

Abstract
Background
Copy number variants (CNVs) may play an important part in the development of common birth defects such as oral clefts, and individual patients with multiple birth defects (including clefts) have been shown to carry small and large chromosomal deletions. In this paper we investigate de novo deletions defined as DNA segments missing in an oral cleft proband but present in both unaffected parents. We compare de novo deletion frequencies in children of European ancestry with an isolated, non-syndromic oral cleft to frequencies in children of European ancestry from randomly sampled trios.
Results
We identified a genome-wide significant 62 kilo base (kb) non-coding region on chromosome 7p14.1 where de novo deletions occur more frequently among oral cleft cases than controls. We also observed wider de novo deletions among cleft lip and palate (CLP) cases than seen among cleft palate (CP) and cleft lip (CL) cases.
Conclusions
This study presents a region where de novo deletions appear to be involved in the etiology of oral clefts, although the underlying biological mechanisms are still unknown. Larger de novo deletions are more likely to interfere with normal craniofacial development and may result in more severe clefts. Study protocol and sample DNA source can severely affect estimates of de novo deletion frequencies. Follow-up studies are needed to further validate these findings and to potentially identify additional structural variants underlying oral clefts.
Keywords: Oral clefts, DNA copy numbers, de novo deletions, Case-parent trios
Background
Oral clefts are among the most common birth defects, and include three anatomical defects: cleft lip (CL), cleft lip and palate (CLP) and cleft palate (CP). Because there are similarities in embryological and epidemiological evidence [1,2], CL and CLP are often grouped together as cleft lip with/without cleft palate (CL/P), although debate remains about whether all three groups may have distinct etiologies [3,4]. Collectively, oral clefts represent the most common type of craniofacial malformations [5] and create a major public health burden for both affected children and their families. The overall birth prevalence of oral clefts is estimated at 1 per 700 live births worldwide, but there is dramatic variation across populations and between racial and ethnic groups, in particular for CL/P [2]. Oral clefts show strong familial aggregation, and the recurrence risk among first degree relatives is approximately 32 times greater than the general population risk for CL/P, and approximately 56 times greater for CP [6]. Twin studies also suggest a major role for genes controlling risk of oral clefts with monozygotic twins showing much higher concordance rates than dizygotic twins: 31% versus 2% for CL/P, and 43% versus 7% for CP [7]. Normal development of craniofacial features is a complex process and disruption of any of numerous steps can lead to development of oral clefts [8]. This etiologic complexity is further supported by mounting evidence that multiple genes or their regulatory genetic elements, in addition to environmental influences, play a role in the etiology of oral clefts, although supporting evidence for relatively few genes would be considered definitive [9-14].
Assessment of chromosomal anomalies such as microdeletions and translocations have played an important role in identifying genes and genomic regions underlying craniofacial disorders [15-23]. In particular, high throughput technologies such as comparative genomic hybridization (CGH) and single nucleotide polymorphism (SNP) arrays have gained popularity in identifying chromosomal alterations [24,25]. Sivertsen et al. assessed the prevalence of duplications and deletions in the 22q11 region (DiGeorge syndrome region) among Norwegian offspring with CP, but did not detect any association [26]. Shi et al. used SNP genotyping, DNA sequencing, high-resolution DNA microarray analysis, and long-range PCR to characterize chromosomal deletions in 333 candidate genes for orofacial clefting in 2,823 samples from 725 two and three generation families ascertained through a proband with a CL/P [27]. These authors confirmed several de novo deletions (defined as DNA segments missing in an oral cleft proband but present in both parents in two copies) in some of these candidate genes, in particular SUMO1, TBX1, and TFAP2A, raising the possibility that genes or regulatory elements contained within deleted regions might play a role in the etiology of oral clefts. Further, high rates of Mendelian inconsistencies were observed in 11 different genes, suggesting the existence of additional micro-deletions among oral cleft cases.
Family-based study designs as used by Shi et. al [27] are a popular alternative to the more common population-based designs (e.g. case-control studies) to assess associations between copy number variants (CNVs) and a disorder of interest [28-32], since investigating parents and offspring simultaneously enables the researcher to infer structural variants that occur de novo in the offspring (typically through a germline deletion). However, while numerous methods for CNV delineation in individual samples [33-38] or multiple independent samples [39-42] are available, only relatively few statistical approaches for detecting de novo CNVs have been proposed, and these are limited to offspring-parent trios. PennCNV[43] is based on a hidden Markov model (HMM), jointly modeling the unknown copy number states in all three trio members. Maximum likelihood methods are then employed to identify the most likely copy number states in the father, mother and offspring, which includes de novo deletions in the proband as a special case. MinimumDistance[44] on the other hand was specifically developed for detecting de novo deletions in case-parent trios, since the computational demands of the PennCNV joint HMM are substantial, and false positive calls of de novo deletions remain a concern even when the recommended quality control corrections are employed [44]. MinimumDistance captures differences in copy number estimates between the offspring and each parent at each locus before smoothing and posterior calling are carried out (see Methods), which greatly reduces technical and experimental sources of noise such as genomic waves, probe effects and batch effects [45,46], which are the major sources of false positive identifications in copy number analyses. Here, we employ both MinimumDistance and PennCNV to estimate and compare frequencies of de novo deletions in cleft probands and unaffected children from trios of European ancestry.
Results
We compared the frequencies of de novo deletions in cleft probands and control children from trios. We identified de novo deletions in 467 cleft and 391 control trios, and found a 62 kilo base (kb) non-coding region on chromosome 7p14.1 where de novo deletions occurred significantly more often among the cleft trios. Two different algorithms were employed to delineate de novo deletions in the probands – MinimumDistance[44] and PennCNV[43] – and yielded a total of 190 and 455 CNV regions, respectively, where at least five de novo deletions occurred in both sets of trios combined. A significantly higher rate of de novo deletions in the cleft trios compared to control trios was observed near the 38.3 MB region on chromosome 7p14.1 (Figure 1; p=4.3×10-2 and p=1.1×10-3 respectively, corrected for multiple comparisons). This exact genomic region has been previously identified as a region with high structural variation (projects.tcag.ca/variation/), and deletions in this area have been associated with developmental problems including craniofacial abnormalities [47-51].
Figure 1.
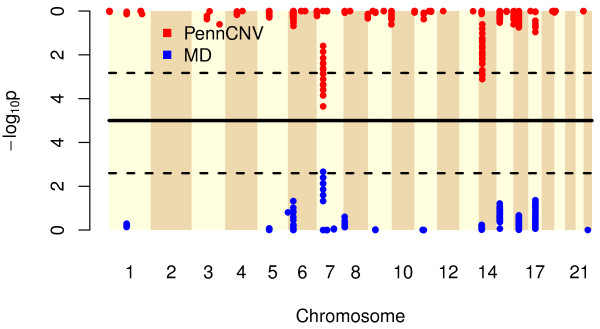
The -log10 p-values (y-axis) derived from testing associations of inferred de novo deletions and oral clefts, shown by chromosomal location (x-axis). Each point represents a de novo deletion CNV segment, delineated through MinimumDistance[44] (lower half) or PennCNV[43] (upper half). The dashed lines represent the genome-wide significance levels for a family-wise error rate of 5%, derived via permutation tests. The striped vertical bands indicate the 22 autosomes interrogated.
The most significant association was observed in a sub-region where MinimumDistance (PennCNV) identified 10 (20) cleft cases with an apparent de novo deletion, and none (one) among the control trios (Figure 2). The 10 (20) case probands with de novo deletions in this region included 6 (9) CL, 3 (6) CP, and 1 (5) CLP cases. The nearest gene to this 7p14.1 region, about 20 kb upstream from the peak of this signal, is the T cell receptor gamma alternate reading frame protein (TARP). While this particular gene to our knowledge has not been previously associated with craniofacial abnormalities per se, copy number changes in T cell receptors (including those on 7p14) have been strongly associated with developmental problems [51]. For the 44 probes contained in this segment of 7p14.1, the signal intensities show a clear reduction among the 10 cases identified by MinimumDistance, indicating hemizygous deletions. These lower log R ratios were not observed in their parents, indicating a normal DNA copy number state (Figure 3). Sufficient DNA was available for three of the cleft trios with an inferred hemizygous de novo deletion at this region. Quantitative real-time PCR confirmed a clear copy number decrease in the child relative to his/her parents (Additional file 1). While TARP is not a very strong candidate for a causal gene per se, HOXA2 on 7p14.2 is a functional candidate just over 1Mb away. A mutation in HOXA2 causes microtia (deformity of the external ear), hearing impairment and cleft palate (http://www.omim.org/entry/604685). Though purely speculative, it might be possible that a copy number variant involving a distal enhancer might cause clefting similar to the way an enhancer 1 Mb from SHH (sonic hedgehog) produces preaxial polydactyly [52].
Figure 2.

The count of CNV components (y-axis) delineated via MinimumDistance in the cleft lip/palate trios (green) and control trios (yellow), as a function of genomic location (x-axis) near 7p14.1, plus the corresponding numbers delineated via PennCNV in the cleft lip/palate trios (red) and control trios (blue). Segments are defined as collections of probes where CNV composition does not change in the combined trio sets. Short lines thus represent small differences in width among de novo deletions. The most significant association was observed in a sub-region where MinimumDistance (PennCNV) identified 10 (20) cleft lip/palate subjects with a de novo deletion, and none (one) in the control trios. The nearest gene is the T cell receptor gamma alternate reading frame protein TARP, about 20 kb away from the de novo deletions.
Figure 3.
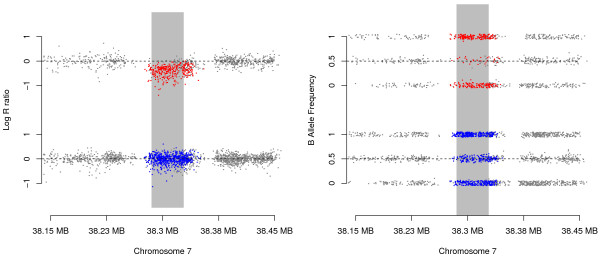
The log R ratios (left) and B allele frequencies (right) near the identified 7p14.1 locus. The upper panels (containing the red dots) represent the 10 oral cleft probands with MinimumDistance inferred de novo deletions at this locus, the lower panels (containing the blue dots) show the data for the parents of these probands. For each subject (parent or proband), color was used for the markers within the inferred de novo deletions (which differ in length between trios), gray dots were used for markers outside the deletions. The vertical gray bar indicates the segment of markers that yielded the most significant association test. For visualization, slight horizontal jitter was applied for both plots, and vertical jitter was applied for the B allele frequency plot.
A second region of potential interest was identified by PennCNV on chromosome 14, however, upon manual inspection of signal intensities, this region appeared to be a false positive result (see Additional file 1). An analysis of de novo deletions called by both MinimumDistance and PennCNV also yielded the chromome 7p14.1 locus as the only significant finding among 90 CNV components from 11 distinct loci that had at least 5 de novo deletions called by both methods, with nine de novo deletions in cleft trios compared to none in the controls (p=0.032, corrected for multiple comparisons). It is also noteworthy that among the oral cleft candidate genes examined by Jugessur et al [11] and Shi et al [27], we only detected one inherited deletion (in UGT1A7), and no de novo deletions in these trios.
Another technique to infer or confirm de novo deletions, based solely on genotypes, is to search for clusters of Mendelian inconsistencies between genotypes of the trio [27,53]. In our study however, the identified regions on chromosome 7 were small and the corresponding SNPs interrogated by these probes had low minor allele frequency in our population, so no Mendelian inconsistencies were observed among the trios with an inferred de novo deletion in the proband.
Comparing the overall widths of MinimumDistance and PennCNV inferred de novo deletions in cleft cases and controls revealed that the estimated deletions were substantially larger in cases than in controls. The median deletion width inferred from MinimumDistance (PennCNV) was 71.7 kb (61.3 kb) among controls and 102.7 kb (70.5 kb) among cases, corresponding to an increase of 43% (15%) in median width of a deletion among cases (Table 1). These observed differences in widths were statistically significant (Kolmogorov-Smirnov p-values of 1.2×10-4 and 2.9×10-3 respectively, Wilcoxon rank-sum p-values of 1.0×10-4 and 5×10-2 respectively; see Methods). Compared to the controls, the MinimumDistance inferred de novo deletions were also larger when cleft types were considered individually, increasing from CL (median 87.8 kb) to CP (95.2 kb) to CLP (128.5 kb). For inferred de novo deletions identified by PennCNV, we did not observe any trend of increasing size by cleft type, as CP deletions (median 52.5 kb) were smaller than apparent deletions in controls (Table 1). However, this observation may reflect an excess number of false positive (and mostly short) PennCNV identifications among controls, as discussed in more detail below.
Table 1.
The median width in kilo bases (kb) of de novo deletions, stratified by case status and cleft type for both discovery methods
| Controls | Clefts | p | CL | p | CP | p | CLP | p | |
|---|---|---|---|---|---|---|---|---|---|
|
MD |
71.7 |
102.7 |
1e-4 |
87.8 |
0.084 |
95.2 |
0.067 |
128.5 |
1e-5 |
| PennCNV | 61.3 | 70.5 | 0.051 | 81.1 | 0.006 | 52.5 | 0.923 | 85.7 | 0.041 |
MD: de novo deletions with coverage of at least ten markers inferred by MinimumDistance[44]; PennCNV: de novo deletions with coverage of at least ten markers inferred by PennCNV[43]. CL: cleft lip; CP: cleft palate; CLP: cleft lip and palate. The p-values were derived using a one-sided Wilcoxon rank-sum test to assess a potential increase in width of de novo deletions in the cleft offspring versus controls.
Discussion
Even though all trios with at least one sample of poor data quality were excluded (see Methods), the probe intensity signal used to identify regions of copy number changes was somewhat noisy, and substantially more variable among control trios than in the oral cleft trios, resulting in an inflated rate of called de novo deletions (i. e. likely false positives) in the control group (Figure 4). This effect was much more prominent in the set of de novo deletions identified by PennCNV, consistent with a previous observation that MinimumDistance might be more robust to false positive identifications (see Figure 2 in [44]). When delineated via PennCNV, the control group had more than a three-fold de novo deletion rate, and less than a two-fold rate when de novo deletions were inferred with MinimumDistance (Table 2). However, our statistical procedure for inferring de novo deletions employed in this study guards against spurious associations, and thus type I error inflation, due to higher rates of false de novo deletions called in the control trios, since we performed a one-sided test with the alternative hypothesis that the de novo deletion rate was larger among the cases than controls. In contrast, a two-sided test would not protect against this type I error inflation due to excessive false positives among the controls (see Additional file 1). We also note a one-sided hypothesis test would not guard against type I error inflation if higher variability in the control group would mask deletions. Thus, all significant findings should be carefully inspected, and validated if possible.
Figure 4.
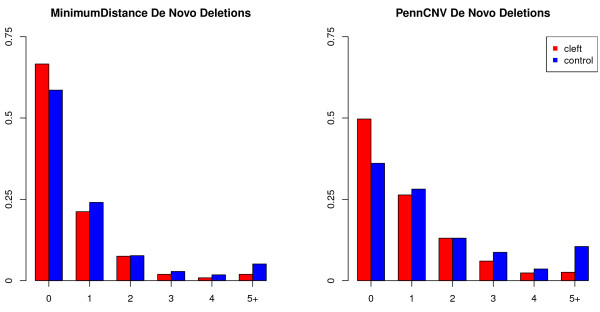
The frequency of inferred de novo deletions per subject, inferred via MinimumDistance (left) and PennCNV (right), shown separately for oral cleft probands (red) and controls (blue). Subjects with five or more called de novo deletions were grouped together.
Table 2.
The total number of de novo deletions inferred (“count”) and the average number of inferred de novo deletions per child (“average”), stratified by case status and cleft type for both discovery methods
| Controls | Oral clefts | CL | CP | CLP | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
MD |
438 |
1.12 |
286 |
0.61 |
89 |
0.64 |
80 |
0.62 |
117 |
0.59 |
|
PennCNV |
1,422 |
3.64 |
518 |
1.11 |
185 |
1.32 |
144 |
1.12 |
189 |
0.95 |
| count | average | count | average | count | average | count | average | count | average |
MD: de novo deletions with coverage of at least ten markers inferred by MinimumDistance; PennCNV: de novo deletions with coverage of at least ten markers inferred by PennCNV. CL: cleft lip; CP: cleft palate; CLP: cleft lip and palate. All differences between cleft and control counts are highly significant, assuming independence of deletions (p<10-6 for all MD comparisons, p<10-40 for all PennCNV comparisons).
As DNA source is correlated with sample quality and affects all CNV call rates, we assessed and found substantial differences in proportions of DNA sources between cases and controls. Around 36% of the control samples were collected either by buccal swab, mouthwash or saliva, while only 17% of the cleft cases were extracted this way (Table 3). Among samples passing quality control (see Methods for details), the rate of inferred de novo deletions was much higher among samples where DNA was extracted from anything other than whole blood (see Additional file 1). We conjecture that the increased rate of called de novo deletions in the control group is likely driven by the differences in the DNA sample collections, with MinimumDistance being more robust to this artifact than PennCNV. Thus, for this particular study, the MinimumDistance based statistics and comparisons should be more reliable. We also note that false identifications tend to involve very short segments of DNA, based on fewer markers from the array. In short, false positive identifications can skew the distribution of CNV lengths, therefore we report the median deletion widths here.
Table 3.
The total number (“count”) and relative frequencies (“proportion”) of DNA source types among all subjects, grouped by proband cleft/control status
| Controls | Oral clefts | |||
|---|---|---|---|---|
| blood |
581 |
0.644 |
1139 |
0.828 |
| buccal |
64 |
0.071 |
125 |
0.091 |
| mouthwash |
50 |
0.055 |
21 |
0.015 |
| saliva |
207 |
0.229 |
90 |
0.065 |
| count | proportion | count | proportion |
DNA source types differ significantly between cases and controls (p<10-35).
Conclusions
We identified a genome-wide significant 62 kb non-coding region on chromosome 7p14.1 where de novo deletions occurred more frequently in oral cleft cases than control probands, adding to the evidence that structural variants are involved in the etiology of oral clefts. This region has been previously identified as a genomic region containing high structural variation, and large deletions in this region have been reported to result in developmental problems including craniofacial abnormalities [47-51]. Only 20 kb upstream from the signal peak lies the gene coding for the T cell receptor gamma alternate reading frame protein (TARP), adding to the existing literature that T cell receptors can play a role in human development. We also observed an overall increase in the width of de novo deletions among oral cleft probands, with CLP exhibiting wider de novo deletions than CL and CP cases. Study protocol and sample DNA source affect estimated frequencies of de novo deletions, and the problem of false positive identifications remains a concern when examining the role of structural variants from genomic array data.
Methods
Samples
Case-parent trios were collected as part of an international collaborative study in the GENEVA Consortium [54]. These trios were ascertained through probands with an isolated, non-syndromic oral cleft (either cleft lip, cleft palate or cleft lip and palate) from 13 different recruitment sites in the United States, Europe, Southeast and East Asia [12]. Control trios were derived from small pedigrees collected from rural Appalachia as part of a genome-wide study of dental caries [55]. The DNA sources for cleft trio samples included whole blood, buccal brush/swab, saliva, mouthwash and dried blood spots, and varied by recruitment site. DNA sources for control trios also included whole blood, buccal brush/swab, saliva, and mouthwash. All samples were hybridized to the Illumina Human610-Quad Beadchip and typed at the Center for Inherited Disease Research (CIDR) at Johns Hopkins University (http://www.cidr.jhmi.edu/). This research project complies with the Helsinki Declaration and all participating institutions provided their own institutional review board (IRB) review and approval, in addition to the review and approval of the Johns Hopkins School of Public Health IRB for the collaborative analysis of genome-wide marker data. Written informed consent was obtained from parents of children ascertained through an oral cleft, as well as their own consent or assent when the proband could appropriately give such. To avoid potential confounding due to ethnic differences (i.e. genetic background), we restricted our analysis to subjects of European ancestry only.
Both MinimumDistance and PennCNV utilize the log R ratios (LRRs) and B allele frequencies (BAFs) from the Illumina Human610-Quad Beadchip probes to infer de novo deletions. The LRR is a standardized estimate of the probe intensity, quantifying the total number of allele copies at each locus of interest. The BAF is a standardized estimate for the proportion of the B allele’s contribution to the total probe intensity, assessing the genotype at the locus of interest. The BAF is standardized so homozygous genotypes in copy neutral states (two allele copies) have BAFs of approximately zero or one (for AA and BB genotypes, respectively), and heterozygous AB genotypes yield BAFs roughly equal to 0.5. Following previously established guidelines for quality control [43,44] devised particularly to avoid excessive false positive identifications due to poor data quality, we excluded trios for which any sample had whole genome amplified DNA or a LRR median absolute deviation (MAD) above 0.3. We also excluded trios with members flagged by CIDR’s internal quality control pipeline. These data cleaning procedures yielded 467 oral cleft trios composed of 1,375 subjects, and 391 trios composed of 902 subjects as controls. Aside from the CNV discovery via PennCNV, all analyses were carried out in the statistical environment R (http://cran.r-project.org/) using the packages DNACopy, GenomicRanges, GWASTools, IRanges, MinimumDistance, all available as free software via the Bioconductor project (http://www.bioconductor.org/) [56].
Algorithms
The PennCNV algorithm for detection of de novo DNA copy number aberrations is based on a hidden Markov model (HMM), jointly modeling the (unknown) copy number states in all three trio members. The state transition probabilities are based on the observed LRRs and BAFs in the samples, and the population BAF. Maximum likelihood methods are employed to identify the most likely copy number states in the father, mother and offspring, and these are encoded as a three-digit numerical code. A normal DNA copy number (two alleles) is designated as a 3, a hemizygous deletion (one allele copy) is indicated as a 2, and a homozygous deletion (zero allele copies) is indicated as a 1. Thus, de novo deletions in offspring with genotypic normal parents are encoded as trio state ‘332’ (loss of one allele copy in the child) or ‘331’ (loss of both alleles). PennCNV addresses genomic waves by incorporating the population GC content at each marker into the HMM.
While the joint PennCNV HMM considers all possible copy number states including inherited deletions (e.g. ‘322’ or ’232’), MinimumDistance was developed specifically for detecting de novo copy number changes since the computational demands of the joint PennCNV HMM are substantial, and false positive identifications of de novo deletions remain a concern even when the recommended quality control procedures (including genomic wave correction) are employed [44]. This approach, freely available as a Bioconductor package (http://www.bioconductor.org/), is based on the “minimum distance” statistic, capturing differences in copy number estimates between the offspring and each parent at each locus, making it robust to genomic waves by design. In particular when the samples of the trio members are hybridized on the same plate (which is the highly recommended and commonly employed approach), MinimumDistance is an effective approach for reducing technical and experimental sources of noise which can generate false positives in experimental data sets. Following genome-wide segmentation of these minimum distances by circular binary segmentation [34,57] (an extremely fast procedure), final inference regarding de novo copy number events is based on a posterior calling step on the inferred candidate regions. MinimumDistance uses the same code for the trio copy number states as PennCNV, where ‘332’ and ‘331’ represent de novo loss of alleles in the child.
Inference
To test for association with oral cleft status, we compared CNV components among cases and controls. Since inferred deletions (required to span at least 10 probes on the array for our analysis) typically only partially overlap between trios, we used the IRanges package to delineate the CNV components into sets of markers where no change in copy number state occurred among any of the cleft or control trios, defining homogeneous sets of CNV states (see Additional file 1). For all CNV components with a total of at least five observed de novo deletions in the cleft and control trios combined, we performed a one-sided Fisher’s exact test, where the alternative hypothesis was a higher de novo deletion frequency in the cleft probands. To correct for multiple comparisons while simultaneously taking correlations between component statistics into account, we performed a permutation test by shuffling case and control status across all probands. This procedure, based on over 100,000 permutations, established the genome-wide significance level for a 5% family-wise error rate at the nominal values of 2.60 and 2.83 for the – log10 p-values for MinimumDistance and PennCNV, respectively. We also performed simulations to compare the widths of de novo deletions in cleft and control trios. More specifically, we simulated 10,000 quantile-quantile plots under the assumption that the cleft and control samples came from the same distribution (see Additional file 1), and used a one sided two-sample Kolmogorov-Smirnov test to assess a potential increase in width of de novo deletions in the cleft offspring. Since non-parametric mean comparisons might be less sensitive to subtle batch effects on deletion width, we also carried out a one-sided Wilcoxon rank-sum test on the observed deletion widths in the case and control trios.
Competing interests
The authors declare no competing interests.
Authors’ contributions
SGY, RBS, HS and MMP wrote all code, performed all analyses, and generated all tables and figures. THB and IR conceived of the study. All authors participated in its design, coordination, and drafting of the manuscript. All authors read and approved the final version of the manuscript.
Supplementary Material
Supplementary material.
Contributor Information
Samuel G Younkin, Email: syounkin@jhsph.edu.
Robert B Scharpf, Email: rscharpf@jhsph.edu.
Holger Schwender, Email: holger.schwender@udo.edu.
Margaret M Parker, Email: mmparker1@gmail.com.
Alan F Scott, Email: afscott@jhmi.edu.
Mary L Marazita, Email: marazita@pitt.edu.
Terri H Beaty, Email: tbeaty@jhsph.edu.
Ingo Ruczinski, Email: iruczins@jhsph.edu.
Acknowledgements
We thank the families who participated in the studies and gratefully acknowledge the invaluable assistance of clinical, field, and laboratory staff who contributed to this study, in particular the Center for Oral Health Research in Appalachia. We also gratefully acknowledge the financial support provided by the National Institute of Health grants R03 DE021437 (SGY, IR), R01 DE016148 (MLM), and Deutsche Forschungsgemeinschaft grant SCHW 1508/3-1 (HS). The consortium for GWAS genotyping and analysis was supported by the National Institute for Dental and Craniofacial Research through U01 DE018993 and U01 DE018903 (THB, MLM). The International Cleft Consortium involved many recruitment sites directed by separate investigators: Jeffrey C. Murray (University of Iowa), Rolf Terje Lie (University of Bergen), Allen Wilcox (NIEHS), Kare Christensen (University of Southern Denmark), Yah-Huei Wu-Chou (Chang Gang Memorial Hospital), Vincent Yeow (KK Women’s & Children’s Hosptial), Xiaoqian Ye (Wuhan University), Bing Shi (Sichaun University), Samuel Chong (National University of Singapore). Part of the original recruitment of Norwegian case–parent trios was supported by the Intramural Research Program of the National Institute of Health, National Institute of Environmental Health Sciences.
References
- Mossey P, Little J. Cleft lip palate: from origin to treatment, Epidemiology of oral clefts: an international perspective, 1st edition. New York: Oxford University Press; 2002. [Google Scholar]
- Mossey PA, Little J, Munger RG, Dixon MJ, Shaw WC. Cleft lip and palate. Lancet. 2009;374(9703):1773–1785. doi: 10.1016/S0140-6736(09)60695-4. [DOI] [PubMed] [Google Scholar]
- Harville EW, Wilcox AJ, Lie RT, Vindenes H, Abyholm F. Cleft lip and palate versus cleft lip only: are they distinct defects? Am J Epidemiol. 2005;162(5):448–453. doi: 10.1093/aje/kwi214. [DOI] [PubMed] [Google Scholar]
- Forrester MB, Merz RD. Comparison of cleft lip only and cleft lip and palate, Hawai’i, 1986-2003. Hawaii Med J. 2007;66(11):300–302. [PubMed] [Google Scholar]
- Mossey P. Epidemiology underpinning research in the aetiology of orofacial clefts. Orthod Craniofac Res. 2007;10(3):114–120. doi: 10.1111/j.1601-6343.2007.00398.x. [DOI] [PubMed] [Google Scholar]
- Sivertsen A, Wilcox AJ, Skjaerven R, Vindenes HA, Abyholm F, Harville E, Lie RT. Familial risk of oral clefts by morphological type and severity: population based cohort study of first degree relatives. BMJ. 2008;336(7641):432–434. doi: 10.1136/bmj.39458.563611.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell LE. Cleft lip palate: from origin to treatment, Twin, Studies in Oral Cleft Research. USA: Oxford University Press; 2002. [Google Scholar]
- Stanier P, Moore GE. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum Mol Genet. 2004;13 Spec No 1:R73–R81. doi: 10.1093/hmg/ddh052. [DOI] [PubMed] [Google Scholar]
- Farrall M, Holder S. Familial recurrence-pattern analysis of cleft lip with or without cleft palate. Am J Hum Genet. 1992;50(2):270–277. [PMC free article] [PubMed] [Google Scholar]
- Schliekelman P, Slatkin M. Multiplex relative risk and estimation of the number of loci underlying an inherited disease. Am J Hum Genet. 2002;71(6):1369–1385. doi: 10.1086/344779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugessur A, Shi M, Gjessing HK, Lie RT, Wilcox AJ, Weinberg CR, Christensen K, Boyles AL, Daack-Hirsch S, Trung TN, Bille C, Lidral AC, Murray JC. Genetic determinants of facial clefting: analysis of 357 candidate genes using two national cleft studies from Scandinavia. PLoS One. 2009;4(4):e5385. doi: 10.1371/journal.pone.0005385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty TH, Murray JC, Marazita ML, Munger RG, Ruczinski I, Hetmanski JB, Liang KY, Wu T, Murray T, Fallin MD, Redett RA, Raymond G, Schwender H, Jin SC, Cooper ME, Dunnwald M, Mansilla MA, Leslie E, Bullard S, Lidral AC, Moreno LM, Menezes R, Vieira AR, Petrin A, Wilcox AJ, Lie RT, Jabs EW, Wu-Chou YH, Chen PK, Wang H. et al. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet. 2010;42(6):525–529. doi: 10.1038/ng.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011;12(3):167–178. doi: 10.1038/nrg2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig KU, Mangold E, Herms S, Nowak S, Reutter H, Paul A, Becker J, Herberz R, AlChawa T, Nasser E, Boehmer AC, Mattheisen M, Alblas MA, Barth S, Kluck N, Lauster C, Braumann B, Reich RH, Hemprich A, Poetzsch S, Blaumeiser B, Daratsianos N, Kreusch T, Murray JC, Marazita ML, Ruczinski I, Scott AF, Beaty TH, Kramer FJ, Wienker TF. et al. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat Genet. 2012;44(9):968–971. doi: 10.1038/ng.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocian M, Walker AP. Lip pits and deletion 1q32–41. Am J Med Genet. 1987;26(2):437–443. doi: 10.1002/ajmg.1320260223. [DOI] [PubMed] [Google Scholar]
- Sander A, Schmelzle R, Murray J. Evidence for a microdeletion in 1q32-41 involving the gene responsible for Van der Woude syndrome. Hum Mol Genet. 1994;3(4):575–578. doi: 10.1093/hmg/3.4.575. [DOI] [PubMed] [Google Scholar]
- Sander A, Murray JC, Scherpbier-Heddema T, Buetow KH, Weissenbach J, Zingg M, Ludwig K, Schmelzle R. Microsatellite-based fine mapping of the Van der Woude syndrome locus to an interval of 4.1 cM between D1S245 and D1S414. Am J Hum Genet. 1995;56:310–318. [PMC free article] [PubMed] [Google Scholar]
- Brewer C, Holloway S, Zawalnyski P, Schinzel A, FitzPatrick D. A chromosomal deletion map of human malformations. Am J Hum Genet. 1998;63(4):1153–1159. doi: 10.1086/302041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer C, Holloway S, Zawalnyski P, Schinzel A, FitzPatrick D. A chromosomal duplication map of malformations: regions of suspected haplo- and triplolethality–and tolerance of segmental aneuploidy–in humans. Am J Hum Genet. 1999;64(6):1702–1708. doi: 10.1086/302410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte BC, Murray JC. The many faces and factors of orofacial clefts. Hum Mol Genet. 1999;8(10):1853–1859. doi: 10.1093/hmg/8.10.1853. [DOI] [PubMed] [Google Scholar]
- FitzPatrick DR, Carr IM, McLaren L, Leek JP, Wightman P, Williamson K, Gautier P, McGill N, Hayward C, Firth H, Markham AF, Fantes JA, Bonthron DT. Identification of SATB2 as the cleft palate gene on 2q32-q33. Hum Mol Genet. 2003;12(19):2491–2501. doi: 10.1093/hmg/ddg248. [DOI] [PubMed] [Google Scholar]
- Alkuraya FS, Saadi I, Lund JJ, Turbe-Doan A, Morton CC, Maas RL. SUMO1 haploinsufficiency leads to cleft lip and palate. Science. 2006;313(5794):1751. doi: 10.1126/science.1128406. [DOI] [PubMed] [Google Scholar]
- Benko S, Fantes JA, Amiel J, Kleinjan DJ, Thomas S, Ramsay J, Jamshidi N, Essafi A, Heaney S, Gordon CT, McBride D, Golzio C, Fisher M, Perry P, Abadie V, Ayuso C, Holder-Espinasse M, Kilpatrick N, Lees MM, Picard A, Temple IK, Thomas P, Vazquez MP, Vekemans M, Crollius HR, Hastie ND, Munnich A, Etchevers HC, Pelet A, Farlie PG, Fitzpatrick DR, Lyonnet S. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet. 2009;41(3):359–364. doi: 10.1038/ng.329. [DOI] [PubMed] [Google Scholar]
- Milunsky JM, Maher TA, Zhao G, Roberts AE, Stalker HJ, Zori RT, Burch MN, Clemens M, Mulliken JB, Smith R, Lin AE. TFAP2A mutations result in branchio-oculo-facial syndrome. Am J Hum Genet. 2008;82(5):1171–1177. doi: 10.1016/j.ajhg.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osoegawa K, Vessere GM, Utami KH, Mansilla MA, Johnson MK, Riley BM, L’Heureux J, Pfundt R, Staaf J, van der Vliet WA, Lidral AC, Schoenmakers EFPM, Borg A, Schutte BC, Lammer EJ, Murray JC, de Jong PJ. Identification of novel candidate genes associated with cleft lip and palate using array comparative genomic hybridisation. J Med Genet. 2008;45(2):81–86. doi: 10.1136/jmg.2007.052191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivertsen A, Lie RT, Wilcox AJ, Abyholm F, Vindenes H, Haukanes BI, Houge G. Prevalence of duplications and deletions of the 22q11 DiGeorge syndrome region in a population-based sample of infants with cleft palate. Am J Med Genet A. 2007;143(2):129–134. doi: 10.1002/ajmg.a.31445. [DOI] [PubMed] [Google Scholar]
- Shi M, Mostowska A, Jugessur A, Johnson MK, Mansilla MA, Christensen K, Lie RT, Wilcox AJ, Murray JC. Identification of microdeletions in candidate genes for cleft lip and/or palate. Birth Defects Res A Clin Mol Teratol. 2009;85:42–51. doi: 10.1002/bdra.20571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS. et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320(5875):539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CEJ, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, Summers A, Gibbons CA, Teebi A, Chitayat D, Weksberg R, Thompson A, Vardy C, Crosbie V, Luscombe S, Baatjes R. et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82(2):477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor A, Gianakopoulos PJ, Fernandez B, Marshall CR, Szatmari P, Roberts W, Scherer SW, Vincent JB. Copy number variation analysis and sequencing of the X-linked mental retardation gene TSPAN7/TM4SF2 in patients with autism spectrum disorder. Psychiatr Genet. 2009;19(3):154–155. doi: 10.1097/YPG.0b013e32832a4fe5. [DOI] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BHY, Cochrane L, Corsello C. et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, Robson S, Vukcevic D, Barnes C, Conrad DF, Giannoulatou E, Holmes C, Marchini JL, Stirrups K, Tobin MD, Wain LV, Yau C, Aerts J, Ahmad T, Andrews TD, Arbury H, Attwood A, Auton A, Ball SG, Balmforth AJ, Barrett JC, Barroso I, Barton A, Bennett AJ, Bhaskar S. Wellcome Trust Case Control Consortium et al. Genome-wide association study of, CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464(7289):713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, Bassett AS, Seller A, Holmes CC, Ragoussis J. QuantiSNP: an objective bayes Hidden-Markov model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35(6):2013–2025. doi: 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23(6):657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SFA, Hakonarson H, Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17(11):1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharpf RB, Parmigiani G, Pevsner J, Ruczinski I. Hidden Markov models for the assessment of chromosomal alterations using high-throughput SNP arrays. Ann Appl Stat. 2008;2(2):687–713. doi: 10.1214/07-AOAS155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pique-Regi R, Monso-Varona J, Ortega A, Seeger RC, Triche TJ, Asgharzadeh S. Sparse representation and Bayesian detection of genome copy number alterations from microarray data. Bioinformatics. 2008;24(3):309–318. doi: 10.1093/bioinformatics/btm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau C, Papaspiliopoulos O, Roberts GO, Holmes C. Bayesian nonparametric Hidden Markov models with application to the analysis of copy-number-variation in mammalian genomes. J R Stat Soc Series B Stat Methodol. 2011;73:37–57. doi: 10.1111/j.1467-9868.2010.00756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pique-Regi R, Ortega A, Asgharzadeh S. Joint estimation of copy number variation and reference intensities on multiple DNA arrays using GADA. Bioinformatics. 2009;25(10):1223–1230. doi: 10.1093/bioinformatics/btp119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zöllner S. CopyMap: localization and calling of copy number variation by joint analysis of hybridization data from multiple individuals. Bioinformatics. 2010;26(21):2776–2777. doi: 10.1093/bioinformatics/btq515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang NR, Siegmund DO, Ji H, Li JZ. Detecting simultaneous changepoints in multiple sequences. Biometrika. 2010;97(3):631–645. doi: 10.1093/biomet/asq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Lebarbier E, Hoebeke M, Rigaill G, Thiam B, Robin S. Joint segmentation, calling, and normalization of multiple CGH profiles. Biostatistics. 2011;12(3):413–428. doi: 10.1093/biostatistics/kxq076. [DOI] [PubMed] [Google Scholar]
- Wang K, Chen Z, Tadesse MG, Glessner J, Grant SFA, Hakonarson H, Bucan M, Li M. Modeling genetic inheritance of copy number variations. Nucleic Acids Res. 2008;36(21):e138. doi: 10.1093/nar/gkn641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharpf RB, Beaty TH, Schwender H, Younkin SG, Scott AF, Ruczinski I. Fast detection of de novo copy number variants from SNP arrays for case-parent trios. BMC Bioinformatics. 2012;13:330. doi: 10.1186/1471-2105-13-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin SJ, Li M, Hou C, Yang S, Glessner J, Hakonarson H, Bucan M, Maris JM, Wang K. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008;36(19):e126. doi: 10.1093/nar/gkn556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K, Irizarry RA. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11(10):733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K, Kroisel PM, Rosenkranz W. Molecular and cytogenetic analysis in two patients with microdeletions of 7p and Greig syndrome: hemizygosity for PGAM2 and TCRG genes. Genomics. 1990;8(3):487–491. doi: 10.1016/0888-7543(90)90035-S. [DOI] [PubMed] [Google Scholar]
- Chotai KA, Brueton LA, van Herwerden L, Garrett C, Hinkel GK, Schinzel A, Mueller RF, Speleman F, Winter RM. Six cases of 7p deletion: clinical, cytogenetic, and molecular studies. Am J Med Genet. 1994;51(3):270–276. doi: 10.1002/ajmg.1320510320. [DOI] [PubMed] [Google Scholar]
- Schwarzbraun T, Windpassinger C, Ofner L, Vincent JB, Cheung J, Scherer SW, Wagner K, Kroisel PM, Petek E. Genomic analysis of five chromosome 7p deletion patients with Greig cephalopolysyndactyly syndrome (GCPS) Eur J Med Genet. 2006;49(4):338–345. doi: 10.1016/j.ejmg.2005.10.133. [DOI] [PubMed] [Google Scholar]
- Bilguvar K, Bydon M, Bayrakli F, Ercan-Sencicek AG, Bayri Y, Mason C, DiLuna ML, Seashore M, Bronen R, Lifton RP, State M, Gunel M. A novel syndrome of cerebral cavernous malformation and Greig cephalopolysyndactyly. Laboratory investigation. J Neurosurg. 2007;107(6 Suppl):495–499. doi: 10.3171/PED-07/12/495. [DOI] [PubMed] [Google Scholar]
- Shih B, Tassabehji M, Watson JS, Bayat A. DNA copy number variations at chromosome 7p14.1 and chromosome 14q11.2 are associated with dupuytren’s disease: potential role for MMP and Wnt signaling pathway. Plast Reconstr Surg. 2012;129(4):921–932. doi: 10.1097/PRS.0b013e3182442343. [DOI] [PubMed] [Google Scholar]
- Lettice LA, Heaney SJH, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. 2003;12(14):1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett JC, Dallaire S, Gabriel SB, Lee C, Daly MJ, Altshuler DM, Consortium IH. Common deletion polymorphisms in the human genome. Nat Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- Cornelis MC, Agrawal A, Cole JW, Hansel NN, Barnes KC, Beaty TH, Bennett SN, Bierut LJ, Boerwinkle E, Doheny KF, Feenstra B, Feingold E, Fornage M, Haiman CA, Harris EL, Hayes MG, Heit JA, Hu FB, Kang JH, Laurie CC, Ling H, Manolio TA, Marazita ML, Mathias RA, Mirel DB, Paschall J, Pasquale LR, Pugh EW, Rice JP, Udren J. et al. The Gene, Environment Association Studies consortium (GENEVA): maximizing the knowledge obtained from GWAS by collaboration across studies of multiple conditions. Genet Epidemiol. 2010;34(4):364–372. doi: 10.1002/gepi.20492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polk DE, Weyant RJ, Crout RJ, McNeil DW, Tarter RE, Thomas JG, Marazita ML. Study protocol of the Center for Oral Health Research in Appalachia (COHRA) etiology study. BMC Oral Health. 2008;8:18. doi: 10.1186/1472-6831-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5(4):557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.