

Abstract
Following infection with Human immunodeficiency virus 1 (HIV-1) there is a remarkable variation in virus replication and disease progression. Both host and viral factors have been implicated in the observed differences in disease status. Here, we focus on understanding the contribution of HIV-1 viral protein R (Vpr) by evaluating the disease-associated Vpr polymorphism and its biological functions from HIV-1 positive rapid progressor (RP) and long-term nonprogressor (LTNP) subjects. Results presented here show distinct variation in phenotypes of Vpr alleles from LTNP and RP subjects. Most notably, the polymorphism of Vpr at R36W and L68M associated with RP shows higher levels of oligomerization, and increased virus replication, whereas R77Q exhibits poor replication kinetics. Interestingly, we did not observe correlation with cell cycle arrest function. Together these results indicate that polymorphisms in Vpr in part may contribute to altered virus replication kinetics leading to the observed differences in disease progression in LTNP and RP groups.
Introduction
Human immunodeficiency virus type 1 (HIV-1) infection results in the destruction of several target cell types, including cells of the immune system, which leads to overt disease (Levy, 2006). However, disease progression within the infected individuals varies. Based on the disease status, patients are classified as normal/rapid progressor (RP), long-term nonprogressor (LTNP) or elite controller (EC). LTNP patients maintain their CD4+ T cell count (>500) and control viraemia more efficiently either with or without antiviral therapy, whereas RPs exhibit a rapid decline in CD4+ T cell counts, that are susceptible to other infections, and develop AIDS (in the absence of highly active anti-retroviral therapy, HAART) within 5–8 years of seroconversion. Both host cellular factors and viral proteins have been implicated in HIV-1 disease progression (Fang et al., 2001; Shioda & Nakayama, 2006). Deletions and premature stop codons in HIV-1 accessory genes have been suggested as the viral factors responsible for a lack of disease progression (Lum et al., 2003b; Rhodes et al., 2000; Rodés et al., 2004; Saksena et al., 1996; Somasundaran et al., 2002). Vpr is a highly conserved accessory protein in primate lentiviruses including HIV-1, HIV-2 and Simian immunodeficiency virus (SIV) that has a pivotal role in virus replication and pathogenesis (Cohen et al., 1990; Majumder et al., 2009). HIV-1 Vpr is known to exist in multiple forms (cell-associated, virion-associated and free Vpr) both in vitro and in vivo (Tungaturthi et al., 2003). Though Vpr is synthesized as a late protein, the presence of Vpr as part of the incoming virion suggests that Vpr may play an important role during early events associated with infection (Hrimech et al., 1999; Poon & Chen, 2003). Virion-associated Vpr enables the translocation of the pre-integration complex (PIC) into the nucleus of non-dividing cells such as macrophages (Zhao & Zhu, 2004). Although Vpr is not required for virus replication in T cells, the presence of Vpr enhances virus production in primary T cells and latently infected cells (Levy et al., 1995). Collectively, these studies implicate a role for Vpr in viral pathogenesis.
Several studies have attempted to delineate the role of Vpr in HIV-1 pathogenesis and further identified potential Vpr signature motifs and defective Vpr protein in relation to HIV-1 disease progression in LTNP and mother–child nontransmitter pairs (Caly et al., 2008; Lum et al., 2003a; Mologni et al., 2006; Wang et al., 1996; Zhao et al., 2002). It has been reported that a mutation in residue 77 of Vpr (R77Q) was present in several LTNP patients and has been linked to an apoptosis defective Vpr phenotype (Lum et al., 2003b; Mologni et al., 2006). However, other studies did not find an association between the R77Q mutation or C-terminal mutations and disease nonprogression (Cavert et al., 2004; Chui et al., 2006). Recently, Caly et al. (2008) have reported a role for F72L mutation in LTNPs. Collectively, these results are controversial and do not allow us to draw any definitive conclusions regarding the relationship between specific sequences and/or domains in Vpr and viral pathogenesis.
In an effort to identify signature sequences specific to LTNP or RP groups, we assessed the Vpr sequences deposited in the HIV-1 Los Alamos database (www.hiv.lanl.gov). Analysing sequence data available from hundreds of patients is likely to reveal motifs and/or specific residues that are linked to a particular disease progression group. We have analysed the amino acid sequences of Vpr from HIV-1 isolates from individuals with well defined characteristics (LTNP versus RP). The results from our analysis identified several residues in Vpr that are unique for LTNP and RP phenotypes. Next, we evaluated the effects of these Vpr variants on oligomerization, subcellular localization, cell cycle arrest function, and virus replication. Results from these analyses indicate that Vpr variants alter the functionality of Vpr in a pattern that correlates with the two clinical populations of HIV-1 infected individuals, the LTNP and RP groups.
Results
Identification and selection of variant Vpr residues associated with RPs or LTNPs
Studies have reported on the amino acid polymorphisms in sequences generated from HIV-1 infected individuals (Andreoni, 2004; Bimber et al., 2009; Holzmayer et al., 2009; Reinis et al., 2007). Here, we have utilized a strategy in which the frequency of each amino acid is compared between LTNPs and RPs from subtype B. Of the 1112 Vpr sequences, the data yielded 192 LTNP and 102 RP Vpr sequences. These sequences were used to derive phylogenetic tree analyses (Fig. 1). Results indicate that most of the RP sequences formed a major cluster with the exception of four sequences that are segregated. Interestingly, the sequences from LTNPs clustered in small groups and spread across the tree (Fig. 1), suggesting a more diverse sequence variation that could lead to nonprogression in vivo. These sequences were concatenated and aligned via the clustal w algorithm using VprNL43 as the reference sequence. The frequencies of polymorphisms at each of the 96 residues of Vpr in the LTNP and RP sequences were calculated as a percentage and the results are presented in Table 1. We also performed Bonferroni analysis to identify the significance of the residue variation between LTNPs and RPs. Residues with significant differences are indicated as magnitude Δp in Table 1. If there was no significant change from the reference at a specific residue, the position was marked as ‘absent’. Residues were selected for further biological analysis using the following criteria. The residue must have only one or two mutations at a position associated with the LTNP group and no change of residue in the RP group or vice versa, and the significance of the residue variation must have a magnitude greater than 0.1.
Fig. 1.
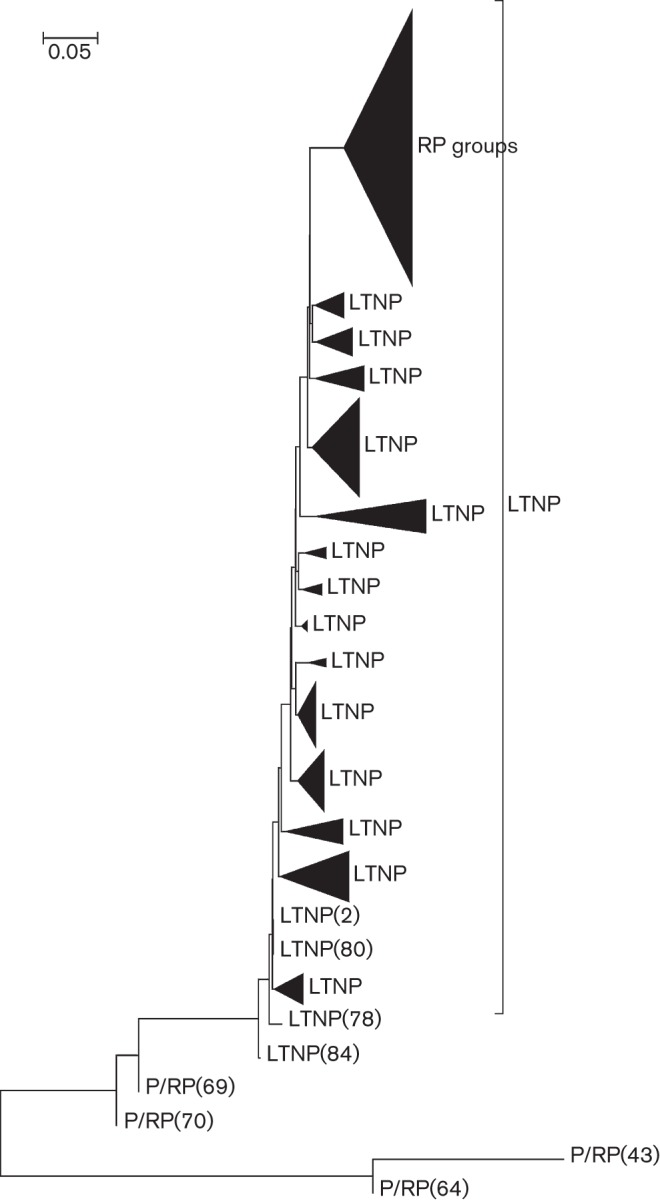
Phylogenetic tree analysis of HIV-1 vpr genes from LTNPs and P/RPs. HIV-1 Vpr sequences obtained from the database were assessed for donor information and classified as LTNP and RP. The phylogenetic tree was generated using the mega program.
Table 1. Frequency analysis of Vpr variants from long-term nonprogressor (LTNP) and rapid progressor (RP) subjects.
| Residue no. | Amino acid in NL43 (WT) | Amino acid (frequency, %) | Magnitude Δp | |
| LTNP (n = 192)* | RP (n = 102)* | |pLTNP−pNP/RP| | ||
| 3 | Q | H (2.54), R (4.57) | Absent | 0.073 |
| 16 | N | H (5.58) | Absent | 0.055 |
| 19 | T | A (24.48) | Absent | 0.281† |
| 28 | S | T (7.65) | Absent | 0.204† |
| 32 | R | K (18.87) | K (2.96) | 0.149† |
| 36 | R | Absent | W (17.64) | 0.156† |
| 37 | I | L (15.3), V (12.2) | Absent, V (41.16) | 0.02 |
| 42 | L | Absent | F (2.94) | 0.024 |
| 55 | A | Absent, T (32.64) | V (4.9), T (20.58) | 0.065 |
| 63 | I | Absent, T (3.06) | S (6.86), T (31.36) | 0.265† |
| 68 | L | Absent | M (15.68) | 0.157† |
| 84 | T | S (0.51) | S (12.74) | 0.326† |
| 85 | R | Absent | Y (6.36) | 0.083 |
| 86 | Q | R (33.66), absent | R (5.88), P (9.8) | 0.183† |
| 87 | R | G (4.59) | Absent | 0.08 |
| 88 | R | P (14.28), T (4.98), G (0.51) | Absent, absent, G (4.9) | 0.149† |
| 89 | A | R (23.97) | R (4.9) | 0.222 |
| 90 | R | N (18.36), A(4.59) | Absent, absent | 0.266† |
| 91 | N | R (6.59), G (13.77) | R (34.3), absent | 0.202† |
| 94 | S | R (22.44), A (7.68) | Absent, A (34.3) | 0.014 |
| 96 | S | R (4.59) | R (35.28) | 0.264† |
Sequences were obtained from the HIV sequences database at Los Alamos National Laboratories.
The number of sequences (n) for each group (LTNP and RP) is indicated.
Significance was assessed by Bonferroni analysis using the magnitude Δp.
Five variant residues matched the selection criteria: T19, R36, L68, R85 and R90. Mutations that were found to be associated with RPs include R36W, L68M and R85Y. Mutations that were found to be associated with LTNPs include T19A and R90N. Residue T19 is found in helical domain I and shows a significant difference with a frequent variation to alanine in the LTNP group (24.5 %) and no change in the RP group (Table 1). Residue R36 is found in the loop between helical domains I and II and shows a significant difference with a frequent variation to tryptophan (17.7 %) in RP and no change in the LTNP group. Residue L68 is found in helical domain III and shows a significant difference with a frequent variation to methionine (15.7 %), which is a conservative change. Residue R85 is found in the C terminus and shows a significant difference with a frequent variation to tyrosine (6.9 %). Residue R90 is found in the C terminus and shows a significant difference with a frequent variation to asparagine (18.8 %). These results suggest that there are detectable and significant differences between LTNP and RP Vpr alleles. In addition to the above-mentioned residues, we also included R77Q, as R77Q is found in a number of Vpr sequences from LTNPs (Lum et al., 2003a; Mologni et al., 2006). Residue R77 is found in helical domain III and shows a significant difference with a frequent variation to glutamine (46.4 % in LTNPs, 29.3 % in RPs).
Generation and characterization of Vpr containing variant residues in bimolecular fluorescence complementation (BiFC) constructs
The sequences corresponding to variant residues were introduced in BiFC haemagglutinin (HA)-tagged Vpr expression constructs as a reporter system to facilitate further assays and are depicted in Fig. 2(a). To understand the consequence of these mutations in Vpr structure, structure analysis was performed and the non-conserved and structurally critical residues were mapped onto the three-dimensional structure (Fig. 2b). Residues in helical domain I (T19), helical domain III (L68), and the C terminus (R85 and R90) did not alter the overall structure of Vpr. However, R36W mutation in the loop is likely to stabilize Vpr and/or promote oligomerization. To establish that the variant residues do not affect the expression of Vpr, steady state level was measured by transient transfection followed by immunoblot. Results indicate that expression levels of chimeric Vpr containing variant residues are maintained at high levels comparable to the expression levels of wild-type Vpr, suggesting that expression and stability are not affected (Fig. 2c).
Fig. 2.

Construction and characterization of expression plasmids representing HIV-1 Vpr variants from LTNP and RP subjects. (a) Schematic depicting Vpr variants selected for analysis from the frequency analysis shown in Table 1. Mutations introduced in the VprNL43 background are shown in their respective amino acid positions as Venus split constructs with an HA-tag. (b) Three dimensional structure of Vpr representing residues from LTNP and RP subjects. The overall structure of Vpr consists of three helices (H1–H3) and the N- and C-terminal regions (shown in green). Vpr variants used in this study are shown in red. (c) Expression of Venus-C (VC-) Vpr and Venus-N (VN-) Vpr variants was assessed by transfecting HEK293T cells with VN-Vpr and VC-Vpr variant expression plasmids or control plasmid. Forty eight hours post-transfection cell lysates were harvested and Vpr expression levels verified by immunoblot against the HA antibody. Actin was probed as loading control. The figure represents one of five independent experiments.
Vpr oligomerization is enhanced by RP-associated polymorphism
While several studies have identified residues that are essential for Vpr oligomerization (Fritz et al., 2008; Venkatachari et al., 2010), it is important to note that these have been identified through in silico means using structural analysis and molecular modelling approaches. To determine the potential role of the five selected Vpr variants in disease progression, a comparison of the oligomerization potential of the LTNP and RP variants was performed using flow cytometry. Residue R36W, which was found in RP, exhibited a 45.67 % (P = 0.0287) increase in mean fluorescence intensity (MFI) compared with wild-type Vpr (Fig. 3a), suggesting an increased capacity of oligomerization. In contrast, none of the LTNP-associated variants showed a difference in MFI, indicating that the oligomerization function of Vpr was not affected. To assess whether the observed difference is due to transfection efficiency, the percentage of BiFC positive cells was measured and no significant change was observed (Fig. 3b). Together these results suggest that, among the five selected variant residues, only one of the RP-associated variants, R36W shows increased oligomerization, while none of the other Vpr variants alters this function. Similar results were observed in other cell lines such as HeLa (data not shown).
Fig. 3.
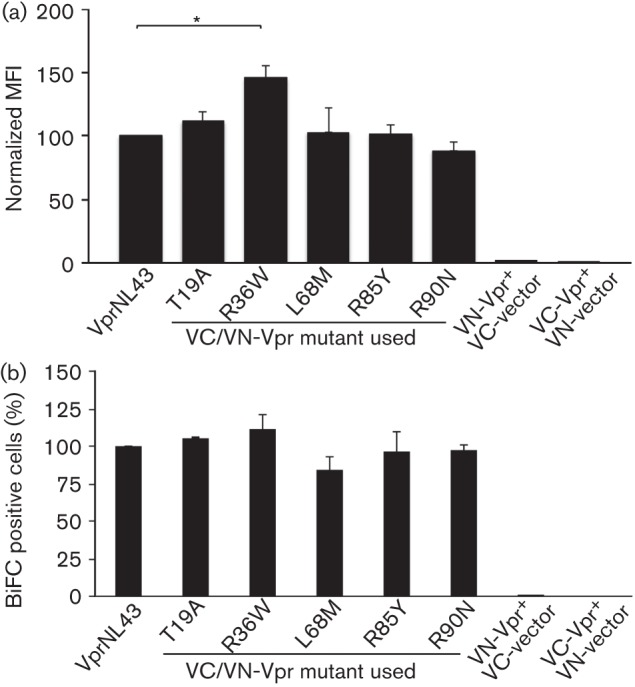
Visualization of Vpr variants’ oligomerization by (a) flow cytometry and (b) fluorescence microscopy. (a) Quantitative analysis of Venus fragment complementation in HEK293T cells was performed by co-transfecting cells with VC-Vpr and VN-Vpr or with VC/VN control plasmids. Thirty six hours post-transfection, cells were harvested and analysed by flow cytometry to determine the percentage of cells positive for BiFC fluorescence. The mean fluorescence intensity (MFI) of each mutant is normalized to wild-type, considered as 100 %. Results represent the means of five independent experiments. (b) The flow data were also analysed for transfection efficiency by the percentage of BiFC positive cells. BiFC (%) of each Vpr mutant is normalized to wild-type. The vector represents BiFC positive cells in VC and VN control plasmid co-transfection. Each graph represents the mean of five independent experiments. Error bars represent standard error of the mean. *P<0.05.
Vpr variants show altered subcellular localization of Vpr oligomers
Subcellular localization of Vpr plays a role in Vpr-mediated host cellular functions such as cell cycle arrest, apoptosis and transactivation (Mahalingam et al., 1997; Sherman et al., 2001). To analyse whether Vpr variants from LTNPs and RPs exhibit altered localization pattern, transfected cells were evaluated by microscopy. HIV-1 VprNL43, as well as LTNP-associated variants T19A and R90N, localized to the nucleus whereas two of the three RP-associated variants exhibited differences in their localization (Fig. 4a). Vpr R36W and R85Y showed an increased signal in the cytoplasm as well as in the nucleus of the cells (Fig. 4a). Furthermore, variant R36W exhibited aggregates in both the nuclear and cytoplasmic compartments with puncta throughout the cell, while R85Y exhibited a more diffuse pattern in the cytoplasm. L68M did not affect the subcellular localization of Vpr. These results indicate that RP-associated variants affect the nucleocytoplasmic shuttling of Vpr and may do so through different mechanisms as implied by the different pattern of altered subcellular localization between the variants R36W and R85Y.
Fig. 4.
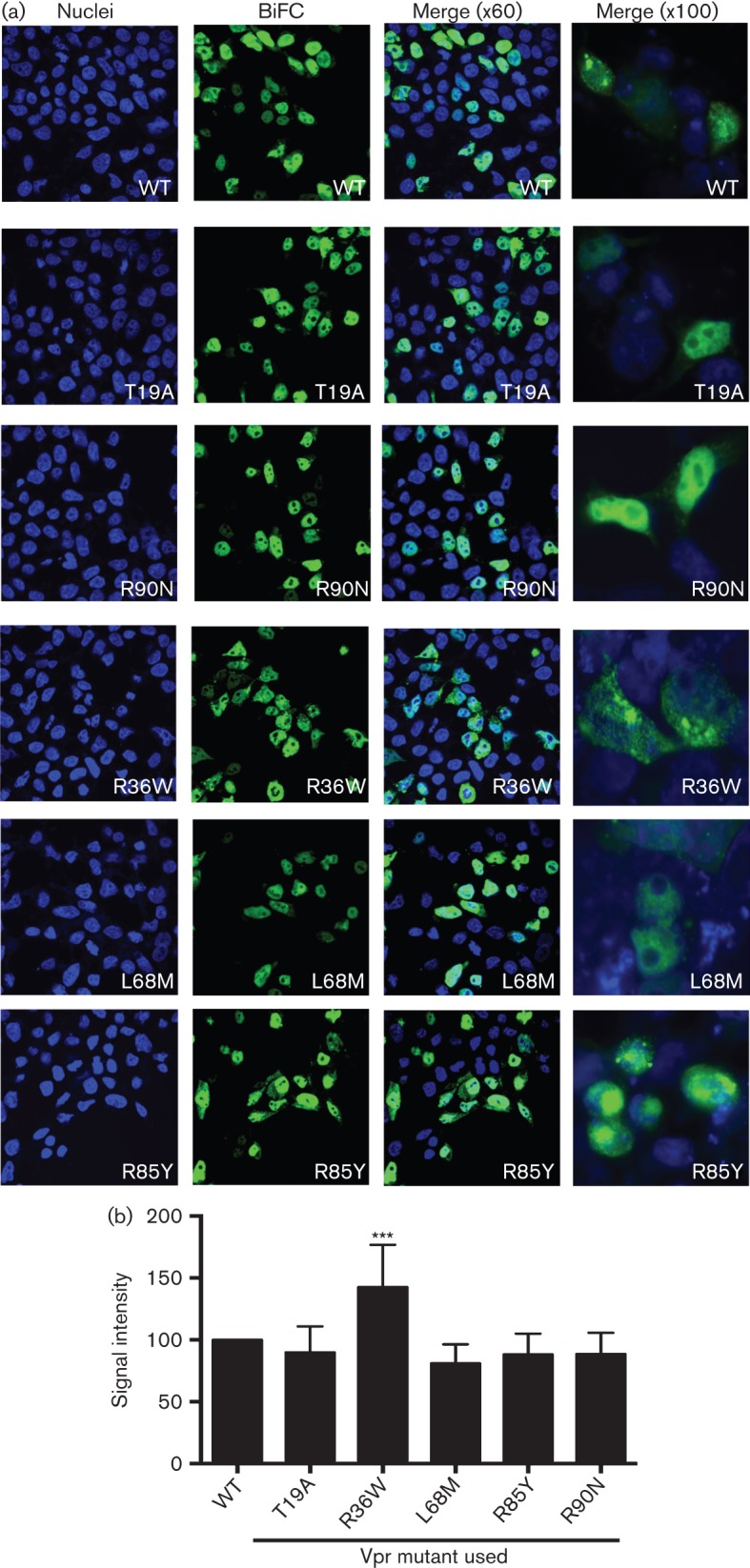
Subcellular localization of Vpr variants by microscopy. (a) The subcellular localization of the Vpr variant oligomers was assessed by co-transfection of the VC-Vpr and VN-Vpr mutant constructs in HEK293T cells. Thirty-six hours post-transfection cells were fixed, stained with DAPI for visualization of nuclei, and visualized under fluorescence microscopy at magnifications of ×60 and ×100 to assess the localization of the BiFC signal within the cell. The figure represents one of six independent experiments. Blue, DAPI, represents nuclei; green, BiFC, represents Vpr oligomers. (b) BiFC signal was quantified using NIS Elements Ar Microscope Imaging Software. BiFC positive cells (n = 12) per field were quantified from multiple fields and intensity was measured in total cells, cytoplasmic and nuclear compartments. Signal in wild-type (VprNL43) was normalized as 100 % and the mutants were calculated accordingly.
Further, quantitative analysis of Vpr oligomers was performed by measuring the intensity of BiFC signals in cells expressing Vpr oligomers within the nucleus and cytoplasmic compartment compared with total BiFC (Fig. 4b). Results indicate that compared with VprNL43 (considered as 100 %), VprR36W showed the highest order of oligomers with a signal intensity of 175 % (P<0.0001). However, L68M and R85Y showed 71 % and 64 %, respectively; T19A and R90N remained at 100 %, i.e. did not show any difference, further supporting that R36W exhibits significant oligomerization. However, these changes could be due to structural changes induced by R36 rather than the disease status of the subjects.
LTNP- and RP-associated Vpr variants alter G2 cell cycle arrest differentially
The localization of Vpr to the nucleus is thought to be associated with Vpr induced G2 cell cycle arrest (Gummuluru & Emerman, 1999; Morellet et al., 2009). CD4+ T cell line Jurkat was infected with 0.1 m.o.i. of HIV-1 EGFP reporter Vpr variant viruses along with HIV-1 WT and HIV-1 ΔVpr viruses and was assessed for cell cycle arrest function by flow cytometry. The polymorphisms associated with rapid progression (R36W, L68M and R85Y) showed changes in our previous panel of assays whereas the LTNP-associated mutants chosen (T19A and R90N) have thus far shown no difference from wild-type Vpr. This indicated that T19A and R90N may not have functional relevance in the context of disease progression; thus these mutants were excluded from further analysis. In place of T19A and R90N, we decided to use the mutant R77Q, which is also associated with LTNPs and has been reported to have functional alterations (Lum et al., 2003b; Mologni et al., 2006). Infected cells were gated based on EGFP expression and cell cycle arrest (G2/G1 ratio) was calculated using the gating strategy shown in Fig. 5(a). Overall, the infection rate (percentage EGFP positive) in Jurkat cells remained ~23–25 %, suggesting that Vpr variants did not alter the infectivity (data not shown). Jurkat cells infected with HIV-1 NL43 (WT) exhibited a G2/G1 ratio of 2.01 whereas the cells infected with HIV-1 ΔVpr showed a G2/G1 of 0.64, corroborating the positive cell cycle arrest function of Vpr (Fig. 5b). In the case of cells infected with HIV-1 expressing Vpr variants, R36W, L68M and R85Y showed a slight reduction in cell cycle compared with HIV-1 wild-type. Interestingly R77Q showed a significantly higher level of G2/G1 ratio (4.1 with P<0.001), suggesting an enhanced cell cycle arrest function (Fig. 5c). These results indicate a slight increase in G2 cell cycle arrest induction in R77Q, whereas two of the three RP variants reduced G2 cell cycle arrest. However, the correlation is not significantly different.
Fig. 5.

Evaluation of cell cycle arrest induced by HIV-1 expressing Vpr variants. (a) Gating strategy used to assess Vpr induced cell cycle arrest by flow cytometry in Jurkat cells infected with HIV-1 EGFP reporter virus expressing representative Vpr variants from LTNPs and RPs. Seventy-two hours post-infection cells were collected, fixed and stained with propidium iodide, and analysed by flow cytometry. (b) Cell cycle analysis of HIV-1 wild-type, HIV-1 ΔVpr and HIV-1 Vpr variant virus infected Jurkat cells. The EGFP positive cells were analysed for their cell cycle profiles, which were then fitted to a peak distribution for the relative proportion of cells in the G0 and G2 cell phases. Results represent the cell cycle distributions for each of the Vpr variants tested, and the figure represents one of three independent experiments. (c) Significance testing was performed using Student’s t-test comparing each Vpr variant with the wild-type (NL43) control. The graph represents the mean of three independent experiments. *P<0.05, **P<0.01, ***P<0.001. Error bars represent ±se/sd.
Effect of RP and LTNP Vpr variants on virus replication in primary cells
To assess the replication kinetics of virus encoding these Vpr variants, mutations (R36W, L68M, R77Q and R85Y) were introduced in the context of HIV-1 NL43 EGFP reporter virus and replication kinetics were evaluated using normal human donor peripheral blood mononuclear cells (PBMCs) (Fig. 6a, b). First, the percentage of productively infected cells (EGFP+) in PBMC culture was monitored by flow cytometry and results indicate that viruses containing either the wild-type or Vpr mutants infected PBMCs in a similar manner (1.3 %) and did not show any significant differences within different viruses in each donor (Fig. 6a), suggesting that viruses with different Vpr mutants did not affect the process of initiation of infection. However, virus replication kinetics results indicate that HIV-1 NL43 virus containing full-length Vpr showed the highest replication kinetics on day 4 post-infection, followed by HIV-1 VprL68M, HIV-1 ΔVpr and HIV-1 VprR36W viruses, whereas R85Y and R77Q showed the least replication (Fig. 6b). Virus replication monitored by p24 values showed a four-fold reduction in HIV-1 R85Y and seven-fold reduction in HIV-1 R77Q, compared with HIV-1 wild-type on day 4 post-infection. Together these results suggest that HIV-1 virus containing Vpr variants exhibits distinct replication kinetics. Based on the differential replication kinetics observed in HIV-1 virus with Vpr variants, we next assessed whether this defect could be due to altered expression of Vpr mutants. Therefore, HIV-1 viruses harbouring the Vpr variant residues were tested for the expression of Vpr and Gag (p24). All viruses expressed detectable levels of viral proteins Vpr and Gag, except HIV-1 ΔVpr for Vpr expression in the producer cells, as well as in the virus particles (Fig. 6c). Thus, Vpr proteins containing variant residues were expressed in the context of viral replication and the integrity of the mutant virus was not altered, suggesting the differences in replication are not due to lack of Vpr expression.
Fig. 6.

Replication kinetics of HIV-1 containing Vpr variations associated with LTNPs and RPs compared with wild-type Vpr. (a) PBMCs infected with NL43 EGFP reporter virus expressing various Vpr mutants were assessed by flow cytometry to quantify the productively infected cells by gating the EGFP positive cells. EGFP positive cells (%) are gated and marked. ‘Untreated’ represents uninfected (UI) cells and Vpr mutants are marked on the x-axis. The figure represents results from one of three experiments using different donors. (b) Replication kinetics in PBMCs infected with NL43 virus containing Vpr mutations were measured. The Vpr mutants (R36W, L68M, R77Q and R85Y) are mutations introduced in the VprNL43 background. PBMCs were infected with 0.1 m.o.i. virus and the supernatants were collected at 2 day intervals and assessed for p24. Representative viral replication kinetics are presented from two independent donors. A similar replication pattern was observed in multiple donors (n = 4). (c) Virus containing Vpr variants were tested for Vpr expression in virus particles and cell lysates by transfecting HEK293T cells with WT and mutant proviral DNA constructs. Supernatants and cell lysates harvested 72 h post-transfection were subjected to immunoblot against Vpr and Gag. Panels represent Vpr and Gag expression in cell lysates and virus particles.
Discussion
HIV-1 exhibits genetic heterogeneity like other RNA viruses. While analyses have revealed a specific pattern of polymorphisms in env and nef genes with respect to the disease stage, the reports on Vpr are based on a single residue (R77Q and F72L) using a limited number of patients. Based on this, we reason that a comprehensive analysis of Vpr alleles from LTNPs and RPs from subtype B sequences may reveal the underlying differences. Considering the differences reported between Vpr alleles derived from different subtypes (Romani et al., 2009; Shen et al., 2008; Tzitzivacos et al., 2009), we limited our analysis to HIV-1 subtype B sequences. The selected Vpr variants span the three helices and the C-terminal region. All four of these domains have important structural and functional determinants as previously reported (Morellet et al., 2009).
Except R77Q, none of the above noted residues has been reported in the literature. This could be due to analyses being performed by individual investigators working with a small number of samples from a single cohort. We believe that our analyses identified these additional residues owing to the large number of samples from multiple cohorts spanning across clade B. To our knowledge this is the first comprehensive analysis of Vpr polymorphisms in LTNP and RP groups utilizing all the available sequences. We have identified RP-associated variant R36W, which shows a higher level of oligomerization than wild-type. The original residue at this position is arginine, a positively charged basic residue, but it is altered to tryptophan in the RP group. As this change results in a large difference in the side chain of the amino acid, it is reasonable to predict that this would result in large structural and functional changes, given the crucial location of the residue. It likely results in exposure of the hydrophobic faces of the first two helices, which would lead to stronger binding of the Vpr monomers to shield the hydrophobic faces from the environment. This is further supported by the structural studies presented here. Interestingly, none of the other mutations studied showed differences in oligomerization compared with wild-type Vpr. Although it is the only RP-associated variant that showed higher levels of oligomerization out of the three studied (R36W, L68M, R85Y), it does indicate that oligomerization may have a role in pathogenesis. Further analyses are needed to identify whether additional mutations flanking the identified in vivo mutants have any impact on oligomerization function.
Although Vpr does not contain canonical nuclear localizing signal motifs, it has been shown to contain various residues across its three alpha-helices and the C terminus that enable Vpr to translocate into the nucleus (Sherman et al., 2001). Comparing the Vpr variants’ subcellular distribution pattern, we have found differences with the RP-associated VprR36W and VprR85Y. VprR36W localized across the entire cell, and exhibited the formation of puncta, as expected with the higher order of oligomerization. This indicates the possibility of an alteration in the nucleocytoplasmic shuttling mechanism of Vpr and the formation of higher-level oligomers. It is reported that the nuclear localization feature of Vpr enhances virus replication in both macrophages and lymphocytes, although through two different means; in the former, the localization function of Vpr enhances nuclear entry of the PIC, and in the latter, the localization function of Vpr enables Vpr to act as a transactivator (Iijima et al., 2004; Nitahara-Kasahara et al., 2007; Popov et al., 1998). The difference in the pattern of localization of VprR36W and VprR85Y implies that perhaps two separate mechanisms are involved. It has been noted that the localization of Vpr to the nucleus is a necessary, but not sufficient, criterion of G2 arrest (Morellet et al., 2009). Our data corroborate this claim, as the mutant VprR36W is deficient in this function and also is unable to induce G2 cell cycle arrest. Though the significance of this mutation is not fully understood, based on the increased oligomerization (aggregation) and drastic loss of G2 cell cycle arrest function, it is possible to conceive that the higher order of oligomerization may hinder Vpr interaction with host cellular proteins that are necessary for cell cycle arrest function. Interestingly, we observed a higher level of G2 cell cycle arrest with R77Q mutant virus. This also could be due to lack of apoptosis so that cells survive longer when R77Q is expressed than other Vpr mutants. These correlations imply that the LTNP mutations seem to be associated with higher levels of G2 arrest, and RP mutations with reduced levels of G2 arrest. These results are counter-intuitive given that G2 arrest enhances viral replication and would be expected to increase in RP mutations rather than be abolished. One possible explanation is that this could be due to Vpr induced immune evasion. Recent data suggest that the induction of G2 cell cycle arrest upregulates the natural killer cell ligand NKG2D on infected cells, targeting them for cytolysis, and that Vpr mutants unable to induce G2 blockade are able to block and evade detection by NK cells (Pham et al., 2011). However, given the small sample size (two LTNP mutations versus three RP mutations), a more comprehensive analysis of Vpr sequences is needed to draw final conclusions.
For assessing the effect of selected variants on virus replication, we incorporated Vpr variants R36W, L68M, R77Q and R85Y in HIV-1 NL43 and assessed viral replication. As the backbone of Vpr is common to all the constructs, gain or loss of function noted with Vpr can be linked to a single residue. Results from the virus replication studies indicate that virus containing RP-associated Vpr mutations (VprR36W and L68M) showed similar level of virus production compared with wild-type (VprNL43) virus, whereas R77Q and R85Y showed the least virus replication. However, the exact mechanism by which the Vpr containing the variant residues brings about the changes is unclear. None of these mutations is defective in virion association, indicating its presence during early infection, yet R77Q fails to establish infection. It is also possible that there is a cell specific effect that is confounded by the use of total PBMCs and Vpr variants in an NL43 (CXCR4 co-receptor utilizing virus) background as the target cells are primarily CD4+ T cells. Interestingly, we did not observe a high level of virus replication in mutants derived from RP, which is expected based on the rapid disease progression, suggesting that multiple changes within Vpr as well as other genes might be involved to enhance disease development rather than a single point mutation. Results from these studies as well as similar studies on other accessory genes such as Nef, Vif and Vpu should be considered cautiously. While the residues chosen do show some significant differences between LTNP and RP, their frequencies within these groups are not 100 %, suggesting other viral factors may be involved. Perhaps it is not a single residue or a domain, but rather the changes that work collectively or compensate for each other within Vpr as well as other genes that influence pathogenesis. It is possible to predict that HIV-1 induced disease progression might be regulated by multiple HIV-1 proteins working together rather than by a single gene. Studying the entire viral genome from these subjects will provide a better understanding on viral factors contributing to HIV-1 pathogenesis.
Methods
Multiple sequence alignment of Vpr sequences from LTNPs and RPs.
The predicted amino acid sequences of Vpr used in this study were taken from the HIV database (www.hiv.lanl.gov, accessed 2009) and belong to subtype B (total of 1223). In order to perform multiple sequence alignment, with fewer or no gaps, Vpr sequences with deletions (>than 20 amino acids) and premature stop codons were excluded from our analysis. Next, we separated the Vpr sequences that are marked with information about the disease status and grouped them as LTNP and RP. Of the 1112 full-length Vpr sequences available in the database, 192 were listed as LTNP and 102 were from patients with rapid progression and from patients with terminal AIDS. Based on this scenario, we have considered the following parameters for our analysis. (i) Variation among Vpr alleles is noted at the level of individual amino acids (HIV-1 NL43 used as a reference). (ii) The variant amino acid in Vpr is calculated for its frequency in the respective group such as RP, LTNP and overall subtype B. This enables us to compare the changes in specific residues between LTNP and RP. Phylogenetic tree analysis was performed using the mega program (http://www.megasoftware.net/).
Structure based predictions of Vpr alleles derived from LTNP and RP subjects.
A three-dimensional model of the Vpr was obtained using sybyl8.0 (Tripos). A previously determined NMR model of Vpr (Pdb code 1M8L) by Morellet et al. (2003) was used as a template. The solution structure shows that Vpr is made of three major helices, H1, H2 and H3, connected by flexible loops. In this model, the loop segments between helices were placed in aqueous solutions and were generated randomly. The initial and selected mutant structures were energy minimized and refined by molecular dynamics at 900 K for 60 ps, with the backbone of helical segments fixed during the simulation. After this, the simulation was continued at 600 K for 5 ps, and at 300 K for 5 ps. The resulting structure was energy minimized to obtain the final model for analysis.
Generation and expression of Vpr expression plasmids.
Vpr mutations were introduced in BiFC constructs as described previously (Venkatachari et al., 2010). Sequences encoding the amino (residues 1 to 173, VN) or carboxyl (residues 155 to 238, VC) fragments of Venus fluorescence protein were fused to the N terminus of Vpr via a six-alanine linker and HA-tag for detection. Vpr mutants T19A, R36W, L68M, R85Y and R90N were introduced using the QuikChange II Site-Directed Mutagenesis kit (Agilent) and confirmed by sequencing. HEK293T cells co-transfected with 0.5 µg each (1 µg total) of VC-Vpr and VN-Vpr mutant constructs using Polyjet (Signagen). Post-transfection (48 h) cells were lysed with RIPA buffer (50mM Tris; 150 mM NaCl; 1 % Triton-X 100; 1 % IGPAL; 0.1 % SDS; 1X PIC (proteinase inhibitor complex); 1mM PMSF) and immunoblotted with anti-HA (Vpr) and anti-actin antibodies from Sigma, and anti-mouse from Cell Signalling.
Cells.
HEK293T, HeLa and COS cells were maintained at 37 °C, 5 % CO2 in DMEM (Mediatech) supplemented with 10 % fetal bovine serum, 1 % glutamine and 1 % penicillin/streptomycin. Jurkat cells were maintained at 37 °C, 5 % CO2 in RPMI (Mediatech) supplemented with 10 % fetal bovine serum, 1 % glutamine and 1 % penicillin/streptomycin. PBMCs were isolated by gradient centrifugation using lymphocyte separation media (Mediatech) from healthy donors and then maintained in RPMI supplemented with 10 % fetal bovine serum, 1 % glutamine and 1 % penicillin/streptomycin. PBMCs were activated for three days prior to infection with PHA (5 ng ml−1) and then incubated with IL-2 (1 U ml−1) throughout the infection phase.
BiFC analyses by fluorescence microscopy and flow cytometry.
HEK293T cells grown on coverslips in a 24-well plate were transfected with BiFC-Vpr plasmids for 48 h, fixed and stained with DAPI. The slides were viewed under the Olympus Fluoview 500 upright confocal microscope at ×100 magnification. Vpr oligomerization was quantified using NIS Elements Ar Microscope Imaging Software (Nikon). Briefly, BiFC positive cells (n = 12 per slide) from different experiments were measured for total BiFC intensity as well as cytoplasmic and nuclear signal. Signal intensity was measured and normalized using VprNL43 as 100 % and the Vpr mutants were calculated accordingly. BiFC interaction was assessed via flow cytometry. HEK293T cells transfected with BiFC-Vpr constructs were collected 48 h post-transfection, fixed with 4 % paraformaldehyde for 20 min at room temperature, and analysed immediately for BiFC signal. The percentage of positive cells and MFI were calculated using FlowJo software.
Vpr induced cell cycle analysis in infected cells.
Cell cycle analysis was performed in infected Jurkat, CD4+ T cells. Cells (1×106) were infected with 0.1 m.o.i. HIV-1 (NL43), HIV-1 ΔVpr or HIV-1 Vpr mutant EGFP reporter viruses. Seventy-two hours post-infection cells were fixed with 70 % ethanol for 1 h at room temperature, and then stained with cell cycle solution (propidium iodide, 50 µg ml−1; RNase A, 50–100 µg ml−1) at 37 °C for 30–40 min. Stringent gating strategy was used to eliminate dead cells followed by EGFP positive infected single cells. Cell cycle was assessed by quantifying the DNA content in G0 and G2 phase of the infected (EGFP+) and uninfected (EGFP−) cells by flow cytometer (BDFACSCanto). Data analysis was performed using BDFACSDiva software or Weasel software (obtained from http://www.wehi.edu.au/, website of the Walter and Eliza Hall Institute of Medical Research, VA, Australia).
Vpr variant virus characterization and replication kinetics.
HIV-1 proviral constructs expressing the above specific Vpr mutations were generated using HIV-1 NL43 EGFP reporter virus backbone. HIV-1 NL43 EGFP reporter virus was constructed as described before (Venkatachari et al., 2007). Virus stocks were generated by transfecting HEK293T cells with proviral constructs. The supernatants (72 h post-transfection) were then collected, clarified by centrifugation, and then filtered through a 0.22 µm membrane. Virus supernatants were titred by TZM-bl assay. Supernatants and cell lysates were subjected to Western blot to determine integrity of virus particles. PBMCs were infected at an m.o.i. of 0.1 for 16 h. Following infection, supernatants were collected at different time intervals specified in Results and subjected to p24 ELISA.
Statistical analysis.
For analysis of all biological assays, statistics were performed using the Graphpad Prism software suite. Comparisons of samples with control were done separately using one-way ANOVA or two-tailed Student’s t-test using a significance level of P<0.05 (* indicates P<0.05; ** indicates P<0.01; *** indicates P<0.001).
Acknowledgements
This work was in part supported by funding from NIMH, NIH (R01 MH087247) to V. A. We thank Callen Wallace, CBI, for his technical assistance with the imaging studies.
References
- Andreoni M. (2004). Viral phenotype and fitness. New Microbiol 27 (Suppl. 1), 71–76 [PubMed] [Google Scholar]
- Bimber B. N., Chugh P., Giorgi E. E., Kim B., Almudevar A. L., Dewhurst S., O’Connor D. H., Lee H. Y. (2009). Nef gene evolution from a single transmitted strain in acute SIV infection. Retrovirology 6, 57 10.1186/1742-4690-6-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caly L., Saksena N. K., Piller S. C., Jans D. A. (2008). Impaired nuclear import and viral incorporation of Vpr derived from a HIV long-term non-progressor. Retrovirology 5, 67 10.1186/1742-4690-5-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavert W., Webb C. H., Balfour H. H., Jr (2004). Alterations in the C-terminal region of the HIV-1 accessory gene vpr do not confer clinical advantage to subjects receiving nucleoside antiretroviral therapy. J Infect Dis 189, 2181–2184 10.1086/420788 [DOI] [PubMed] [Google Scholar]
- Chui C., Cheung P. K., Brumme C. J., Mo T., Brumme Z. L., Montaner J. S., Badley A. D., Harrigan P. R. (2006). HIV VprR77Q mutation does not influence clinical response of individuals initiating highly active antiretroviral therapy. AIDS Res Hum Retroviruses 22, 615–618 10.1089/aid.2006.22.615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E. A., Dehni G., Sodroski J. G., Haseltine W. A. (1990). Human immunodeficiency virus vpr product is a virion-associated regulatory protein. J Virol 64, 3097–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G., Burger H., Chappey C., Rowland-Jones S., Visosky A., Chen C. H., Moran T., Townsend L., Murray M., Weiser B. (2001). Analysis of transition from long-term nonprogressive to progressive infection identifies sequences that may attenuate HIV type 1. AIDS Res Hum Retroviruses 17, 1395–1404 10.1089/088922201753197060 [DOI] [PubMed] [Google Scholar]
- Fritz J. V., Didier P., Clamme J.-P., Schaub E., Muriaux D., Cabanne C., Morellet N., Bouaziz S., Darlix J.-L. & other authors (2008). Direct Vpr–Vpr interaction in cells monitored by two photon fluorescence correlation spectroscopy and fluorescence lifetime imaging. Retrovirology 5, 87 10.1186/1742-4690-5-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gummuluru S., Emerman M. (1999). Cell cycle- and Vpr-mediated regulation of human immunodeficiency virus type 1 expression in primary and transformed T-cell lines. J Virol 73, 5422–5430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzmayer V., Aitken C., Skinner C., Ryall L., Devare S. G., Hackett J., Jr (2009). Characterization of genetically diverse HIV type 1 from a London cohort: near full-length genomic analysis of a subtype H strain. AIDS Res Hum Retroviruses 25, 721–726 10.1089/aid.2009.0033 [DOI] [PubMed] [Google Scholar]
- Hrimech M., Yao X. J., Bachand F., Rougeau N., Cohen E. A. (1999). Human immunodeficiency virus type 1 (HIV-1) Vpr functions as an immediate-early protein during HIV-1 infection. J Virol 73, 4101–4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima S., Nitahara-Kasahara Y., Kimata K., Zhong Zhuang W., Kamata M., Isogai M., Miwa M., Tsunetsugu-Yokota Y., Aida Y. (2004). Nuclear localization of Vpr is crucial for the efficient replication of HIV-1 in primary CD4+ T cells. Virology 327, 249–261 10.1016/j.virol.2004.06.024 [DOI] [PubMed] [Google Scholar]
- Levy J. A. (2006). HIV pathogenesis: knowledge gained after two decades of research. Adv Dent Res 19, 10–16 10.1177/154407370601900104 [DOI] [PubMed] [Google Scholar]
- Levy D. N., Refaeli Y., Weiner D. B. (1995). Extracellular Vpr protein increases cellular permissiveness to human immunodeficiency virus replication and reactivates virus from latency. J Virol 69, 1243–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum J. J., Cohen O. J., Nie Z., Weaver J. G., Gomez T. S., Yao X.-J., Lynch D., Pilon A. A., Hawley N. & other authors (2003a). Vpr R77Q is associated with long-term nonprogressive HIV infection and impaired induction of apoptosis. J Clin Invest 111, 1547–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum J. J., Cohen O. J., Nie Z., Weaver J. G., Gomez T. S., Yao X. J., Lynch D., Pilon A. A., Hawley N. & other authors (2003b). Vpr R77Q is associated with long-term nonprogressive HIV infection and impaired induction of apoptosis. J Clin Invest 111, 1547–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam S., Ayyavoo V., Patel M., Kieber-Emmons T., Weiner D. B. (1997). Nuclear import, virion incorporation, and cell cycle arrest/differentiation are mediated by distinct functional domains of human immunodeficiency virus type 1 Vpr. J Virol 71, 6339–6347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder B., Venkatachari N. J., Srinivasan A., Ayyavoo V. (2009). HIV-1 mediated immune pathogenesis: spotlight on the role of viral protein R (Vpr). Curr HIV Res 7, 169–177 10.2174/157016209787581445 [DOI] [PubMed] [Google Scholar]
- Mologni D., Citterio P., Menzaghi B., Poma B. Z., Riva C., Broggini V., Sinicco A., Milazzo L., Adorni F. & other authors (2006). Vpr and HIV-1 disease progression: R77Q mutation is associated with long-term control of HIV-1 infection in different groups of patients. AIDS 20, 567–574 10.1097/01.aids.0000210611.60459.0e [DOI] [PubMed] [Google Scholar]
- Morellet N., Bouaziz S., Petitjean P., Roques B. P. (2003). NMR structure of the HIV-1 regulatory protein VPR. J Mol Biol 327, 215–227 10.1016/S0022-2836(03)00060-3 [DOI] [PubMed] [Google Scholar]
- Morellet N., Roques B. P., Bouaziz S. (2009). Structure–function relationship of Vpr: biological implications. Curr HIV Res 7, 184–210 10.2174/157016209787581490 [DOI] [PubMed] [Google Scholar]
- Nitahara-Kasahara Y., Kamata M., Yamamoto T., Zhang X., Miyamoto Y., Muneta K., Iijima S., Yoneda Y., Tsunetsugu-Yokota Y., Aida Y. (2007). Novel nuclear import of Vpr promoted by importin α is crucial for human immunodeficiency virus type 1 replication in macrophages. J Virol 81, 5284–5293 10.1128/JVI.01928-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham T. N. Q., Richard J., Gerard F. C. A., Power C., Cohen É. A. (2011). Modulation of NKG2D-mediated cytotoxic functions of natural killer cells by viral protein R from HIV-1 primary isolates. J Virol 85, 12254–12261 10.1128/JVI.05835-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon B., Chen I. S. (2003). Human immunodeficiency virus type 1 (HIV-1) Vpr enhances expression from unintegrated HIV-1 DNA. J Virol 77, 3962–3972 10.1128/JVI.77.7.3962-3972.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov S., Rexach M., Ratner L., Blobel G., Bukrinsky M. (1998). Viral protein R regulates docking of the HIV-1 preintegration complex to the nuclear pore complex. J Biol Chem 273, 13347–13352 10.1074/jbc.273.21.13347 [DOI] [PubMed] [Google Scholar]
- Reinis M., Weiser B., Kuiken C., Dong T., Lang D., Nachman S., Zhang Y., Rowland-Jones S., Burger H. (2007). Genomic analysis of HIV type 1 strains derived from a mother and child pair of long-term nonprogressors. AIDS Res Hum Retroviruses 23, 309–315 10.1089/aid.2006.0180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes D. I., Ashton L., Solomon A., Carr A., Cooper D., Kaldor J., Deacon N., Australian Long-Term Nonprogressor Study Group (2000). Characterization of three nef-defective human immunodeficiency virus type 1 strains associated with long-term nonprogression. J Virol 74, 10581–10588 10.1128/JVI.74.22.10581-10588.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodés B., Toro C., Paxinos E., Poveda E., Martinez-Padial M., Benito J. M., Jimenez V., Wrin T., Bassani S., Soriano V. (2004). Differences in disease progression in a cohort of long-term non-progressors after more than 16 years of HIV-1 infection. AIDS 18, 1109–1116 10.1097/00002030-200405210-00004 [DOI] [PubMed] [Google Scholar]
- Romani B., Glashoff R., Engelbrecht S. (2009). Molecular and phylogenetic analysis of HIV type 1 vpr sequences of South African strains. AIDS Res Hum Retroviruses 25, 357–362 10.1089/aid.2008.0251 [DOI] [PubMed] [Google Scholar]
- Saksena N. K., Ge Y. C., Wang B., Xiang S. H., Dwyer D. E., Randle C., Palasanthiran P., Ziegler J., Cunningham A. L. (1996). An HIV-1 infected long-term non-progressor (LTNP): molecular analysis of HIV-1 strains in the vpr and nef genes. Ann Acad Med Singapore 25, 848–854 [PubMed] [Google Scholar]
- Shen C., Gupta P., Wu H., Chen X., Huang X., Zhou Y., Chen Y. (2008). Molecular characterization of the HIV type 1 vpr gene in infected Chinese former blood/plasma donors at different stages of diseases. AIDS Res Hum Retroviruses 24, 661–666 10.1089/aid.2007.0270 [DOI] [PubMed] [Google Scholar]
- Sherman M. P., de Noronha C. M. C., Heusch M. I., Greene S., Greene W. C. (2001). Nucleocytoplasmic shuttling by human immunodeficiency virus type 1 Vpr. J Virol 75, 1522–1532 10.1128/JVI.75.3.1522-1532.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioda T., Nakayama E. E. (2006). Human genetic polymorphisms affecting HIV-1 diseases. Int J Hematol 84, 12–17 10.1532/IJH97.06100 [DOI] [PubMed] [Google Scholar]
- Somasundaran M., Sharkey M., Brichacek B., Luzuriaga K., Emerman M., Sullivan J. L., Stevenson M. (2002). Evidence for a cytopathogenicity determinant in HIV-1 Vpr. Proc Natl Acad Sci U S A 99, 9503–9508 10.1073/pnas.142313699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tungaturthi P. K., Sawaya B. E., Singh S. P., Tomkowicz B., Ayyavoo V., Khalili K., Collman R. G., Amini S., Srinivasan A. (2003). Role of HIV-1 Vpr in AIDS pathogenesis: relevance and implications of intravirion, intracellular and free Vpr. Biomed Pharmacother 57, 20–24 10.1016/S0753-3322(02)00328-1 [DOI] [PubMed] [Google Scholar]
- Tzitzivacos D. B., Tiemessen C. T., Stevens W. S., Papathanasopoulos M. A. (2009). Viral genetic determinants of nonprogressive HIV type 1 subtype C infection in antiretroviral drug-naive children. AIDS Res Hum Retroviruses 25, 1141–1148 10.1089/aid.2009.0080 [DOI] [PubMed] [Google Scholar]
- Venkatachari N. J., Majumder B., Ayyavoo V. (2007). Human immunodeficiency virus (HIV) type 1 Vpr induces differential regulation of T cell costimulatory molecules: direct effect of Vpr on T cell activation and immune function. Virology 358, 347–356 10.1016/j.virol.2006.08.030 [DOI] [PubMed] [Google Scholar]
- Venkatachari N. J., Walker L. A., Tastan O., Le T., Dempsey T. M., Li Y., Yanamala N., Srinivasan A., Klein-Seetharaman J. & other authors (2010). Human immunodeficiency virus type 1 Vpr: oligomerization is an essential feature for its incorporation into virus particles. Virol J 7, 119 10.1186/1743-422X-7-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Ge Y. C., Palasanthiran P., Xiang S. H., Ziegler J., Dwyer D. E., Randle C., Dowton D., Cunningham A., Saksena N. K. (1996). Gene defects clustered at the C-terminus of the vpr gene of HIV-1 in long-term nonprogressing mother and child pair: in vivo evolution of vpr quasispecies in blood and plasma. Virology 223, 224–232 10.1006/viro.1996.0471 [DOI] [PubMed] [Google Scholar]
- Zhao L. J., Zhu H. (2004). Structure and function of HIV-1 auxiliary regulatory protein Vpr: novel clues to drug design. Curr Drug Targets Immune Endocr Metabol Disord 4, 265–275 10.2174/1568008043339668 [DOI] [PubMed] [Google Scholar]
- Zhao Y., Chen M., Wang B., Yang J., Elder R. T., Song X. Q., Yu M., Saksena N. K. (2002). Functional conservation of HIV-1 Vpr and variability in a mother–child pair of long-term non-progressors. Virus Res 89, 103–121 10.1016/S0168-1702(02)00127-2 [DOI] [PubMed] [Google Scholar]