314). Excessive 4-HNE adduct formation in the myocardium during ischemia-reperfusion injury impairs cardiac contractility (1), mitochondrial bioenergetics (116), and redox balance (154) while impairing also protein quality control (31, 55) by targeting key metabolic enzymes (313) and the 26S proteasome (Figure 3; Ref. 75).
Accumulation of 4-HNE is also associated with the establishment of cardiomyopathies from different etiologies and with progression to heart failure (192, 218, 219). 4-HNE-modified proteins are significantly increased in myocardial biopsies from patients with hypertrophic and dilated cardiomyopathy compared with the levels in control subjects who do not have cardiac disease (218, 219). Of interest, sustained treatment with a beta-blocker (carvedilol) for 9 months, which improves cardiac functions, is associated with a 40% reduction in the levels of cardiac 4-HNE-modified proteins in patients with dilated cardiomyopathy (218). Mak et al. (192) reported that 4-HNE levels are consistently elevated in the plasma of congestive heart failure patients and are inversely correlated with left ventricular contractility. Moreover, animal models that present increased circulating levels of 4-HNE due to alcohol dehydrogenase overexpression or ALDH2 silencing are more sensitive to ischemia and reperfusion injury and are more prone to develop cardiomyopathy and heart failure (40, 101, 138, 185, 186, 259, 298, 377). Exposure of isolated adult rat cardiomyocytes to 4-HNE (0.5–400 μM) disrupts the cellular redox balance, induces calcium overload, causes contractile dysfunction, and elicits pro-arrhythmic effects (15, 220). Thus increased aldehydic load is detrimental to the heart.
Elevated 4-HNE-modified proteins have also been reported in patients with hypertension or with peripheral artery disease (189, 251, 252, 299). Gastrocnemius muscle biopsy obtained from peripheral artery disease patients presents higher levels of protein carbonyls, lipid peroxides, and 4-HNE compared with control muscles (251). These changes are accompanied by reduced mitochondrial respiration and reduced antioxidant activity (251). Although 4-HNE accumulation negatively associates with peripheral artery diseases, the mechanisms involved in this process, including the contribution of ALDH2 to the pathophysiology of peripheral artery disease, remain to be elucidated.
Acetaldehyde, the first oxidized metabolic product of ethanol, has been considered another important candidate for the pathogenesis of cardiovascular diseases. Accumulation of acetaldehyde is very harmful to the heart, causing cardiac hypertrophy and contractile dysfunction in mouse models of alcohol dehydrogenase overexpression (67, 117). Acetaldehyde directly impairs cardiac excitation-contraction coupling and inhibits sarco(endo)plasmic reticulum calcium-ATPase (SERCA) function in rodents (117, 261). However, the molecular basis of acetaldehyde cardiotoxicity has remained elusive since acetaldehyde has very high chemical reactivity, it is volatile and it is metabolized quite quickly (67).
The cardioprotective effects of mitochondrial ALDH2 during cardiac ischemia and reperfusion injury, in models of ethanol-mediated cardiomyopathy and in heart failure, have been demonstrated by the use of a variety of ALDH2 transgenic mice (96, 97, 173, 185, 186, 259, 377). Overexpression of ALDH2 wild-type enzyme attenuates infarct size and improves fractional shortening in an in vivo mouse model of acute myocardial infarction (185). Conversely, ALDH2 null mice display exacerbated cardiac damage following ischemia-reperfusion along with increased formation of mitochondrial reactive oxygen species and endothelial dysfunction (185, 334). In agreement with the cardioprotective role ALDH2, Endo et al. (69) reported that mice overexpressing the inactive ALDH2*2 exhibited impairment in mitochondrial bioenergetics along with increased oxidative stress, and elevated levels of 4-HNE-protein adducts.
Although not tested for resistance to acute myocardial infarction injury, adult transgenic mice overexpressing ALDH2, which results in increased ALDH2 enzymatic activity by fourfold, are more resistant to acute ethanol cardiotoxicity (186). Acute intraperitoneal ethanol injection (3 g/kg) in mice with wild-type ALDH2 expression drastically increases cardiac acetaldehyde levels along with reduced ex vivo left ventricular developed pressure and mitochondrial uncoupling (186, 187). This cardiac toxicity was attenuated and accentuated in ALDH2 overexpressing and ALDH2 knockout mice, respectively (186, 187). Mice overexpressing ALDH2 exhibited a significantly reduced ethanol-mediated cardiac damage by decreasing acetaldehyde levels, thus improving myocardial contractility and restoring mitochondrial membrane potential (186).
The sensitivity of the myocardium to aldehydic load has also been examined in another animal model, in which chronic ingestion of a 4% ethanol diet for 12 wk was found to induce cardiac hypertrophy, myocardial fibrosis, reduced fractional shortening, cell shortening, impaired intracellular Ca2+, and homeostasis in wild-type mice. These hallmarks of alcohol cardiomyopathy, described first in humans (79, 317), were associated with elevated blood acetaldehyde levels, increased oxidative stress, lipid peroxidation, and protein carbonyl formation in the wild-type, nontransgenic mice (66, 259). Hyperphosphorylation of oxidative stress related kinases, glycogen synthase kinase-3β (GSK-3β) and apoptosis signaling regulated kinase (ASK-1), and increased phosphorylation of the transcription factors, GATA4 and CREB cAMP-response element binding protein (CREB), are reported as the mediators of ethanol-induced cardiac cardiomyopathy (66, 376). Chronic administration of ethanol also induced impaired glucose tolerance, cardiac insulin insensitivity, cardiomyopathy, and contractile defects (173), similar to those observed in diabetic cardiomyopathy from streptozotocin-induced diabetic mice (375). In both animal models of ethanol-induced cardiomyopathy and streptozotocin-induced cardiomyopathy, reduced phosphorylation of Akt, GSK-3β, and forkhead transcription factor of the O subtype (Foxo3a) was the key signaling cascade that led to apoptosis and mitochondrial dysfunction. Importantly, both molecular and pathological abnormalities were prevented in the ALDH2 overexpressing mice. In addition, in an acute model of ethanol challenge (3 g/kg), ALDH2 overexpression reduced blood acetaldehyde levels and prevented myocardial and cardiomyocyte contractile dysfunction (186). Therefore, in both acute and chronic ethanol-induced myocardial injuries, a common protective mechanism that is mediated by ALDH2 appears to exist. As illustrated in Figure 5, ALDH2 conferred its protection against ethanol toxicity by the regulation of two key kinases, Akt and AMPK. Akt and AMPK, in turn, regulate transcription factor Foxo3a phosphorylation by ALDH2-mediated decreased phosphatases, PP2A and PP2C (138, 186, 376). Simultaneously, it was demonstrated that ALDH2 overexpression also conferred its protection by the regulation of autophagy and apoptosis through the balance between Akt and AMPK and their downstream substrates mTOR, STAT3, and Notch1 (95, 96). Detoxification of acetaldehyde in ALDH2 overexpressing mice also led to attenuation of chronic ethanol exposure-induced damages in brain and liver by a similar mechanism involving Akt and apoptotic regulation (Figure 5; Refs. 107, 260). More recently, Zhang et al. (372) described that genetic or pharmacological ALDH2 activation protects against endoplasmic reticulum stress-induced cardiomyocyte contractile dysfunction possibly through reduction of autophagy (317).
FIGURE 5.

Ethanol-induced cardiotoxicity. ALDH2 confers protection against ethanol toxicity by affecting the activity of key proteins in cardiac myocytes. The scheme summarizes data from transgenic mice that elucidated the potential pathways that are regulated by ALDH2 and acetaldehydes. Activation of ALDH2 or ALDH2 overexpression were found to confer cardiac protection by regulating autophagy and apoptosis through the balance between Akt and AMPK and their downstream substrates, such as mTOR, STAT3, Notch1, PP2A, and PP2C (93, 98, 112).
Consistent with the role of ALDH2-mediated cardioprotection against ischemia-reperfusion injury, ALDH2 overexpression prevents 4-HNE-protein adduct formation and attenuates myocardial damage from ischemia-reperfusion also via the Akt- and AMPK-mTOR signaling cascades (185). Hence, ethanol toxicity and ischemia-reperfusion appear to trigger a common molecular signaling event that leads to mitochondrial dysfunction, apoptosis, and autophagy. These studies illustrate the intricacy of signaling networks that are elicited by acetaldehyde and/or oxidative stress-derived 4-HNE. These rodent models of cardiac damage associated with increased aldehydic load recapitulate well the pathological findings in humans (see in the following segment), and the data therefore suggest that ALDH2 activation may benefit patients with any of these cardiac diseases.
The ALDH2 isozyme-selective activator Alda-1 enabled a direct examination of whether ALDH2 activation is sufficient to produce cardiac protection. It was shown that pharmacological ALDH2 activation by Alda-1 treatment is sufficient to protect hearts against ischemia-reperfusion injury in a rat model of myocardial infarction (40). Administration of 20 μM Alda-1 for 10 min prior to 35 min of ischemia followed by 60 min of reperfusion increases ALDH2 activity by twofold and significantly reduced cardiac damage by 60% in an ex vivo model of ischemia-reperfusion in rats (40). Alda-1-induced cardioprotection is also effective in an open-chest model of acute myocardial infarction in rats (40, 41). Injection of Alda-1 (8 mg/kg) into the left ventricle 5 min before ligating the left anterior descending coronary artery decreases the extent of myocardial infarction and creatine kinase release into the blood by 50% (40). Conversely, ALDH2 inactivation increases infarct size with a tight inverse correlation between infarct size and ALDH2 activity (40). In another study, it was demonstrated that selective ALDH2 activation using an Alda-1 analog, Alda-44, circumvents the requirement of ϵPKC to induce cardioprotection (25). In an independent study, Lagranha et al. (158) reported that Alda-1 reduces infarct size and post-ischemic contractile dysfunction in male but not in female rats (because females already have phosphorylated/activated ALDH2) after ischemia-reperfusion injury (158). More recently, in another study, Gong et al. (102) demonstrated that addition of Alda-1 to cardioplegic solution (histidine-tryptophan-ketoglutarate) significantly reduces cardiac ischemia-reperfusion injury in an ex vivo rat model. Alda-1 supplementation improves the cardioprotective effects of the cardioplegic solution by increasing ALDH2 activity, which results in reduction of cardiac 4-HNE levels and better myocardial contractility depicted by improved left ventricular developed pressure (102). Such applications of Alda-1 as an adjuvant in cardioplegic solutions for scheduled open-heart surgery may have a wide utility for organ preservation in open-heart surgery or other solid organ transplantations.
In addition to its direct effect on cardiomyocytes, Koda et al. (150) have shown that Alda-1 plays a key role in cardiac mast cell biology during ischemia-reperfusion injury. In this ex vivo study using a guinea pig model, it was found that ALDH2 activation prevents degranulation of cardiac mast cells and subsequent local renin-angiotensin activation during ischemia-reperfusion injury (150). Alda-1 treatment also drastically reduces mast cell degranulation and release of renin (the rate-limiting step in renin/angiotensin system activation) elicited by acetaldehyde, 4-HNE, or during cardiac ischemia-reperfusion. Preincubation with ALDH2 inhibitors, such as cyanamide or high levels of nitroglycerin, prevents the benefits of Alda-1 (150). Arrhythmias in patients with myocardial infarction are common and a major cause for sudden death in this patient population (267). Thus, in addition to protecting the heart muscle functions, activation of ALDH2 by Alda-1-like compounds may reduce the damage resulting from reperfusion-induced arrhythmias via enhanced aldehyde detoxification (265). Together, these results uncover novel basic mechanisms of pharmacological cardioprotection induced by ALDH2 activation.
Finally, Ge et al. (96) demonstrated the use of the ALDH2 activator, Alda-1, in protecting cardiac cells from ethanol-induced contractile dysfunction and exacerbated autophagy. Administration of Alda-1 to ethanol-treated adult mouse cardiomyocytes significantly improves intracellular calcium handling, abolishes the ethanol-mediated cardiomyocyte mechanical abnormalities characterized by depressed peak shortening and relaxation, and reduces caspase-3 activity, a key player in programmed cell death (96). Together, these findings highlight the potential use of ALDH2 activators and the critical role of activating ALDH2 to protect against cardiac damage caused by toxic aldehydes that accumulate following myocardial infarction, or under conditions leading to alcoholic cardiomyopathy and to heart failure.
The ALDH2-mediated protection also extends to ischemic brain injury, or ischemic stroke (155). Okun et al. (234) found elevated levels of 4-HNE-protein adducts in the ischemic cerebral cortex when measured within 2 h of stroke induction in mice. More recently, Lee et al. (168) showed that plasma 4-HNE levels are elevated in patients with stroke and in two different animal models of cerebral ischemia. Indeed, intravenous administration of 4-HNE before stroke not only enlarged the cerebral ischemia-induced infarct area, but also increased oxidative stress in rats (168). Furthermore, ALDH2 overexpression increases neuronal survival in the presence of 4-HNE toxicity in PC12 cells. Cerebral damage and neuronal cell death are also reduced in ALDH2 transgenic mice after 12 wk of ethanol consumption (260). Two studies have evaluated the association between ALDH2*2 allele and stroke, a common cause of death in East Asia, where no correlation between the incidence of stroke and the genotype was found (215, 353). However, considering that heavy alcohol consumption increases the risk of stroke (125, 247), further large cohort studies are needed to better understand the contribution of ALDH2 mutation to stroke.
2. Epidemiological human studies suggesting a role for ALDH2 in the health of the myocardium
Over the past decade, the reduced ALDH2 activity due to the ALDH2*2 mutation has been shown to be an independent risk factor for acute coronary syndrome in humans. Takagi et al. (306) demonstrated that the ALDH2*2 allele is a risk factor for myocardial infarction in Japanese men. More recently, Xu et al. (348) showed that Chinese male carriers of the ALDH2*2 inactivating mutation have a significantly higher incidence of acute coronary syndrome compared with noncarriers. Similar results were found in a subgroup analysis of patients with primary ST-segment elevation myocardial infarction (STEMI) (348). However, this ALDH2 mutation is not associated with the number of coronary arteries with significant stenosis (348). [In this latter study ALDH2*1/*2 and ALDH2*2/*2 were pooled into a single group for the analysis.] A case control study of an elderly Korean male population found that the ALDH2*2 allele was significantly linked to increased risk of developing acute coronary syndrome (134). Furthermore, the ALDH2 mutation along with the abnormal high-density lipoprotein, cholesterol, and elevated body index mass is an independent risk marker for myocardial infarction in these elderly Korean males. A multivariant regression analysis showed that subjects carrying the ALDH2*2 allele displayed elevated plasma levels of inflammatory markers (i.e., high C-sensitive protein, hs-CRP) after the onset of acute myocardial infarction, suggesting higher myocardial injuries following ischemic insult (16).
The ALDH2*2 allele has also been linked to chronic cardiovascular pathophysiologies. In a large scale meta-analysis of genome-wide association studies from 19,608 subjects, ALDH2*2 (G to A exchange, which is also designated as SNP rs671) was identified as the most prominent SNP that is associated with blood pressure variation in East Asians (139). Other studies confirmed that ALDH2*2 is a strong risk factor for elevated blood pressure and hypertension among males who consume ethanol excessively (118, 217, 305). Chang et al. (38) recently reported that homozygous carriers of the ALDH2*2 allele were more likely to progress to hypertension compared with noncarriers over a follow-up period of 5.7 yr. The risk associated with the ALDH2*2 mutation is the strongest in alcohol drinkers relative to nondrinkers, providing further evidence for an association between ALDH2 genetic variants and alcohol intake on the risk of hypertension (38). Chen et al. (43) carried out a meta-analysis to assess the association between the ALDH2 genotype and blood pressure (5 studies, n = 7,658) and hypertension (3 studies, n = 4,219). Unlike the previous publication, they found that individuals carrying the ALDH2*1/*1 genotype, with average alcohol intake of 25 g/day (or 2 alcoholic beverages), had strikingly higher systolic and diastolic blood pressure than those with the ALDH2*2/*2 genotype. Systolic blood pressure was 7.44 mmHg greater for ALDH2*1/*1 than for ALDH2*2/*2 and 4.24 mmHg greater among ALDH2*1/*2 than among ALDH2*2/*2. Moreover, the risk for hypertension among ALDH2*1/*1 was found to be ∼2.5-fold higher than that among ALDH2*2/*2. Since, unlike the study of Chang et al. (38), this latter study did not examine heavy drinkers (e.g., >4 alcoholic beverages/day), the data in these two studies may not be inconsistent. Together, these epidemiological findings highlight an association between the ALDH2 inactivating mutation and alcohol intake on blood pressure and on the risk of developing hypertension. To date, there are no epidemiological studies that examine the association between ALDH2 mutation and heart failure.
3. Nitroglycerin tolerance
Another aspect relevant to cardiac disease and ALDH2 relates to nitroglycerin metabolism. Nitroglycerin has been widely used as a vasodilator to treat ischemic and congestive cardiac diseases, including angina pectoris, myocardial infarction, and heart failure (82, 242). Chen et al. (48) demonstrated that ALDH2 plays an essential role for the bioconversion of nitroglycerin to NO to achieve its vasodilating effects. However, nitroglycerin is a potent ALDH2 inhibitor, and continued treatment of nitroglycerin leads to insensitivity to its vasodilating effect in patients with angina pectoris (82). As expected, in East Asian patients carrying the inactivating ALDH2*2 mutation, nitroglycerin is less effective in eliciting a cardiovascular response compared with ALDH2 wild-type subjects receiving the same treatment (106, 174, 374). Yet nitroglycerin use in East Asia for patients with acute myocardial infarction and in congestive heart failure is very common.
Because nitroglycerin is widely used in patients with acute myocardial infarction, its inactivating effect on the cytoprotective enzyme ALDH2 may be of concern. Chen et al. (40) examined this question using a rat model of myocardial infarction. It was found that 16 h use of nitroglycerin (7.2 mg·kg−1·day−1) just prior to acute myocardial infarction resulted in an infarct size that was 31% bigger than in nontreated rats (40). Furthermore, a worse cardiac function was observed in rats treated with nitroglycerin overnight, at clinically relevant concentrations (298). Importantly, we found that a sustained treatment with Alda-1 (16 mg·kg−1·day−1) during nitroglycerin use blocked nitroglycerin-induced inhibition of ALDH2 activity and prevented the nitroglycerin-induced decrease in cardiac function and nitroglycerin-induced increase in infarct size (298). Sustained treatment with nitroglycerin also increased protein carbonylation in the infarct region of the myocardium. In agreement with the protective role of Alda-1 on ALDH2, cotreatment of Alda-1 and nitroglycerin greatly reduced the level of protein carbonylation after myocardial infarction, in vivo (298).
Other studies have shown that NO-induced nitrosylation on critical cysteines can lead to ALDH2 inactivation (175). It is likely that the protection offered by Alda-1 against increased myocardial infarction by nitroglycerin is mediated by a mechanism which provides a protective shield for Cys302 from nitrosylation, similar to the protection mediated by Alda-1 against carbonylation by 4-HNE. Regardless of the exact mechanism, these data suggest that protecting ALDH2 from nitroglycerin-induced inactivation will benefit patients treated with nitroglycerin in a sustained fashion. The therapeutic value of an Alda-like drug which can enhance nitroglycerin bioconversion and prevent the inhibitory effect of nitroglycerin has been reviewed recently (82).
B. Diabetes Mellitus
Generally considered a disease of the Western world, diabetes is now a global epidemic, predicted to affect 380 million people by 2025, and Asian countries are predicted to make up 60% of the diabetic world's population (36). Unlike the Western countries, the diabetic population in Asian countries is proportionally younger (36). A number of potential genes identified in the Asian population may link to developing diabetes. These include ALDH2*2, which may be a risk factor for non-insulin-dependent diabetes mellitus (NIDDM).
Initial observations by Pyke and Leslie (171, 254) in 1978 for a Caucasian population suggested that chlorpropamide, a sulfonylurea anti-diabetic agent, taken prior to consumption of alcohol, caused a high percentage of NIDDM to have facial flushing. The initial studies identified this effect to be specific for chlorpropamide, and in a study where patients first received a placebo instead of chlorpropamide prior to alcohol consumption, facial flushing was not observed. The researchers also found that this flushing effect of chlorpropamide did not occur in insulin-dependent diabetics. Although chlorpropamide is no longer used clinically, in 2001, chlorpropamide was found to inhibit ALDH, suggesting a possible link between ALDH and diabetes.
Another study investigating the correlation between ethanol and diabetes concluded that, among those with a normal ALDH2 genotype, ethanol consumption greater than 20 drinks per week may increase insulin resistance. Among those with the ALDH2*2 genotype, less ethanol consumption was required to develop insulin resistance (135). Hemoglobin A1C, a long-term marker of glycemic control, was also higher for those ALDH2*2 diabetics who drank a light to moderate amount of ethanol compared with carriers of the wild-type enzyme (212). These studies further lead to determining whether ALDH2*2, independent of alcohol consumption, is a risk factor for diabetes.
Several studies have suggested that the maternal ALDH2 genotype may contribute to NIDDM (213, 301). An association between ALDH2*2 genotype and diabetes prevalence is greater in people with diabetic mothers (32.6%) compared with those whose mothers have the wild-type ALDH2 (19.2%) (301). A follow-up study found that those with an ALDH2*2 genotype were five times more likely to have a diabetic mother compared with those with wild-type ALDH2 (300). The authors of these studies suggest the ALDH2*2 genotype may lead to mitochondrial DNA damage, which in turn may contribute to diabetic complications in the offspring of diabetic mothers (302). In 542 coronary artery disease patients, all Han Chinese, a genetic link was investigated between the ALDH2*2 genotype and NIDDM. Interestingly, the ALDH2*2 genotype was suggested to be a risk factor for NIDDM only in females, and not males (347).
Preclinical studies in rats also suggest that ALDH2 activity may affect diabetic pathology (326, 375). Furthermore, in transgenic mice, overexpression of ALDH2 has been shown to be protective against streptozotocin-induced diabetic cardiomyopathy. In this setting of diabetic complications, Alda-1 was able to recapture the protective effect against high glucose-induced mitochondrial damage and contractile function in cardiomyocytes (375). Together, this work suggests an association between diabetes and ALDH2 activity. However, more studies are needed to further examine the underlying pathology associated with ALDH2 and diabetes.
C. Neurodegenerative Diseases
Brain is an organ that is rich in polyunsaturated fatty acid and, at the same time, harbors a relatively high concentration of oxygen in the lipid bilayer (257). Oxidative stress-induced lipid peroxidation, mitochondria dysfunction, and accumulation of aldehydic products are key features in aging, memory loss, cognitive decline, and neurodegeneration (133, 258, 379). The role of ALDH2 and aldehydic load in a variety of neurodegenerative diseases has been suggested based on animal studies, human pathological findings, and data from epidemiological studies. Here we focus on two neurodegenerative diseases, Parkinson's disease and Alzheimer's disease.
1. Parkinson's disease
Parkinson's disease is a progressive neurodegenerative disease manifested as a loss of movement control and varying degree of dementia. Parkinson's disease afflicts ∼1% of the population at age 65, and the incidence increases to 4–5% of the population at age 85 (74, 76). The disease is associated with a progressive loss of neurons in deep nuclei in the brain, in particular dopaminergic neurons in the substantia nigra. Both genetic and environmental toxins have been associated with the disease. Relevant to this review, a number of years ago, Burke and collaborators proposed and have since accumulated substantial evidence that toxic aldehydes play a causal role in the disease (27–29, 153, 238). In particular, a very reactive aldehyde, 3,4-dihydroxyphenylacetaldehyde (DOPAL), has been implicated in the disease. DOPAL is a dopamine metabolite and a product of monoamine oxidase in dopaminergic neurons of the brain (29). Studies in animals demonstrated that DOPAL is a neutoxin, and its injection induces Parkinsonism (238, 336). As DOPAL is converted to its nontoxic metabolite 3,4-dihydroxyphenylacetic acid (DOPAC) by ALDH2, increasing the activity of ALDH2 may provide a means to reduce dopaminergic neuronal cell death. Indeed, accumulation of DOPAL in the substantia nigra is documented in animal models of and in humans with Parkinson's disease (28, 29, 93, 193, 335, 336).
Other aldehydes contribute to neurotoxicity in general and to the loss of dopaminergic neurons in particular. Culture studies demonstrate that 4-HNE induces neuronal cell apoptosis (155). Post mortem analysis of brain samples from patients with Parkinson's disease and control patients demonstrated that ∼60% of substantia nigra neurons are positively stained for 4-HNE-modified proteins in brains of the patients with Parkinson's disease; in contrast, only 9% of the control subjects showed such staining (365). Furthermore, physiologically relevant concentrations of 4-HNE (low micromolar) inhibit biotransformation of the toxic DOPAL to the inactive DOPAC (86). As discussed earlier, even though 4-HNE is a substrate of ALDH2, it can also inactivate ALDH2 by covalently adducting to the Cys in the catalytic site of the enzyme (65). Thus increased accumulation of 4-HNE in neurons may, in turn, increase the levels of DOPAL, thus linking oxidative stress to the uncontrolled production of an endogenous neurotoxin that has been linked to Parkinsonism (86).
Epidemiological studies show a correlation between Parkinson's disease with living in a rural area and/or exposure to agricultural pesticides (9, 62, 84, 308). Many of these pesticides, such as maneb, mancozeb, and benomyl, were found to directly inhibit the activity of purified rat ALDH2 (and ALDH5A), thus decreasing the detoxification of lipid peroxidation-derived aldehydes, such as 4-HNE and DOPAL (170). For example, Benomyl is a benzimidazole fungicide and a potent inhibitor of ALDH2 in vitro and in vivo. Benomyl was found to have an IC50 of 24 μM/kg when injected intraperitoneally to animals (151, 293, 294). Recently, Fitzmaurice et al. (85) provided substantial supportive evidence for the inhibition of mitochondria ALDH by benomyl as a plausible pathogenic mechanism of Parkinson's disease. In that study, an in vivo metabolite of benomyl which inhibits mitochondria ALDH activity at nanomolar concentrations was identified. This inhibitor led to accumulation of DOPAL and to selective damage of dopaminergic neurons in primary cell cultures and in a zebrafish model system (85). Epidemiological studies also confirmed the association between exposure to benomyl and Parkinsonism; there is a twofold increase of Parkinson's disease risk following chronic exposure to ambient benomyl at the workplace (85). Genetic studies on the association of ALDH2*2 mutation and Parkinson's disease has been sparse. Only one case-control study has been reported, comparing 93 genotyped Parkinson's disease patients with 297 healthy controls. In this study, no association between ALDH2 genotype and Parkinson's disease was found (90). Finally, in animals, diminished capacity for toxic biogenic aldehyde removal in double knock-out mice of ALDH1A1−/− and ALDH2−/− was found to lead to 4-HNE and DOPAL accumulation in the substantia nigra. Importantly, increased neurodegeneration and age-dependent motor dysfunction were also found in this transgenic model compared with wild-type mice (336).
2. Alzheimer's disease
Alzheimer's disease is a degenerative disease for which there is currently no cure. It is the most common cause of dementia, and although a less prevalent early-onset form exists, it is the most common form of dementia afflicting people over the age of 65. More than 4.5 million Americans are believed to have Alzheimer's disease. By 2050, because of the increase in life span, the number is expected to increase to 13.2 million (22). Currently, the only therapeutic treatment of Alzheimer's disease involves managing the symptoms, with limited success.
Progressive loss of mitochondrial function or defects in mitochondrial metabolism in the later stages of Alzheimer's disease can lead to the generation of reactive oxygen species resulting in oxidation of membrane lipids and accumulation of 4-HNE in the brain (232) and blood (205). Additionally, 4-HNE accumulates in the hippocampal regions of patients with mild cognitive impairment and patients with early Alzheimer's disease (342). 4-HNE adduction was found on amyloid proteins (277), and this 4-HNE adduction is thought to contribute to amyloid plaque formation in a later stage of the disease (5, 205). The accumulation of amyloid plaques over time correlates with neurodegeneration and subsequent atrophy of the affected regions of the brain. It is therefore possible that diminishing 4-HNE accumulation during the early stages of Alzheimer's disease may reduce or prevent the disease progression.
As we discussed earlier (see also Figure 3), ALDH2 protects mitochondrial functions through the detoxification of 4-HNE that accumulate in this organelle. Recent human epidemiological studies correlate a higher incidence of Alzheimer's disease in Asian patients with the inactivating ALDH2*2 mutation (110, 136, 324). In a Japanese study involving more than 2,200 subjects between the ages of 40 and 70 yr, levels of serum lipid peroxides were clearly higher in female ALDH2*2 carriers, after exclusion of alcohol drinking behavior (230). There is also a synergistic risk increase of Alzheimer disease between ALDH2*2 and APOE-ϵ4 allele (136, 324). Apolipoprotein E protein, APOE-ϵ4, is the least effective 4-HNE-eliminating apolipoprotein, compared with APOE-ϵ2 and APOE-ϵ3 proteins (245). This may explain the increased risk of Alzheimer's disease associated with the APOE-ϵ4 allele and its combined vulnerability with ALDH2*2 to this disease.
Relative to wild-type mice, transgenic mice overexpressing ALDH2*2 exhibit accelerated neurodegeneration in the hippocampus as well as diminished 4-HNE clearance as the mice age (231, 233). Therefore, agents that accelerate 4-HNE removal may provide a possible therapeutic mode of intervention to prevent or reduce disease progression. The same study showed that primary cortical neuronal cells derived from the ALDH2*2 transgenic mice are more susceptible to 4-HNE-induced cell death in culture (231). Concomitantly, elevated 4-HNE accumulation and premature brain degeneration were observed in these animals. Furthermore, accumulation of hyperphosphorylated tau protein, which is found also in human patients, was increased in the transgenic mice compared with wild-type mice (231). Importantly, these ALDH2*2 transgenic mice developed marked age-related neuronal loss in the brain with cognitive impairment of memory loss, premature aging, and shortened life span (221, 231, 232). These phenotypes of neurodegeneration mimic those seen in patients with Alzheimer's disease and were enhanced by knocking-out the ApoE gene, a well-known risk factor in Alzheimer's disease in humans (179). Therefore, agents that increase the activity of ALDH and thereby increase 4-HNE detoxification hold a great potential for slowing down or preventing Alzheimer's disease progression. This was demonstrated in a recent pilot study, using the ALDH2 activator Alda-1, to enhance 4-HNE detoxification; Alda-1 treatment prevented amyloid β peptide-induced impairment of angiogenesis in cultured endothelial cells (290).
D. Alcohol-Induced Pathophysiology
An estimated 1.9 billion adults (=15 yr old) worldwide consume at least a daily average of one alcoholic beverage (13 g of ethanol) regularly (11, 338). About 140 million people consume ethanol in excess, which results in alcoholism or alcohol dependence (337, 339). The mechanism by which ethanol addiction (alcoholism) relates to activation of the reward system in the brain, the genetic predisposition to alcoholism, its direct effect on the functions of the central nervous system, and the mechanism of end organ injuries due to alcohol consumption have all been topics of active research (23, 225, 367). Here we focus on the roles of acetaldehydes in these pathologies.
1. Alcohol intoxication and Asian acetaldehyde toxicity syndrome
Much of the damage by ethanol can be attributed to acetaldehyde accumulation. However, acetaldehyde toxicity has attracted much less public awareness than the emphasis on the dangers of alcohol. For example, in 2007, The International Agency for Research on Cancer (IARC) classified ethanol in alcoholic beverages as a group 1 carcinogen in humans based on epidemiological studies (11). In fact, it is well known that ethanol is not a carcinogen (142); rather, it is acetaldehyde that causes malignancy. This highlights the under-recognized cytotoxicity and genotoxicity risks of acetaldehyde.
Ingested ethanol is quickly metabolized to acetaldehyde (by alcohol dehydrogenase) and then to acetate, by the mitochondrial enzyme ALDH2 (Figure 1). In ALDH2-deficient subjects, blood acetaldehyde concentrations are significantly higher even after a moderate amount of ethanol (46, 47, 246, 318, 364). One to two alcoholic beverages of any kind (or an equivalent of 0.2–0.6 g/kg ethanol) results in peak blood acetaldehyde levels of 73.7 μM ∼30 min after consumption in ALDH2*2 heterozygotes compared with 4.4 μM blood acetaldehyde levels in subjects with wild-type enzyme. Roh et al. (268) used the technique of “alcohol clamp” and demonstrated the contribution of acetaldehyde to intoxicated behaviors in social drinkers. In this study, volunteers with either wild-type ALDH2*1/*1 or ALDH2*1/*2 genotypes were infused with ethanol intravenously to maintain an identical stable breath ethanol concentration of 50 mg/dl for 165 min. Significant differences in subjective physiological responses and stimulant and sedative effects of ethanol were clearly distinguishable between the two genotypes, including increased central stimulation sensation, anesthetic sensation, and impaired function of skills and abilities in the ALDH2*1/*2 relative to wild-type subjects (268). Therefore, the physiological and behavioral consequences of ethanol consumption are due to effects of both ethanol and acetaldehyde (60).
In 6- to 8-wk free-choice ethanol and water experiments, ALDH2 knockout mice drank much less ethanol compared with the wild-type control mice (80, 128), thus mimicking the phenotype of ALDH2*2 East Asians (24). Studies in mice demonstrated that oral administration of increasing doses of ethanol from 0.5 to 5.0 g/kg in wild-type and ALDH2 knockout mice leads to significantly higher blood acetaldehyde levels in the knockout mice (127). At high ethanol doses, a significant delay in ethanol clearance was also observed in the ALDH2 knockout mice relative to wild-type mice. Furthermore, increased mortality rate was reported following an 8-day subacute administration of ethanol at 2 g·kg−1·day−1 in the mutant mice compared with the wild-type ALDH2 mice (237), suggesting that ALDH2 is a protective enzyme from ethanol-induced injury.
A single dose of 5 g/kg ethanol did not seem to induce severe oxidative stress to liver tissue in these mice, as measured by the levels of malondialdehyde or glutathione. On the contrary, the deficiency of ALDH2 was suggested to attenuate ethanol-induced oxidative stress in rodent liver tissue (201). The mechanism of this attenuation in ALDH2 knockout mice is likely to be mediated by downregulation of CYP2E1, which is a known oxidative stress generator by ethanol (202). In a separate chronic ethanol study, which subjected the mice to 5 wk of free-drinking ethanol solution, ALDH2 knockout mice showed lower levels of injury markers of malondialdehyde, tumor necrosis factor (TNF)-α, and alanine aminotransferase (ALT) in their serum, compared with wild-type mice (203). However, there was increased ballooning of hepatocytes and DNA adducts of N2-ethylidene-2′-deoxyguanosine in hepatocytes and gastric tissues, indicating a significant increase in the hepatic and gastric damage in the ALDH2 knockout mice (198, 203, 216). Increased levels of DNA adducts were similarly detected in liver, stomach, and kidney of ALDH2 knockout mice compared with wild-type mice when using radioactive-labeled ethanol in the study (229). Therefore, both protective and injurious effects on the liver and the gut appeared to be observed in ALDH2 knockout mice following ethanol administration. It is not known whether different genetic backgrounds of these transgenic mice and/or activation of other enzymes contribute to the differences in these studies.
Hypersensitivity to acetaldehyde vapor was also demonstrated in ALDH2 knockout mice when exposed to vapor inhalation (129). Blood acetaldehyde levels were 2- to 3.2-fold higher in ALDH2 knockout mice compared with wild-type mice, when measured after 4 h of inhaling acetaldehyde at a concentration of 5,000 ppm. Concomitantly, behavioral and physiological symptoms of augmented acute acetaldehyde toxicity, such as staggering gaits, prone position, hypoactivity, abnormal deep breathing, and dyspnea, were observed in the ALDH2 knockout mice relative to wild-type mice (129). These symptoms of acetaldehyde toxicity were reproduced by a single injection of a sublethal dose of acetaldehyde. In a study of intraperitoneal injection of 400 mg/kg acetaldehyde, ALDH2 deficiency delayed the recovery of physiological and behavioral symptoms and the elimination of acetaldehyde compared with wild-type mice (130). Interestingly, mitochondrial liver homogenates derived from these ALDH2 knockout mice also showed a dramatic deficiency in catalytic activity toward 2-methoxyacetaldehyde, a major metabolite of toxic 2-methoxyethanol (ethylene glycol monoethyl ether), which is commonly used as a water-soluble solvent in the chemical industry with known adverse reproductive and central nervous system effects in factory workers (147). These data confirm that ALDH2 is the major acetaldehyde-metabolizing enzyme in mice and that deficiency in ALDH2 leads to elevated acetaldehyde levels after ethanol intake, and to increased susceptibility to prolonged exposure to acetaldehyde and other toxic aldehydes. They also highlight the potential health risks of toxic aldehydes for human carriers of ALDH2*2 relative to the rest of the population.
Acute exposure to acetaldehyde causes much of the unpleasant effects associated with ethanol intoxication, including nausea, palpitations, vomiting, dizziness, and headaches (24). These negative effects of acetaldehyde are also deterrents, preventing reinstatement of alcoholism. After excessive drinking was interrupted, the drug disulfiram, an inhibitor of ALDH2, is used to trigger aversion to ethanol consumption (297), as it causes acute acetaldehyde accumulation and the corresponding unpleasant effects. Disulfiram's effects in the presence of ethanol are not different from the Asian Acetaldehyde (Flushing)-Toxicity Syndrome, which results from the inactivating point mutation in ALDH2 (ALDH2*2) (24). The finding that alcoholism was less common in ALDH2*1/*2 and is nonexistent in ALDH2*2/*2 individuals led to the notion that acetaldehyde is a strong deterrent from ethanol drinking (39, 114, 115). However, social pressure and changes in the work and life-style of urban East Asians led to a greatly increased incidence of alcoholism in this population. Higuchi reported a significant increase of alcoholics in Japan from 2.5 to 13% in the ALDH2*1/*2 heterozygous group from 1979 to 1992 (114). In a more recent study conducted in Japan in 2002, 26% of the heavy drinkers (>400 g ethanol consumption per week) in the Tokyo area were ALDH2*1/*2 heterozygotes (356); ALDH2*1/*2 in the general population is ∼30%. A similar trend was observed in Taiwan, where 17% of alcoholics were found to carry the ALDH2*1/*2 heterozygous genotype in 1999 (39). As will be discussed later, ALDH2*1/*2 subjects that drink excessively are at a much great risk of developing esophageal and other upper aerodigestive track cancers, a likely result of DNA damaging effects of acetaldehyde.
2. Alcoholism
Another mechanism by which ALDH2 inhibitors can be beneficial for the treatment of alcoholism has been recently reported. A study by Yao et al. (354) suggests that alcoholism may be induced, in part, by chemical adduction of dopamine, and its monoamine oxidase aldehyde metabolite DOPAL, to form tetrahydro-papaveroline (THP) in the neurons of the ventral tagmental area. Inhibition of ALDH2 leads to an increase of DOPAL and hence accumulation of THP, which selectively decreased dopamine release and hence led to inhibition of pro-addiction reward signaling through a negative-feedback loop (7, 354). As previously mentioned, CVT-10216 is a potent ALDH2 inhibitor with IC50 of ∼30 nM. In these studies, CVT-10216 prevented the increase of alcohol-induced rewarding dopamine release in brain in the absence of acetaldehyde (7, 235). Because of this unique property, CVT-10216 may therefore be developed as a drug lead for alcoholism. In an animal model of drug addiction, CVT-10216 also suppressed cocaine self-administration and prevented relapse-like behavior, possibly by a similar ALDH2 inhibition-mediated increase of THP via DOPAL and dopamine condensation (354).
These data demonstrate that acetaldehyde has a clear role in the intoxication associated with ethanol ingestion and that a neuronal-derived ALDH2 substrate, DOPAL, may play a direct role in addiction (332). Inhibiting ALDH2 (e.g., by disulfiram) provides ethanol aversion therapy, whereas selective inhibition of ALDH2 by compounds, such as CVT-10216, in the brain may prevent activation of the addictive behavior for ethanol and other drugs of abuse. However, care should be given not to affect ALDH2 in the liver, as acetaldehyde accumulation causes a number of other pathologies, as discussed in this review. Further consideration should be given to the role of ALDH2 in metabolizing aldehydes such as DOPAL and 4-HNE. As we discussed earlier, these aldehydes have been connected with progression of neurodegenerative diseases, such as Parkinson's and Alzheimer's diseases.
3. Fetal alcohol syndrome
Finally, a discussion about alcohol and acetaldehyde must include a very important consequence of acetaldehyde accumulation on early embryonic development. It is well-recognized that exposure to ethanol during the first trimester of embryo development can lead to severe developmental disorders, including craniofacial abnormalities, behavioral and intellectual deficits (collectively referred to as fetal alcohol syndrome), as well as to fetal death (105, 253). Although most of these features were attributed to exposure to ethanol's effects at a critical stage of embryonic development, more recent work suggests that these abnormalities relate directly to acetaldehyde toxicity and DNA damage during this critical developmental period. Exciting new observations highlight a similarity between fetal alcohol syndrome, acute childhood leukemia (164, 190), and Fanconi anemia (161) and demonstrate a role of acetaldehyde in these pathologies. The role of acetaldehyde toxicity in early embryo development will be discussed more in the section related to Fanconi anemia.
E. Upper Aerodigestive Track Cancer
Chronic alcohol (ethanol) consumption is a prominent cause of morbidity and mortality worldwide. The amount of ethanol consumption has remained relatively stable in most parts of the world over the past 40 years; e.g., 6.8 and 6.5 liters of pure ethanol are consumed per capita in the United States and Japan, respectively (344). The only region in which there is a significant increase in ethanol consumption is the Western Pacific; there is about a fivefold increase in ethanol consumption in China alone (11, 338). This rise in numbers in the Western Pacific is alarming, since ∼3.6% of all cancers and ∼3.5% of all cancer death are attributable to ethanol drinking based on data from 2002 (18). As discussed above, alcoholic beverages have been classified as group 1 human carcinogens by IARC, and chronic consumption of ethanol has been causally linked to the development of upper aerodigestive track (UADT) cancers, liver cancer, colorectal cancer, and breast cancer in women (11, 32).
UADT cancers include the cancers of the head and neck region (oral cavity, oropharynx, hypopharynx, larynx) and esophageal cancers. About 600,000 new cases of neck cancer occur each year worldwide. Only 40–50% of these patients survive 5 years after diagnosis, thus leading to annual deaths of 270,000 from these cancers (119, 169). Esophageal cancer alone is the eighth most common and the sixth most deadly cancer in the world (54, 243, 304, 315). Esophageal neoplasms are often undetected. Therefore, more than 50% of the patients have unresectable or metastatic tumors at the time of diagnosis (71). Acetaldehyde, the toxic metabolite of alcohol, forms DNA-DNA and DNA-protein crosslinks. In many experimental animal models, acetaldehyde has been proven unequivocally to be a carcinogen (209, 345, 346) and is the most likely culprit of DNA damage in the development of UADT tumors (17, 278). Yokoyama et al. (357) first reported a dramatic 7- to 12-fold increase of esophageal cancer risk in both alcoholic and nonalcoholic ALDH2*2 carrier drinkers than their respective wild-type ALDH2 controls. Many other studies from Japan, Taiwan, and China have since overwhelmingly confirmed the significant association between ALDH2 enzyme deficiency and UADT cancer risk (8, 64, 167, 172, 214, 352, 361, 362). Reviews on many primary articles and meta-analyses consistently found ALDH2*2 as one of the most common genetic polymorphisms involved in UADT cancers (30, 119). Considering that cigarette smoke is also a source of acetaldehyde, it is not surprising that tobacco use and heavy drinking combined with the ALDH2*2 genotype carry the greatest cancer risk (167, 207); indeed, ALDH2*2 carriers are the youngest patients with esophageal cancer.
Both blood and saliva acetaldehyde concentrations are significantly higher in ALDH2*2 subjects after even a moderate amount (0.2–0.6 g/kg or 1–2 drinks) of ethanol consumption (46, 47, 246, 318, 364). Salaspuro et al. (276) demonstrated the interaction between alcohol drinking and smoking and their effect on the concentration of acetaldehyde in saliva, in vivo. When given the same amount of ethanol, acetaldehyde concentration was found to be two times higher in smokers than in nonsmokers. Furthermore, with concomitant cigarette smoking during ethanol challenge, smokers had seven times higher saliva acetaldehyde concentration compared with nonsmokers (276). A synergistic effect of increased cancer risks associated with ethanol consumption and smoking has been reported for oral cavity, pharynx, larynx, and esophageal cancers (33, 34, 87, 166, 207, 208, 275). Heavy ethanol consumption and smoking in subjects carrying the ALDH2*2 genotype contribute to probably one of the highest known cancer risks (odds ratio of 50.1) (207) and up to a 25-yr earlier onset of carcinoma (45 vs. 70 yr old) compared with nonsmoker, nondrinker wild-type ALDH2 subjects (24, 167, 207). The recognition of the clear association between ALDH2 enzyme deficiency, acetaldehyde toxicity, and the risk of UADT cancer and other cancers cannot therefore be overemphasized (24, 279).
The molecular mechanism underlying acetaldehyde-induced UADT cancers in ALDH2*2 subjects has been actively studied (310, 370). There is direct evidence showing increased DNA modification and chromosomal damage in ALDH2*2 individuals relative to subjects with active ALDH2 after they consume the same amount of alcohol (200). Matusta et al. (200) identified three acetaldehyde-derived DNA adducts that are significantly higher in blood lymphocyte samples of 44 alcoholic patients. One of the acetaldehyde-modified DNA adducts, N(2)-ethyl-2′-deoxyguanosine (200), was identical to its more stable precursor, N(2)-ethylidene-2′-deoxyguanosine, detected in gastric and hepatic samples of ALDH2 knockout mice fed with 20% ethanol for 5 wk (198, 216). N(2)-ethyl-2′-deoxyguanosine is a known blocker of translesion DNA synthesis and may contribute to the hindrance of DNA repair (199, 288, 316). Chromosomal damage or genome instability in ALDH2*2 subjects who consume ethanol were also measured by biomarkers such as cytokinesis-block micronucleus assay (126) or by alkaline comet assay using peripheral blood lymphocytes from the ALDH2*2 subjects (287, 333). Blocked micronuclei frequency was significantly higher in the heavy or moderate alcohol drinkers with the ALDH2*2 allele compared with their ALDH2*1 wild-type counterparts (126). Similarly, using the alkaline comet assay to measure the mobility of DNA as an index of DNA damage in peripheral mononuclear cells in a study of 122 healthy volunteers, increased DNA damage was noted in ALDH2*2 subjects who were frequent alcohol drinkers (333). Although these data paint a clear connection between ethanol consumption, ALDH2 activity, and cancer, much research is still required to identify key genetic and epigenetic changes, which contribute to the high incidence of UADT cancers among the ALDH2*2 populations.
In recent years, UADT cancer prevention has become a public health priority in East Asian countries such as Japan and Taiwan. New measures were taken in these countries. These include identifying high risk groups at the early stage of disease, screening by image-enhanced endoscopy, oral examinations, the use of alcohol flushing questionnaires to identify subjects that carry the ALDH2*2 gene, and intensive surveillance for secondary recurrence in those who were diagnosed and treated for the disease (54, 207, 363). Intensive efforts are also underway to develop better biomarkers and diagnostics for UADT cancers (54). Considering that ALDH2*2 is a very common mutation in all East Asian countries and that ALDH2 genotype is a very strong predictor of multiple squamous cell carcinoma in UADT, it is warranted that the determination of ALDH2 genotype should be included in the overall screening and preventive strategies for UADT cancers. Alternatively, since elimination of primary risk factors (e.g., smoking and drinking) remains a difficult obstacle to overcome, enhancing the ALDH2 activity by its activators in ALDH2*2 enzyme-deficient individuals may help to reduce the exposure of acetaldehyde genotoxicity from saliva and in the UADT region in this very high risk East Asian group.
F. Radiation Dermatitis
Ionized radiation used as a treatment for solid tumors often causes burnlike injury to the skin due to cellular oxidative stress. This painful and debilitating side effect occurs in ∼95% of patients receiving radiation therapy for cancer, and ranges in severity from mild erythema to severe ulceration (274). Although intensity-modulated radiotherapy enables better sparing of normal tissues, this burnlike skin reaction is unavoidable (13). Meta-analysis of 20 clinical trials involving ∼3,000 patients demonstrated that both prophylactic and treatment trials to prevent radiation dermatitis failed when using a variety of topical agents (188). The trials analyzed in that meta-analysis study included use of topical agents such as mometasone furoate, beta-methasone, and beclomethasone (all anti-inflammatory corticosteroids); Gentian violet (a topical antiseptic agent); urea, Lianbai liquid, and aloe vera (traditional medicine remedies with possible antioxidative stress properties); hyaluronic acid, Biafine, dexpanthenol, and Sucralfate (agents that induce wound barrier and/or are moisturizers); and Trolamine (a barrier and analgesic agent) (188). The failure of all these clinical trials emphasizes that radiation dermatitis remains an unmet clinical problem in cancer patients undergoing radiation therapy.
Ionizing radiation causes a robust production of reactive oxygen species (341), which then triggers an inflammatory response as well as a local cell proliferative response, especially keratinocytes. Since anti-inflammatory agents appear to provide little benefit (see above), the source of injury lies elsewhere, perhaps an increased aldehydic load. Early studies attributed the sustained radiation-induced skin injuries to increased levels of aldehydes, such as 4-HNE and MDA, the lipid peroxidation products (2, 58). Therefore, prophylaxis and treatment with agents that prevent aldehyde accumulation, such as activators of ALDH2, may be beneficial for these patients.
In a proof-of-concept mouse model, we found that direct topical application of Alda-1 on the skin of irradiated animals effectively delayed the onset and reduced the severity of radiation dermatitis. This Alda-1-induced protection correlated with increase in ALDH2 activity, a concomitant reduction in epidermal thickness, and a decrease in 4-HNE-adduct formation in irradiated skin biopsy samples from these mice (224). Importantly, we found that Alda-1 did not protect solid tumors in mice against radiation therapy. It is conceivable that the skin protection offered by ALDH2 activation may be extended to other damage caused by reactive oxygen species-generating radiations such as ultraviolet and radioactive nuclear exposure. Furthermore, whether the severity of the response of East Asian carriers of ALDH2*2 to the damaging ionizing radiation is higher than those with wild-type ALDH2 remains to be determined.
G. Fanconi Anemia
Fanconi anemia (FA) is a rare genetic disease, first discovered by Guido Fanconi, a Swiss Physician, in 1927. FA is characterized by a progressive bone marrow failure (BMF) accompanied by increased risk of cancer as well as developmental abnormalities, such as short stature and malformations of the thumbs, forearms, skeletal system, eyes, kidneys, and urinary tract. In addition to hematological abnormalities, FA is clinically diagnosed by the presence of chromosomal breakage, and by hypersensitivity to DNA cross-linking agents such as mitomycin C, diepoxybutane, and cisplatin (140, 141). No effective treatment is currently available for this rare genetic disorder, which occurs in ∼1/350,000 individuals in all ethnic groups (10, 321).
In the United States, ∼31 newborns are diagnosed with Fanconi anemia (FA) each year (270). Based on International Fanconi Anemia Registry, the average life expectancy of FA patients is 29 years. With blood transfusions, many patients can live at times twice as long as life expectancy. FA is caused by homozygous recessive mutations in any of the 15 identified FA genes (FNAC genes), which encode proteins that serve an essential function in DNA repair of damages caused by DNA cross-linking agents (206). The FA gene products play a crucial role in guarding the integrity of the human genome; mutations in any of the FA genes invariably lead to catastrophic consequences of genomic instability due to failure to repair DNA crosslinking damage.
Until recently, the source and chemical agents that cause excessive genomic instability leading to the phenotype of developmental abnormality, BMF, and predisposition to malignancy in FA patients have been elusive. Environmental pollutants, carcinogens, and biogenic reactive chemical species that are capable of attacking DNA and causing genome instability under physiological conditions were the prime suspects of the molecular triggers of FA. Reactive aldehydes are known toxic molecules that can damage DNA by forming DNA-protein or DNA-DNA interstrand crosslinking (53, 61, 156, 296, 309). Ridpath et al. (262) first reported the genotoxic effect of formaldehyde in cells that are deficient in the FANC/BRCA pathway. In a chicken B lymphocyte-derived cell line, DT40, hypersensitivity to low micromolar concentrations of formaldehyde was demonstrated in isogenic DT40 cell lines carrying mutations in several DNA repair genes including FANCD2, FANCD1 (better known as BRCA2, which plays a critical role in genetic forms of breast cancer). In these DT40 mutant cell lines, cell survival was dramatically reduced by exposure to low concentrations of formaldehyde compared with survival of the parental wild-type DT40 cells under the same conditions (262). Hypersensitivity to formaldehyde was similarly shown in human colorectal RKO cancer cells in which the FANCC and FANCG genes were disrupted. These findings suggest that exposure to formaldehyde at levels observed in human plasma (13–97 μM) (113) could cause severe cellular damage due to failure of DNA repair. Interestingly, acetaldehyde, but not other aldehydes such as acrolein, crotonaldehyde, glyoxal, or methylglyoxal, also caused hypersensitivity in a FANCD2-deficient DT40 cell line (262). Endogenous formaldehyde is known to be generated within the nuclei as a by-product of histone demethylation or dealkylation of methylated DNA (210, 272). In vertebrate cells, alcohol dehydrogenase 5 (ADH5) is the major dehydrogenase that metabolizes formaldehyde and protects the toxic effect of formaldehyde (124). With the use of a cell line with ADH5 knockout, it was demonstrated that deficiency in FA DNA-repair pathway became lethal, presumably due the accumulation of endogenous formaldehyde (269).
Chronic exposure to low concentration of acetaldehyde vapor at 125 and 500 ppm for 6–12 days led to a significant elevation of the DNA damage marker 8-hydroxydeoxyguanosine (8-OHdG) in urine samples of the ALDH2 knockout mice (228). As discussed earlier, acetaldehyde, commonly found in many food sources (81), environmental pollutants (121, 311), cigarette smoke (264), or as a metabolite of drinking alcohol, can also react in cells to generate interstrand DNA crosslinks (309). Acetaldehyde can also be generated as a by-product of endogenous metabolism from alanine by myeloperoxidase (112) or from deoxyribose phosphate by aldolase (226). Marietta et al. (196) showed that exposure to acetaldehyde at biologically relevant concentrations (0.1–1 mM) resulted in concentration-dependent FANCD2 mono-ubiquitination, in phosphorylation of the FANC gene product BRCA1, and in appearance of a hallmark of DNA damage, histone γH2AX phosphorylation, in human lymphoblastoid cells.
Using a genetic approach, Langevin et al. (161) showed that maternal ALDH2 is essential to counteract the acetaldehyde toxicity for the normal development of FANCD2 knock-out embryos. Mouse embryos that carried double knockout deficiencies, ALDH2−/− and FANCD2−/−, were not viable, unless they were protected by at least one copy of a functional maternal ALDH2 allele of a ALDH2+/− females. Moreover, these rescued double mutant ALDH2−/− and FANCD2−/− embryos were hypersensitive to developmental deformities in the uterus if the mothers were exposed to ethanol. In the postnatal stage, the ALDH2−/− and FANCD2−/− mice rapidly developed bone marrow failure when ethanol was administered in the drinking water. Incidences of spontaneously acute lymphoblastic leukemia were also observed in mice 3–5 months after weaning in the absence of ethanol exposure. Importantly, in adult ALDH2−/− and FANCD2−/− mice that were spared of leukemia, the hematopoietic stem and progenitor cells, and not the more mature blood precursors, appeared to be the most sensitive to acetaldehyde-induced DNA damage (95). There was a more than 600-fold reduction in the hematopoietic stem cell pool in these mice that are defective in both acetaldehyde detoxification and the FA DNA repair pathway. These ground-breaking studies established a molecular link between toxic acetaldehyde and DNA damage in FA (Figure 6). These findings also underlie the crucial role of ALDH2 activity during embryogenesis and in protecting bone marrow hematopoietic stem cells. Finally, as briefly discussed earlier, because many of the developmental abnormalities observed in FA patients are similar to those with fetal alcohol syndrome, aldehyde-induced DNA damage may underlie both pathologies (Figure 6).
FIGURE 6.
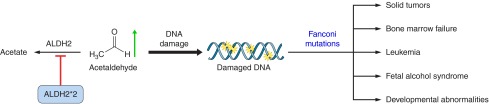
ALDH2 and Fanconi anemia. In a mouse model of Fanconi anemia (FANCD), ALDH2 is essential in counteracting acetaldehyde toxicity. In the absence of a functional ALDH2, DNA damage triggered by elevated acetaldehyde toxicity cannot be repaired efficiently due to Fanconi mutations. Accumulation of such unrepaired damaged DNA leads to the manifestation of multiple pathological conditions and health complications seen also in patients with Fanconi anemia (95, 161, 244).
Exactly what types of xenogenic or biogenic reactive aldehydes that may serve as potent physiological triggers for different pathological features of FA remain to be identified (244). Regardless, the relationship between acetaldehyde toxicity and DNA damage is likely to be especially relevant to the health of East Asian FA patients who carry the ALDH2*2 mutation (Figure 6).
It is of interest to note that in adult FA patients, upper aerodigestive track cancers or head and neck cancers are the most common cancer types (4, 157). These are the very same cancer types that are most prevalent in the ALDH2*2 East Asians who are exposed to the high level of acetaldehyde genotoxicity due to heavy alcohol drinking (359–361). Significant numbers of FA patients have been reported in ALDH2*2 in Japan and Korea (240, 350). However, very few cases of FA have been reported in China or Taiwan, where ALDH2*2 mutation accounts for a very large percentage of the population (i.e., >40% in Taiwan). It is possible that in these ALDH2*2 prevalent countries, germline FA, or non-germline FA mutations are under-diagnosed. Alternatively, human embryos that have both a mutated FA gene and mutated ALDH2 gene may die at early embryonic stage, thus reducing the FA incidence seen in children and adults in these countries.
For both clinical practice and research, it will be important to explore the connection (if any) between ALDH2 genotype status and FA patients who are from the East Asian group. It will be equally important to determine if there is a correlation between ALDH2*2 mutation and FA disease severity and prognosis. Such basic information may help guide future treatment options. Enhancing the function of ALDH2 by either gene regulation or direct enzyme activation may conceivably provide the needed clinical intervention for FA patients who are hypersensitive to reactive aldehydes. This may be especially critical for FA patients who are also ALDH2*2 carriers, but should be relevant to the general population, too.
H. Pain
Therapeutic agents that target ALDH2 may also provide a viable strategy to address the national health problem of pain, resulting in approximately $50 billion annually in health care cost and productivity loss (109). Recent studies suggest that the ability to reduce aldehydic load may address a broad range of pain pathologies, including peripheral neuropathy and perhaps even acute and chronic inflammatory pain.
1. Peripheral neuropathy
Peripheral neuropathy, characterized by allodynia (pain in response to a nonnociceptive stimulus) and hyperalgesia (increased response to painful stimuli), is present in ∼24 million people. More than 100 causes of peripheral neuropathy have been identified, and peripheral neuropathy is idiopathic in ∼33% of the patients (286). Diabetic neuropathy is likely the most common cause of this pathology. Relevant to this review, ethanol, chemotherapeutic agents, and the ALDH2 inhibitor disulfiram are also documented causes of peripheral neuropathy. As discussed in the following, pharmacological and genetic evidence suggests that reducing ALDH2 enzymatic activity may contribute to peripheral neuropathy, whereas ALDH2 activators may perhaps help ameliorate pain.
One side effect of disulfiram in humans is peripheral neuropathy, which can be reversed by stopping or reducing the disulfiram dose (83, 89). The largest case report of 37 patients with disulfiram-induced neuropathy showed that the effect is dose-dependent, with doses greater than 250 mg/day being the greatest factor in causing neuropathic symptoms (89). Since this form of neuropathy is drug induced, given that the disulfiram metabolite S-methyl-N,N-diethylthiocarbamate inhibits ALDH2 (204), these data suggest that decreased ALDH2 activity is sufficient to induce peripheral neuropathy in humans.
Indeed, some reports suggest that individual carriers of the ALDH2*2 mutation are more susceptible to developing alcohol- or diabetic-induced peripheral neuropathy (197, 303). Chronic alcoholism in ALDH2*1/*2 subjects is associated with reduced sensory nerve action potential amplitude, a characteristic of peripheral neuropathy (273), compared with chronic alcoholics with wild-type ALDH2 (197). Although this study included a small number of subjects (21 per group), the findings suggest that further studies are needed to investigate ALDH2 as a genetic cause of alcohol-induced neuropathy. A study of 158 type 2 diabetics investigated how ADH and ALDH2 activity altered diabetic clinical symptoms, including neuropathy (303). Among the patients studied, although hemoglobin A1C, age, and weight were similar between groups, patients with an ALDH2*2 mutation with normal ADH activity had the highest incidence of peripheral neuropathy (16 of 32 people or 50%). The lowest incidence of neuropathy occurred in those patients who had normal ADH and ALDH2 activity (8 of 44 or 18.2%). Although an increase in ADH activity was also considered in this study to contribute to diabetic pathology, reanalyzing the groups in this study without considering ADH activity suggests that an inactive ALDH2 enzyme is correlated with a higher incidence of diabetic-induced neuropathy (36 vs. 26%).
Studies regarding ethnicity and neuropathy prevalence are at best inconclusive (292). Although in both alcohol- and diabetes-induced neuropathy studies the number of patients was small, these findings, in addition to the evidence that disulfiram can induce peripheral neuropathy, suggest that further studies are needed to investigate ALDH2 as a genetic contributor to neuropathy.
2. Inflammatory pain
As compared with other ethnic populations, Asians are more sensitive to painful stimuli such as skin irritation (271), capsaicin treatment (325), exposure to heat (329), cold (122), or mechanical pressure (343). These differences are attributed primarily to social or cultural factors (37, 56). However, the high prevalence of the ALDH2*2 inactivating mutation among East Asians [∼36% on average (24) and in some regions as high as 45% (70)] suggests that increased pain sensitivity in Asians may be attributed to the ALDH2*2 genotype. We recently tested this hypothesis in our laboratory using a C57/BL6 mouse with a knock-in mutation for the ALDH2*2 gene and found that male ALDH2*1/*2 heterozygotic mice subjected to carrageenan-induced inflammation were more hyperalgesic compared with matched male littermates with ALDH2*1/*1 (wild-type) genotype (unpublished data). Although more studies are needed, our findings suggest that ALDH2 activity may regulate inflammatory-induced pain sensation. If correct, restoring the ALDH2 activity in East Asians in particular and increasing ALDH2 activity for subjects with a wild-type ALDH2 could provide a novel means to treat pain.
I. Osteoporosis
Osteoporosis is a disease characterized by decreased bone mineral density, bone mass, and bone strength, which leads to increased risk of bone fracture. Osteoporotic fractures are associated with high morbidity and mortality, as well as a high burden of health care costs, especially with the aging population worldwide (57). There are multiple causes for osteoporosis, including environmental, dietary, and genetic factors. Relevant to this review, excess alcohol consumption and oxidative stress are associated with osteoporosis, decreased bone density, and increased fracture incidence (63, 91, 137, 165, 191); heavy ethanol drinking was found to be a significant risk factor for increased hip fracture and vertebrate deformation in large cohorts of Japanese and Chinese women (91, 165).
In humans, both bone mineral density and osteoporotic fractures have high genetic determinants (180, 349). In a genetic screening of osteoporosis and DNA polymorphism among 403 elderly Japanese, ALDH2*2 was the only mutation that was associated with osteoporosis among 11 other known osteoporosis-related genes (351). This association is particularly pronounced in individuals with the homozygous ALDH2*2/*2 genotype and in women.
In bone marrow cultures derived from rodents, treatment with ethanol and acetaldehyde greatly inhibits early osteoblast progenitor formation (98). The inhibitory effect of ethanol was observed at the physiological concentrations of 0.04–0.6%, similar to those reached in humans. Acetaldehyde exerted an even stronger toxic effect toward osteoblastic progenitor cells, at concentrations as low as 0.004 to 0.02%. At 0.06% acetaldehyde, pluripotent colony-forming units for osteoblast progenitors were completely abolished in cultured murine cells. Acetaldehyde also inhibited cell proliferation in two human osteoblastic cell lines. These observations were also confirmed in human bone marrow cell cultures derived from young, chronic alcoholics (98); the number of colony-forming units for osteoblast progenitors obtained from nine alcoholics was reduced to only ∼30% compared with cells derived from seven age-matched normal control subjects. These data demonstrate a direct effect of both ethanol and acetaldehyde on the formation of osteoblastic progenitor cells and on bone marrow cell proliferation. Taken together, these studies provide strong evidence that the ALDH2*2 mutation in East Asians may be a contributing risk factor to osteoporosis, due to the lack of enzyme protection against aldehyde toxicity for osteoblasts and their progenitor cells. Furthermore, activators of ALDH2 in these patients, and in the general population as well, may prevent or slow down the progression of osteoporosis.
The notion of ALDH2 as a therapeutic target for osteoporosis is supported by two separate animal models of ALDH2 knockout and ALDH2*2 overexpressing transgenic mice. In ALDH2 knockout mice, administration of 5% ethanol for 4 wk resulted in a reduction of trabecular bone and bone volume formation, with a concomitant decrease in gene expression of osteoblast markers, type I collagen, osterix, osteopontin, and osteocalcin (284). Mineralized nodule formation was also decreased in bone marrow cell cultures derived from the ethanol-fed ALDH2 knockout mice. No such changes were observed in ethanol-fed wild-type ALDH2 mice. Independently, in a model of ALDH2*2 overexpressing transgenic mice that exhibited a dominant-negative effect on the endogenous ALDH2, Endo et al. (80) observed phenotypes of small body size, reduced muscle mass and fat, osteopenia, and kyphosis, that are related to osteoporosis. Reduced bone mineral density was confirmed in these ALDH2 transgenic mice by dual-energy X-ray absorptiometry (120). In these ALDH2*2 osteoporotic mice, elevated blood acetaldehyde levels were also detected even in the absence of ethanol ingestion. The severity of osteoporosis also correlated with the level of ALDH2*2 expression, depending on different copy numbers of the inserted gene. Furthermore, acetaldehyde treatment at a concentration as low as 0.04% effectively inhibited proliferation and induced apoptosis in osteoblast primary cultures of wild-type osteoblastic cells or the osteoblast cell line MC3T3-E1. Expression of either ALDH2*2 or treatment with acetaldehyde resulted in excessive accumulation of 4-HNE and increased PPARγ expression, a transcription factor that inhibits osteoblastogenesis. These studies indicated that aldehydic stress resulting from ALDH2*2 mutation leads to impaired osteoblastogenesis and osteoporosis. Independently, severe ethanol-induced suppression of osteoblast differentiation, mineralized nodule formation, and osteogenic gene expressions that led to osteopenia was also observed in ALDH2 knockout mice (284). This is in agreement with the hypothesis that ALDH2 protects normal development and differentiation of osteoblastic stem cells in the bone marrow and maintenance of bone homeostasis.
Interestingly, in a genome-wide association study involving 11,500 individuals from well-defined and homogeneous populations, Guo et al. (108) recently identified a SNP in ALDH7A1 as a highly significant susceptible gene for osteoporosis. These studies provided insights into the crucial function of ALDH in protecting osteoblasts against toxic aldehydes, oxidative stress, and perhaps maintaining proper osmotic pressure and homeostasis for osteoblastic bone marrow cells.
J. Aging
Denham Harman in 1956 proposed the free radical theory of aging, suggesting that oxygen free radicals cause the aging process (111). This theory is also supported by the premature aging disease Hutchinson Gilford progeria, where the average human life span is only ∼13 yr. Fibroblasts of progeria patients have a 1.6-fold higher fluorescence emission measured by intracellular oxidation of 2,7-di-chlorofluorescein (DCF) and a 1.7- to 4-fold increase in carbonylated proteins compared with age-matched control fibroblasts (322). Moreover, in 2007, an inverse correlation was found in 12 species between the rate of hydrogen peroxide (H2O2) generation by the myocardium mitochondria and maximum life span (160). This inverse correlation (R2 = 0.54) existed when measuring H2O2 formation by reverse electron transfer from succinate to complex I in diverse species. In turn, therapeutics that reduce mitochondrial reactive oxygen species generation are considered potential means to reduce aging-induced damage, and mitochondrial targeted anti-oxidants, such as SkQ1, have been under development in an attempt to slow down the aging process (289). As discussed earlier, reactive oxygen generation triggers lipid peroxidation and accumulation of reactive aldehydes (73, 194).
Protecting the mitochondria from reactive oxygen species may increase life span. Decrease in mitochondrial detoxification of lipid peroxidative aldehydes has been observed and proposed as an underlying mechanism of aging in animals (231, 369). It was shown in rats that the capacity of 4-HNE removal by mitochondrial ALDH declines with aging in rats (42). Therefore, ALDH2 activation may slow down, at least to some extent, the aging process. To that end, specifically targeting mitochondrial ALDH2 may reduce or prevent many biological effects associated with free radical-induced aging. Indeed, deleting the ALDH2 homolog in Drosophila reduced survival and longevity when flies were challenged with hyperoxia or with starvation (35). There was also a reduced reproductive rate and increased protein carbonyl level in flies in which ALDH2 was deleted compared with wild-type ALDH2 flies. Consistent with these findings, transgenic mice that overexpress ALDH2*2 (called DAL101) showed a significant reduction in median life span compared with control mice; their life span was 96 wk compared with 126 wk for the wild-type mice. In addition, the ALDH2*2 mice showed a greater muscle weakness and hair discoloration in males (231).
In humans, a high throughput candidate gene study in 137 Koreans over 90 yr old also found that the ALDH2 gene may predict longevity (241). This study suggested, however, that the ALDH2*2 genotype, not the wild-type, was associated with living longer for male Koreans. Contrary to this suggestion, a study conducted in Japan involving more than 2,200 human subjects showed that the frequency of ALDH2*2 homozygous genotype was significantly reduced in females in the 60–70s age group versus 40–50s group (230). No age-related difference in stomach and colon ALDH activity was found in human biopsy samples (159, 355). However, Wang et al. (327) did observe a slight decline in liver ALDH activity in older human subjects. The rate of loss in telomeric repeat sequences in human peripheral leukocytes, as a cellular marker of senescence, has been well correlated with aging (88). It may be warranted for future research to measure the telomere length among the ALDH2*2 individuals and compare that to the wild-type individuals. Whether therapeutic agents that activate ALDH2 are beneficial in diseases of accelerated aging, such as progeria, will help determine the role of aldehydic load in aging. Even if association between ALDH2*2 and life span is not found in humans, we should examine the association between ALDH2 activity and aging-associated damage; in light of the number of studies linking the ADLH2*2 genotype and aldehydic load to many diseases that shorten life expectancy (Table 3), an association between aging-associated damage and ALDH2 function is expected.
Table 3.
Role of ALDH2 in aging
| Findings Supporting An Active and Healthy ALDH2 May Improve Longevity | Reference Nos. |
|---|---|
| Reduces myocardial injury | 25, 40 |
| Reduces cardiac arrhythmias | 149 |
| ALDH2*2 increases cancer risk | 355 |
| ALDH2*2 increases osteoporosis risk | 349 |
| Improves radiation-induced dermatitis | 223 |
| ALDH2*2 increases peripheral neuropathy risk | 302 |
| Increases neuronal survival from reactive aldehyde-induced toxicity | 167 |
| ALDH2*2 increases DNA damage, including mitochondrial DNA | 227, 301 |
| ALDH2 knockout mice have a shortened lifespan | 230 |
V. CONCLUDING REMARKS
As discussed here, ALDH2 is a well-known member of 19 different aldehyde dehydrogenases that are present in humans. Much of what we knew about ALDH2 stemmed from an interest in ethanol metabolism. However, an exciting body of more recent research indicates that this enzyme provides an important shield from oxidative stress and increased aldehydic load, thus contributing to better health. Many human diseases are associated with increased aldehydic load. In fact, positive thiobarbituric acid reactive substance (TBARS) assay, measuring aldehydes in the blood, which was a standard clinical test to determine inflammation and other general stresses, correlates with severity of many human diseases (51, 123, 282). Accordingly, are aldehydes a marker of cellular stress in a variety of diseases or do they actually contribute to these human pathologies?
In this review, we examined the evidence from animal studies and from human epidemiological studies; we believe that an important new picture emerges. It appears that aldehydes are major contributors to the pathology of a variety of human diseases. From developmental abnormalities, such as Fanconi anemia, to diseases often associated with life-style, including UADT cancer, diabetes, and heart disease; as well as diseases associated with aging, including Alzheimer's and Parkinson's disease, reducing aldehydic load appears to correspond to a better outcome. The role of other aldehyde dehydrogenases in a number of human diseases has also been documented recently (194, 195). However, much of the data strongly support a specific role for one aldehyde dehydrogenase, the ubiquitous mitochondrial ALDH2.
Why is there such a central role for the mitochondrial ALDH? Mitochondria are not only critical for energy generation. They are also major determinants in triggering both apoptotic and necrotic cell death (6, 146). Therefore, protecting mitochondrial integrity by removing toxic aldehydes that accumulate in this organelle may be critical for the cell. In particular, emerging data connecting the common ALDH2 inactivating mutation, ALDH2*2, in East Asians with certain diseases suggest that together with other factors, ALDH2 provides an important protective enzyme.
It should be noted that the inactivating mutation of ALDH2*2 is still regarded as a relatively benign mutation. ALDH2*2 was thought to cause a deficiency only in ethanol metabolism, and the resulting acetaldehyde toxicity or “Asian flushing” was thought to be of no medical consequence. Together, this unpleasant flushing and associated phenotypes after ethanol consumption were thought to be positive, as they acted as a deterrent from alcoholism. However, much recent work elucidates the danger of acetaldehyde toxicity. Acetaldehyde is a cytotoxin and carcinogen. Furthermore, ALDH2 inactivating mutation increases the risk of subjects to the damaging effects of many other biogenic and environmental aldehydes. As ALDH2*2 affects 560 million East Asians, public education and awareness are clearly needed to achieve better healthcare of the general public. In one estimate, using population case-control data from Japan for risk factor assessment, it was estimated that if moderate-to-heavy drinkers of the ALDH2*1/*2 heterozygotes were instead only light drinkers, it could reduce 53% of the esophageal cancers in Japanese man (24). With the increased emphasis on personalized medical care, it may also be an interesting question for the medical community to consider whether individual carriers of ALDH2*2 should be treated as a unique subpopulation for disease prevention and treatment.
As a last note, an intriguing biological mystery remains: What is the explanation for the existence of the widespread ALDH2*2 mutation in East Asians? How come a single monogenetic mutation that occurred ∼2,000–3,000 years ago became the most common and uniform genetic enzymopathy, affecting an estimated 8% of the human race? Could there be a selective advantage or added fitness for having this mutation? A few hypotheses have been postulated, but none has been confirmed thus far (100, 176).
Finally, based on the discussion above on the critical role of ALDH2 in cytoprotection and in how aldehydes contribute to the pathology of a variety of human diseases, there may be a role for new therapeutic interventions. One such intervention could be a class of activators of ALDH2, which increase ALDH2 catalytic activity, protect it from inactivation by its toxic substrates, and correct the structural defect in the common human ALDH2*2 mutation. Whether such ALDH2 activators will slow down the progression of some human diseases or actually prevent them and whether this class of drugs will affect life span or only the quality of life of humans will be interesting areas for both basic research and potential pharmaceutical development.
GRANTS
The work in the Mochly-Rosen laboratory was supported by National Institutes of Health (NIH) Grant R37 AA11147. E. R. Gross was supported by NIH K Award HL-109212. J. C. B. Ferreira was supported by FAPESP (2012/05765–2) and CNPq.
DISCLOSURES
D. Mochly-Rosen and C.-H. Chen are founders of ALDEA Pharmaceuticals. C.-H. Chen is also a consultant to the company.
ACKNOWLEDGMENTS
We thank July Lee and Dr. Leslie A. Cruz for providing graphic assistance. Dr. Adrienne Gordon and July Lee are acknowledged for their critical reading of and useful comments for the manuscript.
Address for reprint requests and other correspondence: D. Mochly-Rosen, Dept. of Chemical and Systems Biology, 269 Campus Dr., CCSR Building, Room 3145, Stanford University, School of Medicine, Stanford, CA 94305-5174 (e-mail: mochly@stanford.edu).
REFERENCES
- 1.Aberle NS, 2nd, Picklo MJ, Sr, Amarnath V, Ren J. Inhibition of cardiac myocyte contraction by 4-hydroxy-trans-2-nonenal. Cardiovasc Toxicol 4: 21–28, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Aldini G, Granata P, Orioli M, Santaniello E, Carini M. Detoxification of 4-hydroxynonenal (HNE) in keratinocytes: characterization of conjugated metabolites by liquid chromatography/electrospray ionization tandem mass spectrometry. J Mass Spectrom 38: 1160–1168, 2003 [DOI] [PubMed] [Google Scholar]
- 3.AlQahtani J. Screening of hemoglobin disorders in Najran Schools' Children, KSA. Nature Science 10: 56–62, 2012 [Google Scholar]
- 4.Alter BP, Joenje H, Oostra AB, Pals G. Fanconi anemia: adult head and neck cancer and hematopoietic mosaicism. Arch Otolaryngol Head Neck Surg 131: 635–639, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Ando Y, Brannstrom T, Uchida K, Nyhlin N, Nasman B, Suhr O, Yamashita T, Olsson T, El Salhy M, Uchino M, Ando M. Histochemical detection of 4-hydroxynonenal protein in Alzheimer amyloid. J Neurol Sci 156: 172–176, 1998 [DOI] [PubMed] [Google Scholar]
- 6.Apostolova N, Blas-Garcia A, Esplugues JV. Mitochondria sentencing about cellular life and death: a matter of oxidative stress. Curr Pharm Des 17: 4047–4060, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Arolfo MP, Overstreet DH, Yao L, Fan P, Lawrence AJ, Tao G, Keung WM, Vallee BL, Olive MF, Gass JT, Rubin E, Anni H, Hodge CW, Besheer J, Zablocki J, Leung K, Blackburn BK, Lange LG, Diamond I. Suppression of heavy drinking and alcohol seeking by a selective ALDH-2 inhibitor. Alcohol Clin Exp Res 33: 1935–1944, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asakage T, Yokoyama A, Haneda T, Yamazaki M, Muto M, Yokoyama T, Kato H, Igaki H, Tsujinaka T, Kumagai Y, Yokoyama M, Omori T, Watanabe H. Genetic polymorphisms of alcohol and aldehyde dehydrogenases, and drinking, smoking and diet in Japanese men with oral and pharyngeal squamous cell carcinoma. Carcinogenesis 28: 865–874, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Ascherio A, Chen H, Weisskopf MG, O'Reilly E, McCullough ML, Calle EE, Schwarzschild MA, Thun MJ. Pesticide exposure and risk for Parkinson's disease. Ann Neurol 60: 197–203, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Auerbach AD, Rogatko A, Schroeder-Kurth TM. International Fanconi Anemia Registry: relation of clinical symptoms to diepoxybutane sensitivity. Blood 73: 391–396, 1989 [PubMed] [Google Scholar]
- 11.Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, Altieri A, Cogliano V. Carcinogenicity of alcoholic beverages. Lancet Oncol 8: 292–293, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Bell RG, Smith HW. Preliminary report on clinical trials of antabuse. Can Med Assoc J 60: 286–288, 1949 [PMC free article] [PubMed] [Google Scholar]
- 13.Benomar S, Boutayeb S, Lalya I, Errihani H, Hassam B, El Gueddari BK. Treatment and prevention of acute radiation dermatitis. Cancer Radiother 14: 213–216 [DOI] [PubMed] [Google Scholar]
- 14.Beretta M, Gorren AC, Wenzl MV, Weis R, Russwurm M, Koesling D, Schmidt K, Mayer B. Characterization of the East Asian variant of aldehyde dehydrogenase-2: bioactivation of nitroglycerin and effects of Alda-1. J Biol Chem 285: 943–952, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhatnagar A. Electrophysiological effects of 4-hydroxynonenal, an aldehydic product of lipid peroxidation, on isolated rat ventricular myocytes. Circ Res 76: 293–304, 1995 [DOI] [PubMed] [Google Scholar]
- 16.Bian Y, Chen YG, Xu F, Xue L, Ji WQ, Zhang Y. The polymorphism in aldehyde dehydrogenase-2 gene is associated with elevated plasma levels of high-sensitivity C-reactive protein in the early phase of myocardial infarction. Tohoku J Exp Med 221: 107–112, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Boffetta P, Hashibe M. Alcohol and cancer. Lancet Oncol 7: 149–156, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Boffetta P, Hashibe M, La Vecchia C, Zatonski W, Rehm J. The burden of cancer attributable to alcohol drinking. Int J Cancer 119: 884–887, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Bolli R, Jeroudi MO, Patel BS, DuBose CM, Lai EK, Roberts R, McCay PB. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc Natl Acad Sci USA 86: 4695–4699, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braun T, Bober E, Singh S, Agarwal DP, Goedde HW. Evidence for a signal peptide at the amino-terminal end of human mitochondrial aldehyde dehydrogenase. FEBS Lett 215: 233–236, 1987 [DOI] [PubMed] [Google Scholar]
- 21.Braun T, Bober E, Singh S, Agarwal DP, Goedde HW. Isolation and sequence analysis of a full length cDNA clone coding for human mitochondrial aldehyde dehydrogenase. Nucleic Acids Res 15: 3179, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer's disease. Alzheimers Dement 3: 186–191, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Brooks PJ. Brain atrophy and neuronal loss in alcoholism: a role for DNA damage? Neurochem Int 37: 403–412, 2000 [DOI] [PubMed] [Google Scholar]
- 24.Brooks PJ, Enoch MA, Goldman D, Li TK, Yokoyama A. The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med 6: e50, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Budas GR, Disatnik MH, Chen CH, Mochly-Rosen D. Activation of aldehyde dehydrogenase 2 (ALDH2) confers cardioprotection in protein kinase C epsilon (PKCvarepsilon) knockout mice. J Mol Cell Cardiol 48: 757–764, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Budas GR, Disatnik MH, Mochly-Rosen D. Aldehyde dehydrogenase 2 in cardiac protection: a new therapeutic target? Trends Cardiovasc Med 19: 158–164, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burke WJ. 3,4-Dihydroxyphenylacetaldehyde: a potential target for neuroprotective therapy in Parkinson's disease. Curr Drug Targets CNS Neurol Disord 2: 143–148, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Burke WJ, Kumar VB, Pandey N, Panneton WM, Gan Q, Franko MW, O'Dell M, Li SW, Pan Y, Chung HD, Galvin JE. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol 115: 193–203, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Burke WJ, Li SW, Williams EA, Nonneman R, Zahm DS. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: implications for Parkinson's disease pathogenesis. Brain Res 989: 205–213, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Cadoni G, Boccia S, Petrelli L, Di Giannantonio P, Arzani D, Giorgio A, De Feo E, Pandolfini M, Galli P, Paludetti G, Ricciardi G. A review of genetic epidemiology of head and neck cancer related to polymorphisms in metabolic genes, cell cycle control and alcohol metabolism. Acta Otorhinolaryngol Ital 32: 1–11, 2012 [PMC free article] [PubMed] [Google Scholar]
- 31.Campos JC, Queliconi BB, Dourado PM, Cunha TF, Zambelli VO, Bechara LR, Kowaltowski AJ, Brum PC, Mochly-Rosen D, Ferreira JC. Exercise training restores cardiac protein quality control in heart failure. PloS One 7: e52764, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cancer IAfRo Alcohol drinking. IARC monographs on the evaluation of carcinogenic risks to humans. IARC 44: 153–246, 1988 [PMC free article] [PubMed] [Google Scholar]
- 33.Canova C, Richiardi L, Merletti F, Pentenero M, Gervasio C, Tanturri G, Garzino-Demo P, Pecorari G, Talamini R, Barzan L, Sulfaro S, Franchini G, Muzzolini C, Bordin S, Pugliese GN, Macri E, Simonato L. Alcohol, tobacco and genetic susceptibility in relation to cancers of the upper aerodigestive tract in northern Italy. Tumori 96: 1–10, 2010 [DOI] [PubMed] [Google Scholar]
- 34.Castellsague X, Munoz N, De Stefani E, Victora CG, Castelletto R, Rolon PA, Quintana MJ. Independent and joint effects of tobacco smoking and alcohol drinking on the risk of esophageal cancer in men and women. Int J Cancer 82: 657–664, 1999 [DOI] [PubMed] [Google Scholar]
- 35.Chakraborty M, Fry JD. Drosophila lacking a homologue of mammalian ALDH2 have multiple fitness defects. Chemico-biological Interactions 191: 296–302, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan JC, Malik V, Jia W, Kadowaki T, Yajnik CS, Yoon KH, Hu FB. Diabetes in Asia: epidemiology, risk factors, and pathophysiology. JAMA 301: 2129–2140, 2009 [DOI] [PubMed] [Google Scholar]
- 37.Chan MY, Hamamura T, Janschewitz K. Ethnic differences in physical pain sensitivity: role of acculturation. Pain 154: 119–123, 2013 [DOI] [PubMed] [Google Scholar]
- 38.Chang YC, Chiu YF, Lee IT, Ho LT, Hung YJ, Hsiung CA, Quertermous T, Donlon T, Lee WJ, Lee PC, Chen CH, Mochly-Rosen D, Chuang LM. Common ALDH2 genetic variants predict development of hypertension in the SAPPHIRe prospective cohort: gene-environmental interaction with alcohol consumption. BMC Cardiovasc Disorders 12: 58, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen CC, Lu RB, Chen YC, Wang MF, Chang YC, Li TK, Yin SJ. Interaction between the functional polymorphisms of the alcohol-metabolism genes in protection against alcoholism. Am J Hum Genet 65: 795–807, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 321: 1493–1495, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen CH, Sun L, Mochly-Rosen D. Mitochondrial aldehyde dehydrogenase and cardiac diseases. Cardiovasc Res 88: 51–57, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen JJ, Yu BP. Detoxification of reactive aldehydes in mitochondria: effects of age and dietary restriction. Aging 8: 334–340, 1996 [DOI] [PubMed] [Google Scholar]
- 43.Chen L, Davey Smith G, Harbord RM, Lewis SJ. Alcohol intake and blood pressure: a systematic review implementing a Mendelian randomization approach. PLoS Med 5: e52, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen SH, Zhang M, Scott CR. Gene frequencies of alcohol dehydrogenase2 and aldehyde dehydrogenase2 in Northwest Coast Amerindians. Hum Genet 89: 351–352, 1992 [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Cantrell AR, Messing RO, Scheuer T, Catterall WA. Specific modulation of Na+ channels in hippocampal neurons by protein kinase C epsilon. J Neurosci 25: 507–513, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen YC, Peng GS, Tsao TP, Wang MF, Lu RB, Yin SJ. Pharmacokinetic and pharmacodynamic basis for overcoming acetaldehyde-induced adverse reaction in Asian alcoholics, heterozygous for the variant ALDH2*2 gene allele. Pharmacogenet Genomics 19: 588–599, 2009 [DOI] [PubMed] [Google Scholar]
- 47.Chen YC, Peng GS, Wang MF, Tsao TP, Yin SJ. Polymorphism of ethanol-metabolism genes and alcoholism: correlation of allelic variations with the pharmacokinetic and pharmacodynamic consequences. Chem Biol Interact 178: 2–7, 2009 [DOI] [PubMed] [Google Scholar]
- 48.Chen Z, Foster MW, Zhang J, Mao L, Rockman HA, Kawamoto T, Kitagawa K, Nakayama KI, Hess DT, Stamler JS. An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc Natl Acad Sci USA 102: 12159–12164, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Z, Stamler JS. Bioactivation of nitroglycerin by the mitochondrial aldehyde dehydrogenase. Trends Cardiovasc Med 16: 259–265, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci USA 99: 8306–8311, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cherubini A, Ruggiero C, Polidori MC, Mecocci P. Potential markers of oxidative stress in stroke. Free Radic Biol Med 39: 841–852, 2005 [DOI] [PubMed] [Google Scholar]
- 52.Chhangani D, Joshi AP, Mishra A. E3 ubiquitin ligases in protein quality control mechanism. Mol Neurobiol 45: 571–585, 2012 [DOI] [PubMed] [Google Scholar]
- 53.Cho YJ, Wang H, Kozekov ID, Kurtz AJ, Jacob J, Voehler M, Smith J, Harris TM, Lloyd RS, Rizzo CJ, Stone MP. Stereospecific formation of interstrand carbinolamine DNA cross-links by crotonaldehyde- and acetaldehyde-derived alpha-CH3-gamma-OH-1,N2-propano-2′-deoxyguanosine adducts in the 5'-CpG-3' sequence. Chem Res Toxicol 19: 195–208, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chung CS, Lee YC, Wang CP, Ko JY, Wang WL, Wu MS, Wang HP. Secondary prevention of esophageal squamous cell carcinoma in areas where smoking, alcohol, and betel quid chewing are prevalent. J Formos Med Assoc 109: 408–421, 2010 [DOI] [PubMed] [Google Scholar]
- 55.Churchill EN, Ferreira JC, Brum PC, Szweda LI, Mochly-Rosen D. Ischaemic preconditioning improves proteasomal activity and increases the degradation of deltaPKC during reperfusion. Cardiovasc Res 85: 385–394, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Commitee on Advancing Pain Resarch CaE Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education and Research. Washington, DC: National Acadamies Press, 2011 [PubMed] [Google Scholar]
- 57.Cooper C, Campion G, Melton LJ., 3rd Hip fractures in the elderly: a world-wide projection. Osteoporos Int 2: 285–289, 1992 [DOI] [PubMed] [Google Scholar]
- 58.Cui L, Pierce D, Light KE, Melchert RB, Fu Q, Kumar KS, Hauer-Jensen M. Sublethal total body irradiation leads to early cerebellar damage and oxidative stress. Curr Neurovasc Res 7: 125–135, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dandre F, Cassaigne A, Iron A. The frequency of the mitochondrial aldehyde dehydrogenase I2 (atypical) allele in Caucasian, Oriental and African black populations determined by the restriction profile of PCR-amplified DNA. Mol Cell Probes 9: 189–193, 1995 [DOI] [PubMed] [Google Scholar]
- 60.Deehan GA, Jr, Brodie MS, Rodd ZA. What is in that drink: the biological actions of ethanol, acetaldehyde, and salsolinol. Curr Top Behav Neurosci 13: 163–184, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dellarco VL. A mutagenicity assessment of acetaldehyde. Mutat Res 195: 1–20, 1988 [DOI] [PubMed] [Google Scholar]
- 62.Dhillon AS, Tarbutton GL, Levin JL, Plotkin GM, Lowry LK, Nalbone JT, Shepherd S. Pesticide/environmental exposures and Parkinson's disease in East Texas. J Agromedicine 13: 37–48, 2008 [DOI] [PubMed] [Google Scholar]
- 63.Diamond T, Stiel D, Lunzer M, Wilkinson M, Posen S. Ethanol reduces bone formation and may cause osteoporosis. Am J Med 86: 282–288, 1989 [DOI] [PubMed] [Google Scholar]
- 64.Ding JH, Li SP, Cao HX, Wu JZ, Gao CM, Su P, Liu YT, Zhou JN, Chang J, Yao GH. Polymorphisms of alcohol dehydrogenase-2 and aldehyde dehydrogenase-2 and esophageal cancer risk in Southeast Chinese males. World J Gastroenterol 15: 2395–2400, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Doorn JA, Hurley TD, Petersen DR. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon-2-enal and 4-oxonon-2-enal. Chem Res Toxicol 19: 102–110, 2006 [DOI] [PubMed] [Google Scholar]
- 66.Doser TA, Turdi S, Thomas DP, Epstein PN, Li SY, Ren J. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation 119: 1941–1949, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Duan J, McFadden GE, Borgerding AJ, Norby FL, Ren BH, Ye G, Epstein PN, Ren J. Overexpression of alcohol dehydrogenase exacerbates ethanol-induced contractile defect in cardiac myocytes. Am J Physiol Heart Circ Physiol 282: H1216–H1222, 2002 [DOI] [PubMed] [Google Scholar]
- 68.Ehrig T, Bosron WF, Li TK. Alcohol and aldehyde dehydrogenase. Alcohol Alcohol 25: 105–116, 1990 [DOI] [PubMed] [Google Scholar]
- 69.Endo J, Sano M, Katayama T, Hishiki T, Shinmura K, Morizane S, Matsuhashi T, Katsumata Y, Zhang Y, Ito H, Nagahata Y, Marchitti S, Nishimaki K, Wolf AM, Nakanishi H, Hattori F, Vasiliou V, Adachi T, Ohsawa I, Taguchi R, Hirabayashi Y, Ohta S, Suematsu M, Ogawa S, Fukuda K. Metabolic remodeling induced by mitochondrial aldehyde stress stimulates tolerance to oxidative stress in the heart. Circ Res 105: 1118–1127, 2009 [DOI] [PubMed] [Google Scholar]
- 70.Enomoto N, Takada A, Date T. Genotyping of the aldehyde dehydrogenase 2 (ALDH2) gene using the polymerase chain reaction: evidence for single point mutation in the ALDH2 gene of ALDH2-deficiency. Gastroenterol Japonica 26: 440–447, 1991 [DOI] [PubMed] [Google Scholar]
- 71.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med 349: 2241–2252, 2003 [DOI] [PubMed] [Google Scholar]
- 72.Eriksson CJ. Acetaldehyde metabolism in vivo during ethanol oxidation. Adv Exp Med Biol 85A: 319–341, 1977 [DOI] [PubMed] [Google Scholar]
- 73.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11: 81–128, 1991 [DOI] [PubMed] [Google Scholar]
- 74.Fahn S. Description of Parkinson's disease as a clinical syndrome. Ann NY Acad Sci 991: 1–14, 2003 [DOI] [PubMed] [Google Scholar]
- 75.Farout L, Mary J, Vinh J, Szweda LI, Friguet B. Inactivation of the proteasome by 4-hydroxy-2-nonenal is site specific and dependant on 20S proteasome subtypes. Arch Biochem Biophys 453: 135–142, 2006 [DOI] [PubMed] [Google Scholar]
- 76.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet 7: 306–318, 2006 [DOI] [PubMed] [Google Scholar]
- 77.Farres J, Wang X, Takahashi K, Cunningham SJ, Wang TT, Weiner H. Effects of changing glutamate 487 to lysine in rat and human liver mitochondrial aldehyde dehydrogenase. A model to study human (Oriental type) class 2 aldehyde dehydrogenase. J Biol Chem 269: 13854–13860, 1994 [PubMed] [Google Scholar]
- 78.Ferencz-Biro K, Pietruszko R. Human aldehyde dehydrogenase: catalytic activity in oriental liver. Biochem Biophys Res Commun 118: 97–102, 1984 [DOI] [PubMed] [Google Scholar]
- 79.Fernandez-Sola J, Estruch R, Grau JM, Pare JC, Rubin E, Urbano-Marquez A. The relation of alcoholic myopathy to cardiomyopathy. Ann Intern Med 120: 529–536, 1994 [DOI] [PubMed] [Google Scholar]
- 80.Fernandez E, Koek W, Ran Q, Gerhardt GA, France CP, Strong R. Monoamine metabolism and behavioral responses to ethanol in mitochondrial aldehyde dehydrogenase knockout mice. Alcohol Clin Exp Res 30: 1650–1658, 2006 [DOI] [PubMed] [Google Scholar]
- 81.Feron VJ, Til HP, de Vrijer F, Woutersen RA, Cassee FR, van Bladeren PJ. Aldehydes: occurrence, carcinogenic potential, mechanism of action and risk assessment. Mutat Res 259: 363–385, 1991 [DOI] [PubMed] [Google Scholar]
- 82.Ferreira JC, Mochly-Rosen D. Nitroglycerin use in myocardial infarction patients. Circ J 76: 15–21, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Filosto M, Tentorio M, Broglio L, Buzio S, Lazzarini C, Pasolini MP, Cotelli MS, Scarpelli M, Mancuso M, Choub A, Padovani A. Disulfiram neuropathy: two cases of distal axonopathy. Clin Toxicol 46: 314–316, 2008 [DOI] [PubMed] [Google Scholar]
- 84.Firestone JA, Smith-Weller T, Franklin G, Swanson P, Longstreth WT, Jr, Checkoway H. Pesticides and risk of Parkinson disease: a population-based case-control study. Arch Neurol 62: 91–95, 2005 [DOI] [PubMed] [Google Scholar]
- 85.Fitzmaurice AG, Rhodes SL, Lulla A, Murphy NP, Lam HA, O'Donnell KC, Barnhill L, Casida JE, Cockburn M, Sagasti A, Stahl MC, Maidment NT, Ritz B, Bronstein JM. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc Natl Acad Sci USA 110: 636–641, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Florang VR, Rees JN, Brogden NK, Anderson DG, Hurley TD, Doorn JA. Inhibition of the oxidative metabolism of 3,4-dihydroxyphenylacetaldehyde, a reactive intermediate of dopamine metabolism, by 4-hydroxy-2-nonenal. Neurotoxicology 28: 76–82, 2007 [DOI] [PubMed] [Google Scholar]
- 87.Franceschi S, Talamini R, Barra S, Baron AE, Negri E, Bidoli E, Serraino D, La Vecchia C. Smoking and drinking in relation to cancers of the oral cavity, pharynx, larynx, and esophagus in northern Italy. Cancer Res 50: 6502–6507, 1990 [PubMed] [Google Scholar]
- 88.Frenck RW, Jr, Blackburn EH, Shannon KM. The rate of telomere sequence loss in human leukocytes varies with age. Proc Natl Acad Sci USA 95: 5607–5610, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Frisoni GB, Di Monda V. Disulfiram neuropathy: a review (1971–1988) and report of a case. Alcohol Alcoholism 24: 429–437, 1989 [PubMed] [Google Scholar]
- 90.Fujii C, Harada S, Ohkoshi N, Hayashi A, Yoshizawa K. Study on Parkinson's disease and alcohol drinking. Nihon Arukoru Yakubutsu Igakkai Zasshi 33: 683–691, 1998 [PubMed] [Google Scholar]
- 91.Fujiwara S, Kasagi F, Yamada M, Kodama K. Risk factors for hip fracture in a Japanese cohort. J Bone Miner Res 12: 998–1004, 1997 [DOI] [PubMed] [Google Scholar]
- 92.Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol 2010: 214074, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson's disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis 14: 637–647, 2003 [DOI] [PubMed] [Google Scholar]
- 94.Gao GY, Li DJ, Keung WM. Synthesis of potential antidipsotropic isoflavones: inhibitors of the mitochondrial monoamine oxidase-aldehyde dehydrogenase pathway. J Med Chem 44: 3320–3328, 2001 [DOI] [PubMed] [Google Scholar]
- 95.Garaycoechea JI, Crossan GP, Langevin F, Daly M, Arends MJ, Patel KJ. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 489: 571–575, 2012 [DOI] [PubMed] [Google Scholar]
- 96.Ge W, Guo R, Ren J. AMP-dependent kinase and autophagic flux are involved in aldehyde dehydrogenase-2-induced protection against cardiac toxicity of ethanol. Free Radical Biol Med 51: 1736–1748, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ge W, Ren J. mTOR-STAT3-notch signalling contributes to ALDH2-induced protection against cardiac contractile dysfunction and autophagy under alcoholism. J Cell Mol Med 16: 616–626, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Giuliani N, Girasole G, Vescovi PP, Passeri G, Pedrazzoni M. Ethanol and acetaldehyde inhibit the formation of early osteoblast progenitors in murine and human bone marrow cultures. Alcohol Clin Exp Res 23: 381–385, 1999 [PubMed] [Google Scholar]
- 99.Goedde HW, Agarwal DP, Fritze G, Meier-Tackmann D, Singh S, Beckmann G, Bhatia K, Chen LZ, Fang B, Lisker R. Distribution of ADH2 and ALDH2 genotypes in different populations. Hum Genet 88: 344–346, 1992 [DOI] [PubMed] [Google Scholar]
- 100.Goldman D, Enoch MA. Genetic epidemiology of ethanol metabolic enzymes: a role for selection. World Rev Nutr Diet 63: 143–160, 1990 [DOI] [PubMed] [Google Scholar]
- 101.Gong D, Zhang Y, Zhang H, Gu H, Jiang Q, Hu S. Aldehyde dehydrogenase-2 activation during cardioplegic arrest enhances the cardioprotection against myocardial ischemia-reperfusion injury. Cardiovasc Toxicol 12: 350–358 [DOI] [PubMed] [Google Scholar]
- 102.Gong D, Zhang Y, Zhang H, Gu H, Jiang Q, Hu S. Aldehyde dehydrogenase-2 activation during cardioplegic arrest enhances the cardioprotection against myocardial ischemia-reperfusion injury. Cardiovasc Toxicol 12: 350–358, 2012 [DOI] [PubMed] [Google Scholar]
- 103.Guengerich FP. Cytochrome p450 and chemical toxicology. Chem Res Toxicol 21: 70–83, 2008 [DOI] [PubMed] [Google Scholar]
- 104.Guengerich FP. Cytochrome P450: what have we learned and what are the future issues? Drug Metab Rev 36: 159–197, 2004 [DOI] [PubMed] [Google Scholar]
- 105.Guerri C. Mechanisms involved in central nervous system dysfunctions induced by prenatal ethanol exposure. Neurotox Res 4: 327–335, 2002 [DOI] [PubMed] [Google Scholar]
- 106.Guo R, Chen XP, Guo X, Chen L, Li D, Peng J, Li YJ. Evidence for involvement of calcitonin gene-related peptide in nitroglycerin response and association with mitochondrial aldehyde dehydrogenase-2 (ALDH2) Glu504Lys polymorphism. J Am Coll Cardiol 52: 953–960, 2008 [DOI] [PubMed] [Google Scholar]
- 107.Guo R, Zhong L, Ren J. Overexpression of aldehyde dehydrogenase-2 attenuates chronic alcohol exposure-induced apoptosis, change in Akt and Pim signalling in liver. Clin Exp Pharmacol Physiol 36: 463–468, 2009 [DOI] [PubMed] [Google Scholar]
- 108.Guo Y, Tan LJ, Lei SF, Yang TL, Chen XD, Zhang F, Chen Y, Pan F, Yan H, Liu X, Tian Q, Zhang ZX, Zhou Q, Qiu C, Dong SS, Xu XH, Guo YF, Zhu XZ, Liu SL, Wang XL, Li X, Luo Y, Zhang LS, Li M, Wang JT, Wen T, Drees B, Hamilton J, Papasian CJ, Recker RR, Song XP, Cheng J, Deng HW. Genome-wide association study identifies ALDH7A1 as a novel susceptibility gene for osteoporosis. PLoS Genet 6: e1000806, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hansen RN, Oster G, Edelsberg J, Woody GE, Sullivan SD. Economic costs of nonmedical use of prescription opioids. Clin J Pain 27: 194–202, 2011 [DOI] [PubMed] [Google Scholar]
- 110.Hao PP, Chen YG, Wang JL, Wang XL, Zhang Y. Meta-analysis of aldehyde dehydrogenase 2 gene polymorphism and Alzheimer's disease in East Asians. Can J Neurol Sci 38: 500–506, 2011 [DOI] [PubMed] [Google Scholar]
- 111.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298–300, 1956 [DOI] [PubMed] [Google Scholar]
- 112.Hazen SL, Hsu FF, d'Avignon A, Heinecke JW. Human neutrophils employ myeloperoxidase to convert alpha-amino acids to a battery of reactive aldehydes: a pathway for aldehyde generation at sites of inflammation. Biochemistry 37: 6864–6873, 1998 [DOI] [PubMed] [Google Scholar]
- 113.Heck H, Casanova M. The implausibility of leukemia induction by formaldehyde: a critical review of the biological evidence on distant-site toxicity. Regul Toxicol Pharmacol 40: 92–106, 2004 [DOI] [PubMed] [Google Scholar]
- 114.Higuchi S, Matsushita S, Imazeki H, Kinoshita T, Takagi S, Kono H. Aldehyde dehydrogenase genotypes in Japanese alcoholics. Lancet 343: 741–742, 1994 [DOI] [PubMed] [Google Scholar]
- 115.Higuchi S, Matsushita S, Murayama M, Takagi S, Hayashida M. Alcohol and aldehyde dehydrogenase polymorphisms and the risk for alcoholism. Am J Psychiatry 152: 1219–1221, 1995 [DOI] [PubMed] [Google Scholar]
- 116.Hill BG, Dranka BP, Zou L, Chatham JC, Darley-Usmar VM. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochemical J 424: 99–107, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hintz KK, Relling DP, Saari JT, Borgerding AJ, Duan J, Ren BH, Kato K, Epstein PN, Ren J. Cardiac overexpression of alcohol dehydrogenase exacerbates cardiac contractile dysfunction, lipid peroxidation, and protein damage after chronic ethanol ingestion. Alcohol Clin Exp Res 27: 1090–1098, 2003 [DOI] [PubMed] [Google Scholar]
- 118.Hiura Y, Tabara Y, Kokubo Y, Okamura T, Miki T, Tomoike H, Iwai N. A genome-wide association study of hypertension-related phenotypes in a Japanese population. Circ J 74: 2353–2359, 2010 [DOI] [PubMed] [Google Scholar]
- 119.Hiyama T, Yoshihara M, Tanaka S, Chayama K. Genetic polymorphisms and head and neck cancer risk (Review). Int J Oncol 32: 945–973, 2008 [PubMed] [Google Scholar]
- 120.Hoshi H, Hao W, Fujita Y, Funayama A, Miyauchi Y, Hashimoto K, Miyamoto K, Iwasaki R, Sato Y, Kobayashi T, Miyamoto H, Yoshida S, Mori T, Kanagawa H, Katsuyama E, Fujie A, Kitagawa K, Nakayama KI, Kawamoto T, Sano M, Fukuda K, Ohsawa I, Ohta S, Morioka H, Matsumoto M, Chiba K, Toyama Y, Miyamoto T. Aldehyde-stress resulting from Aldh2 mutation promotes osteoporosis due to impaired osteoblastogenesis. J Bone Miner Res 27: 2015–2023, 2012 [DOI] [PubMed] [Google Scholar]
- 121.Houlgate PR, Dhingra KS, Nash SJ, Evans WH. Determination of formaldehyde and acetaldehyde in mainstream cigarette smoke by high-performance liquid chromatography. Analyst 114: 355–360, 1989 [DOI] [PubMed] [Google Scholar]
- 122.Hsieh AY, Tripp DA, Ji LJ, Sullivan MJ. Comparisons of catastrophizing, pain attitudes, and cold-pressor pain experience between Chinese and European Canadian young adults. J Pain 11: 1187–1194, 2010 [DOI] [PubMed] [Google Scholar]
- 123.Hwang ES, Kim GH. Biomarkers for oxidative stress status of DNA, lipids, and proteins in vitro and in vivo cancer research. Toxicology 229: 1–10, 2007 [DOI] [PubMed] [Google Scholar]
- 124.Iborra FJ, Renau-Piqueras J, Portoles M, Boleda MD, Guerri C, Pares X. Immunocytochemical and biochemical demonstration of formaldhyde dehydrogenase (class III alcohol dehydrogenase) in the nucleus. J Histochem Cytochem 40: 1865–1878, 1992 [DOI] [PubMed] [Google Scholar]
- 125.Ikehara S, Iso H, Yamagishi K, Yamamoto S, Inoue M, Tsugane S. Alcohol consumption, social support, and risk of stroke and coronary heart disease among Japanese men: the JPHC Study. Alcohol Clin Exp Res 33: 1025–1032, 2009 [DOI] [PubMed] [Google Scholar]
- 126.Ishikawa H, Ishikawa T, Yamamoto H, Fukao A, Yokoyama K. Genotoxic effects of alcohol in human peripheral lymphocytes modulated by ADH1B and ALDH2 gene polymorphisms. Mutat Res 615: 134–142, 2007 [DOI] [PubMed] [Google Scholar]
- 127.Isse T, Matsuno K, Oyama T, Kitagawa K, Kawamoto T. Aldehyde dehydrogenase 2 gene targeting mouse lacking enzyme activity shows high acetaldehyde level in blood, brain, and liver after ethanol gavages. Alcohol Clin Exp Res 29: 1959–1964, 2005 [DOI] [PubMed] [Google Scholar]
- 128.Isse T, Oyama T, Kitagawa K, Matsuno K, Matsumoto A, Yoshida A, Nakayama K, Kawamoto T. Diminished alcohol preference in transgenic mice lacking aldehyde dehydrogenase activity. Pharmacogenetics 12: 621–626, 2002 [DOI] [PubMed] [Google Scholar]
- 129.Isse T, Oyama T, Matsuno K, Ogawa M, Narai-Suzuki R, Yamaguchi T, Murakami T, Kinaga T, Uchiyama I, Kawamoto T. Paired acute inhalation test reveals that acetaldehyde toxicity is higher in aldehyde dehydrogenase 2 knockout mice than in wild-type mice. J Toxicol Sci 30: 329–337, 2005 [DOI] [PubMed] [Google Scholar]
- 130.Isse T, Oyama T, Matsuno K, Uchiyama I, Kawamoto T. Aldehyde dehydrogenase 2 activity affects symptoms produced by an intraperitoneal acetaldehyde injection, but not acetaldehyde lethality. J Toxicol Sci 30: 315–328, 2005 [DOI] [PubMed] [Google Scholar]
- 131.Jackson B, Brocker C, Thompson DC, Black W, Vasiliou K, Nebert DW, Vasiliou V. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum Genomics 5: 283–303, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jeng JJ, Weiner H. Purification and characterization of catalytically active precursor of rat liver mitochondrial aldehyde dehydrogenase expressed in Escherichia coli. Arch Biochem Biophys 289: 214–222, 1991 [DOI] [PubMed] [Google Scholar]
- 133.Jenner P. Oxidative stress in Parkinson's disease. Ann Neurol 53 Suppl 3: S26–36, 2003 [DOI] [PubMed] [Google Scholar]
- 134.Jo SA, Kim EK, Park MH, Han C, Park HY, Jang Y, Song BJ, Jo I. A Glu487Lys polymorphism in the gene for mitochondrial aldehyde dehydrogenase 2 is associated with myocardial infarction in elderly Korean men. Clin Chim Acta 382: 43–47, 2007 [DOI] [PubMed] [Google Scholar]
- 135.Jung JG, Kim JS, Oh MK. The role of the flushing response in the relationship between alcohol consumption and insulin resistance. Alcoholism Clin Exp Res 34: 1699–1704, 2010 [DOI] [PubMed] [Google Scholar]
- 136.Kamino K, Nagasaka K, Imagawa M, Yamamoto H, Yoneda H, Ueki A, Kitamura S, Namekata K, Miki T, Ohta S. Deficiency in mitochondrial aldehyde dehydrogenase increases the risk for late-onset Alzheimer's disease in the Japanese population. Biochem Biophys Res Commun 273: 192–196, 2000 [DOI] [PubMed] [Google Scholar]
- 137.Kanis JA. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis: synopsis of a WHO report. WHO Study Group Osteoporos Int 4: 368–381, 1994 [DOI] [PubMed] [Google Scholar]
- 138.Karliner JS. Lessons from the besotted heart. J Am Coll Cardiol 54: 2197–2198, 2009 [DOI] [PubMed] [Google Scholar]
- 139.Kato N, Takeuchi F, Tabara Y, Kelly TN, Go MJ, Sim X, Tay WT, Chen CH, Zhang Y, Yamamoto K, Katsuya T, Yokota M, Kim YJ, Ong RT, Nabika T, Gu D, Chang LC, Kokubo Y, Huang W, Ohnaka K, Yamori Y, Nakashima E, Jaquish CE, Lee JY, Seielstad M, Isono M, Hixson JE, Chen YT, Miki T, Zhou X, Sugiyama T, Jeon JP, Liu JJ, Takayanagi R, Kim SS, Aung T, Sung YJ, Zhang X, Wong TY, Han BG, Kobayashi S, Ogihara T, Zhu D, Iwai N, Wu JY, Teo YY, Tai ES, Cho YS, He J. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet 43: 531–538, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kee Y, D'Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest 122: 3799–3806, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kennedy RD, D'Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev 19: 2925–2940, 2005 [DOI] [PubMed] [Google Scholar]
- 142.Ketcham AS, Wexler H, Mantel N. Effects of alcohol in mouse neoplasia. Cancer Res 23: 667–670, 1963 [PubMed] [Google Scholar]
- 143.Keung WM, Vallee BL. Daidzin: a potent, selective inhibitor of human mitochondrial aldehyde dehydrogenase. Proc Natl Acad Sci USA 90: 1247–1251, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Keung WM, Vallee BL. Kudzu root: an ancient Chinese source of modern antidipsotropic agents. Phytochemistry 47: 499–506, 1998 [DOI] [PubMed] [Google Scholar]
- 145.Khanna M, Chen CH, Kimble-Hill A, Parajuli B, Perez-Miller S, Baskaran S, Kim J, Dria K, Vasiliou V, Mochly-Rosen D, Hurley TD. Discovery of a novel class of covalent inhibitor for aldehyde dehydrogenases. J Biol Chem 286: 43486–43494, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun 304: 463–470, 2003 [DOI] [PubMed] [Google Scholar]
- 147.Kitagawa K, Kawamoto T, Kunugita N, Tsukiyama T, Okamoto K, Yoshida A, Nakayama K. Aldehyde dehydrogenase (ALDH) 2 associates with oxidation of methoxyacetaldehyde; in vitro analysis with liver subcellular fraction derived from human and Aldh2 gene targeting mouse. FEBS Lett 476: 306–311, 2000 [DOI] [PubMed] [Google Scholar]
- 148.Klyosov AA. Kinetics and specificity of human liver aldehyde dehydrogenases toward aliphatic, aromatic, and fused polycyclic aldehydes. Biochemistry 35: 4457–4467, 1996 [DOI] [PubMed] [Google Scholar]
- 149.Klyosov AA, Rashkovetsky LG, Tahir MK, Keung WM. Possible role of liver cytosolic and mitochondrial aldehyde dehydrogenases in acetaldehyde metabolism. Biochemistry 35: 4445–4456, 1996 [DOI] [PubMed] [Google Scholar]
- 150.Koda K, Salazar-Rodriguez M, Corti F, Chan NY, Estephan R, Silver RB, Mochly-Rosen D, Levi R. Aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release from cardiac mast cells. Circulation 122: 771–781, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Koppaka V, Thompson DC, Chen Y, Ellermann M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD, Vasiliou V. Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol Rev 64: 520–539, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kotraiah V, Pallares D, Toema D, Kong D, Beausoleil E. Identification of aldehyde dehydrogenase 1A1 modulators using virtual screening. J Enzyme Inhib Med Chem 2012. [DOI] [PubMed] [Google Scholar]
- 153.Kristal BS, Conway AD, Brown AM, Jain JC, Ulluci PA, Li SW, Burke WJ. Selective dopaminergic vulnerability: 3,4-dihydroxyphenylacetaldehyde targets mitochondria. Free Radic Biol Med 30: 924–931, 2001 [DOI] [PubMed] [Google Scholar]
- 154.Kristal BS, Park BK, Yu BP. 4-Hydroxyhexenal is a potent inducer of the mitochondrial permeability transition. J Biol Chem 271: 6033–6038, 1996 [DOI] [PubMed] [Google Scholar]
- 155.Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci 17: 5089–5100, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kurtz AJ, Lloyd RS. 1,N2-deoxyguanosine adducts of acrolein, crotonaldehyde, and trans-4-hydroxynonenal cross-link to peptides via Schiff base linkage. J Biol Chem 278: 5970–5976, 2003 [DOI] [PubMed] [Google Scholar]
- 157.Kutler DI, Auerbach AD, Satagopan J, Giampietro PF, Batish SD, Huvos AG, Goberdhan A, Shah JP, Singh B. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch Otolaryngol Head Neck Surg 129: 106–112, 2003 [DOI] [PubMed] [Google Scholar]
- 158.Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res 106: 1681–1691, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lai CL, Chao YC, Chen YC, Liao CS, Chen MC, Liu YC, Yin SJ. No sex and age influence on the expression pattern and activities of human gastric alcohol and aldehyde dehydrogenases. Alcohol Clin Exp Res 24: 1625–1632, 2000 [PubMed] [Google Scholar]
- 160.Lambert AJ, Boysen HM, Buckingham JA, Yang T, Podlutsky A, Austad SN, Kunz TH, Buffenstein R, Brand MD. Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan in vertebrate homeotherms. Aging Cell 6: 607–618, 2007 [DOI] [PubMed] [Google Scholar]
- 161.Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475: 53–58, 2011 [DOI] [PubMed] [Google Scholar]
- 162.Larson HN, Weiner H, Hurley TD. Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase “Asian” variant. J Biol Chem 280: 30550–30556, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Larson HN, Zhou J, Chen Z, Stamler JS, Weiner H, Hurley TD. Structural and functional consequences of coenzyme binding to the inactive asian variant of mitochondrial aldehyde dehydrogenase: roles of residues 475 and 487. J Biol Chem 282: 12940–12950, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Latino-Martel P, Chan DS, Druesne-Pecollo N, Barrandon E, Hercberg S, Norat T. Maternal alcohol consumption during pregnancy and risk of childhood leukemia: systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev 19: 1238–1260, 2010 [DOI] [PubMed] [Google Scholar]
- 165.Lau EM, Chan YH, Chan M, Woo J, Griffith J, Chan HH, Leung PC. Vertebral deformity in chinese men: prevalence, risk factors, bone mineral density, and body composition measurements. Calcif Tissue Int 66: 47–52, 2000 [DOI] [PubMed] [Google Scholar]
- 166.Lee CH, Wu DC, Lee JM, Wu IC, Goan YG, Kao EL, Huang HL, Chan TF, Chou SH, Chou YP, Lee CY, Chen PS, Ho CK, He J, Wu MT. Carcinogenetic impact of alcohol intake on squamous cell carcinoma risk of the oesophagus in relation to tobacco smoking. Eur J Cancer 43: 1188–1199, 2007 [DOI] [PubMed] [Google Scholar]
- 167.Lee CH, Wu DC, Wu IC, Goan YG, Lee JM, Chou SH, Chan TF, Huang HL, Hung YH, Huang MC, Lai TC, Wang TN, Lan CC, Tsai S, Lin WY, Wu MT. Genetic modulation of ADH1B and ALDH2 polymorphisms with regard to alcohol and tobacco consumption for younger aged esophageal squamous cell carcinoma diagnosis. Int J Cancer 125: 1134–1142, 2009 [DOI] [PubMed] [Google Scholar]
- 168.Lee WC, Wong HY, Chai YY, Shi CW, Amino N, Kikuchi S, Huang SH. Lipid peroxidation dysregulation in ischemic stroke: plasma 4-HNE as a potential biomarker? Biochem Biophys Res Commun 425: 842–847, 2012 [DOI] [PubMed] [Google Scholar]
- 169.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer 11: 9–22, 2011 [DOI] [PubMed] [Google Scholar]
- 170.Leiphon LJ, Picklo MJ., Sr Inhibition of aldehyde detoxification in CNS mitochondria by fungicides. Neurotoxicology 28: 143–149, 2007 [DOI] [PubMed] [Google Scholar]
- 171.Leslie RD, Pyke DA. Chlorpropamide-alcohol flushing: a dominantly inherited trait associated with diabetes. Br Med J 2: 1519–1521, 1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Li H, Borinskaya S, Yoshimura K, Kal'ina N, Marusin A, Stepanov VA, Qin Z, Khaliq S, Lee MY, Yang Y, Mohyuddin A, Gurwitz D, Mehdi SQ, Rogaev E, Jin L, Yankovsky NK, Kidd JR, Kidd KK. Refined geographic distribution of the oriental ALDH2*504Lys (nee 487Lys) variant. Ann Hum Genet 73: 335–345, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li SY, Gilbert SA, Li Q, Ren J. Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. J Mol Cell Cardiol 47: 247–255, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Li Y, Zhang D, Jin W, Shao C, Yan P, Xu C, Sheng H, Liu Y, Yu J, Xie Y, Zhao Y, Lu D, Nebert DW, Harrison DC, Huang W, Jin L. Mitochondrial aldehyde dehydrogenase-2 (ALDH2) Glu504Lys polymorphism contributes to the variation in efficacy of sublingual nitroglycerin. J Clin Invest 116: 506–511, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res 106: 633–646, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lin YP, Cheng TJ. Why can't Chinese Han drink alcohol? Hepatitis B virus infection and the evolution of acetaldehyde dehydrogenase deficiency. Med Hypotheses 59: 204–207, 2002 [DOI] [PubMed] [Google Scholar]
- 177.Lindahl R, Evces S. Rat liver aldehyde dehydrogenase. I. Isolation and characterization of four high Km normal liver isozymes. J Biol Chem 259: 11986–11990, 1984 [PubMed] [Google Scholar]
- 178.Lipsky JJ, Shen ML, Naylor S. In vivo inhibition of aldehyde dehydrogenase by disulfiram. Chem Biol Interact 130–132: 93–102, 2001 [DOI] [PubMed] [Google Scholar]
- 179.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9: 106–118, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Liu YJ, Shen H, Xiao P, Xiong DH, Li LH, Recker RR, Deng HW. Molecular genetic studies of gene identification for osteoporosis: a 2004 update. J Bone Miner Res 21: 1511–1535, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lowe ED, Gao GY, Johnson LN, Keung WM. Structure of daidzin, a naturally occurring anti-alcohol-addiction agent, in complex with human mitochondrial aldehyde dehydrogenase. J Med Chem 51: 4482–4487, 2008 [DOI] [PubMed] [Google Scholar]
- 182.Luckey SW, Tjalkens RB, Petersen DR. Mechanism of inhibition of rat liver class 2 ALDH by 4-hydroxynonenal. Adv Exp Med Biol 463: 71–77, 1999 [DOI] [PubMed] [Google Scholar]
- 183.Luo HR, Israel Y, Tu GC, Eriksson CJ, Zhang YP. Genetic polymorphism of aldehyde dehydrogenase 2 (ALDH2) in a Chinese population: gender, age, culture, and genotypes of ALDH2. Biochem Genet 43: 223–227, 2005 [DOI] [PubMed] [Google Scholar]
- 184.Luo HR, Wu GS, Pakstis AJ, Tong L, Oota H, Kidd KK, Zhang YP. Origin and dispersal of atypical aldehyde dehydrogenase ALDH2487Lys. Gene 435: 96–103, 2009 [DOI] [PubMed] [Google Scholar]
- 185.Ma H, Guo R, Yu L, Zhang Y, Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur Heart J 32: 1025–1038, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ma H, Li J, Gao F, Ren J. Aldehyde dehydrogenase 2 ameliorates acute cardiac toxicity of ethanol: role of protein phosphatase and forkhead transcription factor. J Am Coll Cardiol 54: 2187–2196, 2009 [DOI] [PubMed] [Google Scholar]
- 187.Ma H, Yu L, Byra EA, Hu N, Kitagawa K, Nakayama KI, Kawamoto T, Ren J. Aldehyde dehydrogenase 2 knockout accentuates ethanol-induced cardiac depression: role of protein phosphatases. J Mol Cell Cardiol 49: 322–329, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ma H, Zhang X, Bai M, Wang X. Clinical effects of lianbai liquid in prevention and treatment of dermal injury caused by radiotherapy. J Tradit Chin Med 27: 193–196, 2007 [PubMed] [Google Scholar]
- 189.Ma R, Yu J, Xu D, Yang L, Lin X, Zhao F, Bai F. Effect of felodipine with irbesartan or metoprolol on sexual function and oxidative stress in women with essential hypertension. J Hypertens 30: 210–216, 2012 [DOI] [PubMed] [Google Scholar]
- 190.MacArthur AC, McBride ML, Spinelli JJ, Tamaro S, Gallagher RP, Theriault G. Risk of childhood leukemia associated with parental smoking and alcohol consumption prior to conception and during pregnancy: the cross-Canada childhood leukemia study. Cancer Causes Control 19: 283–295, 2008 [DOI] [PubMed] [Google Scholar]
- 191.Maggio D, Barabani M, Pierandrei M, Polidori MC, Catani M, Mecocci P, Senin U, Pacifici R, Cherubini A. Marked decrease in plasma antioxidants in aged osteoporotic women: results of a cross-sectional study. J Clin Endocrinol Metab 88: 1523–1527, 2003 [DOI] [PubMed] [Google Scholar]
- 192.Mak S, Lehotay DC, Yazdanpanah M, Azevedo ER, Liu PP, Newton GE. Unsaturated aldehydes including 4-OH-nonenal are elevated in patients with congestive heart failure. J Cardiac Failure 6: 108–114, 2000 [PubMed] [Google Scholar]
- 193.Mandel S, Grunblatt E, Riederer P, Amariglio N, Jacob-Hirsch J, Rechavi G, Youdim MB. Gene expression profiling of sporadic Parkinson's disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann NY Acad Sci 1053: 356–375, 2005 [DOI] [PubMed] [Google Scholar]
- 194.Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol 4: 697–720, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Marchitti SA, Deitrich RA, Vasiliou V. Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol Rev 59: 125–150, 2007 [DOI] [PubMed] [Google Scholar]
- 196.Marietta C, Thompson LH, Lamerdin JE, Brooks PJ. Acetaldehyde stimulates FANCD2 monoubiquitination, H2AX phosphorylation, and BRCA1 phosphorylation in human cells in vitro: implications for alcohol-related carcinogenesis. Mutat Res 664: 77–83, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Masaki T, Mochizuki H, Matsushita S, Yokoyama A, Kamakura K, Higuchi S. Association of aldehyde dehydrogenase-2 polymorphism with alcoholic polyneuropathy in humans. Neurosci Lett 363: 288–290, 2004 [DOI] [PubMed] [Google Scholar]
- 198.Matsuda T, Matsumoto A, Uchida M, Kanaly RA, Misaki K, Shibutani S, Kawamoto T, Kitagawa K, Nakayama KI, Tomokuni K, Ichiba M. Increased formation of hepatic N2-ethylidene-2′-deoxyguanosine DNA adducts in aldehyde dehydrogenase 2-knockout mice treated with ethanol. Carcinogenesis 28: 2363–2366, 2007 [DOI] [PubMed] [Google Scholar]
- 199.Matsuda T, Terashima I, Matsumoto Y, Yabushita H, Matsui S, Shibutani S. Effective utilization of N2-ethyl-2′-deoxyguanosine triphosphate during DNA synthesis catalyzed by mammalian replicative DNA polymerases. Biochemistry 38: 929–935, 1999 [DOI] [PubMed] [Google Scholar]
- 200.Matsuda T, Yabushita H, Kanaly RA, Shibutani S, Yokoyama A. Increased DNA damage in ALDH2-deficient alcoholics. Chem Res Toxicol 19: 1374–1378, 2006 [DOI] [PubMed] [Google Scholar]
- 201.Matsumoto A, Ichiba M, Horita M, Yamashita Z, Takahashi T, Isse T, Oyama T, Kawamoto T, Tomokuni K. Lack of aldehyde dehydrogenase ameliorates oxidative stress induced by single-dose ethanol administration in mouse liver. Alcohol 41: 57–59, 2007 [DOI] [PubMed] [Google Scholar]
- 202.Matsumoto A, Kawamoto T, Horita M, Takahashi T, Isse T, Oyama T, Ichiba M. Single-dose ethanol administration downregulates expression of cytochrome p450 2E1 mRNA in aldehyde dehydrogenase 2 knockout mice. Alcohol 41: 587–589, 2007 [DOI] [PubMed] [Google Scholar]
- 203.Matsumoto A, Kawamoto T, Mutoh F, Isse T, Oyama T, Kitagawa K, Nakayama KI, Ichiba M. Effects of 5-week ethanol feeding on the liver of aldehyde dehydrogenase 2 knockout mice. Pharmacogenet Genomics 18: 847–852, 2008 [DOI] [PubMed] [Google Scholar]
- 204.Mays DC, Ortiz-Bermudez P, Lam JP, Tong IH, Fauq AH, Lipsky JJ. Inhibition of recombinant human mitochondrial aldehyde dehydrogenase by two intermediate metabolites of disulfiram. Biochem Pharmacol 55: 1099–1103, 1998 [DOI] [PubMed] [Google Scholar]
- 205.McGrath LT, McGleenon BM, Brennan S, McColl D, Mc IS, Passmore AP. Increased oxidative stress in Alzheimer's disease as assessed with 4-hydroxynonenal but not malondialdehyde. QJM 94: 485–490, 2001 [DOI] [PubMed] [Google Scholar]
- 206.Moldovan GL, D'Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet 43: 223–249, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Morita M, Kumashiro R, Kubo N, Nakashima Y, Yoshida R, Yoshinaga K, Saeki H, Emi Y, Kakeji Y, Sakaguchi Y, Toh Y, Maehara Y. Alcohol drinking, cigarette smoking, and the development of squamous cell carcinoma of the esophagus: epidemiology, clinical findings, and prevention. Int J Clin Oncol 15: 126–134, 2010 [DOI] [PubMed] [Google Scholar]
- 208.Morita M, Saeki H, Mori M, Kuwano H, Sugimachi K. Risk factors for esophageal cancer and the multiple occurrence of carcinoma in the upper aerodigestive tract. Surgery 131: S1–6, 2002 [DOI] [PubMed] [Google Scholar]
- 209.Morris JB. Dosimetry, toxicity and carcinogenicity of inspired acetaldehyde in the rat. Mutat Res 380: 113–124, 1997 [DOI] [PubMed] [Google Scholar]
- 210.Mosammaparast N, Shi Y. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu Rev Biochem 79: 155–179, 2010 [DOI] [PubMed] [Google Scholar]
- 211.Mukerjee N, Pietruszko R. Human mitochondrial aldehyde dehydrogenase substrate specificity: comparison of esterase with dehydrogenase reaction. Arch Biochem Biophys 299: 23–29, 1992 [DOI] [PubMed] [Google Scholar]
- 212.Murata C, Suzuki Y, Muramatsu T, Taniyama M, Atsumi Y, Matsuoka K, Watanabe T, Okazaki I. Inactive aldehyde dehydrogenase 2 worsens glycemic control in patients with type 2 diabetes mellitus who drink low to moderate amounts of alcohol. Alcoholism Clin Exp Res 24: 5S–11S, 2000 [PubMed] [Google Scholar]
- 213.Murata C, Taniyama M, Kuriyama S, Muramatsu T, Atsumi Y, Matsuoka K, Suzuki Y. Meta-analysis of three diabetes population studies: association of inactive ALDH2 genotype with maternal inheritance of diabetes. Diabetes Res Clin Pract 66 Suppl 1: S145–147, 2004 [DOI] [PubMed] [Google Scholar]
- 214.Muto M, Takahashi M, Ohtsu A, Ebihara S, Yoshida S, Esumi H. Risk of multiple squamous cell carcinomas both in the esophagus and the head and neck region. Carcinogenesis 26: 1008–1012, 2005 [DOI] [PubMed] [Google Scholar]
- 215.Nagasawa H, Wada M, Arawaka S, Kawanami T, Kurita K, Daimon M, Adachi M, Hosoya T, Emi M, Muramatsu M, Kato T. A polymorphism of the aldehyde dehydrogenase 2 gene is a risk factor for multiple lacunar infarcts in Japanese men: the Takahata Study. Eur J Neurol 14: 428–434, 2007 [DOI] [PubMed] [Google Scholar]
- 216.Nagayoshi H, Matsumoto A, Nishi R, Kawamoto T, Ichiba M, Matsuda T. Increased formation of gastric N(2)-ethylidene-2′-deoxyguanosine DNA adducts in aldehyde dehydrogenase-2 knockout mice treated with ethanol. Mutat Res 673: 74–77, 2009 [DOI] [PubMed] [Google Scholar]
- 217.Nakagawa T, Kajiwara A, Saruwatari J, Hamamoto A, Kaku W, Oniki K, Mihara S, Ogata Y, Nakagawa K. The combination of mitochondrial low enzyme-activity aldehyde dehydrogenase 2 allele and superoxide dismutase 2 genotypes increases the risk of hypertension in relation to alcohol consumption. Pharmacogenet Genomics 23: 34–37, 2013 [DOI] [PubMed] [Google Scholar]
- 218.Nakamura K, Kusano K, Nakamura Y, Kakishita M, Ohta K, Nagase S, Yamamoto M, Miyaji K, Saito H, Morita H, Emori T, Matsubara H, Toyokuni S, Ohe T. Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation 105: 2867–2871, 2002 [DOI] [PubMed] [Google Scholar]
- 219.Nakamura K, Kusano KF, Matsubara H, Nakamura Y, Miura A, Nishii N, Banba K, Nagase S, Miyaji K, Morita H, Saito H, Emori T, Ohe T. Relationship between oxidative stress and systolic dysfunction in patients with hypertrophic cardiomyopathy. J Cardiac Failure 11: 117–123, 2005 [DOI] [PubMed] [Google Scholar]
- 220.Nakamura K, Miura D, Kusano KF, Fujimoto Y, Sumita-Yoshikawa W, Fuke S, Nishii N, Nagase S, Hata Y, Morita H, Matsubara H, Ohe T, Ito H. 4-Hydroxy-2-nonenal induces calcium overload via the generation of reactive oxygen species in isolated rat cardiac myocytes. J Cardiac Failure 15: 709–716, 2009 [DOI] [PubMed] [Google Scholar]
- 221.Nakashima Y, Ohsawa I, Konishi F, Hasegawa T, Kumamoto S, Suzuki Y, Ohta S. Preventive effects of Chlorella on cognitive decline in age-dependent dementia model mice. Neurosci Lett 464: 193–198, 2009 [DOI] [PubMed] [Google Scholar]
- 222.Ni L, Zhou J, Hurley TD, Weiner H. Human liver mitochondrial aldehyde dehydrogenase: three-dimensional structure and the restoration of solubility and activity of chimeric forms. Protein Sci 8: 2784–2790, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Niemela O. Acetaldehyde adducts in circulation. Novartis Found Symp 285: 183–192, 2007 [DOI] [PubMed] [Google Scholar]
- 224.Ning S, Budas GR, Churchill EN, Chen CH, Knox SJ, Mochly-Rosen D. Mitigation of radiation-induced dermatitis by activation of aldehyde dehydrogenase 2 using topical alda-1 in mice. Radiat Res 178: 69–74, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Noble EP. Addiction and its reward process through polymorphisms of the D2 dopamine receptor gene: a review. Eur Psychiatry 15: 79–89, 2000 [DOI] [PubMed] [Google Scholar]
- 226.O'Brien PJ, Siraki AG, Shangari N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit Rev Toxicol 35: 609–662, 2005 [DOI] [PubMed] [Google Scholar]
- 227.O'Dowd BF, Rothhammer F, Israel Y. Genotyping of mitochondrial aldehyde dehydrogenase locus of Native American Indians. Alcohol Clin Exp Res 14: 531–533, 1990 [DOI] [PubMed] [Google Scholar]
- 228.Ogawa M, Isse T, Oyama T, Kunugita N, Yamaguchi T, Kinaga T, Narai R, Matsumoto A, Kim YD, Kim H, Uchiyama I, Kawamoto T. Urinary 8-hydoxydeoxyguanosine (8-OHdG) and plasma malondialdehyde (MDA) levels in Aldh2 knock-out mice under acetaldehyde exposure. Ind Health 44: 179–183, 2006 [DOI] [PubMed] [Google Scholar]
- 229.Ogawa M, Oyama T, Isse T, Saito K, Tomigahara Y, Endo Y, Kawamoto T. A comparison of covalent binding of ethanol metabolites to DNA according to Aldh2 genotype. Toxicol Lett 168: 148–154, 2007 [DOI] [PubMed] [Google Scholar]
- 230.Ohsawa I, Kamino K, Nagasaka K, Ando F, Niino N, Shimokata H, Ohta S. Genetic deficiency of a mitochondrial aldehyde dehydrogenase increases serum lipid peroxides in community-dwelling females. J Hum Genet 48: 404–409, 2003 [DOI] [PubMed] [Google Scholar]
- 231.Ohsawa I, Nishimaki K, Murakami Y, Suzuki Y, Ishikawa M, Ohta S. Age-dependent neurodegeneration accompanying memory loss in transgenic mice defective in mitochondrial aldehyde dehydrogenase 2 activity. J Neurosci 28: 6239–6249, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Ohta S, Ohsawa I. Dysfunction of mitochondria and oxidative stress in the pathogenesis of Alzheimer's disease: on defects in the cytochrome c oxidase complex and aldehyde detoxification. J Alzheimers Dis 9: 155–166, 2006 [DOI] [PubMed] [Google Scholar]
- 233.Ohta S, Ohsawa I, Kamino K, Ando F, Shimokata H. Mitochondrial ALDH2 deficiency as an oxidative stress. Ann NY Acad Sci 1011: 36–44, 2004 [DOI] [PubMed] [Google Scholar]
- 234.Okun E, Arumugam TV, Tang SC, Gleichmann M, Albeck M, Sredni B, Mattson MP. The organotellurium compound ammonium trichloro(dioxoethylene-O,O')tellurate enhances neuronal survival and improves functional outcome in an ischemic stroke model in mice. J Neurochem 102: 1232–1241, 2007 [DOI] [PubMed] [Google Scholar]
- 235.Overstreet DH, Knapp DJ, Breese GR, Diamond I. A selective ALDH-2 inhibitor reduces anxiety in rats. Pharmacol Biochem Behav 94: 255–261, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Oyama T, Isse T, Kagawa N, Kinaga T, Kim YD, Morita M, Sugio K, Weiner H, Yasumoto K, Kawamoto T. Tissue-distribution of aldehyde dehydrogenase 2 and effects of the ALDH2 gene-disruption on the expression of enzymes involved in alcohol metabolism. Front Biosci 10: 951–960, 2005 [DOI] [PubMed] [Google Scholar]
- 237.Oyama T, Kim YD, Isse T, Phuong PT, Ogawa M, Yamaguchi T, Kinaga T, Yashima Y, Kim H, Kawamoto T. A pilot study on subacute ethanol treatment of ALDH2 KO mice. J Toxicol Sci 32: 421–428, 2007 [DOI] [PubMed] [Google Scholar]
- 238.Panneton WM, Kumar VB, Gan Q, Burke WJ, Galvin JE. The neurotoxicity of DOPAL: behavioral and stereological evidence for its role in Parkinson disease pathogenesis. PLoS One 5: e15251, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Parajuli B, Kimble-Hill AC, Khanna M, Ivanova Y, Meroueh S, Hurley TD. Discovery of novel regulators of aldehyde dehydrogenase isoenzymes. Chem Biol Interact 191: 153–158, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Park J, Chung NG, Chae H, Kim M, Lee S, Kim Y, Lee JW, Cho B, Jeong D, Park I. FANCA and FANCG are the major Fanconi anemia genes in the Korean population. Clin Genet 84: 271–275, 2013 [DOI] [PubMed] [Google Scholar]
- 241.Park JW, Ji YI, Choi YH, Kang MY, Jung E, Cho SY, Cho HY, Kang BK, Joung YS, Kim DH, Park SC, Park J. Candidate gene polymorphisms for diabetes mellitus, cardiovascular disease and cancer are associated with longevity in Koreans. Exp Mol Med 41: 772–781, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Parker JD, Parker JO. Nitrate therapy for stable angina pectoris. N Engl J Med 338: 520–531, 1998 [DOI] [PubMed] [Google Scholar]
- 243.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 55: 74–108, 2005 [DOI] [PubMed] [Google Scholar]
- 244.Parmar K, D'Andrea AD. Stressed out: endogenous aldehydes damage hematopoietic stem cells. Cell Stem Cell 11: 583–584, 2012 [DOI] [PubMed] [Google Scholar]
- 245.Pedersen WA, Chan SL, Mattson MP. A mechanism for the neuroprotective effect of apolipoprotein E: isoform-specific modification by the lipid peroxidation product 4-hydroxynonenal. J Neurochem 74: 1426–1433, 2000 [DOI] [PubMed] [Google Scholar]
- 246.Peng GS, Chen YC, Tsao TP, Wang MF, Yin SJ. Pharmacokinetic and pharmacodynamic basis for partial protection against alcoholism in Asians, heterozygous for the variant ALDH2*2 gene allele. Pharmacogenet Genomics 17: 845–855, 2007 [DOI] [PubMed] [Google Scholar]
- 247.Peng GS, Yin SJ, Cheng CA, Chiu SW, Lee JT, Lin WW, Lin JC, Hsu YD. Increased risk of cerebral hemorrhage in Chinese male heavy drinkers with mild liver disorder. Cerebrovasc Dis 23: 309–314, 2007 [DOI] [PubMed] [Google Scholar]
- 248.Perez-Miller S, Younus H, Vanam R, Chen CH, Mochly-Rosen D, Hurley TD. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol 17: 159–164, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Peters AL, Van Noorden CJ. Glucose-6-phosphate dehydrogenase deficiency and malaria: cytochemical detection of heterozygous G6PD deficiency in women. J Histochem Cytochem 57: 1003–1011, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radical Biol Med 37: 937–945, 2004 [DOI] [PubMed] [Google Scholar]
- 251.Pipinos II, Judge AR, Zhu Z, Selsby JT, Swanson SA, Johanning JM, Baxter BT, Lynch TG, Dodd SL. Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radical Biol Med 41: 262–269, 2006 [DOI] [PubMed] [Google Scholar]
- 252.Pipinos II, Swanson SA, Zhu Z, Nella AA, Weiss DJ, Gutti TL, McComb RD, Baxter BT, Lynch TG, Casale GP. Chronically ischemic mouse skeletal muscle exhibits myopathy in association with mitochondrial dysfunction and oxidative damage. Am J Physiol Regul Integr Comp Physiol 295: R290–R296, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Pruett D, Waterman EH, Caughey AB. Fetal alcohol exposure: consequences, diagnosis, and treatment. Obstet Gynecol Surv 68: 62–69, 2013 [DOI] [PubMed] [Google Scholar]
- 254.Pyke DA, Leslie RD. Chlorpropamide-alcohol flushing: a definition of its relation to non-insulin-dependent diabetes. Br Med J 2: 1521–1522, 1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Radovanovic S, Savic-Radojevic A, Pljesa-Ercegovac M, Djukic T, Suvakov S, Krotin M, Simic DV, Matic M, Radojicic Z, Pekmezovic T, Simic T. Markers of oxidative damage and antioxidant enzyme activities as predictors of morbidity and mortality in patients with chronic heart failure. J Cardiac Failure 18: 493–501, 2012 [DOI] [PubMed] [Google Scholar]
- 256.Raghunathan L, Hsu LC, Klisak I, Sparkes RS, Yoshida A, Mohandas T. Regional localization of the human genes for aldehyde dehydrogenase-1 and aldehyde dehydrogenase-2. Genomics 2: 267–269, 1988 [DOI] [PubMed] [Google Scholar]
- 257.Raguz M, Mainali L, Widomska J, Subczynski WK. Using spin-label electron paramagnetic resonance (EPR) to discriminate and characterize the cholesterol bilayer domain. Chem Phys Lipids 164: 819–829, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Reed TT. Lipid peroxidation and neurodegenerative disease. Free Radic Biol Med 51: 1302–1319, 2011 [DOI] [PubMed] [Google Scholar]
- 259.Ren J. Acetaldehyde and alcoholic cardiomyopathy: lessons from the ADH and ALDH2 transgenic models. Novartis Found Symp 285: 69–76, 2007 [DOI] [PubMed] [Google Scholar]
- 260.Ren J, Babcock SA, Li Q, Huff AF, Li SY, Doser TA. Aldehyde dehydrogenase-2 transgene ameliorates chronic alcohol ingestion-induced apoptosis in cerebral cortex. Toxicol Lett 187: 149–156, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Ren J, Davidoff AJ, Brown RA. Acetaldehyde depresses shortening and intracellular Ca2+ transients in adult rat ventricular myocytes. Cell Mol Biol 43: 825–834, 1997 [PubMed] [Google Scholar]
- 262.Ridpath JR, Nakamura A, Tano K, Luke AM, Sonoda E, Arakawa H, Buerstedde JM, Gillespie DA, Sale JE, Yamazoe M, Bishop DK, Takata M, Takeda S, Watanabe M, Swenberg JA, Nakamura J. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res 67: 11117–11122, 2007 [DOI] [PubMed] [Google Scholar]
- 263.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol 8: 405–413, 2007 [DOI] [PubMed] [Google Scholar]
- 264.Risner CH, Martin P. Quantitation of formaldehyde, acetaldehyde, and acetone in sidestream cigarette smoke by high-performance liquid chromatography. J Chromatogr Sci 32: 76–82, 1994 [DOI] [PubMed] [Google Scholar]
- 265.Robador PA, Seyedi N, Chan NY, Koda K, Levi R. Aldehyde dehydrogenase type 2 activation by adenosine and histamine inhibits ischemic norepinephrine release in cardiac sympathetic neurons: mediation by protein kinase Cepsilon. J Pharmacol Exp Ther 343: 97–105, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Roede JR, Jones DP. Reactive species and mitochondrial dysfunction: mechanistic significance of 4-hydroxynonenal. Environ Mol Mutagenesis 51: 380–390, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation 125: e2-–e220., 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Roh S, Matsushita S, Hara S, Maesato H, Matsui T, Suzuki G, Miyakawa T, Ramchandani VA, Li TK, Higuchi S. Role of GABRA2 in moderating subjective responses to alcohol. Alcohol Clin Exp Res 35: 400–407, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Rosado IV, Langevin F, Crossan GP, Takata M, Patel KJ. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat Struct Mol Biol 18: 1432–1434, 2011 [DOI] [PubMed] [Google Scholar]
- 270.Rosenberg PS, Tamary H, Alter BP. How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel. Am J Med Genet A 155A: 1877–1883, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Rowell LN, Mechlin B, Ji E, Addamo M, Girdler SS. Asians differ from non-Hispanic Whites in experimental pain sensitivity. Eur J Pain 15: 764–771, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Roy TW, Bhagwat AS. Kinetic studies of Escherichia coli AlkB using a new fluorescence-based assay for DNA demethylation. Nucleic Acids Res 35: e147, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Russell JW, Karnes JL, Dyck PJ. Sural nerve myelinated fiber density differences associated with meaningful changes in clinical and electrophysiologic measurements. J Neurol Sci 135: 114–117, 1996 [DOI] [PubMed] [Google Scholar]
- 274.Ryan JL. Ionizing radiation: the good, the bad, and the ugly. J Invest Dermatol 132: 985–993, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Salaspuro M. Interrelationship between alcohol, smoking, acetaldehyde and cancer. Novartis Found Symp 285: 80–96, 2007 [DOI] [PubMed] [Google Scholar]
- 276.Salaspuro V, Salaspuro M. Synergistic effect of alcohol drinking and smoking on in vivo acetaldehyde concentration in saliva. Int J Cancer 111: 480–483, 2004 [DOI] [PubMed] [Google Scholar]
- 277.Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem 68: 2092–2097, 1997 [DOI] [PubMed] [Google Scholar]
- 278.Seitz HK, Meier P. The role of acetaldehyde in upper digestive tract cancer in alcoholics. Transl Res 149: 293–297, 2007 [DOI] [PubMed] [Google Scholar]
- 279.Seitz HK, Stickel F. Acetaldehyde as an underestimated risk factor for cancer development: role of genetics in ethanol metabolism. Genes Nutr 5: 121–128, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Sekili S, McCay PB, Li XY, Zughaib M, Sun JZ, Tang L, Thornby JI, Bolli R. Direct evidence that the hydroxyl radical plays a pathogenetic role in myocardial “stunning” in the conscious dog and demonstration that stunning can be markedly attenuated without subsequent adverse effects. Circ Res 73: 705–723, 1993 [DOI] [PubMed] [Google Scholar]
- 281.Senior DJ, Tsai CS. Purification and characterization of aldehyde dehydrogenase from rat liver mitochondria. Arch Biochem Biophys 262: 211–220, 1988 [DOI] [PubMed] [Google Scholar]
- 282.Serra JA, Dominguez RO, Marschoff ER, Guareschi EM, Famulari AL, Boveris A. Systemic oxidative stress associated with the neurological diseases of aging. Neurochem Res 34: 2122–2132, 2009 [DOI] [PubMed] [Google Scholar]
- 283.Shen ML, Johnson KL, Mays DC, Lipsky JJ, Naylor S. Determination of in vivo adducts of disulfiram with mitochondrial aldehyde dehydrogenase. Biochem Pharmacol 61: 537–545, 2001 [DOI] [PubMed] [Google Scholar]
- 284.Shimizu Y, Sakai A, Menuki K, Mori T, Isse T, Oyama T, Kawamoto T, Nakamura T. Reduced bone formation in alcohol-induced osteopenia is associated with elevated p21 expression in bone marrow cells in aldehyde dehydrogenase 2-disrupted mice. Bone 48: 1075–1086, 2011 [DOI] [PubMed] [Google Scholar]
- 285.Sinclair D. Deleterious pleiotropic effects of the atypical aldehyde dehydrogenase 2 (ALDH2) allele: comment on Luo et al., 2005. Biochem Genet 44: 385–390, 2006 [DOI] [PubMed] [Google Scholar]
- 286.Singer MA, Vernino SA, Wolfe GI. Idiopathic neuropathy: new paradigms, new promise. J Peripheral Nervous Syst 17 Suppl 2: 43–49, 2012 [DOI] [PubMed] [Google Scholar]
- 287.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 175: 184–191, 1988 [DOI] [PubMed] [Google Scholar]
- 288.Singh R, Sandhu J, Kaur B, Juren T, Steward WP, Segerback D, Farmer PB. Evaluation of the DNA damaging potential of cannabis cigarette smoke by the determination of acetaldehyde derived N2-ethyl-2′-deoxyguanosine adducts. Chem Res Toxicol 22: 1181–1188, 2009 [DOI] [PubMed] [Google Scholar]
- 289.Skulachev VP. A biochemical approach to the problem of aging: “megaproject” on membrane-penetrating ions. The first results and prospects. Biochem Biokhimiia 72: 1385–1396, 2007 [DOI] [PubMed] [Google Scholar]
- 290.Solito R, Chen CH, Mochly-Rosen D, Giachetti A, Ziche M, Donnini S. Mitochondrial aldehyde dehydrogenase-2 activation prevents beta amyloids induced endothelial cell dysfunction and restores angiogenesis. J Cell Sci 126: 1952–1961, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Sophos NA, Vasiliou V. Aldehyde dehydrogenase gene superfamily: the 2002 update. Chem Biol Interact 143–144: 5–22, 2003 [DOI] [PubMed] [Google Scholar]
- 292.Sosenko JM. The prevalence of diabetic neuropathy according to ethnicity. Curr Diabetes Reports 9: 435–439, 2009 [DOI] [PubMed] [Google Scholar]
- 293.Staub RE, Quistad GB, Casida JE. Mechanism for benomyl action as a mitochondrial aldehyde dehydrogenase inhibitor in mice. Chem Res Toxicol 11: 535–543, 1998 [DOI] [PubMed] [Google Scholar]
- 294.Staub RE, Quistad GB, Casida JE. S-methyl N-butylthiocarbamate sulfoxide: selective carbamoylating agent for mouse mitochondrial aldehyde dehydrogenase. Biochem Pharmacol 58: 1467–1473, 1999 [DOI] [PubMed] [Google Scholar]
- 295.Stewart MJ, Malek K, Crabb DW. Distribution of messenger RNAs for aldehyde dehydrogenase 1, aldehyde dehydrogenase 2, and aldehyde dehydrogenase 5 in human tissues. J Invest Med 44: 42–46, 1996 [PubMed] [Google Scholar]
- 296.Stone MP, Cho YJ, Huang H, Kim HY, Kozekov ID, Kozekova A, Wang H, Minko IG, Lloyd RS, Harris TM, Rizzo CJ. Interstrand DNA cross-links induced by alpha,beta-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc Chem Res 41: 793–804, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Suh JJ, Pettinati HM, Kampman KM, O'Brien CP. The status of disulfiram: a half of a century later. J Clin Psychopharmacol 26: 290–302, 2006 [DOI] [PubMed] [Google Scholar]
- 298.Sun L, Ferreira JC, Mochly-Rosen D. ALDH2 activator inhibits increased myocardial infarction injury by nitroglycerin tolerance. Sci Transl Med 3: 107ra111, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Suzuki K, Nakazato K, Kusakabe T, Nagamine T, Sakurai H, Takatama M. Role of oxidative stress on pathogenesis of hypertensive cerebrovascular lesions. Pathol Int 57: 133–139, 2007 [DOI] [PubMed] [Google Scholar]
- 300.Suzuki Y, Kuriyama S, Atsumi Y, Murata C, Matsuoka K, Suzuki Y, Taniyama M, Muramatsu T, Suzuki Y, Ohta S. Maternal inheritance of diabetes is associated with inactive ALDH2 genotype in diabetics with renal failure in Japanese. Diabetes Res Clin Pract 60: 143–145, 2003 [DOI] [PubMed] [Google Scholar]
- 301.Suzuki Y, Muramatsu T, Taniyama M, Atsumi Y, Kawaguchi R, Higuchi S, Hosokawa K, Asahina T, Murata C, Matsuoka K. Association of aldehyde dehydrogenase with inheritance of NIDDM. Diabetologia 39: 1115–1118, 1996 [DOI] [PubMed] [Google Scholar]
- 302.Suzuki Y, Muramatsu T, Taniyama M, Atsumi Y, Suematsu M, Kawaguchi R, Higuchi S, Asahina T, Murata C, Handa M, Matsuoka K. Mitochondrial aldehyde dehydrogenase in diabetes associated with mitochondrial tRNA(Leu(UUR)) mutation at position 3243. Diabetes Care 19: 1423–1425, 1996 [DOI] [PubMed] [Google Scholar]
- 303.Suzuki Y, Taniyama M, Muramatsu T, Higuchi S, Ohta S, Atsumi Y, Matsuoka K. ALDH2/ADH2 polymorphism associated with vasculopathy and neuropathy in type 2 diabetes. Alcohol Clin Exp Res 28: 111S–116S, 2004 [DOI] [PubMed] [Google Scholar]
- 304.Szymanska K, Levi JE, Menezes A, Wunsch-Filho V, Eluf-Neto J, Koifman S, Matos E, Daudt AW, Curado MP, Villar S, Pawlita M, Waterboer T, Boffetta P, Hainaut P, Brennan P. TP53 and EGFR mutations in combination with lifestyle risk factors in tumours of the upper aerodigestive tract from South America. Carcinogenesis 31: 1054–1059, 2010 [DOI] [PubMed] [Google Scholar]
- 305.Takagi S, Baba S, Iwai N, Fukuda M, Katsuya T, Higaki J, Mannami T, Ogata J, Goto Y, Ogihara T. The aldehyde dehydrogenase 2 gene is a risk factor for hypertension in Japanese but does not alter the sensitivity to pressor effects of alcohol: the Suita study. Hypertens Res 24: 365–370, 2001 [DOI] [PubMed] [Google Scholar]
- 306.Takagi S, Iwai N, Yamauchi R, Kojima S, Yasuno S, Baba T, Terashima M, Tsutsumi Y, Suzuki S, Morii I, Hanai S, Ono K, Baba S, Tomoike H, Kawamura A, Miyazaki S, Nonogi H, Goto Y. Aldehyde dehydrogenase 2 gene is a risk factor for myocardial infarction in Japanese men. Hypertens Res 25: 677–681, 2002 [DOI] [PubMed] [Google Scholar]
- 307.Tang MS, Wang HT, Hu Y, Chen WS, Akao M, Feng Z, Hu W. Acrolein induced DNA damage, mutagenicity and effect on DNA repair. Mol Nutr Food Res 55: 1291–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR, Comyns K, Richards MB, Meng C, Priestley B, Fernandez HH, Cambi F, Umbach DM, Blair A, Sandler DP, Langston JW. Rotenone, paraquat, and Parkinson's disease. Environ Health Perspect 119: 866–872, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Theruvathu JA, Jaruga P, Nath RG, Dizdaroglu M, Brooks PJ. Polyamines stimulate the formation of mutagenic 1,N2-propanodeoxyguanosine adducts from acetaldehyde. Nucleic Acids Res 33: 3513–3520, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Toh Y, Oki E, Ohgaki K, Sakamoto Y, Ito S, Egashira A, Saeki H, Kakeji Y, Morita M, Sakaguchi Y, Okamura T, Maehara Y. Alcohol drinking, cigarette smoking, and the development of squamous cell carcinoma of the esophagus: molecular mechanisms of carcinogenesis. Int J Clin Oncol 15: 135–144, 2010 [DOI] [PubMed] [Google Scholar]
- 311.TRI Acetaldehyde. In: Toxic Release Inventory. Washington, DC: EPA, 1993 [Google Scholar]
- 312.Tsou PS, Page NA, Lee SG, Fung SM, Keung WM, Fung HL. Differential metabolism of organic nitrates by aldehyde dehydrogenase 1a1 and 2: substrate selectivity, enzyme inactivation, and active cysteine sites. AAPS J 13: 548–555, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Uchida K, Stadtman ER. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase. A possible involvement of intra- and intermolecular cross-linking reaction. J Biol Chem 268: 6388–6393, 1993 [PubMed] [Google Scholar]
- 314.Uchida K, Stadtman ER. Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc Natl Acad Sci USA 89: 4544–4548, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Umar S, Fleischer D. Esophageal cancer: epidemiology, pathogenesis and prevention. Nat Clin Pract Gastroenterol Hepatol 5: 517–526, 2008 [DOI] [PubMed] [Google Scholar]
- 316.Upton DC, Wang X, Blans P, Perrino FW, Fishbein JC, Akman SA. Replication of N2-ethyldeoxyguanosine DNA adducts in the human embryonic kidney cell line 293. Chem Res Toxicol 19: 960–967, 2006 [DOI] [PubMed] [Google Scholar]
- 317.Urbano-Marquez A, Estruch R, Navarro-Lopez F, Grau JM, Mont L, Rubin E. The effects of alcoholism on skeletal and cardiac muscle. N Engl J Med 320: 409–415, 1989 [DOI] [PubMed] [Google Scholar]
- 318.Vakevainen S, Tillonen J, Agarwal DP, Srivastava N, Salaspuro M. High salivary acetaldehyde after a moderate dose of alcohol in ALDH2-deficient subjects: strong evidence for the local carcinogenic action of acetaldehyde. Alcohol Clin Exp Res 24: 873–877, 2000 [PubMed] [Google Scholar]
- 319.Vasiliou V, Bairoch A, Tipton KF, Nebert DW. Eukaryotic aldehyde dehydrogenase (ALDH) genes: human polymorphisms, and recommended nomenclature based on divergent evolution and chromosomal mapping. Pharmacogenetics 9: 421–434, 1999 [PubMed] [Google Scholar]
- 320.Vasiliou V, Thompson DC, Smith C, Fujita M, Chen Y. Aldehyde dehydrogenases: from eye crystallins to metabolic disease and cancer stem cells. Chem Biol Interact 202: 2–10, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Verlander PC, Kaporis A, Liu Q, Zhang Q, Seligsohn U, Auerbach AD. Carrier frequency of the IVS4 + 4 A–>T mutation of the Fanconi anemia gene FAC in the Ashkenazi Jewish population. Blood 86: 4034–4038, 1995 [PubMed] [Google Scholar]
- 322.Viteri G, Chung YW, Stadtman ER. Effect of progerin on the accumulation of oxidized proteins in fibroblasts from Hutchinson Gilford progeria patients. Mech Ageing Dev 131: 2–8, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Vondriska TM, Klein JB, Ping P. Use of functional proteomics to investigate PKC epsilon-mediated cardioprotection: the signaling module hypothesis. Am J Physiol Heart Circ Physiol 280: H1434–H1441, 2001 [DOI] [PubMed] [Google Scholar]
- 324.Wang B, Wang J, Zhou S, Tan S, He X, Yang Z, Xie YC, Li S, Zheng C, Ma X. The association of mitochondrial aldehyde dehydrogenase gene (ALDH2) polymorphism with susceptibility to late-onset Alzheimer's disease in Chinese. J Neurol Sci 268: 172–175, 2008 [DOI] [PubMed] [Google Scholar]
- 325.Wang H, Papoiu AD, Coghill RC, Patel T, Wang N, Yosipovitch G. Ethnic differences in pain, itch and thermal detection in response to topical capsaicin: African Americans display a notably limited hyperalgesia and neurogenic inflammation. Br J Dermatol 162: 1023–1029, 2010 [DOI] [PubMed] [Google Scholar]
- 326.Wang J, Wang H, Hao P, Xue L, Wei S, Zhang Y, Chen Y. Inhibition of aldehyde dehydrogenase 2 by oxidative stress is associated with cardiac dysfunction in diabetic rats. Mol Med 17: 172–179, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Wang RS, Nakajima T, Kawamoto T, Honma T. Effects of aldehyde dehydrogenase-2 genetic polymorphisms on metabolism of structurally different aldehydes in human liver. Drug Metab Dispos 30: 69–73, 2002 [DOI] [PubMed] [Google Scholar]
- 328.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet 8: 735–748, 2007 [DOI] [PubMed] [Google Scholar]
- 329.Watson PJ, Latif RK, Rowbotham DJ. Ethnic differences in thermal pain responses: a comparison of South Asian and White British healthy males. Pain 118: 194–200, 2005 [DOI] [PubMed] [Google Scholar]
- 330.Weiner H, Wang TT, Farres J. Molecular biological studies on liver mitochondrial aldehyde dehydrogenase. Alcohol Alcohol Suppl 1: 91–95, 1991 [PubMed] [Google Scholar]
- 331.Weiner H, Wang X. Aldehyde dehydrogenase and acetaldehyde metabolism. Alcohol Alcohol Suppl 2: 141–145, 1994 [PubMed] [Google Scholar]
- 332.Weinshenker D. Cocaine sobers up. Nat Med 16: 969–970, 2010 [DOI] [PubMed] [Google Scholar]
- 333.Weng H, Weng Z, Lu Y, Nakayama K, Morimoto K. Effects of alcohol-drinking behaviour and ADH1B and ALDH2 polymorphisms on basal DNA damage in human mononuclear cells as determined by the comet assay. Mutat Res 701: 132–136, 2010 [DOI] [PubMed] [Google Scholar]
- 334.Wenzel P, Muller J, Zurmeyer S, Schuhmacher S, Schulz E, Oelze M, Pautz A, Kawamoto T, Wojnowski L, Kleinert H, Munzel T, Daiber A. ALDH-2 deficiency increases cardiovascular oxidative stress–evidence for indirect antioxidative properties. Biochem Biophys Res Commun 367: 137–143, 2008 [DOI] [PubMed] [Google Scholar]
- 335.Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson's disease. Proteome Sci 6: 8, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Wey MC, Fernandez E, Martinez PA, Sullivan P, Goldstein DS, Strong R. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson's disease. PLoS One 7: e31522, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.WHO WHO European Ministerial Conference on Young People and Alcohol 9Your Majesty, http://www.who.int/director%20general/speeches/2001/english/20010219_youngpeoplealcohol.en.html 2001
- 338.WHO WHO Global Alcohol Database, http://www.who.int/globalatlas/default.asp
- 339.WHO WHO to meet beverage company representatives to discuss health-related alcohol issues, http://www.who.int/mediacentre/news/releases/2003/pr6/en/index.html.2003
- 340.Wienkers LC, Heath TG. Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov 4: 825–833, 2005 [DOI] [PubMed] [Google Scholar]
- 341.Williams JP, McBride WH. After the bomb drops: a new look at radiation-induced multiple organ dysfunction syndrome (MODS). Int J Radiat Biol 87: 851–868, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Williams TI, Lynn BC, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer's disease. Neurobiol Aging 27: 1094–1099, 2006 [DOI] [PubMed] [Google Scholar]
- 343.Woodrow KM, Friedman GD, Siegelaub AB, Collen MF. Pain tolerance: differences according to age, sex and race. Psychosom Med 34: 548–556, 1972 [DOI] [PubMed] [Google Scholar]
- 344.World Advertising Research Centre World Drinking Trends. Oxfordshire: WARC, 2005 [Google Scholar]
- 345.Woutersen RA, Appelman LM, Feron VJ, Van der Heijden CA. Inhalation toxicity of acetaldehyde in rats. II. Carcinogenicity study: interim results after 15 months. Toxicology 31: 123–133, 1984 [DOI] [PubMed] [Google Scholar]
- 346.Woutersen RA, Appelman LM, Van Garderen-Hoetmer A, Feron VJ. Inhalation toxicity of acetaldehyde in rats. III. Carcinogenicity study. Toxicology 41: 213–231, 1986 [DOI] [PubMed] [Google Scholar]
- 347.Xu F, Chen Y, Lv R, Zhang H, Tian H, Bian Y, Feng J, Sun Y, Li R, Wang R, Zhang Y. ALDH2 genetic polymorphism and the risk of type II diabetes mellitus in CAD patients. Hypertension Res 33: 49–55, 2010 [DOI] [PubMed] [Google Scholar]
- 348.Xu F, Chen YG, Xue L, Li RJ, Zhang H, Bian Y, Zhang C, Lv RJ, Feng JB, Zhang Y. Role of aldehyde dehydrogenase 2 Glu504lys polymorphism in acute coronary syndrome. J Cell Mol Med 15: 1955–1962, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Xu XH, Dong SS, Guo Y, Yang TL, Lei SF, Papasian CJ, Zhao M, Deng HW. Molecular genetic studies of gene identification for osteoporosis: the 2009 update. Endocr Rev 31: 447–505, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Yagasaki H, Oda T, Adachi D, Nakajima T, Nakahata T, Asano S, Yamashita T. Two common founder mutations of the fanconi anemia group G gene FANCG/XRCC9 in the Japanese population. Hum Mutat 21: 555, 2003 [DOI] [PubMed] [Google Scholar]
- 351.Yamaguchi J, Hasegawa Y, Kawasaki M, Masui T, Kanoh T, Ishiguro N, Hamajima N. ALDH2 polymorphisms and bone mineral density in an elderly Japanese population. Osteoporos Int 17: 908–913, 2006 [DOI] [PubMed] [Google Scholar]
- 352.Yang SJ, Wang HY, Li XQ, Du HZ, Zheng CJ, Chen HG, Mu XY, Yang CX. Genetic polymorphisms of ADH2 and ALDH2 association with esophageal cancer risk in southwest China. World J Gastroenterol 13: 5760–5764, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Yao CT, Cheng CA, Wang HK, Chiu SW, Chen YC, Wang MF, Yin SJ, Peng GS. The role of ALDH2 and ADH1B polymorphism in alcohol consumption and stroke in Han Chinese. Human genomics 5: 569–576, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Yao L, Fan P, Arolfo M, Jiang Z, Olive MF, Zablocki J, Sun HL, Chu N, Lee J, Kim HY, Leung K, Shryock J, Blackburn B, Diamond I. Inhibition of aldehyde dehydrogenase-2 suppresses cocaine seeking by generating THP, a cocaine use-dependent inhibitor of dopamine synthesis. Nat Med 16: 1024–1028, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Yin SJ, Liao CS, Lee YC, Wu CW, Jao SW. Genetic polymorphism and activities of human colon alcohol and aldehyde dehydrogenases: no gender and age differences. Alcohol Clin Exp Res 18: 1256–1260, 1994 [DOI] [PubMed] [Google Scholar]
- 356.Yokoyama A, Kato H, Yokoyama T, Tsujinaka T, Muto M, Omori T, Haneda T, Kumagai Y, Igaki H, Yokoyama M, Watanabe H, Fukuda H, Yoshimizu H. Genetic polymorphisms of alcohol and aldehyde dehydrogenases and glutathione S-transferase M1 and drinking, smoking, and diet in Japanese men with esophageal squamous cell carcinoma. Carcinogenesis 23: 1851–1859, 2002 [DOI] [PubMed] [Google Scholar]
- 357.Yokoyama A, Muramatsu T, Ohmori T, Higuchi S, Hayashida M, Ishii H. Esophageal cancer and aldehyde dehydrogenase-2 genotypes in Japanese males. Cancer Epidemiol Biomarkers Prev 5: 99–102, 1996 [PubMed] [Google Scholar]
- 358.Yokoyama A, Muramatsu T, Ohmori T, Makuuchi H, Higuchi S, Matsushita S, Yoshino K, Maruyama K, Nakano M, Ishii H. Multiple primary esophageal and concurrent upper aerodigestive tract cancer and the aldehyde dehydrogenase-2 genotype of Japanese alcoholics. Cancer 77: 1986–1990, 1996 [DOI] [PubMed] [Google Scholar]
- 359.Yokoyama A, Muramatsu T, Ohmori T, Yokoyama T, Okuyama K, Takahashi H, Hasegawa Y, Higuchi S, Maruyama K, Shirakura K, Ishii H. Alcohol-related cancers and aldehyde dehydrogenase-2 in Japanese alcoholics. Carcinogenesis 19: 1383–1387, 1998 [DOI] [PubMed] [Google Scholar]
- 360.Yokoyama A, Muramatsu T, Omori T, Yokoyama T, Matsushita S, Higuchi S, Maruyama K, Ishii H. Alcohol and aldehyde dehydrogenase gene polymorphisms and oropharyngolaryngeal, esophageal and stomach cancers in Japanese alcoholics. Carcinogenesis 22: 433–439, 2001 [DOI] [PubMed] [Google Scholar]
- 361.Yokoyama A, Omori T. Genetic polymorphisms of alcohol and aldehyde dehydrogenases and risk for esophageal and head and neck cancers. Alcohol 35: 175–185, 2005 [DOI] [PubMed] [Google Scholar]
- 362.Yokoyama A, Omori T. Genetic polymorphisms of alcohol and aldehyde dehydrogenases and risk for esophageal and head and neck cancers. Jpn J Clin Oncol 33: 111–121, 2003 [DOI] [PubMed] [Google Scholar]
- 363.Yokoyama A, Omori T, Yokoyama T. Alcohol and aldehyde dehydrogenase polymorphisms and a new strategy for prevention and screening for cancer in the upper aerodigestive tract in East Asians. Keio J Med 59: 115–130, 2010 [DOI] [PubMed] [Google Scholar]
- 364.Yokoyama A, Tsutsumi E, Imazeki H, Suwa Y, Nakamura C, Mizukami T, Yokoyama T. Salivary acetaldehyde concentration according to alcoholic beverage consumed and aldehyde dehydrogenase-2 genotype. Alcohol Clin Exp Res 32: 1607–1614, 2008 [DOI] [PubMed] [Google Scholar]
- 365.Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA 93: 2696–2701, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Yoshida A, Huang IY, Ikawa M. Molecular abnormality of an inactive aldehyde dehydrogenase variant commonly found in Orientals. Proc Natl Acad Sci USA 81: 258–261, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.You M, Crabb DW. Recent advances in alcoholic liver disease. II. Minireview: molecular mechanisms of alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol 287: G1–G6, 2004 [DOI] [PubMed] [Google Scholar]
- 368.Yoval-Sanchez B, Rodriguez-Zavala JS. Differences in susceptibility to inactivation of human aldehyde dehydrogenases by lipid peroxidation byproducts. Chem Res Toxicol 25: 722–729 [DOI] [PubMed] [Google Scholar]
- 369.Yu BP. Membrane alteration as a basis of aging and the protective effects of calorie restriction. Mech Ageing Dev 126: 1003–1010, 2005 [DOI] [PubMed] [Google Scholar]
- 370.Yu HS, Oyama T, Isse T, Kitagawa K, Pham TT, Tanaka M, Kawamoto T. Formation of acetaldehyde-derived DNA adducts due to alcohol exposure. Chem Biol Interact 188: 367–375, 2010 [DOI] [PubMed] [Google Scholar]
- 371.Yu HS, Oyama T, Isse T, Kitakawa K, Ogawa M, Pham TT, Kawamoto T. Characteristics of aldehyde dehydrogenase 2 (Aldh2) knockout mice. Toxicol Mech Methods 19: 535–540, 2009 [DOI] [PubMed] [Google Scholar]
- 372.Zhang B, Zhang Y, La Cour K, Richmond K, Wang X, Ren J. Mitochondrial aldehyde dehydrogenase obliterates endoplasmic reticulum stress-induced cardiac contractile dysfunction via correction of autophagy. Biochim Biophys Acta 1832: 574–584, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Zhang H, Chen YG, Xu F, Xue L, Jiang CX, Zhang Y. The relationship between aldehyde dehydrogenase-2 gene polymorphisms and efficacy of nitroglycerin. Zhonghua Nei Ke Za Zhi 46: 629–632, 2007 [PubMed] [Google Scholar]
- 375.Zhang Y, Babcock SA, Hu N, Maris JR, Wang H, Ren J. Mitochondrial aldehyde dehydrogenase (ALDH2) protects against streptozotocin-induced diabetic cardiomyopathy: role of GSK3beta and mitochondrial function. BMC Med 10: 40, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 376.Zhang Y, Ren J. ALDH2 in alcoholic heart diseases: molecular mechanism and clinical implications. Pharmacol Ther 132: 86–95, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Zhang Y, Ren J. Autophagy in ALDH2-elicited cardioprotection against ischemic heart disease: slayer or savior? Autophagy 6: 1212–1213, 2010 [DOI] [PubMed] [Google Scholar]
- 378.Zhou J, Weiner H. Basis for half-of-the-site reactivity and the dominance of the K487 oriental subunit over the E487 subunit in heterotetrameric human liver mitochondrial aldehyde dehydrogenase. Biochemistry 39: 12019–12024, 2000 [DOI] [PubMed] [Google Scholar]
- 379.Zhu X, Smith MA, Perry G, Aliev G. Mitochondrial failures in Alzheimer's disease. Am J Alzheimers Dis Other Demen 19: 345–352, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]