

Abstract
Drosophila melanogaster has one of the best characterized metazoan genomes in terms of functionally annotated regulatory elements. To explore how these elements contribute to gene regulation in the context of gene regulatory networks, we need convenient tools to identify the proteins that bind to them. Here, we present the development and validation of a highly automated protein-DNA interaction detection method, enabling the high-throughput yeast one-hybrid-based screening of DNA elements versus an array of full-length, sequence-verified clones containing 647 (over 85%) of predicted Drosophila transcription factors (TFs). Using six well-characterized regulatory elements (82 bp – 1kb), we identified 33 TF-DNA interactions of which 27 are novel. To simultaneously validate these interactions and locate their binding sites of involved TFs, we implemented a novel microfluidics-based approach that enables us to conduct hundreds of gel shift-like assays at once, thus allowing the retrieval of DNA occupancy data for each TF throughout the respective target DNA elements. Finally, we biologically validate several interactions and specifically identify two novel regulators of sine oculis gene expression and hence eye development.
Introduction
Since its adoption over 100 years ago by T.H. Morgan, Drosophila has been a pioneering model organism to study the basic principles underlying many developmental and cellular processes, including transcriptional regulation. For example, Drosophila segmentation, during which the embryonic ectoderm forms visible segments, is one of the best characterized developmental gene regulatory networks given the identification of most of its transcription factor (TF) and regulatory element components and the interactions between them1. In addition, the availability of a high-quality genome sequence 2, a large-scale enhancer trapping assay3, ChIP-chip and ChIP-seq data revealing cis-regulatory modules (CRMs) and specific chromatin states on a genome-wide basis4,5, a convenient transgenesis system to screen the activity of regulatory elements6, as well as powerful comparative genomics methodologies7 has led to the identification of a great number of functional regulatory elements. To explore how these elements contribute to gene regulation and function in the context of gene regulatory networks, we need a technique to identify the TFs binding to these elements. Although several genome-wide techniques exist to determine which DNA elements bind to a specific TF (TF-centered approaches) like ChIP, protein binding microarrays and DamID, techniques that identify the full complement of TFs binding to a specific DNA element (DNA-centered approaches) often suffer from low throughput or high technical complexity (reviewed in 8). Here, we describe the development and validation of a high-throughput Drosophila yeast one-hybrid (Y1H) system that interrogates binding of selected DNA baits versus the majority of predicted Drosophila TFs, making this technique instrumental to construct Drosophila gene regulatory networks.
Results
A comprehensive Drosophila TF open-reading frame library
We developed, building on previous efforts in C. elegans9, a gene-centered, Y1H-based method that allows the high-throughput screening of DNA elements of interest versus the nearly complete Drosophila TF repertoire. To obtain the latter, we first determined, based on bioinformatic analyses10 and manual curation, that the Drosophila genome contains 755 sequence-specific TF-coding genes (Supplementary Table 1). Less than 15% of these have currently been characterized in terms of target genes11. Through incorporation of existing cDNA collections and de novo cloning, we were able to generate 722 (96%) Gateway-compatible Entry clones containing the open-reading frame (ORF) of each TF. To assure that each TF ORF clone encodes a functional protein, we sequence-verified several Entry clones for each TF using a recently developed method based on high-throughput sequencing12, enabling us to confirm the identity of 692 TFs (92%) of which the majority is fully sequence-verified (588 or 78%) (Fig. 1). Successfully cloned TFs were distributed uniformly among all major TF families (Supplementary Fig. 1). Interestingly, 27 distinct TF clones were initially rejected, but were retained after manual curation of their respective assembled ORF sequences because they likely constitute novel TF splice forms. As it constitutes a powerful and versatile resource, the TF ORF clone collection will be made available to the scientific community upon request.
Figure 1.

Workflow underlying the generation of the Drosophila TF ORF clone resource and the Drosophila Y1H AD-TF library. Out of 755 predicted Drosophila TFs, 501 were available as cDNA clones from the Berkeley Drosophila Genome Project. The remaining TFs were targeted for de novo cloning. TF ORFs were PCR amplified and cloned into the pDONR221 ENTRY vector. The resulting Entry clones were sequence verified by high-throughput sequencing and categorized according to the quality and the coverage of the sequencing. All non-rejected clones were subsequently transferred into the Y1H compatible AD vectors pAD-DEST-ARS/CEN and pAD-DEST-2μ by Gateway cloning.
To make the resulting, high-quality TF clone resource Y1H compatible, we simultaneously sub-cloned each accepted TF within the same Gateway reaction to both a high- and low-copy Gal4 activation domain (AD)-containing vector, resulting in an equimolar mix of both AD-ORF plasmids (Supplementary Fig. 2). The former allows higher TF expression than the latter, likely resulting in increased sensitivity. We keep the low-copy plasmid, which was used previously9, as it may allow the detection of interactions involving TFs that are toxic to the yeast when expressed at high levels.
Drosophila high throughput Y1H system
Most Y1H screens have so far been performed using direct transformation of the prey proteins in a haploid yeast strain in whose genome the DNA bait is integrated. Recent efforts demonstrated that this haploid format allows a more comprehensive protein-DNA interaction coverage than when mating, and thus diploid strains are used to pair TFs with DNA baits13. In the same studies, it was shown that matrix assays in which an array of individual TF clones (the “TF array”) is used, also yield more protein-DNA interactions compared to pooling or library-based screening strategies. Such assays have the additional advantage that the identity of the interacting TF can be immediately derived from its position coordinates on the array and thus no sequencing is required. However, haploid transformation comes at the cost of being more laborious and expensive than a mating-based assay as the manual transformation of hundreds of TFs per screen is labor- and reagent-intensive. To pair optimal coverage with higher throughput and lower cost, we engineered a novel robotic platform that completely automates the haploid yeast transformation process (http://www.youtube.com/watch?v=PM8WWXgE1-A). In addition, we significantly decreased overall reagent consumption by scaling down the protocol to enable direct transformation in 384-well format (Fig. 2). Together, this allows us to screen several DNA baits per day in fully automated fashion versus a Drosophila TF array consisting of two 384-well plates currently containing 647 TFs and three negative controls (i.e. empty AD vector). After transformation, individual yeast strains are then grown on a permissive plate and are subsequently replica-plated in 1536-format on selective plates to test the expression of the HIS3 reporter using a second workstation such that each interaction is tested in quadruplicate. We perform two independent screens for each bait using selection reproducibility as a key criterion to filter out potential false positives. This procedure has been shown to be very effective in reducing false positives in yeast two-hybrid (Y2H) screens14 and proved to be reliable for Y1H screens as well (see below). Initially, positives were also evaluated based on the expression of a second reporter, lacZ. We found however that the lacZ reporter was less sensitive than the HIS3 reporter. For example, six interactions were found with the lacZ reporter versus 11 interactions identified by the HIS3 reporter for one of the tested elements (so10). Additionally, the majority of positives from the lacZ screen (five out of six) were also found in both independent HIS3 screens (Supplementary Figs. 3 and 4, Supplementary Table 2), consistent with results obtained previously that positives from one screen are typically positive for both reporters13, prompting us not to include the lacZ screen in further screens. However, the lacZ screen can still be performed if additional stringency in selection of positive interactions is required.
Figure 2.

The Drosophila high-throughput Y1H platform. A yeast DNA bait strain is distributed over a 384-well plate. Each well of this plate is then transformed with a different AD-TF clone from the Drosophila Y1H AD-TF library by a robotic yeast transformation platform which additionally spots the 384 individually transformed yeast strains on a permissive agar plate. A colony pinning robot subsequently transfers the yeast colonies onto a permissive and a selective plate, quadruplicating each colony in a square pattern in the process. TF-DNA bait interactions can be observed as growth on a selective, 3-AT-containing yeast plate.
TIDY, an image analysis program for Transcription factor-DNA Interaction Detection in Yeast
Another source of potential false positives is the identification of positives by eye which is often confounded by a varying background across the same yeast plate. To allow a more objective detection of positives, we generated a Matlab-based image analysis program called TIDY (for Transcription factor-DNA Interaction Detection in Yeast). This program semi-automatically calls positive interactions by convoluting the image with the pattern of four bright spots on a dark background, which has the advantage of ignoring the noisy background of the image and only detecting the yeast array. The intensity value of the convoluted image in the center of each quadrant is used as a measure for the strength of the respective protein-DNA interaction. TIDY then plots the intensity values to determine the upper limit of the background, which we define as the highest intensity value in the largest data point cluster representing most and thus likely negative yeast quadrants (Fig. 3). We conservatively set the positives threshold at 20% above this highest background intensity value as this empirically allowed the most robust detection of strong positives. Importantly, TIDY also takes the uniformity of the quadrant colonies into account when calling positives to filter out high intensity values derived from only one or two contributing quadrant colonies. Thus, uniform yeast quadrants whose resulting intensity values score above the threshold are identified as positives and labeled by their respective names in green color (Fig. 3). TIDY also has the option to perform a separate background normalization for exterior versus interior yeast colonies as we often observe that colonies on the border of the plate grow faster than those in the middle which may introduce an important detection bias (Supplementary Fig. 5a-b). Finally, TIDY allows the user to manually change the default threshold to evaluate the detection stringency. This option was implemented as in some cases, slightly lowering the threshold results in the inclusion of additional positives which clearly still score above the highest background intensity value detected by TIDY. Such interactions are considered “weak” though and labeled in magenta to indicate their distinct status (Supplementary Fig. 5c).
Figure 3.
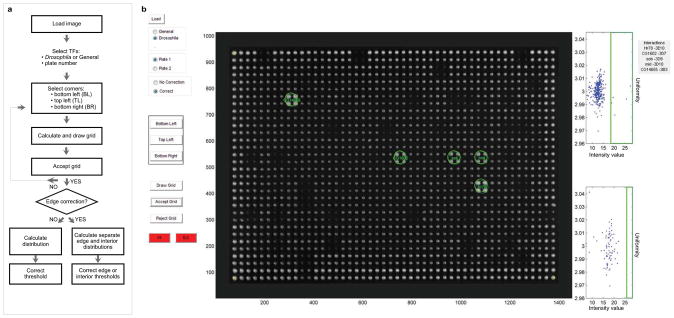
Overview of the TIDY program. (a) Flow chart of the different steps in the TIDY program. (b) Screenshot of the TIDY output upon image analysis of a selective plate from a Y1H screen. In this example, five interactions were observed (green circles). A different threshold was used for interior and exterior yeast colonies.
Drosophila Y1H validation
To stringently validate our approach, we selected 10 well-characterized CRMs ranging from 82 to 1007 bp in size from the REDfly database11 and the literature, based on the criterion of covering as many distinct TFs as possible. We successfully cloned and integrated all 10 DNA elements in yeast. Four baits exhibited high self-activation (data not shown) and were not further considered as initial tests revealed that the interaction reproducibility drops sharply with increasing self-activation. The six remaining elements together contribute 22 reported interactions (Supplementary Table 3). For 19 of these 22 interactions, the interacting TF is present in our library. In total, we detected 33 TF-DNA interactions that overlapped between two independent screens, involving 25 unique TFs (Supplementary Table 2). Representative TIDY-processed images for each screen are shown in Supplementary Figs. 3, 6–10. The detected TFs belong to nine of the 11 main TF families that we defined in Supplementary Figure 1, indicating that the Y1H is not biased against a particular TF family consistent with previous results9,13 and results from Barutcu et al. (co-submitted). We reproducibly detected five of the 19 (26%) positive control interactions, each involving a distinct TF. This percentage falls within the range of Y1H and Y2H screen detection rates9,15.
To evaluate whether some interactions were missed due to the high-throughput nature of the screen, we also tested the 19 reported interactions by manual transformation. Of the 14 interactions that were not detected previously, only two were recovered by performing a manual transformation, showing the robustness of the automated Y1H system (Supplementary Fig. 11, Supplementary Table 4). The 10 remaining interactions may be missed because some TFs may require other proteins or post-translational modifications to bind DNA. Additionally, more than half of the tested positive control interactions were so far only found using one method, most of which are in vitro techniques like electrophoretic mobility shift assays (EMSAs) and DNase I footprinting, and were thus identified using naked DNA. Y1H DNA baits are chromatinized, which may result in different and more biologically relevant DNA binding behavior. Furthermore, some of the positive controls act as repressors in Drosophila (for example GIANT and KRÜPPEL binding to the eve-stripe2 element). It is possible that the repressive function of some of these TFs is able to overcome the activating function of the GAL4 AD that is fused to the TF, thereby preventing the transactivation of the reporter gene, consistent with what was observed for the repressor TRA-1 in C. elegans16 Additionally, the requirement for interactions to be positive in two independent screens may sometimes be too stringent. Indeed, an additional positive control interaction (dpp813–EXD) was found in one replicate. However, this element shares few interactions between two independent screens. To evaluate this further, we first retested the dpp813 element using the lacZ reporter. Consistent with results obtained with the so10 element, the lacZ reporter identified fewer interactions than both HIS3 reporter screens (16 interactions for lacZ, 35 and 30 for the independent HIS3 screens respectively). Three out of six interactions that were found in two independent HIS3 screens, including a positive control, were also identified using the lacZ reporter (Supplementary Fig. 12, Supplementary Table 2). However, no positive control interactions that were missed by the HIS3 screens could be recovered by the lacZ screen, while one positive control interaction that was found with the HIS3 reporter screens was missed on lacZ, indicating that at least in our set-up, the approach of using the overlap of independent screens of the HIS3 reporter produces the most comprehensive results. A final cause of false negatives could be the bait size or orientation. We evaluated this by dividing the dpp813 element in three overlapping elements (dpp813-Frag1-3) or inverting the full-length dpp813 element (dpp813-RC) (Supplementary Table 3). Overall, reducing the size or inverting the element did not improve the positive detection rate. However, for the dpp813-Frag2 element, two out of three interactions found in both replicates and seven out of 30 in single screens (including both positive control interactions) were found at least once with the full-length dpp813 element or the dpp813-RC element, indicating that this fragment is likely the major contributor of the observed protein-DNA interactions with the full-length dpp813 element (Supplementary Figs. 13–16, Supplementary Table 5). Since the size did not have a clear impact on overall coverage but both positive control interactions were found at least once more in the additional screens, we propose for elements showing limited overlap between two independent screens, to perform additional repeats of the screen and use the number of times an interaction is observed as a confidence level to distinguish between spurious and likely true interactions.
Microfluidics-based Y1H validation and binding site mapping
We next aimed to estimate the proportion of interactions found by the Y1H that could be recapitulated with an alternative protein-DNA interaction detection technique. To this end, we used a novel microfluidics-based method based on MITOMI technology17 called MARE (Mitomi-based Analysis of Regulatory Elements) that was initially developed to validate mouse TF-DNA interactions (Gubelmann et al., cosubmitted). MARE allows high-resolution in vitro DNA footprinting, thus enabling us to scan DNA elements for individual TF binding sites in fine-grained fashion, while simultaneously independently validating detected Y1H interactions. In addition, DNA occupancy data for different binding sites within an element are obtained, similar to data obtained when performing an EMSA on small DNA sequences.
First, we analyzed the sine oculis enhancer so10 as DNase I footprinting data for the well-known interactors EYELESS (EY) and TWIN OF EYELESS (TOY) have previously been published18, and can thus be used to benchmark the technique. so10 was divided in 50 fragments of 36 bp with each fragment overlapping the previous one by 24 bp. Each fragment was then tested on-chip for recognition by Y1H-identified TFs and averaged DNA occupancy data were plotted for each 12 bp stretch (Fig. 4, Supplementary Fig. 17). Each TF-specific graph therefore represents data derived from 50 gel shift-like experiments, and each experiment was independently replicated at least twice. Interestingly, EY and TOY reproducibly showed very strong and similar binding patterns, consistent with the fact that they are both homologues of vertebrate PAX619,20, and have been shown to exhibit similar DNA binding properties19. Importantly, the site yielding highest DNA occupancy overlaps with known EY and TOY binding sites18, validating the MARE technology. We further integrated MARE with position weight matrix (PWM) data. Of the six (out of 11) accepted interactors for which PWMs are available, all have a predicted binding site within so10 of which five overlap with a DNA occupancy peak as reproducibly detected by MARE, further demonstrating the power of this technique.
Figure 4.

MARE analysis of DNA elements for recognition by Y1H-identified TFs. Bound DNA levels normalized over surface-immobilized protein amounts are plotted for each 12 bp nucleotide stretch and as an interpolated curve. Significant peaks are indicated with a red line, peak maxima are indicated with a red dot. Peaks found in both replicates are indicated with an asterisk. Where available, DNase I footprinting data and PWM-based binding site predictions are indicated with blue and yellow bars respectively. Overlapping DNase I footprinting data and PWM-based binding site predictions are indicated with grey bars. Note that, as DNA occupancy is plotted as a relative signal normalized for the protein level in the microfluidics chamber, the scale of the Y-axis may vary between replicates. (a) Analysis of the so10 element for binding of EY. (b) Analysis of the so10 element for binding of TOY. (c) Analysis of the so10 element for binding of CG9797. (d) Analysis of the so10 element for binding of TTK. (e) Analysis of the yp1-1 element for binding of DSX. (f) Analysis of the yp1-1 element for binding of TJ.
We similarly tested the yp1-1 element with the Y1H-detected TFs DOUBLESEX (DSX) and TRAFFIC JAM (TJ), which were reproducibly found by Y1H. MARE detected binding sites for both TFs in the yp1-1 element in specific locations. Additionally, PWM-based binding site prediction and DNase I footprinting21 for DSX shows two binding sites in the highest DNA occupancy peak found by MARE (Fig. 4, Supplementary Fig. 18). Overall, we observed site-specific DNA binding for 10 out of 13 (77%) tested TFs, while the remaining three TFs produced mostly non-reproducible background signal, likely reflective of non-specific binding. Thus, MARE proved to be a convenient and sensitive tool enabling us to validate our Y1H data and to pinpoint individual binding sites for the candidate TFs in the DNA elements, which is instrumental to decipher the regulatory mechanisms underlying gene expression.
In vivo relevance of detected Y1H interactions
We chose the so10 element to estimate the proportion of interactions found by the Y1H that could be relevant in vivo since modulation of sine oculis expression levels results in readily observable eye phenotypes22,23. Therefore, knock-down of TFs that regulate so expression in vivo should also result in flies with eye phenotypes. To this end, we knocked down the interacting TFs by crossing distinct UAS-RNAi fly lines obtained from the TRiP24 and VDCR25 collections with so10-GAL4 and ok107 driver lines. As a first approach, we evaluated the effect on eye development by visual inspection of the adult eye. RNAi-mediated knock-down resulted in observable eye phenotypes for EY, TRAMTRACK (TTK), and CG9797 (Fig. 5a-d, Supplementary Fig. 19, Supplementary Table 6). Knock-down of ey and CG9797 resulted in variable but similar eye phenotypes, ranging from completely absent to near wild-type eyes, similar to the phenotype described for hypomorphic ey alleles26. For both genes, the so10-GAL4 driver resulted in more pronounced phenotypes. This may indicate that the so10-GAL4 driver line produces a stronger overall transactivation of the RNAi transgene, but might also be due to a better overlap between expression patterns of so10-GAL4 and the endogenous so gene as compared to OK107. TRiP RNAi lines also showed a more profound effect than the VDRC ones. so10>CG9797-RNAiVDRC flies showed a somewhat distinct phenotype, resulting in a protrusion of the eye coupled to a reduction of the eye perimeter. OK107>ttk-RNAiVDRC and so10>ttk-RNAiVDRC phenotypes are largely overlapping and show ommatidial degeneration consistent with its reported role in promoting photoreceptor cell differentiation at the late stages of eye development27. Interestingly, both so and ttk are expressed in photoreceptor cells, and both mutants of so and ttk display defects in adult photoreceptor rhabdomeres22,27, strengthening the hypothesis that TTK acts through SO in regulating photoreceptor cell differentiation.
Figure 5.

Effect of RNAi-mediated knockdown of Y1H-identified TFs for the so10 element on eye development and so gene expression. (a-b) Bright field microscopy images of adult eyes, lateral view. (a) OK107>CG9797-RNAiTRiP. (b) OK107>UAS-mCD8::GFP. (c-d) Bright field microscopy images of adult eyes, frontal view. (c) OK107>ttk-RNAiVDRC. (d) OK107>UAS-mCD8::GFP. (e) qRT-PCR analysis of so expression in third instar eye-antennal discs of OK107>CG9797-RNAiVDRC and OK107>CG9797-RNAiTRiP. Values are relative to the corresponding controls. The values are mean ± SEM with n = 3. The asterisk indicates a P-value < 0.05. (f) qRT-PCR analysis of so expression in third instar eye-antennal discs of OK107>ttk-RNAiVDRC flies and in so10>ttk-RNAiVDRC adult heads. Values are relative to the corresponding controls. The values are mean ± SEM with n = 3. The asterisk indicates a P-value < 0.05.
To verify that the phenotypes of ttk and CG9797 knock-down are caused by the misregulation of so expression, we quantified so mRNA levels in third instar eye-antennal discs of OK107> CG9797-RNAiTRiP and OK107>ttk-RNAiVDRC flies. As the ttk-RNAi phenotype resembles ommatidial degeneration in the adult stage, we also evaluated so expression in adult heads of so10>ttk-RNAiVDRC flies. We observed a 20% and 30% reduction of so mRNA levels in third instar eye-antennal discs of OK107>CG9797-RNAiVDRC OK107> CG9797-RNAiTRiP flies respectively, but only the latter was statistically significant. Knock-down of TTK resulted in a 30% reduction of so levels in both eye-antennal discs and adult heads, with the difference in adult heads being statistically significant (Fig. 5e-f). These results provide further evidence that the observed phenotypes after RNAi-mediated knock-down of the TFs are caused by a reduction in so expression. Interestingly, although no phenotype was observed upon knock-down of TOY, this TF was previously shown to directly regulate so expression by binding to the so10 element in vivo18. Furthermore, only one transgenic RNAi line for the second positive control interactor EY resulted in the expected eye defect, suggesting that in some cases the knock-down is not sufficient to yield an observable phenotype. It is therefore likely that we underestimate the number of possible in vivo relevant interactions. Taking the results of the knock-down experiments and the previously reported interactions together, we have evidence that at least four out of 11 so10 interactors identified by our Y1H system may be involved in vivo in the regulation of the respective DNA bait.
Discussion
We have established to our knowledge one of the most comprehensive, full-length, fully sequence-verified TF ORF clone collections for a metazoan organism. Since the ORFs were cloned open-ended (without a stop codon) in the versatile Gateway system, this collection should be of significant value to the Drosophila community. Here, we used this resource to develop an automated, yeast-based protein-DNA interaction detection system providing a powerful tool to de-orphanize in a high-throughput manner the many functional Drosophila promoters and CRMs for which the interacting TFs are still unknown. We benchmarked our system using previously characterized CRMs, and identified 26% of positive control interactions. While this detection rate falls, as indicated, within the range of previously reported Y1H and Y2H data9,28,29, we believe that this number is a conservative estimate given the absence of a protein-DNA interaction gold collection comparable to the one available to validate protein-protein interaction assays28. Additionally, we confirmed binding of the TFs found in the Y1H in vitro using a novel technique called MARE. This microfluidics-based approach enables refinement of the identified interactions to the level of individual binding sites. Although other in vitro techniques like DNase I footprinting and EMSAs are available that can identify individual binding sites within a DNA element, these tend to have low-throughput, are technically challenging, and can be difficult to interpret. In contrast and providing an initial investment in microfluidic equipment, MARE allows the relatively straight-forward analysis of multiple TFs across a large panel of individual DNA sequences on one chip while simultaneously providing a quantitative read-out for the observed interactions. Coupled to the high-throughput Y1H system, this pipeline therefore uniquely enables us to identify TFs binding to an uncharacterized CRM, and subsequently locate the specific binding site for each of these TFs within this element. However, although we obtained a high validation rate of Y1H-detected interactions using MARE, not all detected positives showed in vitro site-specific binding. For example, both Y1H data and in vivo validation suggest that CG9797 can directly interact with the so10 DNA-bait, yet it was not recovered by MARE. This may indicate that CG9797 binding to so10 is chromatin-dependent, showing the complementarity of both techniques.
Next to this in vitro validation and presented in vivo data providing support for the Y1H-detected interactions between the so10 element and respectively TTK and CG9797, we have indirect evidence that at least two other observed interactions may also have biological significance. For example, we detected binding of the homeobox TF EXTRADENTICLE (EXD) to the stripe 2 enhancer of the even skipped (eve) gene. Although this interaction is novel for Drosophila, in the cricket Gryllus bimaculatus it was shown that RNAi knock-down of exd leads to reduced eve expression30 suggesting that the regulatory network regulating eve expression may at least be partly conserved in these insect species. A second example involves the binding of the bZIP TF SLOW BORDER CELLS (SLBO) to the fat body enhancer of the Yolk protein 1 (Yp1) gene. Although this interaction was found by DNase I footprinting, it is unlikely that slbo regulates yolk expression in vivo since slbo is not expressed in the fat body of adult flies and yolk haemolymph levels are unchanged in slbo mutant flies31. Our Y1H screen picked up a different bZIP TF, namely TRAFFIC JAM. This TF is involved in female gonad development32 and is therefore a putative candidate to regulate Yp1 expression in vivo. Together, these results indicate that the high-throughput Y1H technique described here is a useful method to uncover previously unknown interactions with putative biological significance.
Methods
Gateway cloning of Drosophila TFs
TF ORFs were PCR-amplified using primers containing the attB1 and attB2 gateway tails at the 5′ end of the forward and reverse primer respectively (for primer sequences, see Supplementary Table 1). The gene-specific part of the primer was designed to have a melting temperature of approximately 60°C and a GC content close to 50%, although these parameters often had to be relaxed to find an appropriate primer. We omitted the stop codon, generating open-ended clones. The PCR was performed using iProof High-Fidelity DNA Polymerase (Bio-Rad) according to manufacturer’s specifications. In a first approach we used BDGP clones as DNA template. We first compared the cDNA clone sequence with the reference sequence for each TF. Clones were rejected if they contained partial ORFs, nonsense mutations, missense mutations in a known functional protein domain or more than 5 missense mutations in total compared to the reference sequence. Applying these criteria reduced the number of acceptable cDNA clones from 656 to 501. When no acceptable cDNA clone was retrieved, an RT-PCR strategy was adopted by extracting total RNA from whole Drosophila embryos, larvae or adult flies using Tri Reagent (Sigma) followed by a clean-up step using the RNeasy mini kit (Qiagen). Five μg of this RNA was used as an input to generate cDNA using the SuperScript III First-Strand Synthesis kit (Invitrogen). The resulting cDNA was subsequently used as a template for PCR amplification. Successfully PCR-amplified TF ORFs were cloned into the pDONR221 vector using Gateway cloning by mixing 100 μg of the pDONR221 vector, 2 μl of the PCR product and 0.5 μl of BP clonase II enzyme mix (Invitrogen). After incubating for 18 h at 25°C, this mix was transformed into competent DH5α cells and single colonies, typically four per TF, were analyzed by colony PCR with M13F and M13R primers using standard protocols. The TFs that were successfully cloned in pDONR221 (further called TF Entry clones) were subsequently analyzed by high-throughput sequencing.
High-throughput sequencing of TF clone ORFs
The TF Entry clones were pooled equimolarly, and subsequently fragmented using a Covaris S2 Adaptive Focused Acoustics instrument (Covaris; Woburn, Massachusetts, USA) using the settings: duty cycle – 20%, intensity – 5, cycles per burst – 200, time – 90 seconds. Five μg of the fragmented plasmid pool was then used for sequencing library preparation using the Illumina DNA Sample Prep Kit (Illumina; San Diego, California, USA) according to the protocol supplied with the reagents. The sequencing library was loaded into one lane of a flow cell, sequencing clusters were generated using the Illumina Single-Read Cluster Generation Kit v2 and the flowcell sequenced on the Illumina Genome Analyzer IIx using Illumina Cycle Sequencing Kit v3 reagents according to the protocol provided by the supplier, producing 76 bp reads. The output data were processed using the Genome Analyzer Pipeline Software v1.4. The resulting file containing the short reads was submitted to the WebPrInSeS server12 together with a file containing the reference sequences for automated assembly of the reads and evaluation of the resulting ORFs in comparison with the respective reference sequences. The TF Entry clones were evaluated for sequencing coverage and quality of the assembled sequence. Clones that are fully covered by sequencing and that meet the criteria used for the evaluation of the cDNA clones described above following the BDGP convention were labeled “Gold” (588 clones or 78%). Clones of which the 5′ and 3′ were covered by sequencing (i.e. standard ORFeome quality), and for which quality criteria were met, were labeled “Silver” (36 or 5%). Clones which were only partially covered by sequencing, but for which the resulting assembled sequence met the quality criteria, were labeled “Bronze” after pooling all clones that were available for a specific TF (typically four) to maximize the chance of having a functional clone in this mix (68 clone mixes or 9%).
Shuttling the TF ORF to Gateway compatible AD vectors
The TF ORFs were subcloned from the TF Entry clones into an equimolar mix of pAD-Dest-ARS/CEN and AD-Dest-2μ by mixing 2 μl of the TF entry clone, 100 ng of the pAD-Dest mix and 0.5 μl of LR clonase II enzyme mix (Invitrogen). After incubating for 18 h at 25°C, this mix was transformed into competent DH5α cells and single colonies were analyzed by colony PCR with the AD primer and a TF-specific reverse primer using standard protocols. All successfully subcloned TFs (647) were maxiprepped and diluted to a final concentration of 100 ng/μl. The plasmid preps were checked again by PCR to verify that no arraying errors were made during prepping.
The AD-TF clones are ordered in a similar way as the TF ORF clone collection, but in a 384-well format. For example, for the TF ORF clones in row A of 96-well plates 1, 2, 3 and 4, the corresponding AD-TF clone would reside in respectively the uneven wells of row A, the even wells of row A, the uneven wells of row B, and the even wells of row B of the 384-well AD-TF plate (Supplementary Table 1). Some of the empty wells in the 384-well AD-TF plates were filled with the original pAD-DEST vectors as negative controls, or with duplicates of some TF clones of specific interest, as indicated in Supplementary Table 1. Interactions that are detected twice with a specific TF are reported only once in Supplementary Table 2.
Cloning of CRMs
CRMs were PCR amplified using primers containing restriction enzyme recognition sites at the 5′ end of the forward and reverse primer respectively and cloned in the pENTRY-5′ vector using standard restriction-ligation techniques. The CRMs were further subcloned in the Y1H-compatible pMW2 (“HIS3”) and pMW3 (“lacZ”) vectors by Gateway LR cloning as described above. Single colonies were selected and verified by Sanger sequencing. A double integration was performed with the resulting CRM destination clones (both pMW2-CRM and pMW3-CRM in a single yeast strain) in Y1H-aS2 (with the exception of element so10 which was integrated in the YM4271 yeast strain) using lithium acetate (LiAc) – polyethylene glycol (PEG) transformation followed by selection on a Sc –His, -Ura plate.
High-throughput yeast transformation
The high-throughput yeast transformation protocol is based on the regular LiAc-PEG yeast transformation protocol but volumes are decreased to allow screening in 384-well format. Briefly, 2 μl of 100 ng/ μl prey plasmid, 5 μl of competent yeast, and 25 μl of TE/LiAc/PEG solution are added in a well of a 384 micro-well plate and resuspended by pipetting. The yeast suspension is incubated for 30 min at 30°C and subsequently heat-shocked for exactly 20 min at 42°C in a hot-air incubator. The yeast is pelleted by centrifugation and the supernatant is removed. The cells are resuspended in 5 μl of sterile water and 1 μl of this suspension is spotted on a Sc –His, -Ura, -Trp plate. We have engineered and programmed a customized robotic system (Tecan Evo) equipped with a 384 pipetting head, incubators and a centrifuge unit to perform the complete transformation and spotting process autonomously. After growing the yeast for 3 days at 30°C, the colonies are transferred to selective Sc –His, -Ura, -Trp plates containing varying 3-Amino-1,2,4-triazole (3-AT) concentrations. To evaluate activation of the lacZ reporter, positive colonies were cherry-picked, re-spotted four times in 384-well format onto permissive yeast plates covered by a nitrocellulose filter to perform a lacZ filter assay as described13.
As a negative control, we also subcloned the multiple cloning site (MCS) of the pENTRY5′ vector into the pMW2 vector and integrated it into the yeast genome. We then transformed this DNA bait yeast strain with all Drosophila TFs as described above. We detected a single, uncharacterized ZF-C2H2 TF, CG14655, which interacted with the control vector (data not shown). This may be due to binding of this TF to the minimal promoter of the HIS3 gene or other vector parts like the Gateway sites or the parts of the MCS present in this vector. Consequently, interactions involving this TF with other DNA baits (e.g. the strongest growing quadrant in the upper left corner of the selective plate in Fig. 2) were considered as false positives and omitted from Supplementary Table 2.
Semi-automated detection of positive interactions
Despite the fact that the transformed yeast colonies were arrayed as quadruplicates to facilitate visual detection, manual inspection can still be inconsistent and subjective. In order to have more objective calls, we developed an image analysis software that allows semi-automatic processing of JPEG images of the Y1H selection plates. This custom-designed tool was written in MATLAB (R2008b, Mathworks, MA, USA) and requires an image in grayscale as input. The user then has to define the three corner colonies (bottom left, top left and bottom right) by clicking on the image. This allows normalizing and reorienting of the image according to the array of yeast colonies. A uniform grid is created to define the position of each yeast colony quadrant. If the grid positioning is not precise, the user can choose to reject the grid and redefine the corners of the image.
The quadruplicated yeast colony pattern was detected by convoluting the image with a pattern of four bright spots on a dark background. The intensity value of the convoluted image in the center of each quadrant is used as a measure for the size of the quadrant colonies with a greater value indicating a stronger interaction. TIDY then groups the intensity values in 10 clusters. We achieved the most robust detection of strong positives when we considered 1) the highest intensity value in the largest of these clusters, representing most and thus likely negative yeast quadrants, as the background threshold; and 2) quadrants scoring at least 20% above this background threshold as positives. Positives that fulfill this criterion have intensity values that typically are at least two standard deviations above the mean or median intensity value of the plate.
In order to avoid detection of interactions where only one or two out of four colonies show strong growth, we also measure the intensity of individual colonies. This is done by dividing the image in 1536 squares, each defining the limit of a single colony, and integrating the intensity over each of these squares. A uniformity coefficient is computed for each colony by subtracting half of the maximal and minimal values from the sum of four intensities and dividing this number by the mean of the four values. Therefore a number close to 3 would indicate little variation in intensity between the four colonies whereas a number greater or lower than 3 would indicate respectively lower or higher growth of one of the quadrant colonies. A second threshold based on this value is empirically set at 2.96 as we specifically want to eliminate quadrants whose intensity values are derived from only one or two large colonies reflecting spotty yeast growth.
The output of the program plots in green the abbreviated names of the TFs corresponding to the interactions scoring 20% above the background threshold. In addition, the TF names are shown in a text box next to the image plot and are returned in the MATLAB command line from where they can be easily copied. A plot visualizing the intensity value distribution also appears beside the image with the intensity values on the horizontal axis and the uniformity coefficient on the vertical axis. The user can modify the area set by the default thresholds by directly clicking on this plot to evaluate the detection stringency. In some cases, this allows the inclusion of weaker interactions that clearly score above background, but below the conservative 20% threshold. The user-defined threshold is drawn in red on the plot and the newly detected interactions appear in magenta indicating their distinct status.
Finally, on some yeast plates, exterior colonies exhibit higher growth than interior ones, potentially biasing the detection threshold. We therefore included an option in TIDY that allows the user to correct for this artifact. In the case where the correction option is selected, we separate the exterior colonies from the interior ones and treat them as two separate distributions. The clustering and definition of the thresholds is done in the same way as explained earlier except that the number of clusters for the exterior distribution is set at six because of the lower number of involved quadrants.
Mitomi-based Analysis of Regulatory Elements (MARE)
MARE analysis was performed essentially as described in Gubelmann et al. (cosubmitted). In brief, a library of 36 bp sequences was designed to cover the whole DNA bait so that each sequence has a 24 bp overlap with the next one in the library and each 12 bp region is covered by three different fragments. Note that the first and last region is only covered by one fragment, and the second and penultimate by two fragments. Each sequence was purchased as a single-strand oligonucleotide (Invitrogen) which served as a template for generating labeled double-stranded oligonucleotides as described17. TFs were subcloned from the ENTRY clones into the pMARE vector by standard Gateway cloning, fusing the eGFP coding sequence to the 3′ end of the TF ORF. Subsequently, linear expression templates containing 5′ end 3′ UTR sequences and the TF-eGFP fusion were generated by PCR using standard techniques. Linear expression templates were printed on top of the DNA baits on an epoxy-coated glass slide using a Qarray (Genetix) microarrayer. Microfluidics device design, fabrication, alignment and surface chemistry was as described (Gubelmann et al., cosubmitted). TF protein was synthesized by loading TNT SP6 High-Yield wheat germ extract mixture (Promega) onto the device. MITOMI was performed and the device was imaged as described17. MARE data analysis was performed as described in Gubelmann et al. (cosubmitted). In brief, for each 12 bp region, the average signal S of the 3 fragments in which it is represented was calculated. For each 12 bp region we defined the mid position as the representative binding event position. Signal values at positions other than representative binding event positions were estimated by cubic interpolation (interp1 function, signal package, R). Specific TF protein-DNA interactions were identified by clustering the signal of each position into two distinct classes, i.e., specific binding positions (SBPs) and non-specific binding positions (NSBPs), using the k-means clustering algorithm (function kmeans, R; settings: centers = 2, algorithm = Hartigan-Wong, nstart = 1000). The center of the NSBP class was defined as the DNA bait-specific mean background signal (MBS). For each SBP, we defined the relative enrichment over non-specific binding as E(SBP)=S(SBP)/MBS and filtered out SBPs that have an E < 2. Specific binding regions (SBRs) were defined by joining consecutive SBPs and SBPs with the largest enrichment within a SBR were defined as the SBR maxima. Each MARE experiment was performed two times. Note that, as DNA occupancy is plotted as a relative signal normalized for the protein level in the microfluidics chamber, the scale of the Y-axis may vary between replicates. Therefore the overall trend of the DNA occupancy signal was compared between replicates. A peak was considered present in both replicates if the SBR maximum of the first replicate overlapped with the SBR of the second replicate and vice versa.
TF binding site analysis
We used the online matrix-scan tool of the RSAT package33. PWMs were from the JASPAR and TRANSFAC databases34,35 (Supplementary Data). The upper detection threshold was set at P <1e-3.
Fly stocks
Flies were maintained at 25°C on standard agar-cornmeal medium. UAS-RNAi lines were from the VDRC25 and TRiP24 collections and are listed in Supplementary Table 6. Additional fly stocks used were OK107, UAS-mCD8::GFP (available from the Bloomington stock centre), y,w[1118];P{attP,y[+],w[3′]} (available for the VDRC stock center) and so10-GAL4 (kind gift from Serge Plaza).
Analysis of phenotypes
Virgin females of the UAS-RNAi lines were crossed with males of the OK107 and so10-GAL4 driver lines. Adult eyes were examined using bright-field microscopy by comparing the size, overall shape and roughness of each knock-down eye to the eye of a control animal (OK107>attP, OK107>mCD8::GFP, so10>attP and so10>mCD8::GFP). Bright-field microscopy images were obtained on a Leica MZ 16 1FA stereomicroscope equipped with a DFC 480 color camera.
RNA extraction, cDNA synthesis and quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from 30 eye-antennal discs per genotype from wandering third-instar larvae or 10 adult heads from adults overexpressing the appropriate RNAi transgene using the Nucleospin RNA XS kit (Macherey-Nagel) according to manufacturer’s specifications. First strand cDNA synthesis was performed using the Superscript VILO cDNA synthesis kit (Invitrogen) starting from 500 ng total RNA. Primer sets were pulled from the GETPrime primer database (Gubelmann et al., in press) (RpL32: 5′-TAA GCT GTC GCA CAA ATG G-3′ and 5′-GGG CAT CAG ATA CTG TCC C-3′; so: 5′-CTG TGT TTG CGA GGT TCT C-3′ and 5′-TTA TCA CAT TGT GGC AGC G-3′). qRT-PCR PCR was performed in 384-well plates with three technical replicates on the ABI-7900HT Real-Time PCR System (Applied Biosystems) using Power SYBR Green Master Mix (Applied Biosystems) using standard procedures. RpL32 expression levels were used as endogenous control and relative expression ratios were calculated using the ΔΔCt method with the expression levels in OK107>mCD8::GFP and so10>mCD8::GFP flies as calibrators. qRT-PCR data were derived from three independent biological replicates and P values were derived using a t-test.
Supplementary Material
Supplementary Figure 1: Distribution of DNA-binding domains within the Drosophila TF repertoire.
Supplementary Figure 2: Distribution between pAD-Dest-ARS/CEN and pAD-Dest-2μ Gateway destination clones after LR cloning.
Supplementary Figure 3: Representative TIDY-processed images for the so10 Y1H screen.
Supplementary Figure 4: lacZ read-out of the so10 Y1H screen.
Supplementary Figure 5: Output of the TIDY program.
Supplementary Figures 6–10: Representative TIDY-processed images for each Y1H screen.
Supplementary Figure 11: Manual screen of positive control interactions.
Supplementary Figure 12: lacZ read-out of the dpp813 Y1H screen.
Supplementary Figures 13–16: Representative TIDY-processed images for dpp813-Frag1–3 and dpp813-RC.
Supplementary Figure 17: MARE analysis of so10 for recognition by Y1H-identified TFs.
Supplementary Figure 18: MARE analysis of yp1-1 for recognition by Y1H-identified TFs.
Supplementary Figure 19: Eye phenotypes observed upon RNAi-mediated knock-down of Y1H-identified TFs.
Supplementary Table 1: Predicted TFs in the Drosophila genome and their cloning status. (Separate excel file)
Supplementary Table 2: Observed interactions in two independent Y1H screens. (Separate excel file)
Supplementary Table 3: CRMs used in this study. (Separate excel file)
Supplementary Table 4: manual screening of reported interactions. (Separate excel file)
Supplementary Table 5: dpp813-Frag1-3 and dpp813-RC interactions. (Separate excel file)
Supplementary Table 6: Effect of RNAi-mediated TF knock-down on eye development (Separate excel file)
Supplementary Data: PWMs used for binding site predictions.
Acknowledgments
We thank the members of the Lausanne DNA array facility (DAFL) for performing the Illumina sequencing, Kenneth H. Wan for managing cDNA sequencing and TF cDNA clone production, John Reece-Hoyes and Marian Walhout for constructive discussions of this work and for kindly providing the Y1H-aS2 strain, and Nele Gheldof for help with the Figures. We thank Nicholas W. Kelley for providing PWMs. We thank Sebastian Waszak for MARE data analysis. We thank the TRiP at Harvard Medical School (NIH/NIGMS R01-GM084947) and the Vienna Drosophila RNAi Center (VDRC) for providing transgenic RNAi fly stocks used in this study. This work was supported by funds from the Swiss National Science Foundation and SystemsX.ch, by a Marie Curie International Reintegration Grant (BD) from the Seventh Research Framework Programme, by the Frontiers in Genetics NCCR Program, and by Institutional support from the Ecole Polytechnique Fédérale de Lausanne (EPFL).
Footnotes
Author Contributions
B.D. supervised the study. K.H. and B.D. designed the study. K.H. and J.B built the TF clone collection. K.H and J.-D.F. performed Y1H screens. K.H. performed in vivo validations. A. Iagovitina developed image analysis software. A. Isakova performed MARE analyses. A.M. analyzed high-throughput sequencing data. P.C. provided cDNA clones. S.E.C. identified TFs with sequence-specific DNA-binding domains used in this study and produced TF cDNA clones. K.H. and B.D. prepared the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Levine M. A systems view of drosophila segmentation. Genome Biol. 2008;9 (2):207. doi: 10.1186/gb-2008-9-2-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams M, et al. The genome sequence of drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 3.O’Kane CJ, Gehring WJ. Detection in situ of genomic regulatory elements in drosophila. Proc Natl Acad Sci USA. 1987;84 (24):9123–9127. doi: 10.1073/pnas.84.24.9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zinzen RP, Girardot C, Gagneur J, Braun M, Furlong EEM. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. 2009;462 (7269):65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- 5.Filion GJ, et al. Systematic protein location mapping reveals five principal chromatin types in drosophila cells. Cell. 2010;143 (2):212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for drosophila using germ-line-specific phic31 integrases. Proc Natl Acad Sci USA. 2007;104 (9):3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stark A, et al. Discovery of functional elements in 12 drosophila genomes using evolutionary signatures. Nature. 2007;450 (7167):219–232. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deplancke B, Simicevic J. DNA-centered approaches to characterize regulatory protein-DNA interaction complexes. Mol Biosyst. 2010;6 (3):462–468. doi: 10.1039/b916137f. [DOI] [PubMed] [Google Scholar]
- 9.Deplancke B, et al. A gene-centered c. Elegans protein-DNA interaction network. Cell. 2006;125 (6):1193–1205. doi: 10.1016/j.cell.2006.04.038. [DOI] [PubMed] [Google Scholar]
- 10.Adryan B, Teichmann SA. Flytf: A systematic review of site-specific transcription factors in the fruit fly drosophila melanogaster. Bioinformatics. 2006;22 (12):1532–1533. doi: 10.1093/bioinformatics/btl143. [DOI] [PubMed] [Google Scholar]
- 11.Gallo SM, et al. Redfly v3.0: Toward a comprehensive database of transcriptional regulatory elements in drosophila. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massouras A, Decouttere F, Hens K, Deplancke B. Webprinses: Automated full-length clone sequence identification and verification using high-throughput sequencing data. Nucleic Acids Res. 2010;38 (Suppl):W378–384. doi: 10.1093/nar/gkq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vermeirssen V, et al. Matrix and steiner-triple-system smart pooling assays for high-performance transcription regulatory network mapping. Nat Methods. 2007;4 (8):659–664. doi: 10.1038/nmeth1063. [DOI] [PubMed] [Google Scholar]
- 14.Koegl M, Uetz P. Improving yeast two-hybrid screening systems. Brief Funct Genomic Proteomic. 2007;6 (4):302–312. doi: 10.1093/bfgp/elm035. [DOI] [PubMed] [Google Scholar]
- 15.Braun P, et al. An experimentally derived confidence score for binary protein-protein interactions. Nat Methods. 2009;6 (1):91–97. doi: 10.1038/nmeth.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deplancke B, Dupuy D, Vidal M, Walhout AJ. A gateway-compatible yeast one-hybrid system. Genome Res. 2004;14 (10B):2093–2101. doi: 10.1101/gr.2445504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maerkl SJ, Quake SR. A systems approach to measuring the binding energy landscapes of transcription factors. Science. 2007;315 (5809):233–237. doi: 10.1126/science.1131007. [DOI] [PubMed] [Google Scholar]
- 18.Punzo C, Seimiya M, Flister S, Gehring WJ, Plaza S. Differential interactions of eyeless and twin of eyeless with the sine oculis enhancer. Development. 2002;129 (3):625–634. doi: 10.1242/dev.129.3.625. [DOI] [PubMed] [Google Scholar]
- 19.Czerny T, et al. Twin of eyeless, a second pax-6 gene of drosophila, acts upstream of eyeless in the control of eye development. Molecular Cell. 1999;3 (3):297–307. doi: 10.1016/s1097-2765(00)80457-8. [DOI] [PubMed] [Google Scholar]
- 20.Quiring R, Walldorf U, Kloter U, Gehring WJ. Homology of the eyeless gene of drosophila to the small eye gene in mice and aniridia in humans. Science. 1994;265 (5173):785–789. doi: 10.1126/science.7914031. [DOI] [PubMed] [Google Scholar]
- 21.Burtis KC, Coschigano KT, Baker BS, Wensink PC. The doublesex proteins of drosophila melanogaster bind directly to a sex-specific yolk protein gene enhancer. Embo J. 1991;10 (9):2577–2582. doi: 10.1002/j.1460-2075.1991.tb07798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serikaku MA, Otousa JE. Sine oculis is a homeobox gene required for drosophila visual-system development. Genetics. 1994;138 (4):1137–1150. doi: 10.1093/genetics/138.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheyette BNR, et al. The drosophila sine oculis locus encodes a homeodomain-containing protein required for the development of the entire visual-system. Neuron. 1994;12 (5):977–996. doi: 10.1016/0896-6273(94)90308-5. [DOI] [PubMed] [Google Scholar]
- 24.Ni JQ, et al. A drosophila resource of transgenic rnai lines for neurogenetics. Genetics. 2009;182 (4):1089–1100. doi: 10.1534/genetics.109.103630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dietzl G, et al. A genome-wide transgenic rnai library for conditional gene inactivation in drosophila. Nature. 2007;448 (7150):151–U151. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 26.Callaerts P, et al. Drosophila pax-6/eyeless is essential for normal adult brain structure and function. J Neurobiol. 2001;46 (2):73–88. doi: 10.1002/1097-4695(20010205)46:2<73::aid-neu10>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 27.Lai ZC, Li Y. Tramtrack69 is positively and autonomously required for drosophila photoreceptor development. Genetics. 1999;152 (1):299–305. doi: 10.1093/genetics/152.1.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braun P, et al. An experimentally derived confidence score for binary protein-protein interactions. Nat Methods. 2009;6 (1):91–98. doi: 10.1038/nmeth.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen YC, Rajagopala SV, Stellberger T, Uetz P. Exhaustive benchmarking of the yeast two-hybrid system. Nat Methods. 2010;7 (9):667–668. doi: 10.1038/nmeth0910-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mito T, et al. Divergent and conserved roles of extradenticle in body segmentation and appendage formation, respectively, in the cricket gryllus bimaculatus. Dev Biol. 2008;313 (1):67–79. doi: 10.1016/j.ydbio.2007.09.060. [DOI] [PubMed] [Google Scholar]
- 31.Hutson SF, Bownes M. The regulation of yp3 expression in the drosophila melanogaster fat body. Dev Genes Evol. 2003;213 (1):1–8. doi: 10.1007/s00427-002-0286-4. [DOI] [PubMed] [Google Scholar]
- 32.Li MA, Alls JD, Avancini RM, Koo K, Godt D. The large maf factor traffic jam controls gonad morphogenesis in drosophila. Nat Cell Biol. 2003;5 (11):994–1000. doi: 10.1038/ncb1058. [DOI] [PubMed] [Google Scholar]
- 33.Turatsinze JV, Thomas-Chollier M, Defrance M, van Helden J. Using rsat to scan genome sequences for transcription factor binding sites and cis-regulatory modules. Nat Protoc. 2008;3 (10):1578–1588. doi: 10.1038/nprot.2008.97. [DOI] [PubMed] [Google Scholar]
- 34.Bryne JC, et al. Jaspar, the open access database of transcription factor-binding profiles: New content and tools in the 2008 update. Nucleic Acids Res. 2008;36 (Database issue):D102–106. doi: 10.1093/nar/gkm955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matys V, et al. Transfac (r) and its module transcompel (r): Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Distribution of DNA-binding domains within the Drosophila TF repertoire.
Supplementary Figure 2: Distribution between pAD-Dest-ARS/CEN and pAD-Dest-2μ Gateway destination clones after LR cloning.
Supplementary Figure 3: Representative TIDY-processed images for the so10 Y1H screen.
Supplementary Figure 4: lacZ read-out of the so10 Y1H screen.
Supplementary Figure 5: Output of the TIDY program.
Supplementary Figures 6–10: Representative TIDY-processed images for each Y1H screen.
Supplementary Figure 11: Manual screen of positive control interactions.
Supplementary Figure 12: lacZ read-out of the dpp813 Y1H screen.
Supplementary Figures 13–16: Representative TIDY-processed images for dpp813-Frag1–3 and dpp813-RC.
Supplementary Figure 17: MARE analysis of so10 for recognition by Y1H-identified TFs.
Supplementary Figure 18: MARE analysis of yp1-1 for recognition by Y1H-identified TFs.
Supplementary Figure 19: Eye phenotypes observed upon RNAi-mediated knock-down of Y1H-identified TFs.
Supplementary Table 1: Predicted TFs in the Drosophila genome and their cloning status. (Separate excel file)
Supplementary Table 2: Observed interactions in two independent Y1H screens. (Separate excel file)
Supplementary Table 3: CRMs used in this study. (Separate excel file)
Supplementary Table 4: manual screening of reported interactions. (Separate excel file)
Supplementary Table 5: dpp813-Frag1-3 and dpp813-RC interactions. (Separate excel file)
Supplementary Table 6: Effect of RNAi-mediated TF knock-down on eye development (Separate excel file)
Supplementary Data: PWMs used for binding site predictions.