

Abstract
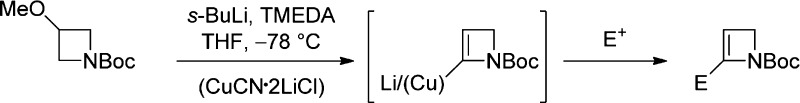
s-BuLi-induced α-lithiation–elimination of LiOMe from N-Boc-3-methoxyazetidine and further in situ α-lithiation generates N-Boc-2-lithio-2-azetine which can be trapped with electrophiles, either directly (carbonyl or heteroatom electrophiles) or after transmetalation to copper (allowing allylations and propargylations), providing a concise access to 2-substituted 2-azetines.
2-Azetidinones (β-lactams) and azetidines are well-studied four-membered azacyclic systems with important biological activities.1 In contrast, the unsaturated and more strained 2-azetine (1,2-dihydroazete) system 1 (Figure 1) is comparatively less explored, which partly reflects issues of ease of access and stability (notably the propensity to undergo 4e electrocyclic ring-opening to 1-azabutadienes), although electron-withdrawing groups on the nitrogen are known to reduce instability.2,3
Figure 1.
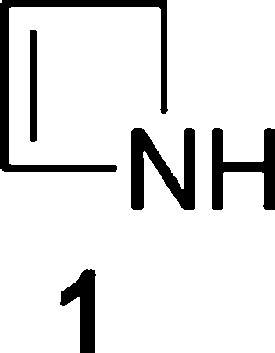
2-Azetine 1.
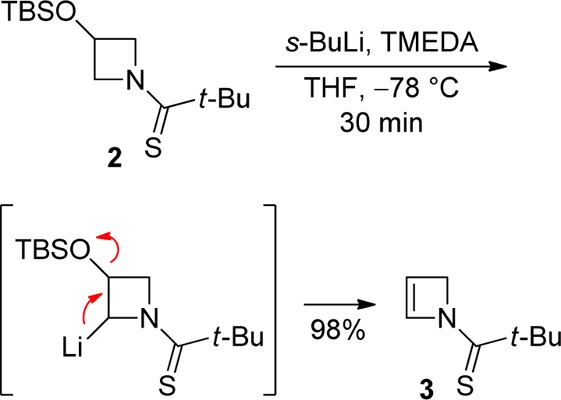
As part of a program with the aim of providing flexible and divergent routes to substituted 4-membered azacycles involving substituent incorporation onto a pre-existing and straightforwardly accessed azetidine core, we recently reported α-lithiation–electrophile trapping of N-thiopivaloylazetidine4 and -azetidin-3-ol5 in enantio- and diastereocontrolled processes, respectively. During the development of the latter work, we found that the unprotected 3-hydroxyl group was essential, as protection (e.g., as the TBS ether 2) only led to efficient elimination on attempted α-lithiation–electrophile trapping, to generate the corresponding stable 2-azetine 3 (98%) (Scheme 1).5 This observation led us to consider the possibility of α-electrophile incorporation using N-protected 2-azetine, in which the unsaturation retained in the adducts could provide a site for further synthetic manipulation. Here we communicate our promising preliminary results in this area.
Scheme 1. Lithiation–Elimination of Silyloxythioamide 2(5).
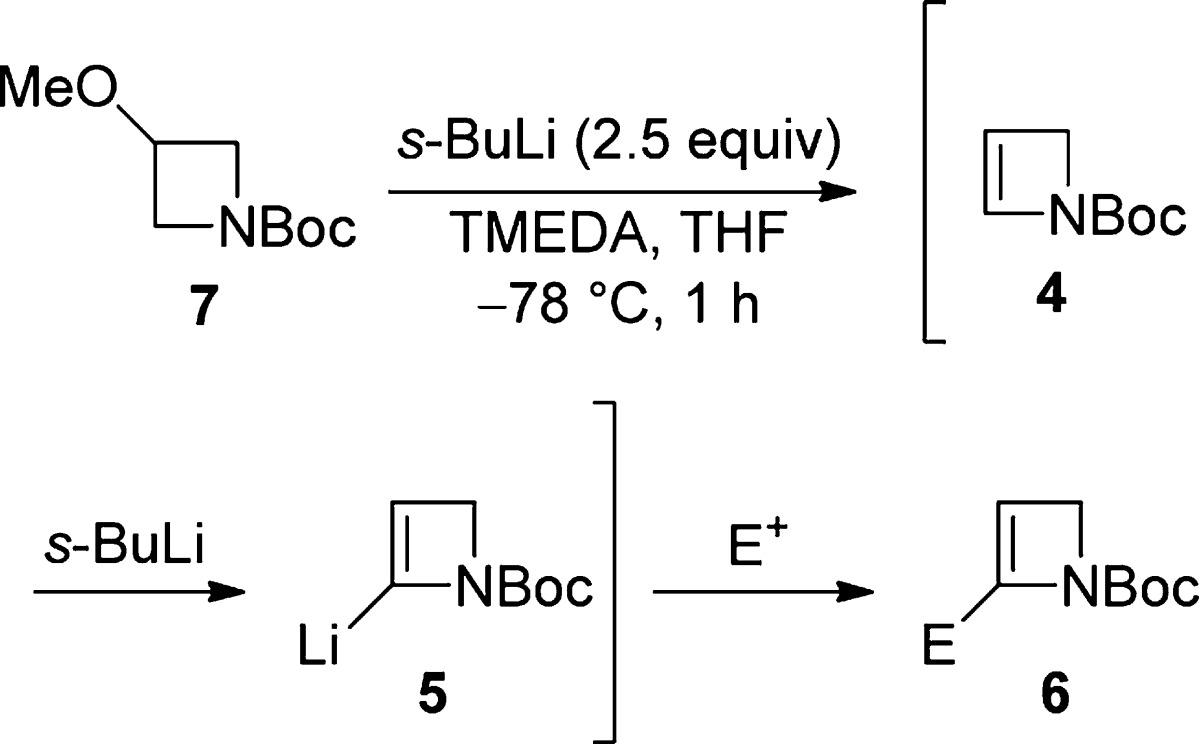
While N-protection/activation with the thiopivaloyl group had been found to be uniquely successful in enabling α-lithiation–electrophile trapping of azetidine and 3-hydroxyazetidine,4,5 with 2-azetine 3 only modest isolated yields of trapped products (∼15% using either MeI or BnBr) could be obtained despite extensive experimental investigations.6 We therefore turned our attention to alternative nitrogen protection. Following pioneering studies by Beak and co-workers, Boc protection now is well-established for facilitating α-lithiation–electrophile trapping of normal-sized azacycles7 (including Csp2 α-lithiation of 1,2,3,4-tetrahydropyridine8), although simple N-Boc aziridines undergo rapid N→C Boc migration following α-lithiation.9 In the present case, we were pleased to find that the known N-Boc-2-azetine (4)10 underwent regioselective lithiation at the α-sp2 position11 followed by deuteration to give 2-D-2-azetine 6a (54% yield, 100% D, Scheme 2) under conditions typically used with N-Boc heterocycles [s-BuLi (1.2 equiv), TMEDA (1.2 equiv), THF, −78 °C, 1 h]. Further experimentation established that n-BuLi was similarly effective (56%); the absence of TMEDA did not affect the yield (54%), and LiTMP (1.4 equiv, THF, −78 °C, 45 min) also delivered sp2 α-lithiation–trapping (70%).
Scheme 2. Lithiation–Deuteration of N-Boc-azetine (4).

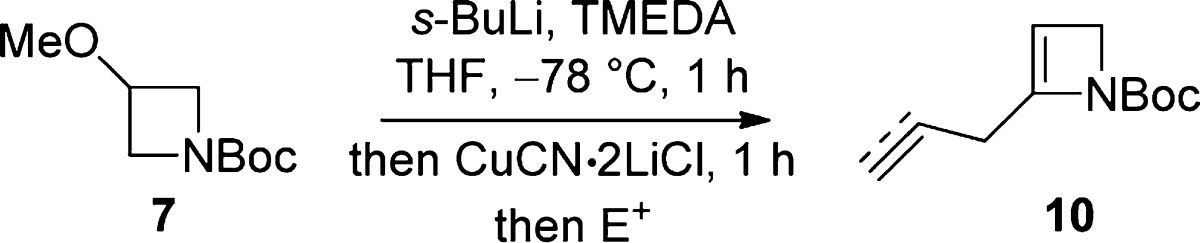
Despite the above encouraging preliminary results, yield variability in duplicate lithiation–deuteration experiments, together with modest yields in the synthesis of N-Boc-azetine (4) [previously prepared (46%) by t-BuOK-induced elimination from the corresponding 3-tosylate;10 in our hands, elimination (66%) by way of the 3-chloride was preferred12] and the instability/tendency of unsubstituted N-Boc-azetine (4) to polymerize when left at rt and slowly during storage at −20 °C, led us to consider a modified strategy7b,8 where the unsubstituted N-Boc-2-azetine (4) was generated and lithiated in situ. This approach resulted in an efficient and reproducible method to 2-D 6a (88% yield, 100% D) when N-Boc-3-methoxyazetidine (7), available commercially or readily prepared by methylation (NaH, MeI, quant)12 of widely available N-Boc-azetidin-3-ol, was reacted with 2.5 equiv of s-BuLi.13 The scope of the reaction with respect to electrophile variation was investigated following these conditions (Table 1).
Table 1. Scope of Carbonyl and Heteroatom Electrophiles with Li-azetine 5.
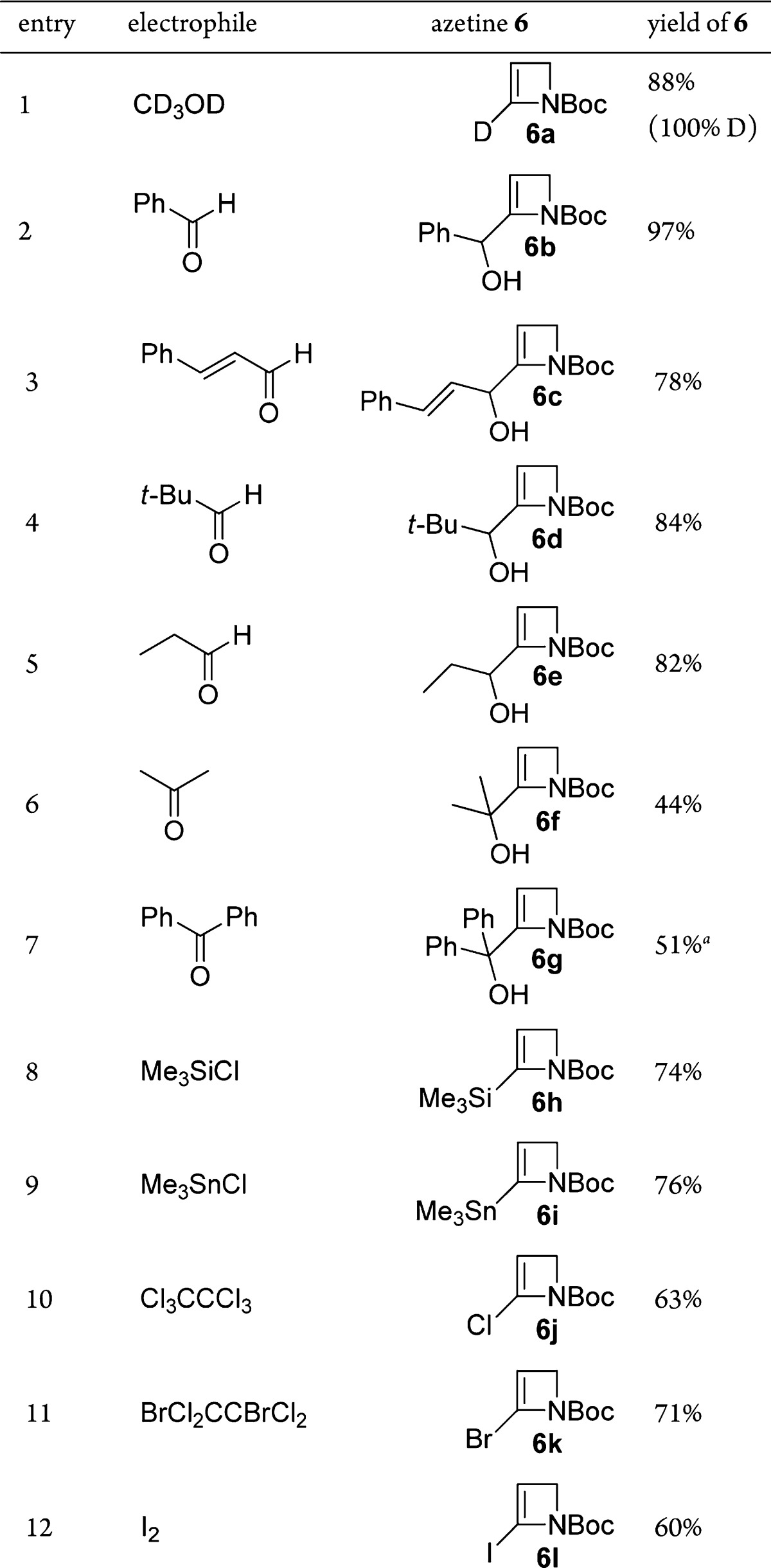
Yield corrected for inseparable N-Boc-azetine 4 (10% by 1H NMR analysis).12
Aldehydes (Table 1, entries 2–5) and ketones (entries 6 and 7), including an enal (entry 3) and potentially enolizable substrates (entries 5 and 6), provided the corresponding secondary and tertiary alcohols 6b–g in generally good yields. Using carbonyl-based electrophiles at a higher oxidation level (BzCl, ethyl chloroformate, Mander’s reagent) or alkylating agents (MeI, BuI, BnBr, allyl bromide) did not prove viable, only resulting in the isolation of N-Boc azetine 4 or decomposition of starting materials. However, heteroatom incorporation could be achieved, generating the corresponding silylated (entry 8), stannylated (entry 9), and halogenated 2-azetines (entries 10–12), although in these latter cases reversed-phase chromatography14 was necessary for satisfactory product isolation.
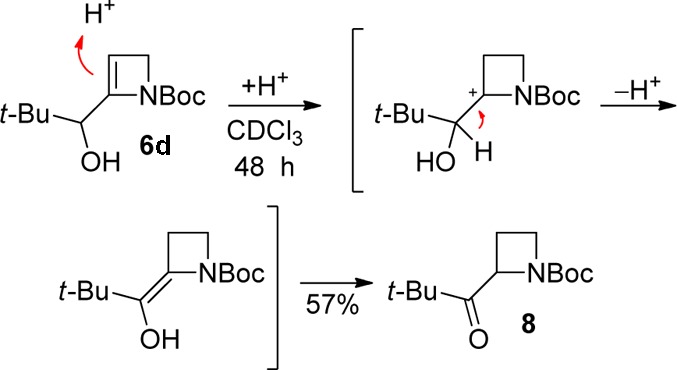
Observations with two electrophiles (pivalaldehyde and diphenyl disulfide) show further reactivity following electrophile trapping. While the hindered alcohol 6d derived from pivalaldehyde could be isolated in good yield as indicated (84%, Table 1, entry 4), exposure to CDCl3 which had not been passed through alumina prior to use resulted in prototropy15 driven by relief of strain, to the corresponding saturated ketone 8 (57%, Scheme 3); other aldehyde-derived adducts did not undergo analogous isomerizations.
Scheme 3. Isomerization of Alcohol 6d to Ketone 8.
Reaction with diphenyl disulfide (2.5 equiv) led, unusually, to N-Boc-α-phenylthio-β-lactam (9) (40%, Scheme 4). The formation of β-lactam 9 may proceed through initial electrophile trapping where the resulting electron-rich azetine 6m undergoes further reaction with the electrophile, followed by hydrolysis (Scheme 4); reduction in the quantity of (PhS)2 used (to 0.9 or 1.0 equiv) did not result in recovery of any identifiable products.
Scheme 4. β-Lactam 9 from 3-Methoxyazetidine 7.

While direct allylation of lithiated azetine 5 had proven problematic, transmetalation to the organocopper16 allowed C–C bond formation by nucleophilic substitution to give allylated and propargylated azetines 10 (Table 2).
Table 2. Scope of Copper-Mediated Allylation and Propargylation.
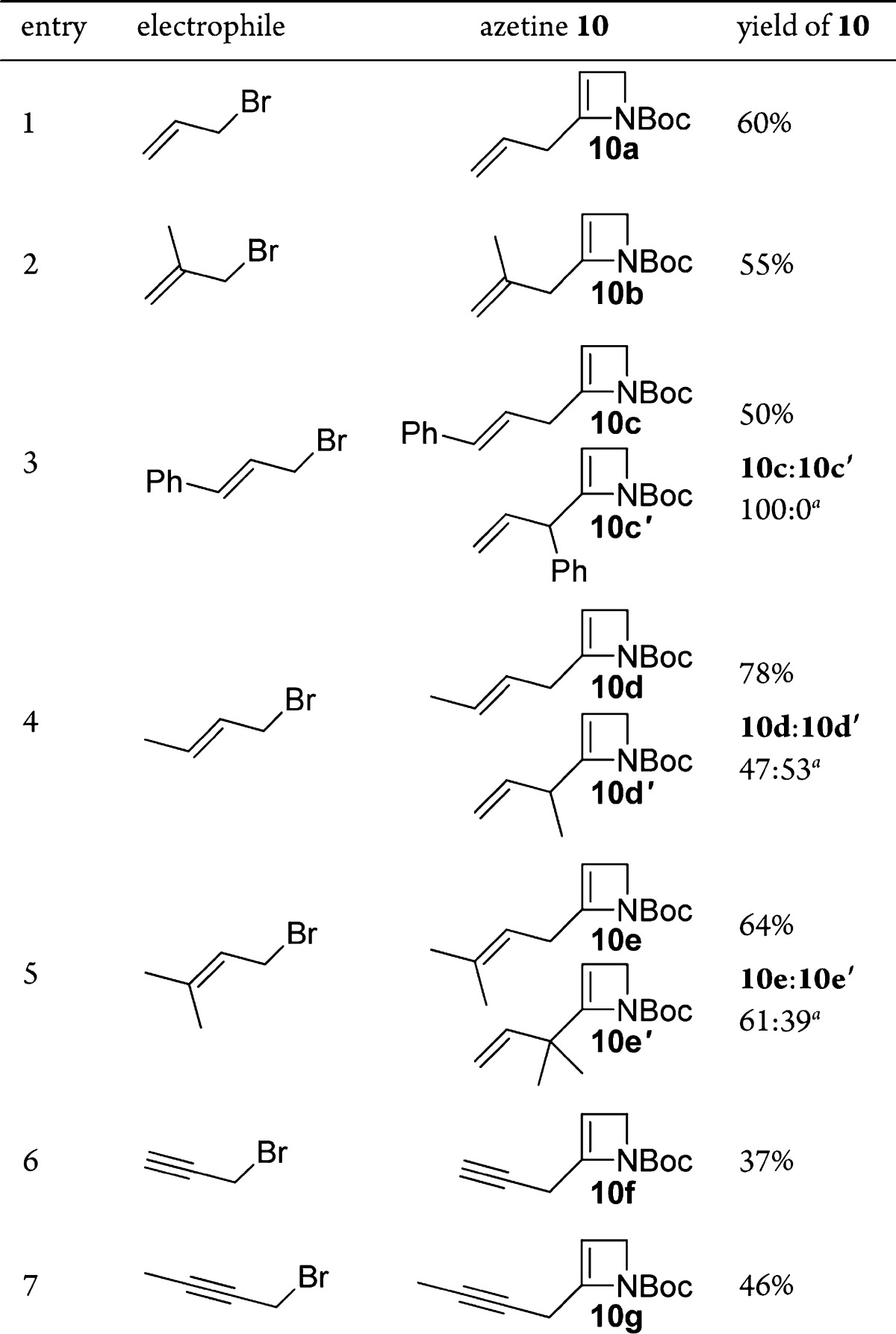
Isolated ratio after chromatography.
Simple allylation and methallylation proceeded satisfactorily (Table 2, entries 1 and 2). With unsymmetrical allylic bromides (cinnamyl, crotyl, and prenyl), mixtures of SN2- and SN2′-derived azetines were observed [SN2:SN2′ by crude 1H NMR analysis, 91:9 (entry 3), 47:53 (entry 4), 66:33 (entry 5)], while propargylation proceeded by SN2 (entries 6 and 7). The latter contrasts with SN2′ regioselectivity giving allenes seen with N-Boc-α-aminoalkylcuprates.17
While attempted Suzuki cross-couplings with bromide 6k did not prove viable, Negishi coupling18,19 gave 2-phenylated azetine 11 in 27% yield (Scheme 5).
Scheme 5. Phenylated Azetine 11 by Negishi Cross-Coupling.

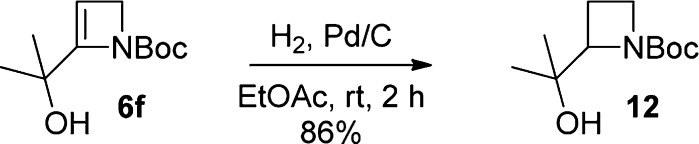
Hydrogenation3b of a carbonyl-derived azetinol 6f provided straightforward access to the corresponding saturated azetidine alcohol 12 (86%, Scheme 6); as noted previously, such adducts are not available by α-lithiation–electrophile trapping of N-Boc-azetidine, although N-thiopivaloylazetidine is a viable substrate.4
Scheme 6. Hydrogenation of a 2-Substituted Azetine.
In summary, commercially available N-Boc-3-methoxyazetidine (7) has been shown to undergo α-lithiation–elimination to form N-Boc-azetine (4) in situ, which can be further α-lithiated regioselectively at the sp2 center8,11 and trapped with a range of electrophiles, including allylic and propargylic halides by way of transmetalation to copper, providing a direct entry to 2-substituted N-Boc azetines. 2-Azetine stability depends significantly on the electron-withdrawing ability of the N substituent (aryl, sulfonyl, acyl, alkoxy carbonyl); our work demonstrates that the comparatively modestly electron-withdrawing Boc group is sufficient to allow isolation of 2-substituted 2-azetines, provided they are handled, and in many cases stored, under basic conditions. These studies indicate that electrophile incorporation can be achieved on simple monocyclic azetines and suggest that further opportunities exist for azetine diversity generation using this strategy, with potential to access substituted azetidines through double bond manipulation.
Acknowledgments
We thank AstraZeneca and the EPSRC for studentship support (to C.I.P.) and the Higher Education Commission of Pakistan for an IRSIP scholarship (to M.K.).
Supporting Information Available
Full experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Title and numbering in Scheme 3 were incorrect in the version published ASAP on January 10, 2014; the correct version reposted on January 13, 2014.
Supplementary Material
References
- Brandi A.; Cicchi S.; Cordero F. M. Chem. Rev. 2008, 108, 3988–4035. [DOI] [PubMed] [Google Scholar]
- a Moore J. A.; Ayers R. S. In Chemistry of Heterocyclic Compounds: Small Ring Heterocycles - Part 2; Hassner A., Ed.; Wiley: New York, 1983; Part 2, pp 1–217. [Google Scholar]; b Jubault P.; Leclerc E.; Quirion J.-C. In Science of Synthesis; Molander G. A., Ed.; Thieme: Stuttgart, 2006; Vol. 33, pp 577–582. [Google Scholar]
- For recent studies, see:; a Mangelinckx S.; Van Speybroeck V.; Vansteenkiste P.; Waroquier M.; De Kimpe N. J. Org. Chem. 2008, 73, 5481–5488. [DOI] [PubMed] [Google Scholar]; b Barluenga J.; Riesgo L.; Lonzi G.; Tomás M.; López L. A. Chem.—Eur. J. 2012, 18, 9221–9224. [DOI] [PubMed] [Google Scholar]
- Hodgson D. M.; Kloesges J. Angew. Chem., Int. Ed. 2010, 49, 2900–2903. [DOI] [PubMed] [Google Scholar]
- Hodgson D. M.; Pearson C. I.; Thompson A. L. J. Org. Chem. 2013, 78, 1098–1106. [DOI] [PubMed] [Google Scholar]
- Chromatographic purification of these substituted thioamide azetines proved challenging, usually resulting in significant loss of material, and with products typically eluting with nonpolar byproducts.
- a Beak P.; Lee W.-L. Tetrahedron Lett. 1989, 30, 1197–1200. [Google Scholar]; b Beak P.; Lee W. L. J. Org. Chem. 1993, 58, 1109–1117. [Google Scholar]; c Gawley R. E.; O’Connor S.; Klein R. In Science of Synthesis; Snieckus V., Majewski M., Eds.; Thieme: Stuttgart, 2006; Vol. 8a, pp 677–757. [Google Scholar]
- Wilkinson T. J.; Stehle N. W.; Beak P. Org. Lett. 2000, 2, 155–158. [DOI] [PubMed] [Google Scholar]
- a Capriati V.; Florio S.; Luisi R.; Musio B. Org. Lett. 2005, 7, 3749–3752. [DOI] [PubMed] [Google Scholar]; b Hodgson D. M.; Humphreys P. G.; Xu Z.; Ward J. G. Angew. Chem., Int. Ed. 2007, 46, 2245–2248. [DOI] [PubMed] [Google Scholar]; c Hodgson D. M.; Humphreys P. G.; Miles S. M.; Brierley C. A. J.; Ward J. G. J. Org. Chem. 2007, 72, 10009–10021. [DOI] [PubMed] [Google Scholar]
- McDonald R. I.; Wong G. W.; Neupane R. P.; Stahl S. S.; Landis C. R. J. Am. Chem. Soc. 2010, 132, 14027–14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tae J.; Paquette L. A. Can. J. Chem. 2000, 78, 689–696. [Google Scholar]; b Herzon S. B.; Myers A. G. J. Am. Chem. Soc. 2005, 127, 5342–5344. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for details.
- s-BuLi without TMEDA gave 2-D-2-azetine 6a (74% 100% D). LiTMP (2.5 equiv, THF −78 °C) gave 2-D-2-azetine 6a (62%, 100% D) after 5 h (48%, 100% D after 30 min).
- Farina V. J. Org. Chem. 1991, 56, 4985–4987. [Google Scholar]
- Green M. B.; Hickinbottom W. J. J. Chem. Soc. 1957, 3270–3274. [Google Scholar]
- Dieter R. K.; Oba G.; Chandupatla K. R.; Topping C. M.; Lu K.; Watson R. T. J. Org. Chem. 2004, 69, 3076–3086. [DOI] [PubMed] [Google Scholar]
- Coldham I.; Leonori D. J. Org. Chem. 2010, 75, 4069–4077. [DOI] [PubMed] [Google Scholar]
- a Campos K. R.; Klapars A.; Waldman J. H.; Dormer P. G.; Chen C. J. Am. Chem. Soc. 2006, 128, 3538–3539. [DOI] [PubMed] [Google Scholar]; b Barker G.; O’Brien P.; Campos K. R. Org. Lett. 2010, 12, 4176–4179. [DOI] [PubMed] [Google Scholar]; c Barker G.; McGrath J. L.; Klapars A.; Stead D.; Zhou G.; Campos K. R.; O’Brien P. J. Org. Chem. 2011, 76, 5936–5953. [DOI] [PubMed] [Google Scholar]
- No 2-phenylated azetine 11 was isolated if TMEDA was present; see also ref (18c).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.