

Abstract
It has been shown that iron progressively accumulates in the brain with age. Calorie restriction (CR) may allay many of the adverse effects of aging on the brain, yet the underlying mechanisms, in particular in relation to brain iron metabolism, remain unclear. This study aimed to investigate the role of CR in the regulation of cerebral cellular iron homeostasis. C57BL/6 mice were randomly divided into four groups of eight. The control group was fed a conventional diet ad libitum; the CR group received 70% of the calories of the control mouse intake per day; the d-galactose (d-gal) group received subcutaneous injection of d-gal at a dose of 100 mg/kg once daily to produce mouse model of aging; the d-gal plus CR group received both of the two interventions for 14 weeks. The Morris water maze (MWM) was employed to test the cognitive performance of all animals, and the expression of iron regulatory genes, ferroportin and hepcidin, in the cortex and hippocampus were detected by quantitative real-time PCR. Compared to the controls, the d-gal group mice showed significant spatial reference memory deficits in the MWM test, whereas the d-gal-CR group mice exhibited almost normal cognitive function, indicating that CR protects against d-gal–induced learning and memory impairment. Hepcidin mRNA expression was increased in the d-gal group, decreased in the CR group, and was basically unchanged in the d-gal-CR group. There was no statistical difference in the transmembrane iron exporter ferroportin expression between control and any of the experimental groups. The results suggest that the anti-aging effects of CR might partially lie in its capacity to reduce or avoid age-related iron accumulation in the brain through down-regulating expression of brain hepcidin—the key negative regulator for intracellular iron efflux—and that facilitating the balance of brain iron metabolism may be a promising anti-aging measure.
Introduction
With a worldwide demographic shift toward aging populations, brain aging, and age-related neurodegenerative diseases have become more and more important social and medical problems. Investigating the mechanisms of aging and searching for effective measures of anti-aging will be beneficial to the healthy aging and longevity of humans. Naturally old animals are usually used in aging studies; however, old animals have high a mortality rate and high incidence of age-related diseases, such as tumor, hypertension, diabetes, and other diseases. Therefore, an artificial aging model has been increasingly used for studying aging mechanisms and anti-aging agents. d-galactose (d-gal) is a normal reducing sugar in the body and is metabolized by d-galactokinase and galactose-1-phosphate uridyltransferase. Over-supply of d-gal results in abnormal metabolism due to conversion of the excess galactose to galactitol, which is not metabolized by the enzymes listed above but accumulates in the cell, leading to osmotic stress and production of reactive oxygen species (ROS).1
The d-gal–induced brain aging model was first reported 18 years ago.2 Chronic systemic exposure to d-gal causes an acceleration of senescence in different animal species and produces symptoms similar to those of natural aging, especially the decline in cognitive functions.3–8 Various hypotheses have been put forward to explain the underlying mechanisms for d-gal–induced brain aging, including glycometabolism block, formation of advanced glycation end products (AGE), and free radical damage with the evidence of excessive production of ROS and significant reduction of anti-oxidant activities in brain tissue.9,10 In addition to causing metabolism disorders and oxidative stress, growing evidence revealed that here were neurodegenerative changes in the brain of rodents treated with d-gal.4,8,11 Given all of this, d-gal–induced senescence is an ideal model for studying the basic mechanisms involved in brain aging and age-associated neurodegeneration.
Iron is an essential co-factor for many proteins involved in the normal function of neuronal tissue, such as the non-heme iron enzyme tyrosine hydroxylase, which is required for dopamine synthesis. Lack of iron may lead to cognitive impairment in children, adults, and the elderly. The problem is that iron has a dual nature in the central nervous system (CNS). Excessive iron accumulation in the CNS is believed to be responsible for neuronal injury and cell death and subsequent neurodegenerative disorders.12,13 The importance of adequate amounts of iron for brain health is well established. Mounting evidence suggests that iron progressively accumulates in the brain with age,13–15 and it has become evident that brain iron accumulation is one of the initial events in many age-related neurodegenerative diseases and perhaps aging itself.16–18 However, little is known about the mechanism of how an excess of iron accumulation occurs in the brain. One hypothesis holds that dysfunction of the blood–brain barrier, which limits iron entry to the brain via highly regulated transport systems, might play a role in brain iron overload.19–21 Another hypothesis with strong experimental support proposes that brain iron accumulation is a consequence of the dysregulation of proteins that govern cellular iron homeostasis. Mammalian cellular iron efflux is mediated by the sole iron exporter ferroportin 1 (Fpn1), a transmembrane protein.22 The abundance of Fpn1 on the plasma membrane can be modulated by interaction with its ligand hepcidin, an iron regulatory hormone produced in the liver in response to inflammatory stimuli, iron, and hypoxia.23 Hepcidin binds to Fpn1 and induces its internalization and degradation, resulting in decreased cellular iron export and subsequent iron acuumulation.24,25 Recently, it has been demonstrated that hepcidin is widely expressed in the brain,26,27 indicating hepcidin might have a key role in brain iron homeostasis.
Calorie restriction (CR) is the only non-genetic intervention that has been consistently shown to slow the intrinsic rate of aging and increase both median and maximum life span in a variety of species.28,29 CR may increase the resistance of the nervous system to neurodegenerative disorders and allay many of the adverse effects of aging on the brain.30–32 In 1998, Cook and Yu reported that age-related iron accumulation in liver, brain, and kidney was markedly attenuated by CR in rodent,34 and a more recent study found that CR decreases brain iron accumulation in old Rhesus monkeys as measured in vivo using magnetic resonance imaging.35 However, the specific mechanisms underlying CR's protection of the brain from age-associated iron dyshomeostasis remain to be determined.
We hypothesized that CR might play its role by facilitating removal of excess iron from brain cells via preserving proper expression levels of two critical proteins—the brain-derived hepcidin and its receptor, iron exporter Fpn1. In this study, d-gal–induced brain aging mouse models were established with or without CR intervention. The total iron and the mRNA levels of hepcidin and Fpn1 in the cortex and hippocampus were determined to get a preliminary understanding of CR in improving brain iron homeostasis. Our data demonstrated that CR can down-regulate brain hepcidin mRNA expression in normal mice and reverse the elevated hepcidin mRNA levels in brain of aging model mice.
Materials and Methods
Animals and treatment
Six-week-old female C57BL/6 mice (15±1 gram) were obtained from the Experimental Animal Center of Capital Medical University and were housed individually in a specific pathogen-free (SPF) facility on a 12-hr light/dark cycle. After 1 week of acclimation, the mice were randomized into four groups (eight in each group), i.e., d-gal group, CR group, d-gal+CR (d-gal-CR) group, and normal control group, for a 14-week intervention, respectively. d-gal and control groups were fed a standard diet ad libitum. CR and d-gal-CR groups received a gradually reduced (over 2 weeks) daily food portion until their food intake was reduced to 70% of that consumed by ad libitum–fed mice, and they were maintained on this aliquot until the end of the experiment. The diet for CR was fortified with minerals and vitamins, as shown in Table 1, to ensure an equivalent intake of iron and vitamins by CR mice as by ad libitum mice. d-gal (Sigma-Aldrich, MO) was dissolved in sterile saline (0.9% NaCl) and administered by subcutaneous injection at a dose of 100 mg/kg per day for 10 weeks prior to laboratory test, whereas a daily subcutaneous saline injection was given to the non-involved mice. All mice were fed daily, had free access to water, and were weighed weekly.
Table 1.
Composition of Experimental Diets
| Standard diet (grams/kg)a | Restricted diet (grams/kg)b | |
|---|---|---|
| Ingredientc | ||
| Cornstarch | 315 | 315 |
| Sucrose | 314.5 | 314.5 |
| Casein | 200 | 200 |
| Soybean oil | 70 | 70 |
| Cellulose | 50 | 30.7 |
| Mineral mix | 35 | 50 |
| Vitamin mix | 10 | 14.3 |
| l-Cystine | 3 | 3 |
| Choline bitartrate | 2.5 | 2.5 |
The standard diet was based on the AIN-93G recommendation.33
Restricted diet was mineral- and vitamin-fortified to ensure an equivalent intake of iron and vitamins when feeding amount reduced to 70% of standard diet.
All dietary components were prepared by Research Diets.
The procedures for both feeding and experiments complied with the national guidelines for the care and use of animals for scientific purposes in China, and approved by the ethics committee of the Capital Medical University.
Morris water maze test
The spatial learning and memory ability was evaluated by using Morris water maze (MWM), which consisted of a circular pool (120 cm in diameter, 60 cm in height filled to a depth of 30 cm with water at 24±2°C) and a video monitoring system (Chinese Academy of Medical Sciences). The maze was placed in illuminated light room, surrounded by several visual clues external to the maze (e.g., the experimenter, ceiling fan, lights, racks, etc.), which were visible from within the pool and could be used by the mice for spatial location. These external clues remained unchanged throughout the experimental period. The pool was divided into four imaginary quadrants with equal areas, and the water was rendered opaque by the addition of milk powder.
Acquisition trial
In one of the quadrants, the target quadrant, a circular platform (6 cm diameter) was submerged in a fixed position (0.5 cm below water surface and 28 cm from the wall). In each trial, an individual mouse was released gently into the water facing the wall with the drop location in one of three quadrants except that containing the hidden platform. The mouse swam to find the hidden platform within 120 sec. After finding the submerged platform, the mouse was allowed to rest on the platform for 15 sec. If the animal was unable to escape to the platform within 120 sec, the trial was terminated and the animal was guided to reach the platform and stay on it for 15 sec. The task was conducted two sessions a day for 4 consecutive days; each session was comprised of two trials with 5-min inter-trial intervals during which the animal was kept in a dry cage. The latency of escape onto the platform, considered a measure of spatial learning and reference memory, was recorded by the video monitoring system in each trial, and the mean daily escape latency was calculated thereafter.
Probe trial
The retrieval of spatial memory was tested by probe trial. On the fifth day of the MWM test, the platform was removed from the pool. Each mouse was placed into the water as in the acquisition trial for a 90-sec test. The following parameters were recorded for each animal: Platform crossing, i.e., the number of crossings through the place where the platform had been located, and target quadrant preference, i.e., time spent in the target quadrant.
Inductively coupled plasma mass spectroscopy measurement
The total iron in brain tissues was determined using inductively coupled plasma mass spectroscopy (ICP-MS; Thermo Fisher, FL). Mice were sacrificed after the MWM test, at 22 weeks of age, by cervical translocation. The hippocampal and cortical tissues were dissected from an isolated brain on ice, and the left and right hemispheric tissues were harvested and weighed separately, frozen immediately in liquid nitrogen, and stored at −80°C until use. Before the experiments, all of the containers were soaked with 15% nitric acid for 24 hr, washed in deionized water, followed by rinsing with ultrapure water, and dried.
A known weight of each sample (one hemispheric cortex and hippocampus, approximately 100 mg total) was soaked in a 3:1 ratio of ultra-pure nitric acid (70%) and hydrogen peroxide (35%) for 30 min in a Teflon beaker, then digested in microwave digestion system (EZ digester, Milestone Scientific, USA) according to the procedure described elsewhere.36 The completely digested samples were cooled to room temperature, diluted to 10 mL with ultra-pure water, and analyzed by ICP-MS for total iron content. All samples were read in triplicate. The calibration curve was obtained using four iron standard solutions (Sigma-Aldrich) in the range 0.2–0.05 μg/mL. A sample containing only the digested reagents was used for blank subtraction purposes.
Real-time PCR assay
The other half of the cortical/hippocampal combined sample was used for analysis of gene expression. Total RNA was extracted using TRIzol reagent (Invitrogen, CA). Synthesis of cDNA was performed using 1 μg of total mRNA using a First Strand cDNA Synthesis kit (CWBio, Beijing, China) according to the manufacturer's instructions. Real-time PCR quantification was conducted in a LightCycler® 480 system (Roche, Penzberg, Germany) using Ultra SYBR Mixture (CWBio). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. Primer sequences of hepcidin, ferroportin, and GAPDH were as follows: Hepcidin, forward, 5′-CCTATCTCCATCAACAGATG-3′, reverse, 5′-AACAGATACCACACTGGGAA-3′; Fpn1, forward, 5′- GATGGGAGCATGAGCAAT-3′, reverse, 5′- GGCTTCCAGGCATGAATAC-3′; GAPDH, forward, 5′-CTGCCCAGAACATCATCCCT-3′, reverse, 5′-GGTCCTCAGTGTAGCCCAAG-3′. Reactions were performed in triplicate according to the following PCR cycling profile: 95°C for 10 min,40 cycles of 95°C for 15 sec, and 60°C for 60 sec. Relative quantification of mRNA was determined by the comparative Ct method.37 The mRNA level of tested gene was expressed as the amount relative to that of GAPDH, and was calculated as 2−ΔCt, whereas the experimental/control mRNA ratio was calculated as 2−ΔΔCt.
Statistical analysis
Data were expressed as mean±standard error of the mean (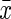 ±SEM). Differences between groups were assessed using repeated-measures analysis of variance (ANOVA) followed by a least significant difference (LSD) post hoc test. For non-normally distributed data, differences were assessed using the Mann–Whitney U test. p<0.05 was considered statistically significant.
±SEM). Differences between groups were assessed using repeated-measures analysis of variance (ANOVA) followed by a least significant difference (LSD) post hoc test. For non-normally distributed data, differences were assessed using the Mann–Whitney U test. p<0.05 was considered statistically significant.
Results
General conditions of the animals
No mice showed obvious health problems (hypo-motility, illness, death) throughout the whole experimental procedure. The results for body weight are shown in Fig. 1. All ad libitum mice gained weight over time, whereas those on the CR regimen gained little weight throughout the feeding period. By the end of the experiment, the average weight gain of CR mice was markedly less than that of the control group (1.25 grams vs. 11.53 grams, p<0.01). Similar results were observed for the difference between d-gal-CR group and d-gal group (−1.43 grams vs. 9.12 grams, p<0.01). In other words, CR-treated (CR and d-gal-CR) mice weighed significantly less than other groups (p<0.01), differing by nearly 11 grams from the control mice, and ≈9 grams from d-gal mice at the end of the study. No statistically significant differences in the weight gain were observed between d-gal and control groups, or between d-gal-CR and CR groups (p>0.05, respectively).
FIG. 1.

Body weight (BW) changes in differently treated mice (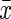 ±standard error of the mean, n=8). Initial weights of the mice did not differ between groups. At the end of the experiment, the mean weight for calorie restriction (CR)-treated but not d-gal–treated mice showed significant differences as compared with normal control mice. d-gal, d-galactose. (*) p<0.01 vs. control group, (#) p<0.01 vs. the d-gal group.
±standard error of the mean, n=8). Initial weights of the mice did not differ between groups. At the end of the experiment, the mean weight for calorie restriction (CR)-treated but not d-gal–treated mice showed significant differences as compared with normal control mice. d-gal, d-galactose. (*) p<0.01 vs. control group, (#) p<0.01 vs. the d-gal group.
Spatial reference memory performance
As seen in Table 2, the d-gal group mice showed a longer escape latency to the platform compared with normal controls (p<0.05), indicating impaired spatial learning and memory. The d-gal-CR group mice had a shorter latency than d-gal mice (p<0.05). No significant difference in escape latency was observed between d-gal-CR and control animals, or between the CR and control groups. In the probe trial, the d-gal mice not only made fewer platform crossings but also exhibited a decreased spatial preference for the target quadrant as compared with the control or the d-gal-CR group (p<0.05, respectively), whereas the CR, the control, and the d-gal-CR group mice showed similar numbers of platform crossings and time spent in the target quadrant (p>0.05, respectively).
Table 2.
Results of the Morris Water Maze Test (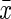 ±SEM, n=8)
±SEM, n=8)
| Escape latency (sec) | Platform crossings (times) | Quadrant preference (time ratio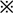 ) ) |
|
|---|---|---|---|
| d-gal | 59.33±25.54* | 1.87±0.42* | 0.25±0.02* |
| d-gal+CR | 29.58±10.92# | 3.94±0.26 # | 0.39±0.05# |
| CR | 30.00±13.79 | 3.90±0.37 | 0.37±0.04 |
| Control | 32.58±8.99 | 4.38±0.25 | 0.41±0.04 |
Note: *p<0.05 vs. control group; #p<0.05 vs. d-gal group; time spent in the target quadrant/total time.
SEM, standard error of the mean; d-gal, d-galactose; CR, calorie restriction.
Brain iron content
The amount of total iron in brain tissue samples assayed by ICP-MS was expressed as micrograms of iron per gram of wet tissue. As Fig. 2 shows, there were no significant differences in brain iron concentration between the three experimental groups and control group (p>0.05). That is, no iron accumulation was detected in the cortical/hippocampal tissues in d-gal group mice, and meanwhile, reduced level of total iron in the brain of CR mice was not found either.
FIG. 2.

Effect of calorie restriction (CR) and d-gal on total iron content in the brain (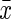 ±standard error of the mean, n=8). Total iron was measured by inductively coupled plasma mass spectroscopy (ICP-MS). Brain tissues prepared from differently treated mice display no difference in total iron content. d-gal, d-galactose.
±standard error of the mean, n=8). Total iron was measured by inductively coupled plasma mass spectroscopy (ICP-MS). Brain tissues prepared from differently treated mice display no difference in total iron content. d-gal, d-galactose.
Expression of hepcidin and Fpn1 in brain
The hepcidin gene expression was up-regulated in the d-gal group and down-regulated in the CR group, as compared with the control group (p<0.05, respectively). The d-gal-CR group had significantly less expression of hepcidin than the d-gal group and was comparable to the level of control group. (Fig. 3). There was no statistically significant divergence in the expression of Fpn1 between experimental and control groups (Fig. 4).
FIG. 3.

Influence of calorie restriction (CR) and d-gal on hepcidin expression in mouse brain (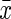 ±standard error of the mean, n=8). The relative expression level of hepcidin was assessed by quantitative real-time PCR. Hepcidin expression in the control mice was defined as 1. The results showed that d-gal enhanced, whereas CR reduced, hepcidin mRNA expression in the brain. Moreover, the d-gal–induced increase in hepcidin expression was significantly attenuated by CR treatment. d-gal, d-galactose. (*) p<0.05 vs. control group; (#) p<0.05 vs. the d-gal group.
±standard error of the mean, n=8). The relative expression level of hepcidin was assessed by quantitative real-time PCR. Hepcidin expression in the control mice was defined as 1. The results showed that d-gal enhanced, whereas CR reduced, hepcidin mRNA expression in the brain. Moreover, the d-gal–induced increase in hepcidin expression was significantly attenuated by CR treatment. d-gal, d-galactose. (*) p<0.05 vs. control group; (#) p<0.05 vs. the d-gal group.
FIG. 4.

Influence of calorie restriction (CR) and d-Gal on Fpn1 expression in mouse brain (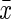 ±standard error of the mean, n=8). Fpn1 expression in the control mice was defined as 1. No statistically significant difference was found between groups. d-gal; d-galactose.
±standard error of the mean, n=8). Fpn1 expression in the control mice was defined as 1. No statistically significant difference was found between groups. d-gal; d-galactose.
Discussion
Aging negatively influences cognitive function. Memory decline is a characteristic of aging and age-related neurodegenerative disorders. The results of the present study showed that the d-gal group mice had significant spatial reference memory deficits in the MWM test, indicating that the d-gal induced brain aging mouse model was established successfully.
The brain is particularly susceptible to oxidative damage. As people age, there is a significant and progressive increase in the level of oxidatively damaged DNA and lipids in the brain. Over time, this free radical damage leads to the death of neurons. Numerous studies have implicated oxidative stress and free radical damage in the pathology of age-related cognitive decline and dementia.38 Modifying oxidative stress could provide a basis for preventive and therapeutic approaches. CR is the most promising anti-aging and life-extending dietary strategy. CR has been postulated to decrease the rate of intellectual decay and potentially reverse age-related cognitive decline through reducing oxidative stress, increasing synaptic plasticity, and so on.39,40 d-gal–induced senescence acceleration may be due to the excessive formation of ROS and a significant reduction of anti-oxidant activities and consequent oxidative damage.3–8 In this study, the implementation of a CR protocol, which proved to be logical and effective by the fact that all animals subjected to CR showed no obvious health problems except having little to no weight gain throughout the experimental period, prevents memory deficits against d-gal–induced senescence in mice. After chronic exposure to d-gal, animals on CR regimen exhibited shorter escape latencies to the hidden platform, and more platform site crossings and strong preferences for the target quadrant than those on ad libitum regimen, indicating that CR had a potential effect for reserving the ability in learning and memory that would otherwise be impaired by d-gal–induced oxidative stress. However, no positive effect of CR on spatial memory performance was observed in normal mice in our study, even though many studies have reported that CR enhances learning and memory in animals and humans.41–43
Various biological systems work in conjunction to maintain optimal brain function and cognitive ability. Perturbations in the harmony of these systems, caused by such age-associated insults as oxidative stress, chronic inflammation, and changed hormone levels, result in physical deterioration of the brain and subsequent cognitive decline. Iron is an essential element for normal cellular functions and plays specific roles in the CNS. Control of iron homeostasis is essential for healthy CNS function. In the present study, we chose the cortical hemisphere and hippocampus, the cognition-related and iron invasion–vulnerable regions,44 as target tissues for investigating the potential anti-iron accumulation effect of CR with the d-gal–induced aging mouse model. Contrary to what we had expected, no significant differences in the total iron content of the brain were observed between experimental and control groups. In other words, direct evidence of iron accumulation in d-gal–induced aging mice or of anti-iron accumulation in CR treatment mice was not detected. One reason for this probably lies in the chronic, long-term process of brain iron accumulation; that is, noticeable changes in intracellular iron content may occur in late-stage pathology, with misregulation of iron metabolism being the primary event preceding such changes,45 as demonstrated by this study. Another reason may be that, throughout the experiment, the animals had not been treated with an iron-rich diet, which was a prerequisite for experimental iron accumulation to build up.
Iron enters the brain mainly by transport through the blood–brain barrier (BBB), a tightly regulated process that under normal circumstances protects the brain from being affected by fluctuations in systemic iron. Within the brain, there are still more sophisticated regulatory systems governing the cellular iron level stability to ensure normal function of neural cells and protect them from toxicity as well.12 In view of the fact that iron accumulates with aging in specific brain regions, we suggested dysregulation of iron metabolism in specific areas of the brain, rather than disturbance of iron uptake across the BBB, is more likely to play a dominant role in the pathogenesis of age-related brain iron deposition. Cerebral intracellular iron homeostasis can be divided into iron influx and iron efflux. Hepcidin and Fpn1 are linked to cellular iron efflux and widely expressed in the brain.12,25,26 As in the periphery, Fpn1 is the sole known cellular iron exporter and plays a key role in brain iron release.22,46 The turnover of Fpn1 is controlled by hepcidin. Injection of hepcidin into the lateral cerebral ventricle resulted in decreased Fpn1 protein content in the cerebral cortex, the hippocampus, and the striatum.46 Functionally, hepcidin acts to internalize Fpn1, leading to its degradation and thereby blocking iron release from cells into extracellular fluid. Thus, facilitation of iron efflux by hepcidin–Fpn pathway would be able to protect cells from excessive accumulation of iron. Our study found that the mRNA expression of Fpn1 was not altered in d-gal–induced mouse aging models, nor was it in CR-treated animals; however, the expression of hepcidin, the ligand of Fpn1 protein, was significantly affected by both d-gal and CR interventions. On one hand, elevated hepcidin mRNA contents in cerebral cortex and hippocampus was observed in d-gal–induced aging model mice, which were characterized by cognitive deficits in the MWM task. On the other hand, proactive treatment with CR largely offset the d-gal–induced increase in hepcidin mRNA levels along with its protective effect on the animals' cognitive performance. In normal control mice, CR intervention could also reduce brain hepcidin expression markedly, although no synchronized enhancement in learning and memory was displayed.
These results suggest that CR might guard against age-related declines in cognitive function partially by down-regulating the expression of hepcidin in the brain, thereby making it possible for the cellular iron exporter Fpn1 to play its role efficiently on the cell membrane to resist or prevent iron accumulation in cells in the CNS. One limitation of the study was lack of determination of hepcidin and Fpn1 protein concentrations. Although hepcidin was mainly regulated at the transcriptional level, Fpn1 was mainly regulated post-transcriptionally.47 So, although the Fpn1 mRNA levels were unaltered across the groups, it would be unreasonable to infer that d-gal, CR intervention, and even the elevated hepcidin have no influence on Fpn1 protein abundance/effectiveness.
Consistent with the evidence of age-related iron accumulation in the brain, a new study found that hepcidin mRNA levels increased with aging in the cerebral cortex, hippocampus, and striatum.26 We have presented here for the first time that, in both aging model and normal mice, CR has the potential to down-regulate the mRNA expression of hepcidin in the cortex and hippocampus. Iron dysregulation is considered to be an early event in the pathogenesis of iron deposition. Once deposited in cells, iron was difficult to remove. In this regard, it is better to prevent iron accumulation than to have to treat it, and the value and significance of our present study lies in revealing that CR may be a way to lessen or avoid age-related iron accumulation in the brain. In fact, a recent study did observe CR attenuates brain iron accumulation in old rhesus monkeys measured in vivo using magnetic resonance imaging.35 Yet, how hepcidin gene expression is affected in d-gal and CR conditions remains unclear. Further studies are necessary to clarify the molecular mechanisms underlying CR's modulation of hepcidin and other genes involved in brain iron homeostasis, and the mechanisms for age-related, as well as d-gal–induced up-regulation of hepcidin expression.
In summary, the present study demonstrated the existence of dysregulation of iron metabolism in the brains of d-gal–induced aging model mice and the potential of CR as an anti-aging dietary regimen against the imbalance of brain iron homeostasis. CR could down-regulate the expression of hepcidin mRNA in cerebral cortex and hippocampus in normal mice, and suppress the up-regulation of hepcidin expression in brains of d-gal–induced mice. Taking into account the involvement of progressive brain iron accumulation in the natural aging process, with hepcidin being the key negative regulator for cellular iron release, we assume CR may possess anti-aging benefits by reducing or avoiding abnormal accumulation of iron in the brain.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 81373020) and the Beijing Natural Science Foundation (grant no. 7112014).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Kumar A, Prakash A, Dogra S. Centella asiatica attenuates D-galactose-induced cognitive impairment, oxidative and mitochondrial dysfunction in mice. Int J Alzheimers Dis 2011;2011:347569–347577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li W, Wei F, Fan M, Zhang J, Zhang B, Ma X, Yang W, Wei W. Mimetic brain aging effect induced by D-galactose in mice. Chin J Pharmacol Toxicol 1995;9:93–95 [Google Scholar]
- 3.Cui X, Wang L, Zuo P, Han Z, Fang Z, Li W, Liu J. D-galactose-caused life shortening in Drosophila melanogaster and Musca domestica is associated with oxidative stress. Biogerontology 2004;5:317–325 [DOI] [PubMed] [Google Scholar]
- 4.Cui X, Zuo PP, Zhang Q, Li XK, Hu YZ, Long JG, Packer L, Liu JK. Chronic systemic D-galactose exposure induces memory loss, neurodegeneration, and oxidative damage in mice: Protective effects of R-α-lipoic acid. J Neurosci Res 2006;83:1584–1590 [DOI] [PubMed] [Google Scholar]
- 5.Chen CF, Lang SY, Zuo PP, Yang N, Wang XQ, Xia C. Effects of D-galactose on the expression of hippocampal peripheral-type benzodiazepine receptor and spatial memory performances in rats. Psychoneuroendocrinology 2006;31:805–811 [DOI] [PubMed] [Google Scholar]
- 6.Hua XD, Lei M, Zhang YJ, Ding J, Han QY, Hu G, Xiao M. Long-term D-galactose injection combined with ovariectomy serves as a new rodent model for Alzheimer's disease. Life Sci 2007;80:1897–1905 [DOI] [PubMed] [Google Scholar]
- 7.Sun SW, Yu HQ, Zhang H, Zheng YL, Wang JJ, Luo L. Quercetin attenuates spontaneous behavior and spatial memory impairment in D-galactose-treated mice by increasing brain antioxidant capacity. Nutr Res 2007;27:169–175 [Google Scholar]
- 8.Wei SG, He XJ, Yun SJ, Zhang SH, Xiao ZX. Dietary restriction to accompany the aging process in mice: Can it be neuroprotective? Neural Regen Res 2010;5:789–795 [Google Scholar]
- 9.Ho SC, Liu JH, Wu RY. Establishment of the mimetic aging effect in mice caused by D-galactose. Biogerontology 2003;4:15–18 [DOI] [PubMed] [Google Scholar]
- 10.Shen YX, Xu SY, Wei W, Sun XX, Yang J, Liu LH, Dong C. Melatonin reduces memory changes and neural oxidative damage in mice treated with D-galactose. J Pineal Res 2002;32:173–178 [DOI] [PubMed] [Google Scholar]
- 11.Shang YZ, Gong MY, Zhou XX, Li ST, Wang BY. Improving effects of SSF on memory deficits and pathological changes of neural and immunological systems in senescent mice. Acta Pharmacologica Sinica 2001;22:1078–1083 [PubMed] [Google Scholar]
- 12.Ke Y, Qian ZM. Brain iron metabolism: Neurobiology and neurochemistry. Prog Neurobiol 2007;83:149–173 [DOI] [PubMed] [Google Scholar]
- 13.Salvador GA, Uranga RM, Giusto NM. Iron and mechanisms of neurotoxicity. Int J Alzheimer's Dis 2011;2011:720658–720666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zecca L, Youdim MBH, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nature Rev Neurosci 2004;5:863–873 [DOI] [PubMed] [Google Scholar]
- 15.Sullivan EV, Adalsteinsson E, Rohlfing T, Pfefferbaum A. Relevance of iron depositon in deep gray matter brain structures to cognitive and motor performance in healthy elderly men and women: Exploratory findings. Brain Imaging Behav 2009;3:167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaur D, Andersen J. Does cellular iron dysregulation play a causative role in Parkinson's disease?. Ageing Res Rev 2004;3:327–343 [DOI] [PubMed] [Google Scholar]
- 17.Ke Y, Qian ZM. Iron misregulation in the brain: A primary cause of neurodegenerative disorders. Lancet Neurol 2003;2:246–253 [DOI] [PubMed] [Google Scholar]
- 18.Thomas M, Jankovic J. Neurodegenerative disease and iron storage in the brain. Curr Opin Neurol 2004;17:437–442 [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Liu H, Wu J, Zhang X, Liu M, Wang Y. Metabonomic alterations in hippocampus, temporal and prefrontal cortex with age in rats. Neurochem Int 2009;54:481–487 [DOI] [PubMed] [Google Scholar]
- 20.Morita T, Mizutani Y, Sawada M, Shimada A. Immunohistochemical and ultrastructural findings related to the blood-brain barrier in the blood vessels of the cerebral white matter in aged dogs. J Comp Pathol 2005;133:14–22 [DOI] [PubMed] [Google Scholar]
- 21.Wallace M, Frankfurt M, Arellanos A, Inagaki T, Luine V. Impaired recognition memory and decreased prefrontal cortex spine density in aged female rats. Ann NY Acad Sci 2007;1097:54–57 [DOI] [PubMed] [Google Scholar]
- 22.Aguirre P, Mena N, Tapia V, Arredondo M, Núñez MT. Iron homeostasis in neuronal cells: A role for IREG1. BMC Neurosci 2005;6:3. 10.1186/1471-2202-6-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002;110:1037–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Domenico I, Ward DM, Kaplan J. Hepcidin and ferroportin: The new players in iron metabolism. Semin Liver Dis 2011;31:272–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Domenico I, Ward DM, Langelier C, Vaughn MB, Nemeth E, Sundquist WI, Ganz T, Musci G, Kaplan J. The molecular mechanism of hepcidin-mediated Ferroportin down-regulation. Mol Biol Cell 2007;18: 2569–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang SM, Fu LJ, Duan XL, Crooks DR, Yu P, Qian ZM, Di XJ, Li J, Rouault TA, Chang YZ. Role of hepcidin in murine brain iron metabolism. Cell Mol Life Sci 2010;67:123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zechel S, Huber-Wittmer K, von Bohlen , Halbach O. Distribution of the iron-regulating protein hepcidin in the murine central nervous system. J Neurosci Res 2006;84:790–800 [DOI] [PubMed] [Google Scholar]
- 28.Spindler SR. Caloric restriction: From soup to nuts. Ageing Res Rev 2010;9:324–353 [DOI] [PubMed] [Google Scholar]
- 29.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009,325:201–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung K, Lee E, Kim J, Zou Y, Sung B, Heo HS, Kim MK, Lee J, Kim ND, Yu BP, Chung HY. Effect of short term calorie restriction on pro-inflammatory NF-kB and AP-1 in aged rat kidney. Inflamm Res 2009;58:143–150 [DOI] [PubMed] [Google Scholar]
- 31.Dal-Pan A, Pifferi F, Marchal J, Picq JL, Aujard F. Cognitive performances are selectively enhanced during chronic caloric restriction or resveratrol supplementation in a primate. PLoS One 2011;6: e16581. 10.1371/journal.pone.0016581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009;325:201–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.American Institute of Nutrition AIN-93 purified diets for laboratory rodents: Final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of AIN-76A rodent diet. J Nutr 1993;123:1939–1951 [DOI] [PubMed] [Google Scholar]
- 34.Cook CI, Yu BP. Iron accumulation in aging: Modulation by dietary restriction. Mech Ageing Dev 1998;102:1–13 [DOI] [PubMed] [Google Scholar]
- 35.Kastman EK, Willette AA, Coe CL, Bendlin BB, Kosmatka KJ, McLaren DG, Xu G, Canu E, Field AS, Alexander AL, Voytoko ML, Beasley TM, Colman RJ, Weindruch RH, Johnson SC. A calorie-restricted diet decreases brain iron accumulation and preserves motor performance in old rhesus monkeys. J Neurosci 2012;32:11897–11904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Zhang J, Zhao L, Hu S, Piao F. Effect of subchronic exposure to arsenic on levels of essential trace elements in mice brain and its gender difference. Biometals 2013;26:123–131 [DOI] [PubMed] [Google Scholar]
- 37.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008;3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 38.Mangialasche F, Polidori MC, Monastero R, Ercolani S, Camarda C, Cecchetti R, Mecocci P. Biomarkers of oxidative and nitrosative damage in Alzheimer's disease and mild cognitive impairment. Ageing Res Rev 2009;8:285–305 [DOI] [PubMed] [Google Scholar]
- 39.Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science 1996;273:59–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gillette-Guyonnet S, Vellas B. Caloric restriction and brain function. Curr Opin Clin Nutr Metab Care 2008;11:686–692 [DOI] [PubMed] [Google Scholar]
- 41.Witte AV, Fobker M, Gellner R, Knecht S, Floel A. Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci USA 2009;106:1255–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin CK, Anton SD, Han H, York-Crowe E, Redman LM, Ravussin E, Williamson DA. Examination of cognitive function during six months of calorie restriction: Results of a randomized controlled trial. Rejuvenation Res 2007;10:179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bryan J, Tiggemann M. The effect of weight-loss dieting on cognitive performance and psychological well-being in overweight women. Appetite 2001;36:147–156 [DOI] [PubMed] [Google Scholar]
- 44.Jellinger K, Paulus W, Grundke-Iqbal I, Riederer P, Youdim MB. Brain iron and ferritin in Parkinson's and Alzheimer's diseases. J Neural Transm Park Dis Dement Sect 1990;2:327–340 [DOI] [PubMed] [Google Scholar]
- 45.Smith MA, Zhu XW, Tabaton M, Liu G, McKeel DW, Cohen ML, Wang XL, Siedlak SL, Dwyer BE, Hayashi T, Nakamura M, Nunomura A, Perry G. Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis 2010;19:363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding H, Yan CZ, Shi H, Zhao YS, Chang SY, Yu P, Wu WS, Zhao CY, Chang YZ, Duan XL. Hepcidin is involved in iron regulation in the ischemic brain. PLoS One 2011;6: e25324. 10.1371/journal.pone.0025324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang DL, Senecal T, Ghosh MC, Ollivierre-Wilson H, Tu T, Rouault TA. Hepcidin regulates ferroportin expression and intracellular iron homeostasis of erythroblasts. Blood 2011;118: 2868–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]