

Summary
The incretin hormone Glucagon-like peptide 1 (GLP-1) requires delivery by injection for the treatment of Type 2 diabetes mellitus. Here, we test if the properties of glycosphingolipid trafficking in epithelial cells can be applied to convert GLP-1 into a molecule suitable for mucosal absorption. GLP-1 was coupled to the extracellular oligosaccharide domain of GM1 species containing ceramides with different fatty acids and with minimal loss of incretin bioactivity. When applied to apical surfaces of polarized epithelial cells in monolayer culture, only GLP-1 coupled to GM1-ceramides with short-or cis-unsaturated fatty acids trafficked efficiently across the cell to the basolateral membrane by transcytosis. In vivo studies showed mucosal absorption after nasal administration. The results substantiate our recently reported dependence on ceramide structure for trafficking the GM1 across polarized epithelial cells and support the idea that specific glycosphingolipids can be harnessed as molecular vehicles for mucosal delivery of therapeutic peptides.
Keywords: GM1, hGLP-1, drug delivery, transcytosis
Introduction
One of the major difficulties in effectively treating Type II diabetes and other chronic diseases involves patient compliance for life-long management by frequent injections of therapeutic peptides or proteins. Two of the most potent therapeutic peptides used to control blood glucose in Type II diabetes, insulin and analogues of the incretin hormone GLP-1, are impermeant to epithelial barriers at mucosal surfaces and require delivery by subcutaneous (SC) injection. Optimal control of blood sugar levels by these agents entails frequent daily injections, which can be sometimes missed by patients due to lifestyle and the social stigma of self-injection in public. Thus, much effort has gone into the development of technologies to render these therapeutic peptides absorbable across mucosal surfaces for efficient delivery without self-injection and with improved patient compliance. Here, we test if our recent discoveries on glycosphingolipid trafficking in polarized epithelial cells [1, 2] can be applied to convert GLP-1 into a molecule suitable for mucosal absorption.
The gastrointestinal, respiratory, and genitourinary tracts represent vast mucosal surfaces where host tissues are separated from the environment only by a delicate but highly effective single layer of columnar epithelial cells, joined by tight junctions that are impermeable to proteins and even small peptides. Proteins non-specifically taken up into the epithelial cell by endocytosis are generally transported to lysosomes for degradation, establishing an effective barrier to uncontrolled entry of these materials into the body and the maintenance of systemic homeostasis. So far, the lack of rational and efficient methods to circumvent this barrier has prevented the application of most therapeutic proteins for efficient mucosal drug delivery.
Glucagon-like peptide-1 (GLP-1) is an incretin hormone derived from the transcription product of the pro-glucagon gene and is primarily released from L-cells of the intestinal mucosa [3]. This hormone has lasting and potent antihyperglycemic actions by inducing glucose-dependent stimulation of insulin secretion while suppressing glucagon secretion, as well as restoring glucose sensitivity of pancreatic β-cells while it inhibits gastric secretion and motility to protract carbohydrate absorption and contribute to satiety [4]. Once in the circulation, GLP-1 is rapidly degraded (with a half-life of less than two minutes). Thus, the majority of pharmaceutical approaches have been focused on developing longer-acting analogues of GLP-1 [5]. Exenatide, a synthetic form of the peptide exendin-4 from lizard saliva, is a potent GLP-1 receptor (GLP-1 R) agonist on the market for Type 2 diabetes [6] and even has been developed into a once-weekly formulation by encapsulation in Poly-(D,L-Lactide-Co-Glycolide) Microspheres [7, 8]. Another marketed example is liraglutide, an acylated human GLP-1 analogue [9]. Although these degradation-resistant GLP-1 R agonists are excellent agents for the treatment of diabetes, they still require daily to weekly delivery by injection. Developing an oral long-acting GLP-1 formulation would allow patients to manage their blood glucose levels by a non-parenteral form of administration.
We recently discovered that the structure of the ceramide domain of the glycosphingolipid GM1, the receptor for cholera toxin that mediates cell entry, dictates intracellular trafficking and the subcellular localization of this lipid in mammalian cells [1, 2]. GM1 is comprised of two major domains: the hydrophilic oligosaccharide head group that protrudes from the cell membrane into the extra-cellular space; and the hydrophobic ceramide lipid tail that anchors the molecule within the membrane bilayer. Native GM1 ceramides in human cells show heterogeneity in structure, most typically in length of the fatty acid carbon chain and degree of saturation [10].
When exogenously applied to epithelial cells, the GM1-ceramide species that contain a short chain C12:0 or an unsaturated C16:1 fatty acid in the ceramide domain enter the early sorting/recycling endosome from where they are sorted to other destinations [1]. The GM1-ceramide species with long, saturated C18:0 fatty acyl chains are transported instead to the lysosome, presumably for degradation. In simple polarized epithelial cells that form the single-cell thick monolayer lining mucosal surfaces, exogenously added GM1 species with short or unsaturated ceramide domains are transported from the apical (luminal) membrane across the cell to the basolateral (serosal) membrane by transcytosis, following a transcellular pathway leading to absorption [2].
Here, we test if GM1 can be used as a molecular carrier to deliver analogues of GLP-1 across epithelial barriers by harnessing these properties of endogenous sphingolipid trafficking. We linked the GLP-1 peptide to the extracellular domain of different GM1 species with ceramide domains of known structure, synthesized as described before [1]. We found that ceramide-directed trafficking remained intact after GM1 attachment to a therapeutic analogue of GLP-1. Fusion molecules with short- or cis-unsaturated ceramide domains entered epithelial cells by endocytosis, trafficked to the recycling endosome, and reached the basolateral membrane of polarized cells by transcytosis; suggesting that specific glycosphingolipids may, indeed, be harnessed as molecular vehicles for mucosal delivery of therapeutic peptides.
Materials and Methods
Materials and Reagents
Defatted bovine serum albumin (df-BSA), fetal bovine serum (FBS), Hank's balanced salts solution (HBSS) and 2-nitrophenyl β-D-galactopyranoside were from Sigma-Aldrich (St. Louis, MO). Steadylite reagent was from PerkinElmer (Boston, MA), the Cytotox ONE™ kit from Promega (Madison, WI). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from Invitrogen (Eugene, OR) and wheat germ agglutinin labeled Alexa594 (WGA-Alexa594) from Molecular Probes (Eugene, OR). The plasmids encoding the human GLP-1 receptor (GLP-1 R), the CRE6X-LUC reporter gene and β-galactosidase were kind gifts of Alan S. Kopin (Tufts, Boston, MA), and have been described previously [11, 12].
GM1 containing C18:0 fatty acids were extracted from bovine brains using the method of Folch [13] and purified by ion exchange and reversed-phase chromatography. GM1 variants were provided by Prof. S. Sonnino (Italy) and were prepared essentially as described in [1]. Synthesis of the GLP-1 analogue (GLP-1* Figure 1) was performed by New England Peptide NEP (MA, USA) by solid phase peptide synthesis.
Figure 1. Characterization of GLP-1*-GM1 fusion molecules.
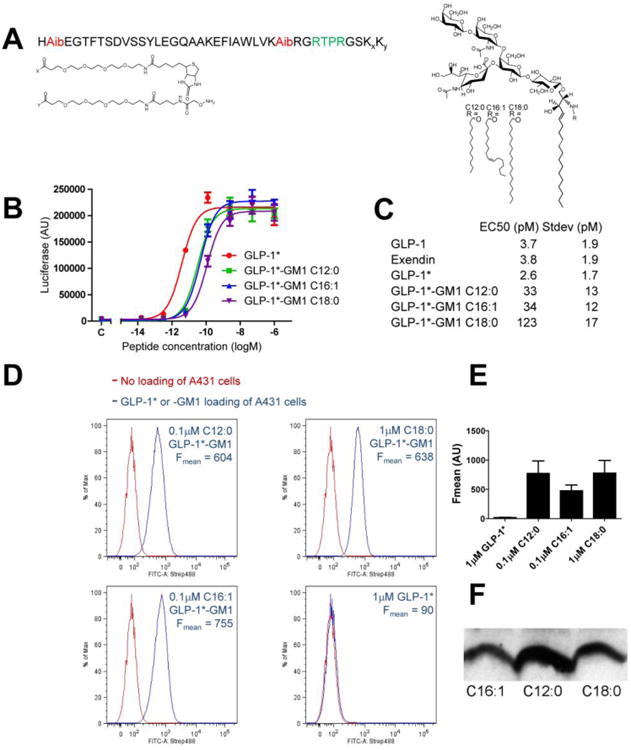
(A) Structures of GLP-1* (left panel) and GM1 (right panel). The different GM1 acyl chains (R) and functionalized lysyl-PEG linkers (Kx, Ky) are indicated. (B) GLP-1*-GM1 fusion molecules retain functional activity. The activities were determined by an in vitro assay for peptide-induced cAMP using HEK293 cells expressing the hGLP-1 receptor measured in triplicate (error bars represent mean ± SEM). (C) Table of the EC50 values as determined in (B) as mean ± SD for at least 4 separate experiments. (D) Optimizing conditions to obtain equally loaded GLP-1*-GM1 cell membranes. FACS analysis of A431 cells loaded with GLP-1*-GM1 as indicated for 10 min, post-trypsin wash (blue trace). Cells were incubated with streptavidin-Alexa488 and fluorescence was measured in the FITC-A channel. Control cells not loaded with GM1 are shown in red (Fmean = 75.4). At the indicated concentrations, the cells loaded with C12:0, C16:1 or C18:0 GM1 fusions showed equal fluorescence (blue traces), while the free peptide did not bind detectably to the cells (quantification from at least two separate experiments corrected for background are shown in (E) as mean ± SEM). (F) Immunoblot of lysates from polarized MDCK monolayers incubated for 10 min with GLP-1*-GM1 fusion molecules at 0.25 μM C16:1, 0.2 μM C12:0 and 2 μM C18:0 (post-trypsin wash). Immunoblots were probed with neutravidin-HRP and a representative of two independent experiments is shown.
Synthesis of GLP-1* -GM1 Structural Variants
Gangliosides were labeled via the oligosaccharide head group following a modified method from [1]. The glycerol side chain of the sialic acid of GM1 (2 μmoles) was oxidized with 2 mM sodium periodate in 100 mM sodium acetate pH 5.5, 150 mM sodium chloride for 30 min on ice, and the reaction quenched with glycerol. The oxidized ganglioside was then desalted by Bond Elut reverse phase cartridge as previously described [1].
Conjugation of GM1 to GLP-1* and subsequent purification was performed by New England Peptide NEP (MA, USA). Briefly, 2 μmoles oxidized ganglioside were resuspended in phosphate buffered saline pH 7.4 (PBS) and reacted with 6 μmol (1:3 ratio) GLP-1* in PBS containing 100 mM aniline for 16 hours. The coupled GLP-1*-GM1 analogues were purified by preparative RP-HPLC on a YMC C-18 column (250 × 4 mm, 5 μm, Shiseido, Japan) using a mobile phase of 10 mM aqueous ammonium acetate (eluent A) and 10 mM ammonium acetate in 80% acetonitrile and 20% water (eluent B). Products were confirmed using MALDI-TOF-MS (Biosystems Voyager-DE; PE Biosystems, Foster City, CA) using a matrix solution of 10 mg/mL alpha cyano 4-hydroxycinnamic acid in 50/50 eluent A/B. Mass spectra were recorded in linear mode, with either positive-ion or negative-ion TOF detection using an accelerating voltage of 20 kV. Purity was assessed by SDS-PAGE analysis as described in the Supplemental Materials and Methods.
Bioactivity assay
Bioactivity of GLP-1* and all GLP-1*-GM1 fusion molecules was assayed in HEK293 cells expressing the hGLP-1 receptor and a luciferase-CRE reporter for cAMP described in [11, 12, 14, 15]. In brief, HEK293 cells plated at a density of 2000–3000 cells per well in clear-bottom, white 96-well plates were transiently transfected using Lipofectamine® reagent (Invitrogen, Carlsbad, CA) with cDNAs encoding (1) hGLP-1-R, (2) the CRE6X-LUC reporter gene and (3) β-galactosidase. Twenty four hours after transfection, cells were incubated with different concentrations of free peptide or a fusion molecule in serum-free medium with df-BSA for 4 h. Following ligand stimulation, cells were lysed and luciferase activity was quantified after addition of Steadylite reagent using a Victor ×3 2030 Multilabel Reader (PerkinElmer, Boston, MA). A β-galactosidase assay was performed after adding the enzyme substrate 2-Nitrophenyl β-D-galactopyranoside, and incubating at 37°C for 15–45 minutes. Substrate cleavage was quantified by measurement of optical density at 420 nm using a SpectraMax 250 microplate reader (Molecular Devices, Sunnyvale, CA). Corresponding values were used to normalize the luciferase data for transfection efficiency. Results were analyzed using non-linear regression techniques with GraphPad Prism v5.00 (San Diego, California, USA), which was used to determine the ED50.
Trafficking in A431 Cells
Stable A431 cell lines expressing EGFP markers were plated on 18 mm coverslips two days prior to the incubation. Cells were washed in 37 °C Hank's Balanced Salt Solution (HBSS) and equilibrated with df-BSA in HBSS for 10 min. Cells were incubated for 1 - 2 h at 37 °C with GLP-1*-GM1 lipids at 0.2-2.0 μM concentrations in a molar ratio of 1:10 (lipid:df-BSA). Cells were then washed with 37 °C HBSS with 1% df-BSA and then washed with PBS before fixation with 4% paraformaldehyde (Electron Microscopy Sciences) for 20 min at room temperature. All remaining steps were performed at room temperature in a humidified chamber. After quenching the paraformaldehyde with 50 mM ammonium chloride and several washes with PBS, cells were permeabilized with 0.05% saponin in PBS for 30 min. The cells were then incubated with streptavidin-Alexa568 (Invitrogen) diluted at 1:200 in 0.01% saponin in PBS for 30 minutes. Preparations were mounted in anti-fade medium Mowiol and imaged by confocal microscopy.
Polarized Epithelial Trafficking
MDCK II cells plated on 24-well Transwell® inserts (0.4 μm, polyester membranes, Costar) were washed and equilibrated in DMEM containing df-BSA prior to apical addition of GLP-1*- GM1 fusion molecules at 0.2 - 4.0 μM complexed to df-BSA in a 1:10 ratio or free peptide. In continuous studies, cells were incubated with the compounds for 3 h at 37 °C. In the pulse-chase studies, cells were incubated for 1 h with GLP-1*-GM1 compounds at 37 °C prior to exchange with DMEM followed by a 3 h chase. Control cells were incubated for 10 min at 37 °C, after which they were placed at 4 °C for the remaining incubation time. A similar protocol was performed for T84 cells, which were plated on 24-well Transwell® inserts (3.0 μm, polyester membranes, Costar) and all incubations were performed in T84 medium (without serum). Only high electrical resistance T84 monolayers or MDCK II cells that kept their resistance during the experiments were included in this study.
For transcellular detection of GLP-1*-GM1 fusion molecules in polarized epithelial cells, an excess of BSA (1% wt/vol) was added basolaterally to aid in the extraction of lipids from the basolateral surface. At the end of incubations, the basolateral media was collected and the amount of GLP-1* or GLP-1*-GM1 fusion molecule in the media were determined by the bioactivity assay described above with concentrations calculated from a standard curve.
For cell imaging, cells were washed after the incubation with 37 °C HBSS with 1% df-BSA and then PBS before fixation with 4% paraformaldehyde (Electron Microscopy Sciences, PA) for 20 min at room temperature. All remaining steps were performed in a humidified chamber. After several washes with PBS, the cells were incubated with streptavidin- Alexa 568 (Invitrogen) diluted at 1:100 in PBS and incubated overnight at 4 °C. After extensive washes with PBS, the cells were mounted in the anti-fade medium Mowiol.
In vivo studies
WT C57BL/6 mice (male, 6-9 weeks old) were purchased from Jackson Laboratory (Maine, USA). These mice have a naturally occurring deletion in the nicotinamide nucleotide transhydrogenase gene, which results in impaired glucose-stimulated insulin secretion [16]. Animals were acclimatized for 2 weeks and housed under a 12 h light/dark cycle and allowed food and water ad libitum. The nasal in vivo activity was evaluated using an oral glucose tolerance test (OGTT). Briefly, mice that had been fasted for 18 h, were lightly anesthetized with isoflurane and 20 μL of GLP-1* or GLP-1*-GM1 C16:1 (various concentrations) or saline was slowly dripped into the nasal cavity. After 1 h, mice were injected a 2 g/kg dose of glucose (n = 2 each). Blood glucose levels were determined at given times in approximately 5 μL whole blood obtained from tail nick using a one-touch blood glucose meter (Contour, Bayer Healthcare, IN, USA) and area-under-the-curve (AUC) values were calculated using GraphPad Prism v5.00 (San Diego, California, USA).
Results and Discussion
Synthesis and characterization of GLP-1 analogues and GLP-1-GM1 fusion variants
We synthesized a stable GLP-1 analogue containing α-amino-isobutyric acids (Aib) at residues 8 and 33 to allow for increased half-life [17] (Figure 1A). The peptide was extended at the C-terminus to incorporate a short linker sequence followed by two modified lysine residues. The penultimate amino acid contained a biotin molecule linked via a 0.5 kDa PEG spacer, allowing us to track the molecule biochemically and by microscopy. The terminal amino acid contained the same PEG spacer ending with an aminooxy group for coupling to the extracellular oligosaccharide domain of GM1 (Figure 1A).
To allow, in theory, for cleavage and release of the GLP-1 analogue from GM1 after transcytosis, we designed the peptide to include one of the several subtilisin-related endoprotease furin cleavage motifs [18, 19] inserted between GLP-1 and the terminal amino acids of a linker-sequence. The linker sequence contained the biotin and aminoxy reactive group for fusion to GM1 (illustrated in green, Figure 1A). Furin processes a wide range of bioactive proteins and localizes, among other intracellular organelles, to the basolateral surface of polarized epithelial and endothelial cells [20, 21]. When tested in vitro, however, the GLP-1 linker peptide was resistant to cleavage by the furin protease (Figure S1); perhaps due to interference with the enzyme by the more stably coiled structure of the GLP-1 analogue with its Aib residues [17], or due to the additional PEG groups and/or biotin blocking the furin recognition site. Thus, despite our intentions, the peptide and GM1 sphingolipid domains in this design remained stably fused. Throughout this paper, we refer to the PEGylated analogue peptide as GLP-1* and to the fusion molecule as GLP-1*-GM1.
GM1 was oxidized on the sialic acid to generate a reactive aldehyde group to conjugate, via oxime ligation, to the aminooxy side chain engineered into the peptide. GLP-1* was fused to GM1 species synthesized to contain either a short saturated 12-carbon fatty acyl chain (C12:0), a single unsaturated 16-carbon acyl chain (C16:1), or a long saturated 18-carbon acyl chain in the ceramide domain (C18:0) (Figure 1A).
The structures of all fusion molecules were confirmed by mass spectrometry (Figure S2) and their purity by gel analysis and densitometry (Figure S3 A and B). With the exception of the C12:0 molecular species, the GM1 species synthesized are naturally occurring lipids [10, 22]. The molecules did not induce significant cytotoxicity in MDCK II cells after 3 h of continuous incubation as assayed by transepithelial resistance (data not shown) and LDH release (Figure S3 D). Neither was any significant cytotoxicity observed after 24 h observed by LDH release or MTT metabolism (Figure S3 D and E), an incubation time far exceeding the conditions of our assay. All fusion molecules were shown to retain bioactivity of the GLP-1 component as assessed in vitro using HEK293 cells expressing the hGLP-1 receptor, with an ED50 for the GM1 C12:0 and C16:1 fusions of only 10-fold less than the native peptide, and still with picomolar efficacy (Figure 1B-C). The GM1 C18:0 fusion was approximately one and a half-log (40-fold) less active, implicating interference by the more hydrophobic nature of this fusion molecule. Other groups have observed similar decreases in potency when single or dual linked simple fatty acids were attached to a GLP-1 analogue, with very long fatty acids decreasing potency [23].
Conditions for loading cells equally with the different GLP-1*-GM1 fusion molecules were determined in A431 cells by fluorescence-activated cell sorting (FACS) analysis (Figs 1D and E) or in polarized canine kidney MDCK monolayers by Western Blot (Fig 1F). Analysis of GM1 membrane uptake was done after treatment with trypsin, which was used as reported to remove any fusion molecules adhering to the cell but not specifically integrated into the membrane bilayer [1, 24, 25] (Figs 1D-F). In principle, only lipids properly incorporated into the membrane bilayer will traffic according to the structure of the ceramide domains in live cells. Equal membrane incorporation of the GLP-1*-GM1 fusion molecules in A431 cells were observed in a 1:1.25:8 ratio for the C12:0, C16:1, and C18:0 species respectively. Applying the same ratio for loading MDCK cells resulted in a higher incorporation of C12:0 species in comparison to C16:1 or C18:0, but, the levels of incorporation of the C16:1 and C18:0 fusion molecules were closely comparable. A 10-fold molar excess of defatted bovine serum albumin (df-BSA) was used to optimize membrane loading in A431 or MDCK cells (Figure S4).
Influence of peptide coupling on GM1 intracellular trafficking
To test if the different GM1 ceramide domains directed intracellular trafficking of the GLP-1*-GM1 fusion molecule as predicted from our earlier results [1], we applied the fusion proteins to A431 cells stably expressing EGFP-fusions to Rab11a, and Rab7 GTPases that mark the recycling (RE), and late endosomal compartments (LE) respectively. The fraction of GLP-1 *-GM1 occupying each sub-cellular compartment in three dimensions was quantified relative to total GLP-1*-GM1 incorporated into the cell using the Manders colocalization coefficient (termed Mx, as described in supplemental materials and methods [1, 26]). Both GLP-1*-GM1 C12:0 and C16:1 fusion molecules entered the recycling endosome of epithelial cells (Rab11a compartment) (Figure 2A and quantified on plot to right). Less of the C18:0 fusion molecules reached this compartment, and more C18:0 was localized in late endosomes (Rab7, Figure 2B) compared to C12:0 and C16:1. These results are fully consistent with our earlier studies on trafficking Alexa-GM1 [1]. Thus, the biology and influence of ceramide structure on GM1 trafficking remains intact after attachment to a therapeutic analogue of GLP-1.
Figure 2. Intracellular Distribution of the Different GLP-1*-GM1 fusion molecules.
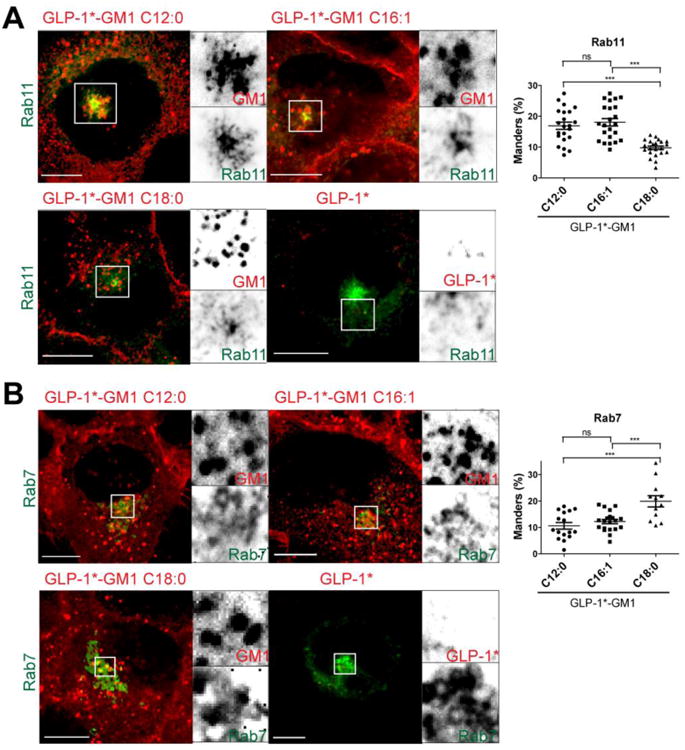
(A and B) Confocal images of fixed A431 cells stably expressing EGFP fusion proteins as indicated. Cells were incubated 2 h with GLP-1* labeled GM1 (0.2 μM C12:0, 0.25 μM C16:1 or 2 μM C18:0) or free peptide (2 μM). After washing, the cells were fixed and stained with streptavidin-Alexa568 to label the fusion molecule via biotin (red). Inverted grayscale images of selected areas (dotted box) for individual fluorescence channels are shown to the right of the merged images. Histograms (far right) show quantitation of GLP-1*-GM1 localized to the respective compartment using Manders (Mx) coefficients for at least three independent experiments. Each dot represents a correlation coefficient calculated from a single field of view containing 2–3 cells on average. Scale bars = 10 μm (ns stands for p > 0.05, *** stands for p ≤ 0.001).
GM1 sphingolipids as transport vehicles across epithelial barriers
To test for transcytosis, GLP-1*-GM1 fusion molecules were introduced into apical membranes of polarized canine kidney MDCK monolayers and analyzed for transport to the basolateral membrane by confocal microscopy. The GLP-1*-GM1 C12:0 and C16:1 fusion molecules were found on basolateral membranes after 3 h at 37°C (Figure 3A, 2nd and 3rd rows). Localization to the basolateral membrane was specified using wheat germ agglutinin (WGA) to stain opposing apical membrane glycoproteins (Supplemental Fig S5), and by using MDCK cells expressing EGFP-tagged FcRn, a Fc-trafficking receptor that localizes at steady state primarily to the cell interior (Supplemental Fig S6). No basolateral membrane localization was detected in control monolayers loaded apically with the free peptide GLP-1 * (Fig 3A top row) or with the GLP-1*-GM1 C12:0 fusion molecule incubated with cell monolayers for 10 min and then shifted to 4°C (Fig 3A bottom row). These results indicate that transcellular glycosphingolipid transport from apical to basolateral cell surfaces was by transcytosis and not by paracellular diffusion through tight junctions. In contrast to the GLP-1*-GM1 C12:0 and C16:1 fusion molecules, we found barely detectable amounts of the GLP-1*-GM1 C18:0 species localized to basolateral membranes (Figure 3A, 4th row); even though comparable levels were initially associated with the apical PM before transport (Figure 1F). Thus, transcellular transport depends on ceramide structure of the GM1 sphingolipid carrier, a result consistent with our earlier studies [2].
Figure 3. Polarized epithelial cells sort the GLP-1*-GM1 fusion molecules into the transcytotic pathway based on structure of the ceramide domain and the amount GLP-1*-GM1 reaching the basolateral surface is concentration dependent.
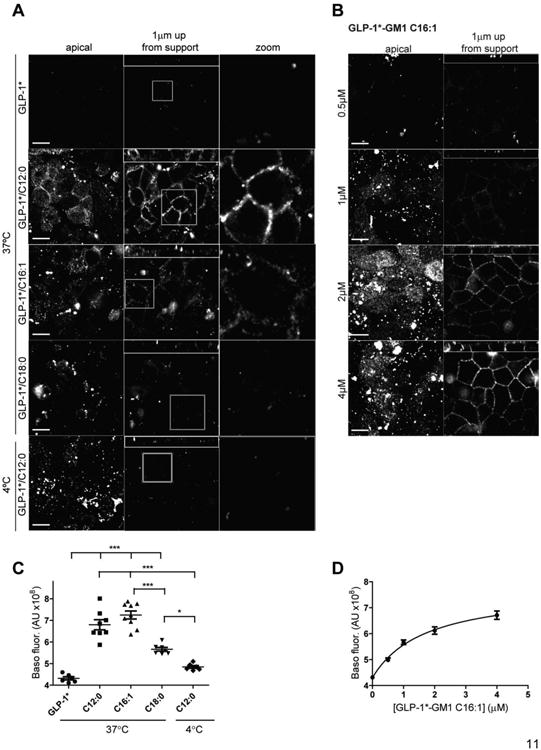
The controls with free peptide or placed at 4°C do not show basolateral membrane staining, nor does GLP-1*-GM1 C18:0. (A) Polarized epithelial monolayers of MDCK cells were continuously incubated apically for 3 h at 37°C with GLP-1* labeled GM1 (0.4 μM C12:0, 0.5 μM C16:1 or 4 μM C18:0), or free peptide (0.4 μM) (B) or pulse labeled apically with GLP-1*-GM1 C16:1 (concentrations as indicated) at 37°C for 1 h and then chased for 3 h. After washing, cells were fixed and stained with streptavidin-Alexa568. Illustrated are confocal images of the apical membrane, basolateral signal at ∼1 μm up from the membrane support (small panels above are XZ reconstructions), and a zoom of the basolateral image. Unlike the GLP-1*-GM1 C12:0 and C16:1 fusion proteins, the GLP-1*-GM1 C18:0 does not visualize at the apical membrane at 3 h incubations because it is trafficked away from the recycling endosomes to the late endosome lysosome pathway. Data are representative of 5 independent experiments for A and 4 for B. Scale bars = 10 μm. (C-D) Quantification of the basolateral fluorescent signal at ∼1 μm up from the membrane support of images obtained in experiment A and B. Each dot represents a single field of view containing 30 cells on average and between 5 - 9 fields were analyzed (mean ± SEM). * stands for p ≤ 0.05 and *** stands for p ≤ 0.001.
We next tested for the membrane topology of GM1 transport to confirm that the GLP-1* peptide was delivered to the extra-cytoplasmic surface of the basolateral membrane and available to the extracellular basolateral (serosal) space after transcytosis. To do this, the basolateral membrane of live MDCK monolayers were stained at 4°C with streptavidin-Alexa568 after their apical 37°C incubation with GLP-1*-GM1 C16:1. At 4°C, membrane traffic is stopped, cell membranes and tight junctions remain intact, and streptavidin can bind only if the GLP-1* peptide containing biotin is exposed on the extracellular surface of the basolateral membrane. The results show that streptavidin bound to basolateral membranes of MDCK monolayers incubated with GLP-1*-GM1 C16:1 (Figure S7). No labeling was observed for monolayers treated with the GLP-1* peptide, or if incubated with the GLP-1*-GM1 C16:1 fusion molecule for just 10 min at 37°C to allow loading of the apical membrane (Fig S7, upper and lower rows).
The amount of GLP-1*-GM1 fusion molecule transported to the basolateral surface was concentration dependent, as assessed using the GLP-1*-GM1 C16:1 species (Figure 3B). Similar results were obtained for the C12:0- and C16:1-containing GLP-1*-GM1 fusion molecules applied to polarized human intestinal T84 cells in monolayer culture, although higher concentrations had to be applied to obtain lipid-uptake and detectable transport in T84 cells (Figure 4, and see [2]). Based on these studies, we conclude that the GM1 sphingolipids with short or unsaturated ceramide domains can act as molecular carriers for transcellular transport of therapeutic peptides across barrier epithelial cells.
Figure 4. GLP-1*-GM1 crosses to the basolateral membrane of T84 monolayers by transcytosis.
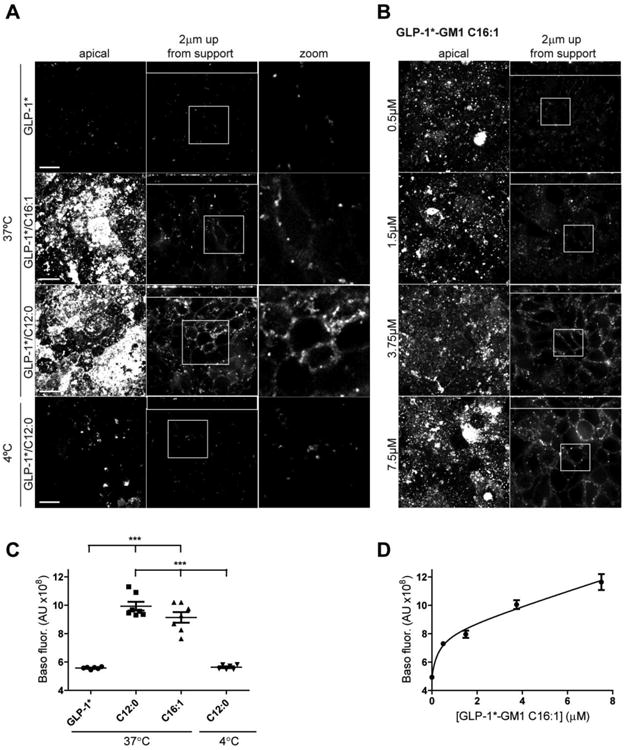
The controls are monolayers incubated with free peptide or with GLP-1*-GM1 at 4°C and do not show a basolateral membrane staining. T84 cells grown on a membrane support were pulse labeled apically at 37°C for 1 h and then chased for 3 h with GLP-1* labeled GM1 C12:0, C16:1 or free peptide at 1 μM (A) or with varying concentrations of GLP-1*-GM1 C16:1 (concentrations as indicated, B). After washing, the cells were fixed and stained with. streptavidin-Al568. Illustrated are a confocal image of the apical membrane, basolateral signal at ∼2 μm up from the membrane support (small panels above are XZ reconstructions), and a zoom of the basolateral image. Data are representative of three independent experiments. Scale bars = 10 μm. (C-D) Quantification of the basolateral fluorescent signal at ∼2 μm up from the membrane support of images obtained in experiment A and B. Each dot represents a single field of view containing 45 cells on average and between 5 - 7 fields were analyzed (mean ± SEM). *** stands for p ≤ 0.001.
To confirm these results, we tested for transcytosis in another way by measuring the amount of GLP-1*-GM1 fusion molecules released from the basolateral membrane of MDCK monolayers to the basolateral media after an apical application. Initial studies showed that both the GLP-1*-GM1 C12:0 and C16:1 fusion molecules and the GLP-1* peptide, crossed the epithelial monolayer to the basolateral reservoir. This result was in contrast to our findings using confocal microscopy and suggested a non-specific paracellular-leak pathway for crossing the monolayer by the GLP-1* peptide. To test this possibility, we studied paired monolayers incubated continuously with GLP-1* or GLP-1*-GM1 peptides (Figure 5A). For the GLP-1 *-GM1 fusion molecules, transport from apical to basolateral reservoirs was strongly inhibited by incubation at 4°C, consistent with transcellular transport by transcytosis. In contrast, transcellular transport of the GLP-1* peptide (when not fused to GM1) remained robust during incubations at 4°C (nearly equal to the levels seen after continuous 37°C incubations) (Figure 5A). This result implicates transport of GLP-1* across the monolayer by paracellular leak. Such paracellular transport for the GLP-1* peptide would not have been detected by confocal microscopy as the free peptide would have been washed away before fixation. How the GLP-1* peptides can traverse intact tight junctions, however, remains unexplained. In principle, tight junctions should be impermeant to peptides the size of GLP-1* [27, 28]. It is possible the appended PEG chains and biotin somehow increases paracellular permeability as previously suggested for the mucosal absorption of PEGylated- or biotinylated-GLP-1 peptides observed in mice [29-32].
Figure 5. Fusion molecules have similar efficacies as the free peptide.
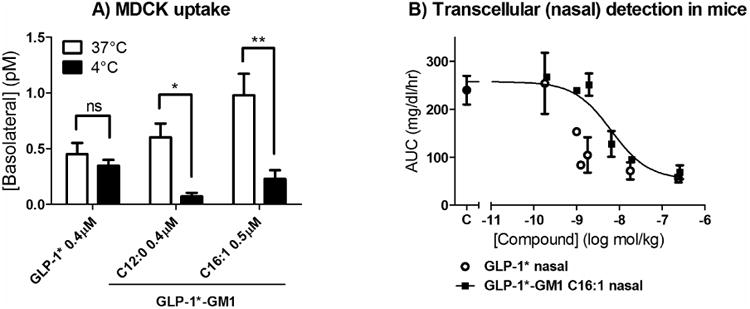
(A) The cells were incubated at 37°C for 3 h with free peptide (0.4 μM) or GLP-1* labeled GM1 C12:0 (0.4 μM) or C16:1 (0.5 μM). The concentration of molecules in the basolateral media was determined based on their activities in the in vitro assay described in Fig. 1B, measured in triplicate (error bars represent mean ± SEM). ns stands for p > 0.05, * stands for p ≤ 0.05 and ** stands for p ≤ 0.01. (B) The 10-fold lower activation of the hGLP-1 receptor by GLP-1*-GM1 fusion molecules compared to GLP-1* (Figure 1B) is preserved in their nasal hypoglycemic efficacy. Dose-response curves of different concentrations of free peptide or GLP-1*-GM1 C16:1 fusion molecules that were nasally applied to mice. Displayed are the areas under the curve of glucose tolerance curves. (n = 2, mean ± SD).
We obtained similar results when testing for proof of principle in vivo by nasal applications of GLP-1* and GLP1*-GM1 C16:1. In these studies, the extent of systemic uptake of the peptide or GM1-fusion molecules was monitored functionally by glucose-tolerance test. Nasal application of GLP-1*-GM1 C16:1 effectively enhanced clearance of blood glucose by 5-fold with apparent EC50 of 5 nanomoles/kg (Figure 5B, closed squares), indicating absorption. But, we also found evidence for absorption of the free GLP-1* peptide (Figure 5B, open circles), presumably by paracellular leak as indicated by our in vitro studies (Figure 5A). Our interpretation is that the GLP-1*-GM1 C16:1 fusion molecule is absorbed at or above background levels. In this assay, we cannot discern the efficiency of absorption between the two because the free-peptide is 10-fold more biologically active, and we lack a measure for delivery to the mucosal surface of the respiratory tract. The GLP-1*-GM1 fusion is larger, more hydrophobic, and likely more interactive with mucus in the extracellular space than the GLP-1*, so it could be less efficient in accessing the nasal epithelium after topical application when administered from simple aqueous formulations. Thus, we can conclude only that the GLP-1*-GM1 fusion molecule can be mucosally absorbed.
Overall, these studies show that GM1 can act as a vehicle for transepithelial transport of a 5 kDa therapeutic peptide across a simple epithelial barrier. We find that trafficking cargo across polarized epithelial cells depends on the specific structure of the GM1 ceramide domain, as predicted by our earlier studies using the same GM1 species labeled with small molecule fluorophores. The underlying cell biology for glycosphingolipid transport remains intact after fusion to the GLP-1 peptide; supporting the feasibility of using specific glycosphingolipids as molecular carriers to deliver biologically active therapeutic agents across mucosal surfaces. This approach is distinct from the use of fatty acid lipidation of a therapeutic peptide to improve metabolic stability or membrane permeability and thus bioavailability [33-35]. Linkage to specific species of GM1 takes advantage of a native ganglioside transcytosis trafficking pathway, providing a novel and previously unappreciated approach of integrating the biology of subcellular sphingolipid trafficking to enhance transmucosal delivery as an element of drug design [36].
Our results, however, also suggest several problems that will need to be overcome for this approach to achieve therapeutic utility. First, we found there was no detectable advantage for transcytosis of the GM1-fusion molecules over paracellular leak of the free-peptide when tested in vitro. PEGylation and biotinylation of the free GLP-1* peptide, in this case, may have allowed for enhanced transport across tight junctions [29-32], strongly confounding our biochemical in vitro results. Other factors may also have been in operation to make the active transcytotic pathway appear only marginally better than non-specific transport by passive paracellular leak. For example, the GM1 sphingolipid acts as a trafficking molecule because it inserts into the membrane as a normal glycosphingolipid, explaining how the structure of the ceramide chain can affect lipid sorting. In doing so, however, the ceramide domain effectively tethers the peptide to the membrane. Release into solution after transcytosis from the basolateral membrane as desired is expected to be restricted – the molecule will be in equilibrium with the membrane bound and soluble state, with a strong affinity to remain membrane associated. Technology to release the peptide from the membrane after transcytosis is predicted to enhance vectorial transport. It is possible that incorporation of furin cleavage motifs into the peptide linker will achieve this aim, though we were not able to test this idea with the current construct. Another potential problem is that incorporation of the GM1-fusion peptides into the apical membrane of epithelial cells lining mucosal surfaces may not have been highly efficient [1]. Here, formulations of the fusion molecules with carrier molecules or with amphipathic hydrogels or emulsions might enhance membrane incorporation.
Our interest in GM1 as a vehicle for drug delivery originated from our studies on the intracellular trafficking of this lipid as a receptor for cholera toxin [1]. We exploit the newly discovered biology of GM1 to achieve this aim, but also note the longstanding interest in utilizing GM1 in drug development. Indirectly, GM1 has been used as a vehicle for delivery of oral vaccines and the induction of mucosal immune responses via coupling antigen to the binding B subunit of cholera toxin [37]. Exendin-4, a GLP-1 analogue, has been expressed as a cholera toxin B subunit–fusion protein to facilitate transmucosal delivery in the gut via toxin binding to GM1 [38]. Vaccine antigens have also been coupled to peptides with affinity for GM1 to enhance uptake with variable success [39, 40]. Other glycosphingolipids have also been used to attach immunoglobulins to liposomes to pursue antibody-mediated targeting of vesicles to cells [41]. Thus, the biology of GM1 sphingolipids has engaged the interest of pharmacologists in the past.
Here, we show that specific GM1 ceramide structures can be used to direct the transcytosis of a biologically active, therapeutic cargo. There are still technological challenges to overcome for translation of this finding into clinical applications. And we do not yet know if GM1 is the only glycosphingolipid exhibiting these properties. It is possible that simplified glycosphingolipids containing ceramides with short or unsaturated fatty acids might traffic through the same pathways, rendering the complex oligosaccharide domain of GM1 dispensable. Nonetheless, our results show it feasible to harness the biology of glycosphingolipid trafficking across simple polarized epithelial cells for mucosal absorption of therapeutic peptides and for other clinical applications.
Supplementary Material
Acknowledgments
We thank J.P. Fortin, A.S. Kopin and M. Beinborn for plasmids required for the hGLP-1 bioactivity assay and their aid in setting up this assay; New England Peptide NEP for peptide bioconjugation to GM1; the entire Lencer lab group for helpful discussions; R. Massol for help with confocal microscopy; and M.A. Wurbel for help with flow cytometry. This study was funded by NIH grants R21 DK090603 and RO1 DK48106, a Technology Innovation and Development grant and a Translational Research grant from Boston Children's Hospital (to W.I.L.); and the Harvard Digestive Diseases Center P30 DK34854.
List of abbreviations
- GLP-1
Glucagon-like peptide 1
- GLP-1*
GLP-1 analogue containing α-amino-isobutyric acids, PEG spacers, biotin and an aminooxy group
- GLP-1*-GM1
fusion molecule of GLP-1 * and GM1
- Aib
α-amino-isobutyric acid
- MDCK II
canine kidney epithelial cells
- T84
Human colon adenocarcinoma cells
- RE
recycling endosomal compartment
- LE
late endosomal compartment
- df-BSA
defatted BSA
- WGA
wheat germ agglutinin
Footnotes
The authors report no conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chinnapen DJ, Hsieh WT, te Welscher YM, Saslowsky DE, Kaoutzani L, Brandsma E, D'Auria L, Park H, Wagner JS, Drake KR, Kang M, Benjamin T, Ullman MD, Costello CE, Kenworthy AK, Baumgart T, Massol RH, Lencer WI. Lipid sorting by ceramide structure from plasma membrane to ER for the cholera toxin receptor ganglioside GM1. Dev Cell. 2012;23:573–586. doi: 10.1016/j.devcel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saslowsky DE, te Welscher YM, Chinnapen DJ, Wagner JS, Wan J, Kern E, Lencer WI. Ganglioside GM1-mediated transcytosis of cholera toxin bypasses the retrograde pathway and depends on the structure of the ceramide domain. J Biol Chem. 2013;288:25804–25809. doi: 10.1074/jbc.M113.474957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kieffer TJ, Habener JF. The glucagon-like peptides. Endocr Rev. 1999;20:876–913. doi: 10.1210/edrv.20.6.0385. [DOI] [PubMed] [Google Scholar]
- 4.Pratley RE, Gilbert M. Targeting Incretins in Type 2 Diabetes: Role of GLP-1 Receptor Agonists and DPP-4 Inhibitors. Rev Diabet Stud. 2008;5:73–94. doi: 10.1900/RDS.2008.5.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 6.Fineman MS, Bicsak TA, Shen LZ, Taylor K, Gaines E, Varns A, Kim D, Baron AD. Effect on glycemic control of exenatide (synthetic exendin-4) additive to existing metformin and/or sulfonylurea treatment in patients with type 2 diabetes. Diabetes Care. 2003;26:2370–2377. doi: 10.2337/diacare.26.8.2370. [DOI] [PubMed] [Google Scholar]
- 7.Kim D, MacConell L, Zhuang D, Kothare PA, Trautmann M, Fineman M, Taylor K. Effects of once-weekly dosing of a long-acting release formulation of exenatide on glucose control and body weight in subjects with type 2 diabetes. Diabetes Care. 2007;30:1487–1493. doi: 10.2337/dc06-2375. [DOI] [PubMed] [Google Scholar]
- 8.Buse JB, Nauck M, Forst T, Sheu WH, Shenouda SK, Heilmann CR, Hoogwerf BJ, Gao A, Boardman MK, Fineman M, Porter L, Schernthaner G. Exenatide once weekly versus liraglutide once daily in patients with type 2 diabetes (DURATION-6): a randomised, open-label study. Lancet. 2013;381:117–124. doi: 10.1016/S0140-6736(12)61267-7. [DOI] [PubMed] [Google Scholar]
- 9.Croom KF, McCormack PL. Liraglutide: a review of its use in type 2 diabetes mellitus. Drugs. 2009;69:1985–2004. doi: 10.2165/11201060-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 10.Hakomori S. Structure, organization, and function of glycosphingolipids in membrane. Curr Opin Hematol. 2003;10:16–24. doi: 10.1097/00062752-200301000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Fortin JP, Chinnapen D, Beinborn M, Lencer W, Kopin AS. Discovery of dual-action membrane-anchored modulators of incretin receptors. Plos One. 2011;6:e24693. doi: 10.1371/journal.pone.0024693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fortin JP, Schroeder JC, Zhu Y, Beinborn M, Kopin AS. Pharmacological characterization of human incretin receptor missense variants. J Pharmacol Exp Ther. 2010;332:274–280. doi: 10.1124/jpet.109.160531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 14.Fortin JP, Zhu Y, Choi C, Beinborn M, Nitabach MN, Kopin AS. Membrane-tethered ligands are effective probes for exploring class B1 G protein-coupled receptor function. Proc Natl Acad Sci U S A. 2009;106:8049–8054. doi: 10.1073/pnas.0900149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beinborn M, Worrall CI, McBride EW, Kopin AS. A human glucagon-like peptide-1 receptor polymorphism results in reduced agonist responsiveness. Regul Pept. 2005;130:1–6. doi: 10.1016/j.regpep.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Freeman H, Shimomura K, Cox RD, Ashcroft FM. Nicotinamide nucleotide transhydrogenase: a link between insulin secretion, glucose metabolism and oxidative stress. Biochem Soc Trans. 2006;34:806–810. doi: 10.1042/BST0340806. [DOI] [PubMed] [Google Scholar]
- 17.Dong JZ, Shen Y, Zhang J, Tsomaia N, Mierke DF, Taylor JE. Discovery and characterization of taspoglutide, a novel analogue of human glucagon-like peptide-1, engineered for sustained therapeutic activity in type 2 diabetes. Diabetes Obes Metab. 2011;13:19–25. doi: 10.1111/j.1463-1326.2010.01313.x. [DOI] [PubMed] [Google Scholar]
- 18.Nakayama K. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem J. 1997;327(Pt 3):625–635. doi: 10.1042/bj3270625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mesonero JE, Gloor SM, Semenza G. Processing of human intestinal prolactase to an intermediate form by furin or by a furin-like proprotein convertase. J Biol Chem. 1998;273:29430–29436. doi: 10.1074/jbc.273.45.29430. [DOI] [PubMed] [Google Scholar]
- 20.Mayer G, Boileau G, Bendayan M. Sorting of furin in polarized epithelial and endothelial cells: expression beyond the Golgi apparatus. J Histochem Cytochem. 2004;52:567–579. doi: 10.1177/002215540405200502. [DOI] [PubMed] [Google Scholar]
- 21.Simmen T, Nobile M, Bonifacino JS, Hunziker W. Basolateral sorting of furin in MDCK cells requires a phenylalanine-isoleucine motif together with an acidic amino acid cluster. Mol Cell Biol. 1999;19:3136–3144. doi: 10.1128/mcb.19.4.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nilsson O, Svennerholm L. Characterization and quantitative determination of gangliosides and neutral glycosphingolipids in human liver. J Lipid Res. 1982;23:327–334. [PubMed] [Google Scholar]
- 23.Madsen K, Knudsen LB, Agersoe H, Nielsen PF, Thogersen H, Wilken M, Johansen NL. Structure-activity and protraction relationship of long-acting glucagon-like peptide-1 derivatives: importance of fatty acid length, polarity, and bulkiness. J Med Chem. 2007;50:6126–6132. doi: 10.1021/jm070861j. [DOI] [PubMed] [Google Scholar]
- 24.Saqr HE, Pearl DK, Yates AJ. A review and predictive models of ganglioside uptake by biological membranes. J Neurochem. 1993;61:395–411. doi: 10.1111/j.1471-4159.1993.tb02140.x. [DOI] [PubMed] [Google Scholar]
- 25.Schwarzmann G. Uptake and metabolism of exogenous glycosphingolipids by cultured cells. Semin Cell Dev Biol. 2001;12:163–171. doi: 10.1006/scdb.2000.0233. [DOI] [PubMed] [Google Scholar]
- 26.Tzaban S, Massol RH, Yen E, Hamman W, Frank SR, Lapierre LA, Hansen SH, Goldenring JR, Blumberg RS, Lencer WI. The recycling and transcytotic pathways for IgG transport by FcRn are distinct and display an inherent polarity. J Cell Biol. 2009;185:673–684. doi: 10.1083/jcb.200809122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donovan MD, Flynn GL, Amidon GL. Absorption of polyethylene glycols 600 through 2000: the molecular weight dependence of gastrointestinal and nasal absorption. Pharm Res. 1990;7:863–868. doi: 10.1023/a:1015921101465. [DOI] [PubMed] [Google Scholar]
- 28.Gedulin BR, Smith PA, Jodka CM, Chen K, Bhavsar S, Nielsen LL, Parkes DG, Young AA. Pharmacokinetics and pharmacodynamics of exenatide following alternate routes of administration. Int J Pharm. 2008;356:231–238. doi: 10.1016/j.ijpharm.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 29.Youn YS, Jeon JE, Chae SY, Lee S, Lee KC. PEGylation improves the hypoglycaemic efficacy of intranasally administered glucagon-like peptide-1 in type 2 diabetic db/db mice. Diabetes Obes Metab. 2008;10:343–346. doi: 10.1111/j.1463-1326.2007.00823.x. [DOI] [PubMed] [Google Scholar]
- 30.Jin CH, Chae SY, Son S, Kim TH, Um KA, Youn YS, Lee S, Lee KC. A new orally available glucagon-like peptide-1 receptor agonist, biotinylated exendin-4, displays improved hypoglycemic effects in db/db mice. J Control Release. 2009;133:172–177. doi: 10.1016/j.jconrel.2008.09.091. [DOI] [PubMed] [Google Scholar]
- 31.Chae SY, Jin CH, Shin HJ, Youn YS, Lee S, Lee KC. Preparation, characterization, and application of biotinylated and biotin-PEGylated glucagon-like peptide-1 analogues for enhanced oral delivery. Bioconjug Chem. 2008;19:334–341. doi: 10.1021/bc700292v. [DOI] [PubMed] [Google Scholar]
- 32.Kim TH, Park CW, Kim HY, Chi MH, Lee SK, Song YM, Jiang HH, Lim SM, Youn YS, Lee KC. Low molecular weight (1 kDa) polyethylene glycol conjugation markedly enhances the hypoglycemic effects of intranasally administered exendin-4 in type 2 diabetic db/db mice. Biol Pharm Bull. 2012;35:1076–1083. doi: 10.1248/bpb.b12-00029. [DOI] [PubMed] [Google Scholar]
- 33.Goodwin D, Simerska P, Toth I. Peptides as therapeutics with enhanced bioactivity. Curr Med Chem. 2012;19:4451–4461. doi: 10.2174/092986712803251548. [DOI] [PubMed] [Google Scholar]
- 34.Bergeon JA, Toth I. Enhancement of oral drug absorption-effect of lipid conjugation on the enzymatic stability and intestinal permeability of l-Glu-l-Trp-NH(2) Bioorg Med Chem. 2007;15:7048–7057. doi: 10.1016/j.bmc.2007.07.040. [DOI] [PubMed] [Google Scholar]
- 35.Zhang L, Bulaj G. Converting peptides into drug leads by lipidation. Curr Med Chem. 2012;19:1602–1618. doi: 10.2174/092986712799945003. [DOI] [PubMed] [Google Scholar]
- 36.Rajendran L, Knolker HJ, Simons K. Subcellular targeting strategies for drug design and delivery. Nat Rev Drug Discov. 2010;9:29–42. doi: 10.1038/nrd2897. [DOI] [PubMed] [Google Scholar]
- 37.Elson CO, Dertzbaugh MT. Mucosal adjuvants. In: Ogra R, editor. Mucosal Immunology. Academic Press; New York: 2005. pp. 967–986. [Google Scholar]
- 38.Kwon KC, Nityanandam R, New JS, Daniell H. Oral delivery of bioencapsulated exendin-4 expressed in chloroplasts lowers blood glucose level in mice and stimulates insulin secretion in beta-TC6 cells. Plant Biotechnol J. 2013;11:77–86. doi: 10.1111/pbi.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montaner AD, De Nichilo A, Elias F, Rodriguez JM, Flo JM, Lopez RA, Zorzopulos J, Frank R. Ganglioside GM1-binding peptides as adjuvants of antigens inoculated by the intranasal route. Vaccine. 2006;24:1889–1896. doi: 10.1016/j.vaccine.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 40.Peachman KK, McLean DM, Tong CT, Alving CR, Rao M. Ganglioside GM1 Binding Peptides: A Potential Adjuvant for Transcutaneous Immunization. The Open Immunology Journal. 2009;2:94–102. [Google Scholar]
- 41.Heath TD, Macher BA, Papahadjopoulos D. Covalent Attachment of Immunoglobulins to Liposomes Via Glycosphingolipids. Biochimica Et Biophysica Acta. 1981;640:66–81. doi: 10.1016/0005-2736(81)90532-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.