

Abstract
Starting from small molecule mTOR inhibitor Torin1, replacement of the piperazine ring with a phenyl ring resulted in a new series of mTOR inhibitors (as exemplified by 10) that showed superior potency and selectivity for mTOR, along with significantly improved mouse liver microsome stability and a longer in vivo half-life.
Keywords: mTOR, PI3K, Torin1
Mammalian target of rapamycin (mTOR) is a key node of the PI3K/Akt/mTOR signal transduction pathway. mTOR pathway has been found to be deregulated in a variety of cancers and has been extensively studied as an oncology drug discovery target.1,2,3,4 Upon activation by external or internal stimuli such as growth factors, nutrients, stress, and energy, mTOR regulates cell growth, proliferation, metabolism, and autophagy through the mTORC1 and mTORC2 complexes by phosphorylation of downstream targets S6K, 4EBP1, and Akt.5 The successful clinical application of rapamycin in renal cell carcinoma has validated mTOR as an anti-cancer drug discovery target. However, a lack of activity against mTORC2, incomplete inhibition of mTORC1 function and negative reactivation of Akt through the S6K/IRS1 pathway may explain the limited clinical efficacy observed with rapamycin related compounds (rapalogs) 6,7,8 Currently, there is significant interest in the development and characterization of ATP-competitive mTOR inhibitors which would result in complete inhibition of both mTORC1 and mTORC2.
mTOR is a member of the PI3K kinase family, which consists of the PI3Ks, DNA-PK, ATR, ATM, and SMG-1.9 PI3Ks, especially PI3Kα, have been found to be hyperactivated in a wide spectrum of cancers, and are currently being evaluated as an anti-cancer drug target in the clinic.1 The structural similarity between PI3Ks and mTOR endowed many early PI3K or mTOR inhibitors, like PI-103 and BEZ-235, with dual activity against both targets.10,11 Because PI3Ks are upstream regulators of Akt, a central hub of many critical cellular processes, it is believed that mTOR-selective inhibitors may demonstrate improved toxicology relative to dual PI3K/mTOR inhibitors. Many of the recently disclosed mTOR inhibitors, including Torin1, have demonstrated that it is possible to selectively inhibit mTOR versus PI3Ks.12–22
We recently prepared a series of tricyclic benzonaphthyridinones, exemplified by Torin1, as highly potent and selective mTOR inhibitors. Torin1 has more than 800-fold selectivity for mTOR relative to PI3Ks in cellular assays and has no other significant protein kinase off-targets among the 450 kinases profiled by KINOMEscan™.12 Torin1 exhibited anti-tumor activity in a U87-MG mouse xenograft that correlated with target inhibition. However, Torin1 exhibits poor mouse microsome stability and a short in vivo half-life which limit its utility as a pharmacological agent in vivo. In this letter, we describe our efforts to improve the pharmacokinetic properties of Torin1 by replacing the metabolically labile phenylpiperazine moiety with a biphenyl system to yield compounds such as 10 (Figure 1).
Figure 1.

Structures of Torin1 and 10.
The preparation of compound 10 and analogs is illustrated in Scheme 1. Ethyl-4,6-dichloroquinoline-3-carboxylate (1) was subjected to nucleophilic substitution with 4-bromo-3-trifluoromethaneaniline (2) to afford compound 3. Reduction of ethyl ester 3 with NaBH4 generated benzyl alcohol 4, which was subjected to benzylic oxidation with MnO2 and olefination/cyclization in situ to furnish general intermediate 5. Pinacol boronic ester 6 was obtained from 4-bromo-benzonic acid (8) via Suzuki coupling and amide formation. A bromine-selective Suzuki coupling was used to install the benzoate side chain (or other substituent in the analogs) to afford the intermediate 7. Finally, the quinoline side chain (or other substituent in the analogs) was attached at the chlorine position using more vigorous Suzuki coupling conditions to afford the final product illustrated by compound 10.
Scheme 1.

Reagents and conditions: (a) 1,4-dioxane, 85 °C, 4 h, 80% yield; (b) NaBH4, EtOH, rt, 4 h, 50% yield; (c) MnO2, CH2Cl2, rt, 2 h; then triethylphosphonoacetate, K2CO3, EtOH, 100°C, 12 h, 60% yield; (d) 6, PdCl2(Ph3P)2, Na2CO3, 1,4-dioxane, 80°C, 2 h, 60% yield; (e) quinoline-3-boronic acid, PdCl2(Ph3P)2, t-Bu-Xphos, Na2CO3, 1,4-dioxane, 100°C, 4 h, 50% yield; (f) PdCl2(dppf), bis(pinacolato)diboron, KOAc, 1,4-dioxane, 80°C, 12 h; crude material was used without purification; (g) HATU, THF, 1-methyl-piperidin-4-amine, diisopropylethylamine, rt, 50% over 2 steps.
Following this general synthetic strategy, a focused library of compounds was generated by varying the benzoate side chain, quinoline side chain and CF3/CH3 moiety. The selected compounds were evaluated in parallel in biochemical assays with mTORC1 complex in cellular assays using a mouse embryonic fibroblast (MEF) cell line by examining the phosphorylation status of mTOR downstream targets such as S6K (T389), and for PI3K activity with the Akt S473D PC-3 cell line by examining the phosphorylation status of AktT308. The results are summarized in Table 1.
Table 1.
Data from biochemical and cellular assays.a
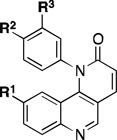 | ||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | mTORC1 biochemical IC50(nM) |
mTOR cellular EC50(nM) |
PI3K cellular EC50(nM) |
| 10 | 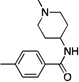 |
CF3 | 21.7 | 5 | 1000 | |
| 11 | CF3 | 10.2 | 15 | >300 | ||
| 12 |  |
CF3 | 188 | 25 | >300 | |
| 13 | CF3 | 17.3 | 30 | >300 | ||
| 14 | CF3 | 22.9 | 20 | >300 | ||
| 15 | CF3 | 299 | 200 | >300 | ||
| 16 | CF3 | 56.3 | 25 | >300 | ||
| 17 | 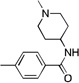 |
CF3 | 43.2 | 10 | 1000 | |
| 18 | 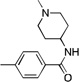 |
CF3 | 14.1 | 10 | 1000 | |
| 19 | 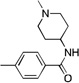 |
CH3 | 14.5 | 1 | 1000 | |
| 20 | 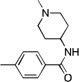 |
CH3 | 38.1 | 150 | >1250 | |
| 21 | 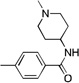 |
CH3 | 283 | 150 | >1250 | |
| 22 | 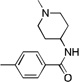 |
CH3 | 18.9 | 25 | 1000 | |
| 23 | H | 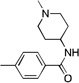 |
CH3 | >3000 | >1000 | >1250 |
| 24 | 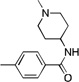 |
CH3 | 39.8 | 50 | >1250 | |
IC50 determinations are the mean of two of independent measurements with a standard error of <20%.
Based on the Torin1 series, we first retained the quinoline side at the R1 position, which was presumably responsible for the selectivity and potency against mTOR. Replacing the phenylpiperazine ring with a biphenyl system provided compounds that demonstrated excellent potency against mTOR and selectivity relative to PI3K. Methyl amide 11, THP-protected benzyl alcohol 12, benzyl alcohol 13, carboxylic acid 14, and primary amide 16 all demonstrated the same level of potency against mTOR (EC50 values between 15–30 nM) and the same selectivity over PI3K (EC50 over 300 nM). The methyl ester 15 lost a significant amount of activity against mTOR, suggesting that the active members of this series of compounds possess hydrogen bond donors through either an –OH, –COOH. or –CONH2 functionality. N-methyl-piperidin-4-amine amide 10 exhibited the best potency against mTOR (EC50 = 5 nM, the same as Torin1) and more than 200-fold selectivity over PI3K. The compounds derived from replacing the quinoline moiety with aminopyrimidine (17) or pyrazole (18) systems were slightly less potent against mTOR, indicating that the inner hydrophobic pocket required a larger substituent. In order to reduce the molecular weight and obtain better solubility, while the R2 fragment was kept as methyl-piperidin-4-amine, the CF3 group was replaced with a CH3 group and the R1 side chain was varied to include a pyrazole (20), methylpyrazole (21), 7-azaindole (22), hydrogen (23) and aminopyridine (24). However, all of these analogs failed to produce the same level of potency against mTOR, although the selectivity over PI3Ks was retained. Compound 19, which bears quinoline side chain at the R1 position, retained the same potency and selectivity against mTOR as Torin1 and compound 10. This reaffirmed that quinoline was the best side chain to occupy the inner hydrophobic pocket, and that the hydrogen bond provided by the –OH or –NH2 was critical to maintain potency.
With the best activity and selectivity, compounds 10 and 19 were chosen for mouse stability study, single point CYP450 inhibition test (Table 2), and in vivo mouse pharmacokinetic analysis (Table 3). In comparison to Torin1, compounds 10 and 19 demonstrated significant improvements in stability in the mouse microsome assay (46 and 42 min, respectively), where both were subjected to NADPH-dependent metabolism. In the single point CYP450 metabolic enzyme inhibitory assay, compound 10 showed more than 60% and 50% inhibition at 10 µM against the major metabolic enzymes CYP3A4 and CYP2D6, respectively, while compound 19 weaker inhibition (34% and 24%, respectively). Further investigation of the ability of these compounds to inhibit metabolism are warranted prior to performing combination studies.
Table 2.
Mouse microsome stability and CYP450 inhibition results.
| Compound | Mouse microsome stability (min) |
NADPH- dependent |
CYP3A4 % inhibition (10 µM) |
CYP2D6 %inhibition (10 µM) |
|---|---|---|---|---|
| 10 | 46 | Y | 61 | 51 |
| 19 | 42 | Y | 34 | 24 |
| Torin1 | 1.3 | N | ND | ND |
Table 3.
In vivo mouse pharmacokinetic data.
| Comp ound |
Cmax (ng/mL) i.v./P.O. |
Tmax (h) i.v./P .O |
AUC(0- ∞) (hr*ng/ ml) i.v./P.O. |
T1/2 (h) i.v./P. O. |
CL (mL/min/k g) i.v./P.O. |
Vss (L/kg) i.v./P.O. |
F (%) i.v./P. O. |
|---|---|---|---|---|---|---|---|
| 10 | 1408/191 | -/4.0 | 1388/1411 | 3.61/- | 11.9/- | 1.95/- | -/10.1 |
| 19 | 1046/115 | -/1 | 737/391 | 1.85/- | 22.6/- | 1.82/- | -/5.4 |
| Torin1 | 2757/223 | -/0.25 | 720/396 | 0.5/0.79 | 23.0/- | 0.59/- | /5.49 |
The improved microsome stability profile of compounds 10 and 19 encouraged us to evaluate their in vivo pharmacokinetic properties. Upon intravenous (7.5% NMP and 40% PEG400 in water) and oral (0.1% v/v Tween-80, 0.5% w/v NaCMC in water) administration, compound 10 demonstrated superior pharmacokinetic properties relative to compound 19, although both were significantly better than Torin1.12 The half-life was improved to 3.6 h (10) and 1.8 h (19) from that of Torin1 (0.5 h). The bioavailability of compound 10 (10.1%) was double that of Torin1 (5.5%), while the bioavailability of compound 19 was 5.4%. Compound 10 also demonstrated much better exposure using both IV and PO delivery routes compared to Torin1(1388/1411 versus 720/396 hr*ng/mL). Other pharmacokinetic properties such as clearance rate (11.9 versus 23.0 mL/min/Kg) and volume of distribution (1.95 versus 0.59 L/Kg) were also superior to those of Torin1. The slower Tmax of compound 10 (4 h) compared to compound 19 (1 h) and Torin1 (0.5 h) was indicative of poor solubility and/or slow absorption.
To evaluate the kinase selectivity of compound 10, it was subjected to the Ambit kinome-wide screen using KINOMEscan™ technology. The assay showed that compound 10 was very selective and did not strongly hit any other protein kinases among the 353 kianses tested, except for several PI3K family lipid kinases (Figure 2, Table 4).
Table 4.
Ambit and Invitrogen profiles of compound 10 against PIKKs
| Kinase symbol | Invitrogen IC50 (nM) | Ambit score (%) |
|---|---|---|
| PI4Kα | >10000 | ND |
| PI4Kβ | 3440 | ND |
| PI3K-C2α | 1110 | ND |
| PI3K-C2β | 437 | 0.65 |
| hVPS34 | 170 | ND |
| P110α/P85α | 922 | 0.1 |
| P110δ/P85α | 1170 | 2 |
| P110γ | 1170 | 0.05 |
| mTOR | 3.01 | 0 |
Compound 10 was evaluated in an in vivo pharmacodynamic study, where it exhibited significant inhibitory activity against the downstream targets of mTOR, S6K, and Akt, and blocked 80–90% phosphorylation of S6K (T389) and pAkt (S473) in liver and lung tissues even after 6h at a dosage of 20 mg/kg
In summary, starting from Torin1, replacement of the metabolically labile 4-amino-phenylpiperazine moiety with a biphenyl system provided a new series of inhibitors that were exemplified by compound 10, which demonstrated significant improvements in mouse microsome stability and in vivo pharmacokinetic properties. Compound 10 is a potent and selective mTOR inhibitor suitable for use in cell culture and in vivo. Further elaboration of this scaffold class to improve the drug-like properties will be reported in due-course.
Acknowledgement
The authors thank Dr. Michael Cameron (Scripps Florida) for the mouse microsome stability studies. The authors also thank the Life Technologies Corporation (Invitrogen) SelectScreen® Kinase profiling service for performing enzymatic biochemical kinase profiling and Ambit Biosciences for performing KINOMEscan™ profiling.
References
- 1.Liu P, Cheng H, Roberts TM, Zhao JJ. Nat. Rev. Drug. Discov. 2009;8:627. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shor B, Gibbons JJ, Abraham RT, Yu K. Cell Cycle. 2009;8:3831. doi: 10.4161/cc.8.23.10070. [DOI] [PubMed] [Google Scholar]
- 3.Meric-Bernstam F, Gonzalez-Angulo AM. J. Clin. Oncol. 2009;27:2278. doi: 10.1200/JCO.2008.20.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Q, Thoreen CC, Wang J, Sabatini DM, Gray NS. Drug Discovery Today: Therapeutic Strategies. 2009;6:47. doi: 10.1016/j.ddstr.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guertin DA, Sabatini DM. Cancer Cell. 2007;12:9. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Mol. Cell. 2006;22:159. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 7.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. J. Biol. Chem. 2009;284:8023. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carracedo A, Pandolfi PP. Oncogene. 2008;27:5527. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- 9.Abraham RT. DNA Repair. 2004;3:883. doi: 10.1016/j.dnarep.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Park S, Chapuis N, Bardet V, Tamburini J, Gallay N, Willems L, Knight ZA, Shokat KM, Azar N, Viguie F, Ifrah N, Dreyfus F, Mayeux P, Lacombe C, Bouscary D. Leukemia. 2008;22:1698. doi: 10.1038/leu.2008.144. [DOI] [PubMed] [Google Scholar]
- 11.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C. Mol. Cancer. Ther. 2008;7:1851. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 12.Liu Q, Chang JW, Wang J, Kang SA, Thoreen CC, Markhard A, Hur W, Zhang J, Sim T, Sabatini DM, Gray NS. J. Med. Chem. 2010;53:7146. doi: 10.1021/jm101144f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Martinez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, Alessi DR. Biochem. J. 2009;421:29. doi: 10.1042/BJ20090489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ, Cole DC, Ellingboe J, Ayral-Kaloustian S, Mansour TS, Gibbons JJ, Abraham RT, Nowak P, Zask A. Cancer. Res. 2009;69:6232. doi: 10.1158/0008-5472.CAN-09-0299. [DOI] [PubMed] [Google Scholar]
- 16.Barr S, Russo S, Buck E, Epstein D, Miglarese M. AACR 101st Annual meeting; April, 2010; Washington DC. p. 1632. [Google Scholar]
- 17.Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, James D, Howard Z, Dudley P, Hughes G, Smith L, Maguire S, Hummersone M, Malagu K, Menear K, Jenkins R, Jacobsen M, Smith GC, Guichard S, Pass M. Cancer Res. 2010;70:288. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 18.Menear KA, Gomez S, Malagu K, Bailey C, Blackburn K, Cockcroft XL, Ewen S, Fundo A, Le Gall A, Hermann G, Sebastian L, Sunose M, Presnot T, Torode E, Hickson I, Martin NM, Smith GC, Pike KG. Bioorg. Med. Chem. Lett. 2009;19:5898. doi: 10.1016/j.bmcl.2009.08.069. [DOI] [PubMed] [Google Scholar]
- 19.Richard DJ, Verheijen JC, Curran K, Kaplan J, Toral-Barza L, Hollander I, Lucas J, Yu K, Zask A. Bioorg. Med. Chem. Lett. 2009;19:6830. doi: 10.1016/j.bmcl.2009.10.096. [DOI] [PubMed] [Google Scholar]
- 20.Yu K, Shi C, Toral-Barza L, Lucas J, Shor B, Kim JE, Zhang WG, Mahoney R, Gaydos C, Tardio L, Kim SK, Conant R, Curran K, Kaplan J, Verheijen J, Ayral-Kaloustian S, Mansour TS, Abraham RT, Zask A, Gibbons JJ. Cancer Res. 2010;70:621. doi: 10.1158/0008-5472.CAN-09-2340. [DOI] [PubMed] [Google Scholar]
- 21.Verheijen JC, Yu K, Toral-Barza L, Hollander I, Zask A. Bioorg. Med. Chem. Lett. 2010;20:375. doi: 10.1016/j.bmcl.2009.10.075. [DOI] [PubMed] [Google Scholar]
- 22.Malagu K, Duggan H, Menear K, Hummersone M, Gomez S, Bailey C, Edwards P, Drzewiecki J, Leroux F, Quesada MJ, Hermann G, Maine S, Molyneaux CA, Le Gall A, Pullen J, Hickson I, Smith L, Maguire S, Martin N, Smith G, Pass M. Bioorg. Med. Chem. Lett. 2009;19:5950. doi: 10.1016/j.bmcl.2009.08.038. [DOI] [PubMed] [Google Scholar]