

Abstract

Histone Nε-methyl lysine demethylases KDM2/7 have been identified as potential targets for cancer therapies. On the basis of the crystal structure of KDM7B, we designed and prepared a series of hydroxamate analogues bearing an alkyl chain. Enzyme assays revealed that compound 9 potently inhibits KDM2A, KDM7A, and KDM7B, with IC50s of 6.8, 0.2, and 1.2 μM, respectively. While inhibitors of KDM4s did not show any effect on cancer cells tested, the KDM2/7-subfamily inhibitor 9 exerted antiproliferative activity, indicating the potential for KDM2/7 inhibitors as anticancer agents.
Introduction
Nε-Methylation of lysine residues on histone tails is a pivotal “epigenetic mark” that is involved in defining both transcriptionally active and inactive chromatin.1 For instance, methylation at lysine 4 of histone 3 (H3K4) is associated with actively transcribed gene loci, whereas methylation at H3K9 and H3K27 leads to reduction in transcription.2 Methylation of histone lysine residues is reversible and is mediated by histone lysine methyltransferases and histone lysine demethylases (KDMs).3
KDMs fall into two classes as defined by their structure and mechanisms.3 One class comprises the KDM1s, which are homologues of the flavin-containing amine oxidases.3a,3b The other class comprises Jumonji C (JmjC)-domain containing demethylases, which belong to the family of Fe(II)/α-ketoglutarate-dependent oxygenases.3c,3d Human JmjC-domain containing demethylases can be divided into five subfamilies, i.e., the KDM2/7, KDM3, KDM4, KDM5, and KDM6 subfamilies, according to sequence and structural similarities.4
Several JmjC-domain containing demethylases such as KDM2, KDM4, KDM5, and KDM7 have been implicated in tumorigenesis.5 For example, it has been reported that KDM7B (also known as JHDM1F, PHF8, KIAA1111) is associated with proliferation of prostate cancer cells and osteosarcoma cells.6 Therefore, KDM2/7-subfamily inhibitors are of interest, both as tools for probing the biological functions of KDM2/7 subfamily and also as candidate anticancer agents. Several types of KDM inhibitors have been found so far by us and others,5a,7 including N-oxalylglycine (NOG, 1), 2,4-pyridinedicarboxylic acid (PCA, 2), NCDM-32 (3), daminozide (4), and GSK-J1 (5) (Chart 1). Among these, daminozide (4) has been reported as a KDM2/7-subfamily selective inhibitor. Daminozide (4) was once widely used as a plant growth retardant but later withdrawn because of genotoxic concerns.8 The genotoxicity of daminozide (4) is proposed to be derived from its 1,1-dimethylhydrazine structure.9 Therefore, it is desirable to identify novel KDM2/7-inhibitors without the 1,1-dimethylhydrazine structure for use as functional probes and for evaluation in cancer therapies. Here we describe the identification of a novel KDM2/7 subfamily inhibitor that shows antiproliferative activity.
Chart 1. Examples of Reported Small Molecule KDM Inhibitors.

Chemistry
The routes used for the synthesis of compounds 7–13 (Chart 2), which were prepared for this study, are shown in Schemes 1 and 2. Scheme 1 shows the preparation of compounds 8–13. Michael addition of O-benzylhydroxylamine to tert-butyl acrylate 14 afforded amine 15. 9-Bromononanoic acid sodium salt 16 was converted to alkyl carboxylic acids 19–22 by treatment with an alkyl magnesium bromide 17 and dilithium tetrachlorocuprate. 9-Phenylnonanoic acid 23 was obtained by the treatment of 16 with phenyl magnesium bromide and ferric acetylacetonate in the presence of tetramethylethylenediamine and hexamethylenetetramine in THF. Carboxylic acids 18–23 were treated with amine 15 in the presence of EDCI and HOBt to give amides 24–29. The benzyl group of compounds 24–29 was removed by hydrogenation to give hydroxamates 30–35. Removal of the tert-butyl group of 30–35 using hydrochloric acid afforded the desired compounds 8–13.
Chart 2. Structures of Compounds 7–13.

Scheme 1. Synthesis of Compounds 8–13.

Reagents and conditions: (a) O-benzylhydroxylamine, Et3N, 1,4-dioxane, reflux, 44%; (b) RMgBr (17), Li2CuCl4, THF, −10 °C, 74–91%; (c) PhMgBr, Fe(acac)3, TMEDA, HMTA, THF, 0 °C, 67%; (d) EDCI, HOBt, DMF, room temp, 24–72%; (e) H2, Pd/C, AcOEt, room temp, 34–95%; (f) HCl, CH2Cl2, room temp, 23–59%.
Scheme 2. Synthesis of Compound 7.

Reagents and conditions: (a) O-benzylhydroxylamine, Et3N, 1,4-dioxane, reflux, 59%; (b) iPrMgBr, Li2CuCl4, THF, −10 °C, 29%; (c) EDCI, HOBt, DMF, room temp, 65%; (d) H2, Pd/C, AcOEt, room temp, 61%.
Scheme 2 shows the synthesis of compound 7. Michael addition of O-benzylhydroxylamine to methyl acrylate 36 afforded amine 37. 10-Methylundecanoic acid 38 was prepared using the procedure described for the synthesis of 19–22. Amine 37 was treated with 38 in the presence of EDCI and HOBt to yield compound 39. Removal of the benzyl group of compound 39 gave compound 7.
Results and Discussion
With the aim of identifying potent KDM2/7 inhibitors, hydroxamate derivatives, which have been previously prepared in work on KDM4 inhibitors,7d were screened for KDM7B-, KDM4A (also known as JMJD2A, JHDM3A)-, KDM4C (also known as JMJD2C, JHDM3C, GASC1)-, and KDM5A (also known as JARID1A, RBP2)-inhibitory activity. We selected KDM4A, KDM4C, and KDM5A for the primary inhibition selectivity studies because they are relatively similar to KDM7B in that they have a plant homeodomain.4 Compounds 1–4 were evaluated as positive controls for inhibition. In our enzyme assays, compound 4 selectively inhibited KDM7B in preference to KDM4 and KDM5 as reported before (Table 1).4 We also found that compounds 1(7a) and 2(7b) did not inhibit KDM7B (at 250 μM), and compound 2 showed selectivity for the KDM4-subfamily over KDM7B as for the reported KDM4 inhibitor 3(7d) under the conditions tested. Among the tested compounds, compound 6 was found to inhibit KDM7B relatively selectively although its potency was moderate (IC50 = 24 μM).
Table 1. In Vitro KDM7B-, KDM4A-, KDM4C-, and KDM5A-Inhibitory Activities of Compounds 1–4 and Screening Hit Compound 6a.

Values are means of two experiments that in all cases varied by 19% or less.
The IC50 of 4 for KDM7B was reported to be 0.55 μM in AlphaScreen assays.4
To improve the potency and selectivity of 6 for KDM7B, we investigated the binding mode of 6 with KDM7B by modeling (Figure 1).10 An inspection of the simulated KDM7B/compound 6 complex indicates that the hydroxamate group of compound 6 coordinates to the Fe(II) in a bidentate manner, via its carbonyl and hydroxyl groups, and that its carboxylate group will bind to form hydrogen bonds/electrostatic interactions with Asn 189, Thr 244, and Lys 264 at the KDM7B active site, in a similar manner to the C-5 carboxylate of 2-oxoglutarate. Most importantly, it is also suggested that the alkyl chain of compound 6 binds to a hydrophobic pocket formed by the side chains of Ile 191, Tyr 234, Leu 236, Phe 250, Val 255, Tyr 257, and Phe 359, which we propose is unique to KDM2 and KDM7 (Supporting Information Figures S1–S6).
Figure 1.
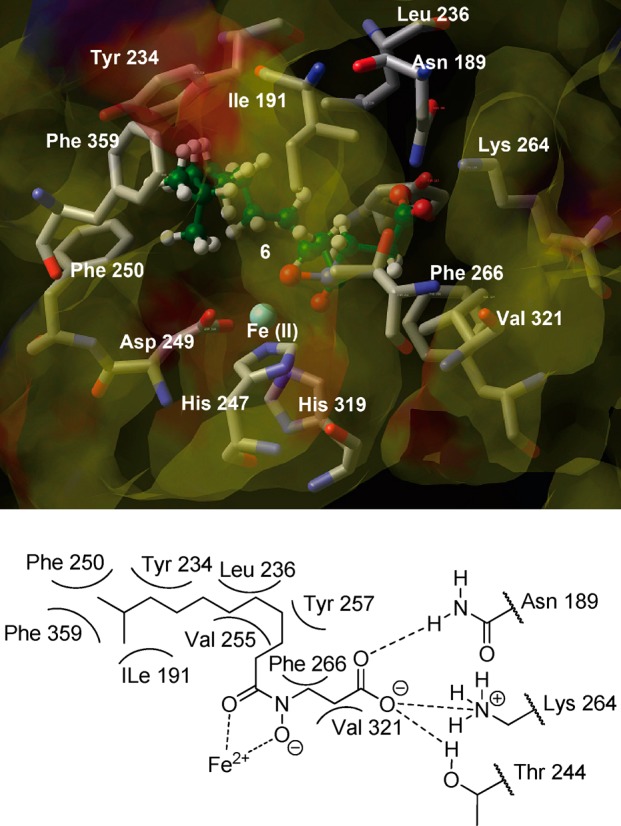
View of the conformation of compound 6 (ball-and-stick) docked into the KDM7B active site.
On the basis of the structure of the modeled KDM7B/compound 6 complex, we designed and synthesized compounds 8–13 (Table 2) in which the isopropyl group of compound 6 was replaced with various hydrophobic groups that might interact with hydrophobic amino acid residues more efficiently than compound 6. Compound 7 was prepared to investigate the importance of the carboxylic acid of the inhibitor.
Table 2. In Vitro KDM7B-, KDM2A-, KDM4A-, KDM4C-, KDM5A-, and KDM6A-Inhibitory Activities of Compounds 4 and 6–13a.

Values are means of two experiments that in all cases varied by 30% or less.
The IC50 of 4 for KDM7B was reported to be 0.55 μM in AlphaScreen assays.4
Not determined.
As shown in Table 2, the conversion of carboxylic acid (6) to a methyl ester (7) significantly reduced the KDM7B-inhibitory activity, supporting the proposed binding mode of compound 6. Compounds 8–13 were found to be KDM7B inhibitors. In particular, a pronounced inhibitory effect (IC50 = 1.2 μM) was observed with the cyclopropyl-containing compound 9, which was 20-fold more active than 6 in KDM7B in MALDI assays. Docking of compound 9 in the active site of KDM7B suggested that the cyclopropyl group of compound 9 interacts with the phenyl-group of Phe 250 through CH−π or cyclopropyl−π interactions (Figure 2). Furthermore, compound 9 inhibited KDM2A (also known as JHDM1A, FBXL11), KDM7A (also known as JHDM1D, KIAA1718), and KDM7B, demonstrating inhibition of all KDM2/7 subfamily members tested (Tables 2 and 3). Compound 9 displayed selectivity for KDM2/7 over KDM4A (IC50 > 120 μM), KDM4C (IC50 = 83 μM), KDM5A (IC50 = 55 μM), and KDM6A (also known as UTX) (IC50 > 100 μM); note however different assays conditions were used (see Experimental Section). Thus, compound 9 was the most potent KDM7B inhibitor identified in the enzyme assays. In addition, compound 13 showed comparatively high selectivity for KDM2A over KDM7B and the other isozymes tested. Because the molecular modeling suggest that the space around the cyclopropane ring of 9 is not so large in the hydrophobic pocket of KDM7B (Figure 2), it may be difficult for compound 13 bearing a phenyl ring to have a conformation which can efficiently interact with Phe of the pocket. On the other hand, KDM2A has a hydrophobic pocket more spacious than KDM7B (Supporting Information Figure S2), in which the phenyl ring can efficiently interact with hydrophobic amino acid residues of KDM2A. These may be the reason that compound 13 shows selectivity for KDM2A over KDM7B.
Figure 2.

View of the conformation of compound 9 (ball-and-stick) docked into the KDM7B active site.
Table 3. Enzyme-Inhibitory Activities and Antiproliferative Activities of Compounds 1–3 and 9a.
| IC50 (μM) |
GI50 (μM) |
|||||
|---|---|---|---|---|---|---|
| compd | KDM2A | KDM7A | KDM7B | KDM4C | KYSE150 | HeLa |
| 1 | 45 | >250 | >250 | >250 | >240 | >710 |
| 2 | 4.1 | 15 | >250 | 8.2 | >240 | >240 |
| 3 | 0.20 | 1.6 | 19 | 2.2 | >470 | >470 |
| 9 | 6.8 | 0.20 | 1.2 | 83 | 16 | 40 |
Values are means of two experiments that in all cases varied by 30% or less.
To investigate whether compound 9 is active as an inhibitor of KDM7A and KDM7B in cells, we performed a cellular assay with Western blot analysis. Because KDM7 is known to catalyze the demethylation of H3K27me2,10,11 the methylation level of H3K27 in cells was analyzed. In this study, we used N2a cells because it has been reported that KDM7 is expressed in the cells.11 As Figure 3 shows, the level of H3K27me2 was dose-dependently elevated in the presence of 9. Although the interpretation of changes in global histone N-methylation status can be complex, the elevation in the H3K27me2 levels is consistent with KDM7 inhibition. These results suggest that H3K27me2 demethylation is inhibited by compound 9 in cells, and it looks to be useful as a tool for probing the biological role of KDM7.
Figure 3.
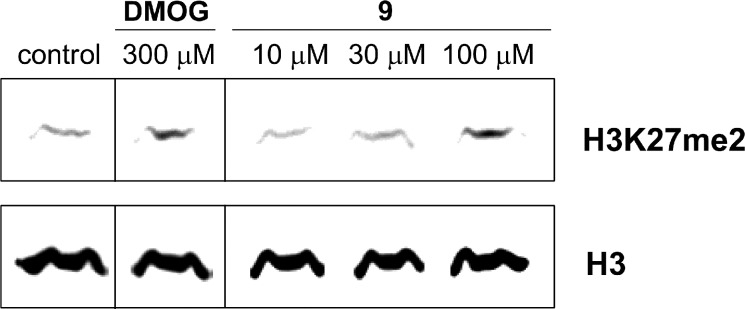
Western blot detection of H3K27me2 levels in N2a cells after 24 h incubation with dimethyl ester prodrug of NOG (DMOG) and compound 9.
Although it has been reported that RNAi-mediated knockdown of KDM7B suppresses the growth of some cancer cells,6 it remains unclear whether the demethylase function of KDM7B is responsible for the suppression because RNAi-mediated knockdown of KDM7B should cause loss of not only the demethylase function but also other functions of KDM7B,12 including those relating to noncatalytic binding domains. Initially, we investigated the N2a cell growth inhibition activity of compound 9. N2a cell growth suppression by compound 9 was observed (GI50 = 86 μM) (Figure 4) at the concentration range in which distinct H3K27 methylation was detected on Western blot analysis (Figures 3). Thus, this may suggest the demethylase function of KDM7 is involved in this cell growth, however, more potent and selective compounds will be needed to fully elucidate this fact. Next, we carried out cell growth inhibition assays of compound 9 as well as prodrugs of 1, 2, and 3 using HeLa cells and KYSE150 cells (Table 3). It has also been reported that knockdown of KDM4C decreases cell proliferation,7a however, the KDM4C inhibitors 2, 3 (Table 3), and their methyl ester prodrugs (GI50 > 500 μM) did not show any effects on the growth inhibition of tested cancer cells although they are cell membrane permeable.7d,13 These results suggest that the demethylase activity of KDM4C is not directly associated with cancer cell growth, at least in some cell types. On the other hand, cell growth suppression by the KDM2/7-subfamily inhibitor 9 was observed for KYSE150 and HeLa cell lines (Figure 5). Furthermore, compound 9 caused H3K27 methylation both in HeLa cells and in KYSE150 cells at the concentration range in which the cell growth inhibition was observed (Figure 6). The data shown in Table 3 indicate that KDM2/7 inhibitors are worthy of evaluation as candidate anticancer agents.
Figure 4.

Growth-inhibitory activity of compound 9 toward N2a cell lines.
Figure 5.
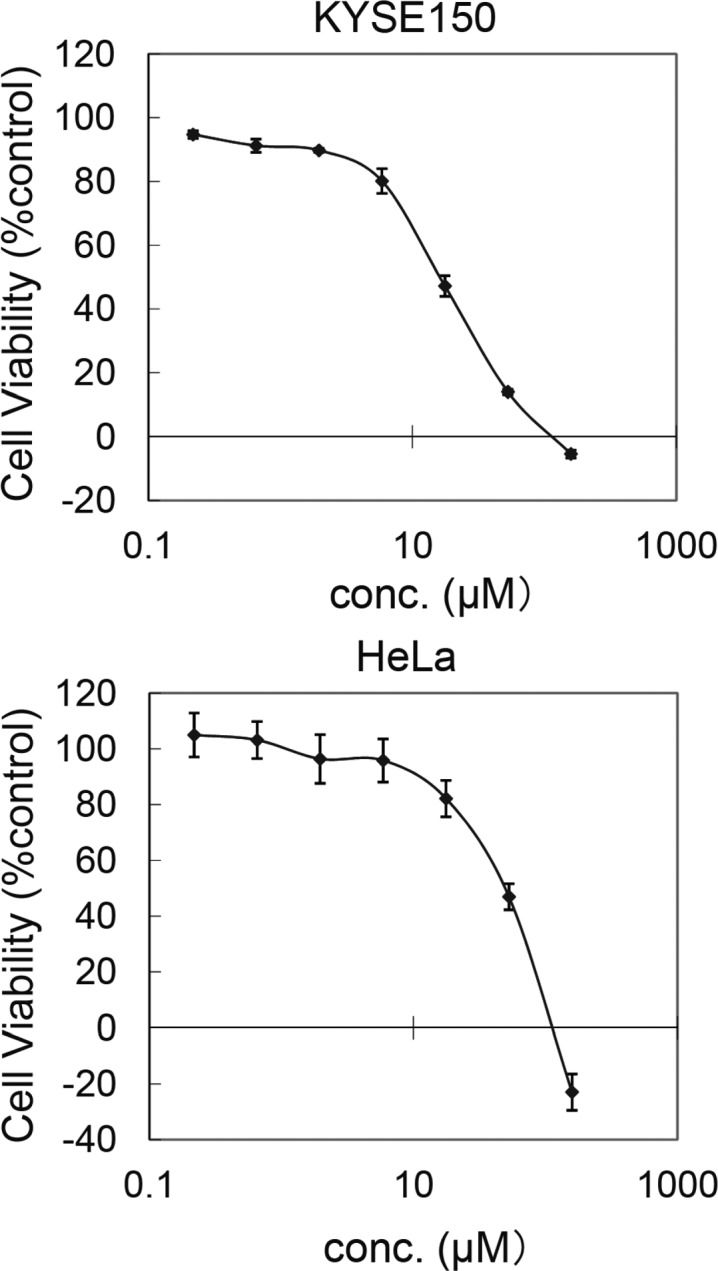
Growth-inhibitory activity of compound 9 toward KYSE150 and HeLa cell lines.
Figure 6.
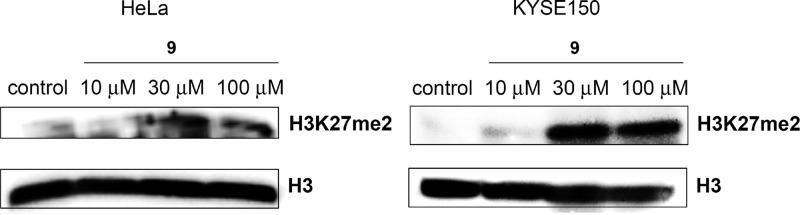
Western blot detection of H3K27me2 levels in HeLa cells and KYSE150 cells after 24 h incubation with compound 9.
A recent study reported that KDM7B activates the transcription of the E2F1 transcription factor in HeLa cells, which promotes cell cycle progression.14 Because compound 9 decreases the growth of HeLa cells with H3K27me2 accumulation (Table 3; Figures 5 and 6), we examined whether compound 9 down-regulates the expression of E2F1 in HeLa cells by quantitative RT-PCR. As shown in Figure 7, compound 9 significantly decreases the mRNA level of E2F1 at 80 μM in which the growth of HeLa cells was affected. These data suggest that the KDM7B-mediated regulation of E2F1 gene expression may be one of the mechanisms of growth regulation in some cancer cells.
Figure 7.
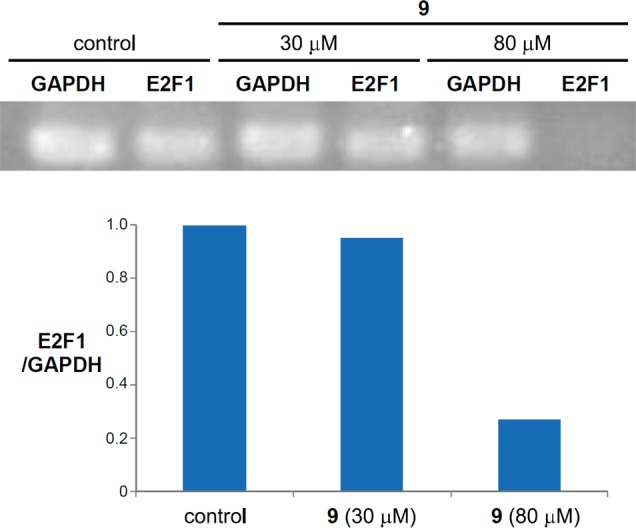
Change of E2F1 gene expression in HeLa cells by compound 9.
We also investigated the effect of compound 9 on cell cycle progression by FACS analysis. HeLa cells and KYSE150 cells incubated with 10 or 100 μM of compound 9 for 24 h showed a dose-dependent reduction in G2–M phase, whereas there was a dose-dependent increase in G0–G1 phase (Figure 8). These results revealed that HeLa cells and KYSE150 cells cultured with compound 9 arrested in the G0/G1 phase of the cell cycle, which is consistent with the down-regulation of E2F1 by compound 9 (Figure 7).15
Figure 8.

Induction of cell cycle arrest in HeLa cells and KYSE150 cells by compound 9.
Conclusions
We have identified a novel KDM2/7 subfamily inhibitor 9, which should be useful as a lead structure in the development of more potent and selective KDM2/7 inhibitors. Such inhibitors are candidates for anticancer agents as well as tools for studying the biological roles of KDM2/7 subfamily in cells.
Experimental Section
Chemistry
Melting points were determined using a Yanagimoto micro melting point apparatus or a Büchi 545 melting point apparatus. Proton nuclear magnetic resonance spectra (1H NMR) and carbon nuclear magnetic resonance spectra (13C NMR) were recorded using a JEOL JNM-LA500, JEOL JNM-A500, or BRUKER AVANCE600 spectrometer in solvent as indicated. Chemical shifts (δ) are reported in parts per million relative to the internal standard tetramethylsilane. Elemental analysis was performed with a Yanaco CHN CORDER NT-5 analyzer, and all values were within ±0.4% of the calculated values, which indicates >95% purity of the tested compounds. High-resolution mass spectra (HRMS) and fast atom bombardment (FAB) mass spectra were recorded on a JEOL JMS-SX102A mass spectrometer. Purity tests by analytical HPLC used a Shimazu instrument equipped with a ODS-3 (4.6 mm × 150 mm, GL Science) and eluted at 1 mL/min with Milli-Q water and CH3CN, and all values ≥95% purity. Preparative HPLC was performed with a Jasco instrument equipped with a Inertsil ODS-3 (20 mm × 250 mm, GL Science) and eluted at 10 mL/min with Milli-Q water and CH3CN. Gradient conditions of HPLC were as follows: (A is CH3CN containing 0.1% TFA (CF3CO2H), B is Milli-Q water containing 0.1% TFA, C is CH3CN containing 0.1% FA, D is Milli-Q water containing 0.1% FA (HCOOH)); gradient (I), A 50% (0–2 min), A 50% to A 80% (2–20 min), A 80% (20– 30 min), A 80% to A 50% (3– 40 min) water B; gradient (II), C 60% (0– 2 min), C 60% to C 80% (2–20 min), C 80% (20–30 min), C 80% to C 50% (30–40 min) water D; gradient (III), C 50% (0–2 min), C 50% to C 80% (2–20 min), C 80% (20–30 min), C 80% to C 50% (30–40 min) water D. Reagents and solvents were purchased from Aldrich, Tokyo Kasei Kogyo, Wako Pure Chemical Industries, and Kanto Kagaku and used without purification. Flash column chromatography was performed using Silica Gel 60 (particle size 0.046–0.063 mm) supplied by Merck.
3-(9-Cyclopropyl-N-hydroxynonanamido)propanoic Acid (9)
Step 1: Preparation of tert-Butyl 3-(benzyloxyamino)propanoate (15)
A solution of tert-butyl acrylate (11.5 g, 90 mmol), benzylhydroxylamine hydrochloride (3.6 g, 26 mmol), and triethylamine (TEA) (6.0 mL) in dioxane (60 mL) was stirred with reflux for 19 h. The reaction mixture was poured into water and extracted with AcOEt. The organic layer was separated and washed with brine and dried over Na2SO4. Filtration, concentration in vacuo, and purification by flash column chromatography (AcOEt/n–hexane = 1/4) gave 3.3 g (44%) of 15 as yellow oil. 1H NMR (CDCl3, 500 MHz, δ; ppm) 7.35 (5H, m), 5.83 (1H, s), 4.70 (2H, s), 3.17 (2H, m), 2.50 (2H, m), 1.44 (9H, s).
Step 2: Preparation of Cyclopropanenonanoic Acid (19)
To the suspension of 9-bromononanoic acid sodium salt (16) (400 mg, 1.5 mmol) in anhydrous THF (3 mL) was added the solution of Li2CuCl4 in THF (200 μL, 0.02 mmol) at −10 °C. Then, cyclopropyl magnesium bromide (4.6 mL, 2.3 mmol) was added dropwise with vigorous stirring. The color of the mixture changed from orange through green and blue to white. After addition of the Grignard reagent, the temperature was kept at −10 °C for another 2 h. The reaction mixture was then diluted with water and extracted three times with Et2O, and the organic extracts were washed by saturated aqueous solution of Na2CO3. The water layer was then acidified by HCl and washed three times by Et2O, and the combined organic layers were washed with water and dried over Na2SO4. Filtration and concentration in vacuo gave 19 (226 mg, 74%) as a white solid. 1H NMR (CDCl3, 500 MHz, δ; ppm) 2.35 (2H, t, J = 7.5 Hz), 1.66–1.59 (2H, m), 1.32–1.21 (12H, m), 1.18–1.14 (2H, m), 0.67–0.60 (1H, m), 0.41–0.35 (2H, m).
Step 3: Preparation of tert-Butyl 3-(N-Benzyloxy-9-cyclopropylnonanamido)propanoate (25)
A mixture of 19 (221 mg, 1.10 mmol), 15 (232 mg, 1.10 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) (320 mg, 1.70 mmol), and HOBt·H2O (226 mg, 1.70 mmol) in DMF (5 mL) was stirred at 80 °C for 15 h. The mixture was poured into water and extracted with AcOEt. The organic layer was washed with brine and dried over Na2SO4. Filtration, concentration in vacuo, and purification by silica gel flash column chromatography (AcOEt/CHCl3 = 1/10) gave 115 mg (24%) of 25 as a colorless oil. 1H NMR (CDCl3, 500 MHz, δ; ppm) 7.38 (5H, m), 4.82 (2H, s), 3.94–3.88 (2H, m), 2.53 (2H, t, J = 7.0 Hz), 2.35 (2H, t, J = 7.0 Hz), 1.60–1.54 (4H, m), 1.41 (9H, s), 1.30–1.24 (10H, m), 1.19–1.15 (2H, m), 0.66–0.63 (1H, m), 0.40–0.36 (2H, m).
Step 4: Preparation of tert-Butyl 3-(9-Cyclopropyl-N-hydroxynonanamido)propanoate (31)
To a solution of 25 (116 mg, 0.27 mmol) in 3 mL of AcOEt was added 50 mg of 5% Pd/C. The mixture was stirred under H2 at room temperature for 1 h and then filtered through Celite. The filtrate was concentrated in vacuo and purified by flash column chromatography (AcOEt/CHCl3 = 3/1) to give 72 mg (78%) of 31 as a white solid. 1H NMR (CDCl3, 500 MHz, δ; ppm) 8.36 (1H, broad s), 8.13 (1H, broad s), 3.89–3.83 (2H, m), 2.65 (2H, t, J = 6.0 Hz), 2.46–2.40 (2H, m), 1.60–1.54 (4H, m), 1.46 (9H, s), 1.40–1.25 (10H, m), 1.19–1.15 (2H, m), 0.65–0.62 (1H, m), 0.40–0.36 (2H, m).
Step 5: Preparation of 3-(9-Cyclopropyl-N-hydroxynonanamido)propanoic Acid (9)
To a solution of 31 (69 mg, 0.20 mmol) in DCM (1 mL) was added 4N HCl in dioxane (1 mL) in a dropwise fashion with ice-bath cooling; the mixture was stirred at room temperature for 16 h. Concentration in vacuo gave a colorless solid. The solid was recrystallized from AcOEt to give 13 mg (23%) of 9 as colorless crystals: mp 86–87 °C. 1H NMR (CDCl3, 500 MHz, δ; ppm) 3.94 (2H, m), 2.79 (2H, m), 2.42 (2H, m), 1.64 (4H, m), 1.37–1.29 (10H, m), 1.19–1.15 (2H, m), 0.64 (1H, m), 0.40–0.36 (2H, m). 13C NMR (CDCl3, 125 MHz, δ; ppm) 176.88, 173.92, 44.62, 44.26, 38.06, 34.75, 29.63, 29.47, 29.36, 26.50, 25.27, 24.63, 10.96, 4.36. MS (FAB) m/z 286 (MH+). HRMS calcd for C15H28O4N, 286.20183, found 286.20242. HPLC tR = 10.27 min (gradient (III), purity 96.1%).
3-(N-Hydroxydecanamido)propanoic Acid (8)
Compound 8 was prepared from 18 using the procedure described for 9 (steps 3–5): yield 14% from 18 (800 mg, 4.6 mmol); a pale-pink solid; mp 84–85 °C. 1H NMR (CDCl3, 500 MHz, δ; ppm) 3.96–3.90 (2H, m), 2.80–2.76 (2H, m), 2.47–2.42 (2H, m), 1.70–1.50 (2H, m), 1.30–1.27 (12H, m), 0.88 (3H, t, J = 7.0 Hz). 13C NMR (CDCl3, 125 MHz, δ; ppm) 177.20, 174.61, 44.50, 32.42, 31.88, 31.60, 29.46, 29.37, 29.28, 25.31, 24.66, 22.67, 14.11. MS (FAB) m/z 260 (MH+). HRMS calcd for C13H26O4N, 260.18618, found 260.18719. HPLC tR = 10.37 min (gradient (I), purity 96.8%).
3-(9-Cyclopentyl-N-hydroxynonanamido)propanoic Acid (10)
Compound 10 was prepared from 16 (400 mg, 1.5 mmol) using the procedure described for 9 (steps 2–5): yield 14%; a white solid; mp 101–102 °C. 1H NMR (CDCl3, 500 MHz, δ; ppm) 3.93 (2H, m), 2.79 (2H, m), 2.42 (2H, m), 1.72 (4H, m), 1.60–1.55 (3H, m), 1.53–1.48 (2H, m), 1.30–1.26 (12H, m), 1.06–1.04 (2H, m). 13C NMR (CDCl3, 125 MHz, δ; ppm) 176.76, 174.28, 44.62, 44.23, 40.18, 36.26, 32.74, 32.44, 32.17, 31.32, 31.05, 29.90, 29.53, 29.38, 28.79, 25.21, 24.62. MS (FAB) m/z 314 (MH+). HRMS calcd for C17H32O4N, 314.23313, found 314.23329; Anal. Calcd for C17H31NO4: C, 65.14; H, 9.97; N, 4.47. Found: C, 64.75; H, 9.95; N, 4.53.
3-(9-Cyclohexyl-N-hydroxynonanamido)propanoic Acid (11)
Compound 11 was prepared from 16 (2.0 g 7.7 mmol) using the procedure described for 9 (steps 2–5): yield 21%; a white solid; mp 94–96 °C. 1H NMR (CDCl3, 500 MHz, δ; ppm) 3.94 (2H, m), 2.81 (2H, m), 2.43 (2H, m), 1.69–1.67 (7H, m), 1.26–1.17 (16H, m), 0.86 (2H, m). 13C NMR (CDCl3, 125 MHz, δ; ppm) 174.35, 168.35, 44.57, 44.33, 37.68, 37.55, 33.47, 32.20, 31.36, 31.04, 29.94, 29.53, 29.37, 26.86, 26.78, 26.47, 25.31, 24.63. MS (FAB) m/z 328 (MH+). HRMS calcd for C18H34O4N, 328.24878, found 328.25015. HPLC tR = 21.86 min (gradient (I), purity 97.8%).
3-(9-Cycloheptyl-N-hydroxynonanamido)propanoic Acid (12)
Compound 12 was prepared from 16 (400 mg, 1.5 mmol) using the procedure described for 9 (steps 2–5): yield 13%; a white solid; mp 86–87 °C. 1H NMR (CDCl3, 500 MHz, δ; ppm) 3.94 (2H, m), 2.79 (2H, m), 2.41 (2H, m), 1.68–1.55 (8H, m), 1.49–1.37 (5H, m), 1.30–1.25 (10H, m), 1.18–1.11 (4H, m). 13C NMR (CDCl3, 125 MHz, δ; ppm) 174.01, 168.27, 60.43, 44.57, 44.25, 39.28, 38.24, 34.68, 32.17, 31.32, 31.04, 29.96, 29.55, 29.37, 28.58, 27.42, 26.59, 25.29, 24.63. MS (FAB) m/z 342 (MH+). HRMS calcd for C19H36O4N, 342.26443, found 342.26507. HPLC tR = 21.64 min (gradient (III), purity 98.9%).
3-(N-Hydroxy-9-phenylnonanamido)propanoic Acid (13)
Step 1: Preparation of 9-Phenylnonanoic Acid (23)
To a solution of 9-bromononanoic acid sodium salt (16) (1.0 g, 3.9 mmol), Fe(acac)3 (67 mg, 0.19 mmol), tetramethylethylenediamine (57 μL, 0.38 mmol), and hexamethylenetetramine (27 mg, 0.19 mmol) in dry THF (6.0 mL) was added phenylmagnesium bromide (4.87 mL, 5.0 mmol) at 0 °C. The reaction mixture was then stirred for 2.5 h, after which it was diluted with water and extracted three times with Et2O. The organic extracts were washed with saturated aqueous solution of Na2CO3. The aqueous layer was then acidified by HCl and extracted three times with Et2O, and the combined organic layers were washed with water and dried over Na2SO4. Filtration and concentration in vacuo gave 23 (610 mg, 67%) as a yellow oil. 1H NMR (CDCl3, 500 MHz, δ; ppm) 7.29–7.25 (2H, m), 7.18–7.16 (3H, m), 2.60 (2H, t, J = 7.3 Hz), 2.34 (2H, m), 1.64–1.61 (4H, m), 1.39–1.28 (8H, m).
Steps 2, 3, and 4: Preparation of 3-(N-Hydroxy-9-phenylnonanamido)propanoic Acid (13)
Compound 13 was prepared from 23 (604 mg, 2.6 mmol) using the procedure described for 9 (steps 3–5): yield 19%; a white solid; mp 87–88 °C. 1H NMR (CDCl3, 500 MHz, δ; ppm) 7.27 (3H, m), 7.18–7.17 (2H, m), 3.93 (2H, m), 2.80 (2H, m), 2.59 (2H, t, J = 7.6 Hz), 2.46–2.41 (2H, m), 1.60 (4H, m), 1.31 (8H, m). 13C NMR (CDCl3, 125 MHz, δ; ppm) 177.15, 174.53, 142.88, 128.40, 128.23, 125.57, 44.40, 35.96, 31.83, 31.47, 29.34, 29.27, 29.16, 25.28, 24.68, 22.65. MS (FAB) m/z 322 (MH+). HRMS calcd for C18H28O4N, 322.20183, found 322.20252. HPLC tR = 15.19 min (gradient (I), purity 96.1%).
Methyl 3-[Hydroxyl(10-methylundecanoyl)amino]propanoate (7)
Step 1: Preparation of Methyl 3-(Benzyloxyamino)propanoate (37)
Compound 37 was prepared from methyl acrylate (36) (4.4 g, 51 mmol) using the procedure described for 9 (step 1): yield 59%; a yellow oil. 1H NMR (CDCl3, 500 MHz, δ; ppm) 7.37–7.28 (5H, m), 5.82 (1H, broad s), 4.68 (2H, s), 3.67 (3H, s), 3.20 (2H, t, J = 6.4 Hz), 2.60 (2H, t, J = 6.4 Hz).
Steps 2, 3, and 4: Preparation of Methyl 3-[Hydroxyl(10-methylundecanoyl)amino]propanoate (7)
Compound 7 was prepared from 37 (3.1 g, 15 mmol) using the procedure described for 9 (steps 2–4): yield 11%; a colorless oil. 1H NMR (CDCl3, 500 MHz, δ; ppm) 3.94 (2H, m), 3.73 (3H, s), 2.75 (2H, m), 2.50–2.44 (2H, m), 1.62–1.56 (4H, m), 1.51 (1H, sep, J = 6.7 Hz), 1.30–1.25 (8H, m), 1.20–1.10 (2H, m), 0.86 (2H, d, J = 6.7 Hz). 13C NMR (CDCl3, 125 MHz, δ; ppm) 52.39, 44.59, 39.04, 32.57, 29.88, 29.54, 29.39, 27.98, 27.39, 25.30, 24.68, 22.66. MS (EI) m/z 301 (M+). HRMS calcd for C16H31O4N, 301.22531, found 301.22442. HPLC tR = 11.81 min (gradient (II), purity 97.9%).
Molecular Modeling
The X-ray structure of KDM7B (PDB code 3KV4) was used as a model for docking. Protein preparation, receptor grid generation, and ligand docking were performed using the software Glide 3.5. Compounds 6 and 9 were docked into the active site of the protein. The standard precision mode of Glide was used to determine favorable binding poses, which allowed the ligand conformation to be flexibly explored while holding the protein as a rigid structure during docking.
Biology
KDM7B Inhibition Assay
KDM7B (0.5 mg/mL) was incubated with 150 mM KCl, 2.5% glycerol, 0.5 mM dithiothreitol, 0.05 mM PMSF, 2.5 mM glutathione reduced form, 20 μM (+)-Fe(II)-l-ascorbic acid, 20 μM ZnCl2, 0.5 mM ascorbic acid, 0.5 mM 2-oxoglutarate, and 5 μM H3K4me3K9me2 (ART(Kme3)QTAR(Kme2)STGGKAPRKQL-Cys) for 1 h at 37 °C in 8 μL of 10 mM Tris-HCl buffer (pH 8.0). The reaction was stopped by adding 75 μL of matrix-solution (5 mg/mL α-cyano-4-hydroxycinnamic acid, 37% acetonitrile, and 0.12% trifluoroacetic acid) and then sonicated for 30 s. Then 1 μL of the reaction mixture was spotted on the sample plate, dried, and analyzed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) using Voyager-DE PRO (Applied Biosystems). The KDM7B inhibition activity of the test compounds was calculated from the remaining amount of H3K9me2. The 50% inhibitory concentration (IC50) of the test compounds was calculated as the concentration at which the half amount of H3K9me2 was removed compared to that removed when the enzyme was added (Supporting Information Figure S7).
KDM4C Inhibition Assay
The KDM4C-inhibitory activity was measured using 0.6 mg/mL enzyme. The compounds were dissolved in DMSO. The final concentrations of DMSO in the reaction mixtures were less than 3.3%, and it was confirmed that 3.3% DMSO did not affect the KDM4C activity. Reaction with DMSO alone was also done as a control. Reaction mixtures (94.6 μL), containing all of the materials except H3K9me3 peptide and 2-oxoglutarate, were preincubated for 5 min. Then the reactions were started by the addition of 5.4 μL of a solution of 0.93 mM H3K9me3 peptide and 3.7 mM 2-oxoglutarate . The enzyme activity was determined as described above. The ratio of the enzyme activity measured in the presence of inhibitor to that of the control was plotted against log [Inhibitor]. To confirm that the reduction of the KDM4C activity by test compounds was not due to inhibition of the coupled enzyme FDH, we examined the effects of the test compounds on FDH activity. The reaction mixture (0.1 mL) contained 20 mM HEPES-KOH, pH 7.5, 50 μM formaldehyde, 1 mM 3-acetylpyridine adenine dinucleotide, 1 mM reduced glutathione, 0.1 mg/mL BSA, 0.1 mg/mL FDH, and a fixed concentration of 123 μM test compound. The FDH activity was measured by monitoring APADH formation as described above. The FDH activity in the presence of test compounds was similar to that in the absence of the compounds.
KDM4A Inhibition Assay
The KDM4A activity was measured by the FDH-coupled assay as described for KDM4C except that reactions were performed in a final volume of 30 μL in 384-well plate (Nunc) and a final concentration of the KDM4A was 0.37 mg/mL.
KDM5A Inhibition Assay
The KDM5A activity was measured by the FDH-coupled assay as described for KDM4C except that reactions were performed with H3K4me3 peptide in a final volume of 30 μL in 384-well plate (Nunc) and a final concentration of the KDM5A was 0.64 mg/mL.
KDM2A Inhibition Assay
The inhibitory activities of test compounds against KDM2A was assayed according to the method reported in ref (4).
KDM6A Inhibition Assay
The Epigenase JMJD3/UTX demethylase activity/inhibition assay kit (Epigentek Group Inc.) was used for KDM6A enzyme assay. The materials supplied with the kit, 100 μM of test compounds and KDM6A (human, recombinant, BPS Bioscience, Inc.) (300 ng/well) were added to wells coating trimethylated histone substrate, according to the supplier’s protocol. The resulting mixtures were incubated at 37 °C for 120 min. After enzyme reaction, each well was reacted with capture antibody for 60 min and with detection antibody for 30 min. Finally, developer solution and stop solution were added to wells in sequence and the absorbance (450 nm) in each well was measured with an ARVO X3 microplate reader. The KDM6A inhibition activity of test compounds was calculated from the absorbance readings.
KDM7A Inhibition Assay
The KDM7A assay was run using MALDI and K27me2 peptide as a substrate under the same conditions as used in ref (4).
Western Blot Analysis
N2a cells, HeLa cells, or KYSE150 cells (5 × 105) were treated for 24 h with inhibitors at the indicated concentrations in OPTI-MEM medium, then collected and extracted with SDS buffer. Protein concentrations of the lysates were determined using a Bradford protein assay kit (Bio-Rad Laboratories); equivalent amounts of proteins from each lysate were resolved in 4–20% SDS-polyacrylamide gel and then transferred onto nitrocellulose membranes (Bio-Rad Laboratories). After having been blocked for 30 min with Tris-buffered saline (TBS) containing 3% skim milk, the transblotted membrane was incubated overnight at 4 °C with dimethylated H3K27 antibody (CST) (1:1000 dilution) or H3 antibody (Abcam) (1:100000 dilution) in TBS containing 3% skim milk or TBS-Tween 20 (TBS-T). The membrane was probed with the primary antibody, then washed twice with TBS or TBS-T, incubated with goat antirabbit IgG-horseradish peroxidase conjugates (diluted 1:2500) for 1.5 h at room temperature, and again washed twice with TBS and once with TBS-T. The immunoblots were visualized by enhanced chemiluminescence.
MTT Assays
The cells were plated at initial densities of 5000 cells/well (100 μL/well) in 96-well plates in RPMI 1640 with 10% fetal bovine serum and allowed to attach overnight. The cells were exposed to inhibitors for 48 h at 37 °C in 5% CO2 incubator. A solution (5 mg/mL) of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) (Sigma) was added (10 μL/well) and incubated with the cells for 3 h before solubilization buffer (0.04 mol/L HCl–2-propanol) was added (100 μL/well) onto the cultured cells. The solubilized dye was quantified by colorimetric reading at 560 nm using a reference wavelength of 750 nm. Absorbance values of control wells (C) and test wells (T) were measured. Moreover, absorbance of the test wells (T0) was also measured at time 0 (addition of compounds). Using these measurements, cell growth inhibition (percentage of growth) by a test inhibitor at each concentration used was calculated as: % growth = 100[(T – T0)/(C – T0)], when T > T0 and % growth = 100[(T – T0)/T], when T < T0. Computer analysis of the % growth values afforded the 50% growth inhibition parameter (GI50). The GI50 was calculated as 100[(T – T0)/(C – T0)] = 50.
RNA Isolation and Semi-qRT-PCR
HeLa cells were treated for 48 h with 0.238% DMSO or compound 9 at the concentration of 30 and 80 μM, respectively. Total RNA was isolated from HeLa cells using RNAzol (Molecular Research Center, Inc.) following the manufacturer’s protocol. First-strand cDNA synthesis from total RNA was carried out using ReverTra Ace (TOYOBO). Resulting cDNA was then analyzed by semiquantitative PCR (semi-qPCR) using 2720 thermal cycler (Applied Biosystems). Primers are specific for genes tested, and their sequences are as follows:
GAPDH 450bp Primer(F): 5′-TCCACCACCCTGTTGCTGTA-3′ (20mer) Primer(R): 5′-ACCACAGTCCATGCCATCAC-3′ (20mer)
E2F1 435bp Primer(F): 5′-ACTCCTCGCAGATCGTCATCATCT-3′(24mer) Primer(R): 5′-GGACGTTGGTGATGTCATAGATGCG-3′(25mer)
Cycle parameters were 94 °C for 2 min, followed by 28 (E2F1), 20 (GAPDH) cycles of 98 °C for 10 s, 60 °C for 30 s, and 68 °C for 30 s, with a final extension at 68 °C for 1 min.
FACS Analysis
Cells (5 × 105) were treated for 24 h with compound 9 at the indicated concentrations in RPMI 1640 with 10% fetal bovine serum, then harvested by trypsinization. The cells were collected by centrifugation, fixed with ice-cold 70% ethanol, washed with phosphate-buffered saline, and resuspended in 0.5 mL of phosphate-buffered saline containing propidium iodide (10 μg/mL) and RNase A (0.2 mg/mL). After a final incubation at 25 °C for 30 min, the cells were analyzed using a JSAN flow cytometer (Bay Bioscience). A total of 30000 events were counted for each sample. Data were analyzed using FlowJo software (Tree Star).
Acknowledgments
We thank Mie Tsuchida and Miho Hosoi for their technical support. This work was supported in part by JST PRESTO program (T.S.), a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (T.S.), Takeda Science Foundation (T.S.), Naito Foundation (T.S.), NOVARTIS Foundation (Japan) for the Promotion of Science, the Wellcome Trust, BBSRC (L.W.), and the Royal Society (A.K.).
Glossary
Abbreviations Used
- KDM
lysine demethylase
- LSD1
lysine-specific demethylase 1
- JmjC
Jumonji C
- NOG
N-oxalylglycine
- PCA
2,4-pyridinedicarboxylic acid
Supporting Information Available
View of the catalytic sites of KDM7B, KDM2A, KDM4A, KDM4C, KDM5A, and KDM6A. KDM7B-inhibitory activity of compound 9. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- a Lee J. S.; Smith E.; Shilatifard A. The language of histone crosstalk. Cell 2010, 142, 682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chi P.; Allis C. D.; Wang G. G. Covalent histone modifications: miswritten, misinterpreted and mis-erased in human cancers. Nature Rev. Cancer 2010, 10, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Margueron R.; Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Carey M.; Workman J. L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [DOI] [PubMed] [Google Scholar]
- a Kubicek S.; Jenuwein T. A crack in histone lysine methylation. Cell 2004, 119, 903–906. [DOI] [PubMed] [Google Scholar]; b Shi Y.; Lan F.; Matson C.; Mulligan P.; Whetstine J. R.; Cole P. A.; Casero R. A.; Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [DOI] [PubMed] [Google Scholar]; c Klose R. J.; Kallin E. M.; Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nature Rev. Genet. 2006, 7, 715–727. [DOI] [PubMed] [Google Scholar]; d Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nature Rev. Genet. 2007, 8, 829–833. [DOI] [PubMed] [Google Scholar]
- Rose N. R.; Woon E. C.; Tumber A.; Walport L. J.; Chowdhury R.; Li X. S.; King O. N.; Lejeune C.; Ng S. S.; Krojer T.; Chan M. C.; Rydzik A. M.; Hopkinson R. J.; Che K. H.; Daniel M.; Strain-Damerell C.; Gileadi C.; Kochan G.; Leung I. K.; Dunford J.; Yeoh K. K.; Ratcliffe P. J.; Burgess-Brown N.; von Delft F.; Muller S.; Marsden B.; Brennan P. E.; McDonough M. A.; Oppermann U.; Klose R. J.; Schofield C. J.; Kawamura A. Plant growth regulator daminozide is a selective inhibitor of human KDM2/7 histone demethylases. J. Med. Chem. 2012, 55, 6639–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Suzuki T.; Miyata N. Lysine demethylases inhibitors. J. Med. Chem. 2011, 54, 8236–8250. [DOI] [PubMed] [Google Scholar]; b Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nature Rev. Drug. Discovery 2012, 11, 384–400. [DOI] [PubMed] [Google Scholar]
- a Björkman M.; Ostling P.; Härmä V.; Virtanen J.; Mpindi J. P.; Rantala J.; Mirtti T.; Vesterinen T.; Lundin M.; Sankila A.; Rannikko A.; Kaivanto E.; Kohonen P.; Kallioniemi O.; Nees M. Systematic knockdown of epigenetic enzymes identifies a novel histone demethylase PHF8 overexpressed in prostate cancer with an impact on cell proliferation, migration and invasion. Oncogene 2012, 31, 3444–3456. [DOI] [PubMed] [Google Scholar]; b Feng W.; Yonezawa M.; Ye J.; Jenuwein T.; Grummt I. PHF8 activates transcription of rRNA genes through H3K4me3 binding and H3K9me1/2 demethylation. Nature Struct. Mol. Biol. 2010, 17, 445–450. [DOI] [PubMed] [Google Scholar]
- a Cloos P. A.; Christensen J.; Agger K.; Maiolica A.; Rappsilber J.; Antal T.; Hansen K. H.; Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006, 442, 307–311. [DOI] [PubMed] [Google Scholar]; b Rose N. R.; Ng S. S.; Mecinović J.; Liénard B. M.; Bello S. H.; Sun Z.; McDonough M. A.; Oppermann U.; Schofield C. J. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J. Med. Chem. 2008, 51, 7053–7056. [DOI] [PubMed] [Google Scholar]; c Rose N. R.; Woon E. C.; Kingham G. L.; King O. N.; Mecinović J.; Clifton I. J.; Ng S. S.; Talib-Hardy J.; Oppermann U.; McDonough M. A.; Schofield C. J. Selective inhibitors of the JMJD2 histone demethylases: combined nondenaturing mass spectrometric screening and crystallographic approaches. J. Med. Chem. 2010, 53, 1810–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hamada S.; Suzuki T.; Mino K.; Koseki K.; Oehme F.; Flamme I.; Ozasa H.; Itoh Y.; Ogasawara D.; Komaarashi H.; Kato A.; Tsumoto H.; Nakagawa H.; Hasegawa M.; Sasaki R.; Mizukami T.; Miyata N. Design, synthesis, enzyme-inhibitory activity, and effect on human cancer cells of a novel series of Jumonji domain-containing protein 2 histone demethylase inhibitors. J. Med. Chem. 2010, 53, 5629–5638. [DOI] [PubMed] [Google Scholar]; e Luo X.; Liu Y.; Kubicek S.; Myllyharju J.; Tumber A.; Ng S.; Che K. H.; Podoll J.; Heightman T. D.; Oppermann U.; Schreiber S. L.; Wang X. A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases. J. Am. Chem. Soc. 2011, 133, 9451–9456. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lohse B.; Nielsen A. L.; Kristensen J. B.; Helgstrand C.; Cloos P. A.; Olsen L.; Gajhede M.; Clausen R. P.; Kristensen J. L. Targeting histone lysine demethylases by truncating the histone 3 tail to obtain selective substrate-based inhibitors. Angew. Chem., Int. Ed. 2011, 50, 9100–9103. [DOI] [PubMed] [Google Scholar]; g Woon E. C.; Tumber A.; Kawamura A.; Hillringhaus L.; Ge W.; Rose N. R.; Ma J. H.; Chan M. C.; Walport L. J.; Che K. H.; Ng S. S.; Marsden B. D.; Oppermann U.; McDomough M. A.; Schofield C. J. Linking of 2-oxoglutarate and substrate binding sites enables potent and highly selective inhibition of JmjC histone demethylases. Angew. Chem., Int. Ed. 2012, 51, 1631–1634. [DOI] [PubMed] [Google Scholar]; h Kruidenier L.; Chung C. W.; Cheng Z.; Liddle J.; Che K.; Joberty G.; Bantscheff M.; Bountra C.; Bridges A.; Diallo H.; Eberhard D.; Hutchinson S.; Jones E.; Katso R.; Leveridge M.; Mander P. K.; Mosley J.; Ramirez-Molina C.; Rowland P.; Schofield C. J.; Sheppard R. J.; Smith J. E.; Swales C.; Tanner R.; Thomas P.; Tumber A.; Drewes G.; Oppermann U.; Patel D. J.; Lee K.; Wilson D. M. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012, 488, 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimm V. J. Alar’s risks. Science 1991, 254, 1276. [PubMed] [Google Scholar]
- Sagelsdorff P.; Lutz W. K.; Schlatter C. DNA methylation in rat liver by daminozide, 1,1-dimethylhydrazine, and dimethylnitrosamine. Fundam. Appl. Toxicol. 1988, 11, 723–730. [DOI] [PubMed] [Google Scholar]
- Horton J. R.; Upadhyay A. K.; Qi H. H.; Zhang X.; Shi Y.; Cheng X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nature Struct. Mol. Biol. 2010, 17, 38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y.; Ishitani T.; Nalayama K. I. KDM7 is a dual demethylase for histone H3 Lys 9 and Lys 27 and functions in brain development. Genes Dev. 2010, 24, 432–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z. A.; Shokat K. M. Chemical genetics: where genetics and pharmacology meet. Cell 2007, 128, 425–430. [DOI] [PubMed] [Google Scholar]
- a Hamada S.; Kim T. D.; Suzuki T.; Itoh Y.; Tsumoto H.; Nakagawa H.; Janknecht R.; Miyata N. Synthesis and activity of N-oxalylglycine and its derivatives as Jumonji C-domain-containing histone lysine demethylase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2852–2855. [DOI] [PubMed] [Google Scholar]; b Kristensen L. H.; Nielsen A. L.; Helgstrand C.; Lees M.; Cloos P.; Kastrup J. S.; Helin K.; Olsen L.; Gajhede M. Studies of H3K4me3 demethylation by KDM5B/Jarid1B/PLU1 reveals strong substrate recognition in vitro and identifies 2,4-pyridine-dicarboxylic acid as an in vitro and in cell inhibitor. FEBS J. 2012, 279, 1905–1914. [DOI] [PubMed] [Google Scholar]
- Liu W.; Tanasa B.; Tyurina O. V.; Zhou T. Y.; Gassmann R.; Liu W. T.; Ohgi K. A.; Benner C.; Garcia-Bassets I.; Aggarwal A. K.; Desai A.; Dorrestein P. C.; Glass C. K.; Rosenfeld M. G. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 2010, 466, 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D. G.; Ohtani K.; Nevins J. R. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycleprogression. Genes Dev. 1994, 8, 1514–1525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.