

Abstract
The clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated (Cas) system has recently been used to engineer genomes of various organisms, but surprisingly, not those of bacteriophages (phages). Here we present a method to genetically engineer the Escherichia coli phage T7 using the type I-E CRISPR-Cas system. T7 phage genome is edited by homologous recombination with a DNA sequence flanked by sequences homologous to the desired location. Non-edited genomes are targeted by the CRISPR-Cas system, thus enabling isolation of the desired recombinant phages. This method broadens CRISPR Cas-based editing to phages and uses a CRISPR-Cas type other than type II. The method may be adjusted to genetically engineer any bacteriophage genome.
Keywords: Escherichia coli, Bacteriophage T7, homologous recombination, positive selection, negative selection, genetic engineering, spacer targeting
Introduction
The abundance of phages and their importance to microbial evolution, and consequently, to major ecological issues, provide an incentive to study their biology.1 Surprisingly, there are only a few published methods for engineering phage genomes.2,3 In bacteria, an antibiotic resistance gene may be inserted in a desired genetic location, and the recombinant bacteria can then be positively selected using the appropriate antibiotics.4 Lytic phages cannot be selected by this method, however, because an antibiotic marker does not confer a selective advantage for them. Other markers, such as genes that are essential for phage growth, have been utilized for genetic engineering. Examples for such genes are the Escherichia coli trxA and cmk genes that are required for phage T7 growth but dispensable for the host’s growth.5,6 T7 phage infecting a host that lacks any of these genes will complete the infection successfully only if it encodes them on its own genome. Thus, it is possible to pair a desired gene with either of the marker genes and then select for the desired gene replacement by plating the manipulated phage population on hosts that lack the marker genes. The only phages that grow will be those that acquired the marker genes. However, using this method does not generate a “clean” deletion because it replaces one genomic location with a marker rather than deleting the desired region without “scars.” In addition, these markers are scarce and have only been identified in well-studied phages. Clearly, genetic engineering of bacteriophages would benefit from additional selection or counter-selection markers.
The CRISPR-Cas system can be used to cleave desired DNA, thus rendering any targeted DNA as a potential counter-selection marker. Recently, the CRISPR-Cas type II system, encoding the Cas9 protein, has been used for genetic engineering by providing a robust and specific selection of recombinant DNA. This system has facilitated construction of recombinant human,7,8 rodent,9,10 fish,11 fly,12 worm,13,14 plant,15,16 yeast,17 and bacterial18 cells, but surprisingly, not of phages, against which the system probably initially evolved. The CRISPR-Cas system comprises of an array of repeated sequences called “repeats” and flanking sequences called “spacers.” Transcribed spacers guide specific proteins to the target nucleic acids, called “protospacers” by virtue of sequence homology, which are then degraded.19-21 The system can specifically target any nucleic acid fragment using an appropriately designed spacer. As a genetic engineering tool, the Cas9 nuclease targets protospacers, generating DNA breaks, which are repaired by non-homologous end joining, occasionally resulting in frameshift, deletion, or insertion mutations (e.g., ref. 7). Alternatively, the breaks are repaired through homologous recombination, by a template supplied in trans, resulting in precisely engineered mutations (e.g., refs. 7 and 18). Here we describe a simple and efficient method to genetically engineer the E. coli phage T7. We use the CRISPR-Cas type I-E system to counter-select non-edited phage genomes, enabling the isolation of desired recombinants. Using this approach, we demonstrate successful selection of two genes of the T7 phage. We believe that this method could be easily adjusted to any phage genome, with minor modifications.
Results
We wished to demonstrate that the E. coli type I-E CRISPR-Cas system could be used to isolate scarless deletions of a specific gene, 1.7, in a T7 phage genome. The 1.7 gene, encoding a nucleotide kinase, is not essential for phage growth under standard laboratory conditions.22 To facilitate deletion of this gene, we constructed a plasmid that encoded 120 bp of the sequences flanking gene 1.7 (60 bp from each side). Propagation of T7 phage on hosts harboring this plasmid resulted in homologous recombination between the plasmid and a small proportion of the progeny phages (Fig. 1A). The progeny thus consisted of phages lacking gene 1.7 along with other wild-type (WT)-T7 phages that did not undergo homologous recombination. To select the desired recombinant phages deleted in gene 1.7, we used a modified version of a plasmid-based type I-E CRISPR-Cas system described by Brouns et al.20 The cascade and cas3 genes, all required for CRISPR-Cas activity, were expressed from two plasmids. A spacer against gene 1.7 was expressed from a third plasmid (Fig. 1B), thus guiding the system’s protein to cleave WT-T7 phages encoding this gene. The efficiency of plating (EOP) of WT-T7 phages on bacteria harboring these three plasmids (termed “targeting bacteria”) was over 10 000 fold lower compared with their growth on “control bacteria” harboring the plasmids encoding the cas genes as well as a plasmid expressing an irrelevant spacer (EOP 1.9 × 10−5 ± 1.7 × 10−5). In contrast, T7 phages lacking gene 1.7 grew on the targeting bacteria as expected, similar to their growth on control bacteria (EOP of 1.08 ± 0.27). This system thus allowed us to select for phages that lacked a specific protospacer encoded within gene 1.7. Indeed, upon plating the phage lysates that underwent homologous recombination on bacteria harboring the targeting CRISPR-Cas system, 17 out of 44 plaques in four independent experiments carried the desired deletion (Fig. 1C), as verified by DNA sequencing. The remaining isolated phages that did not encode the desired deletion escaped the CRISPR-Cas targeting by another deletion of a fragment encoding the corresponding protospacer, as shown in Table S2. The fact that desired recombinants are found at a ratio of approximately 1:2, and escape mutants arise at a frequency of 1.9 × 10−5, indicates that the phages arising by homologous recombination form at an approximate frequency of 1 × 10−5. To demonstrate the wide-range applicability of this method for other DNA segments, we chose to delete another gene in phage T7, gene 4.3. Using similar procedures, but different homologies and spacer (described in the Supplemental Materials), we deleted this gene, achieving an efficiency of 15 out of 36 plaques from three independent experiments.
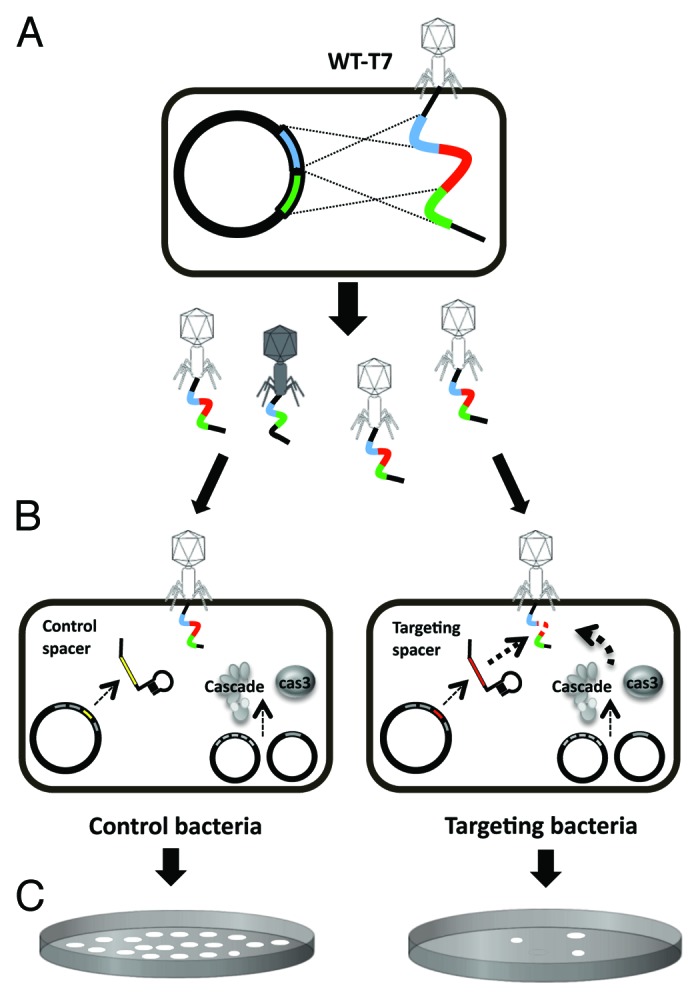
Figure 1. Overview of the procedures for isolating a desired recombinant bacteriophage. (A) Wild-type T7 phages are grown in the presence of a plasmid encoding a 120-bp fragment flanking 60 bp from upstream (green) and downstream (blue) of a desired genetic location. The progeny phages consist mainly of non-recombinant phages, but some recombinant phages also exist. (B) The obtained phage-lysate is plated on E. coli encoding cascade, cas3, and either spacers targeting the region between the flanking homologies (targeting bacteria) or control spacers (control bacteria). (C) Targeting bacteria select for the desired recombinant phages, whereas control bacteria allow growth of all plated phages.
Discussion
The method shown here for genetic engineering of phage genomes is unique in both the targeted DNA (i.e., phage DNA) and the type of CRISPR-Cas system used (i.e., type I-E CRISPR-Cas system). A universal genetic engineering system to engineer phages may prove highly valuable for phage research and for biotechnological use. All hitherto described methods for genetic engineering with the CRISPR-Cas system have used the type II CRISPR-Cas system, which encodes the multifunctional Cas9 protein. In contrast, we used the type I-E endogenous system of E. coli for engineering E. coli phages. The presented system thus provides a unique example that shows that this system may also be used for genome engineering. Similar setups, utilizing endogenous CRISPR systems, can be used for engineering host-specific phages in other biological systems. The T7 phage is capable of homologous recombination without an exogenous supply of recombination-promoting enzymes; therefore, there is no need for exogenous recombination genes in trans. Nevertheless, for phages lacking their own recombination system, either longer homologies can be used for the recombination to render the host’s recombination enzymes effective or exogenous recombination enzymes such as the lambda Red proteins can be provided in trans. We believe that with the suggested adjustments, the system could easily be adapted for manipulating any phage genome.
Supplementary Material
Disclosure of Potential Conflicts
No potential conflicts of interest were disclosed.
Acknowledgments
The research leading to these results has received funding from the European Research Council under the European Community’s Seventh Framework Programme (FP7/207-2013)/ ERC grant agreement no 336079 (EcCRISPR).
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/27766
References
- 1.Suttle CA. Marine viruses--major players in the global ecosystem. Nat Rev Microbiol. 2007;5:801–12. doi: 10.1038/nrmicro1750. [DOI] [PubMed] [Google Scholar]
- 2.Selick HE, Kreuzer KN, Alberts BM. The bacteriophage T4 insertion/substitution vector system. A method for introducing site-specific mutations into the virus chromosome. J Biol Chem. 1988;263:11336–47. [PubMed] [Google Scholar]
- 3.Marinelli LJ, Piuri M, Swigonová Z, Balachandran A, Oldfield LM, van Kessel JC, Hatfull GF. BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS One. 2008;3:e3957. doi: 10.1371/journal.pone.0003957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–5. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qimron U, Marintcheva B, Tabor S, Richardson CC. Genomewide screens for Escherichia coli genes affecting growth of T7 bacteriophage. Proc Natl Acad Sci U S A. 2006;103:19039–44. doi: 10.1073/pnas.0609428103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mark DF, Richardson CC. Escherichia coli thioredoxin: a subunit of bacteriophage T7 DNA polymerase. Proc Natl Acad Sci U S A. 1976;73:780–4. doi: 10.1073/pnas.73.3.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–8. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol. 2013;31:681–3. doi: 10.1038/nbt.2661. [DOI] [PubMed] [Google Scholar]
- 11.Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, Joung JK. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31:227–9. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, Wildonger J, O’Connor-Giles KM. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013;194:1029–35. doi: 10.1534/genetics.113.152710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM, Calarco JA. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods. 2013;10:741–3. doi: 10.1038/nmeth.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lo TW, Pickle CS, Lin S, Ralston EJ, Gurling M, Schartner CM, Bian Q, Doudna JA, Meyer BJ. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 2013;195:331–48. doi: 10.1534/genetics.113.155382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu JL, et al. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol. 2013;31:686–8. doi: 10.1038/nbt.2650. [DOI] [PubMed] [Google Scholar]
- 16.Feng Z, Zhang B, Ding W, Liu X, Yang DL, Wei P, Cao F, Zhu S, Zhang F, Mao Y, et al. Efficient genome editing in plants using a CRISPR/Cas system. Cell Res. 2013;23:1229–32. doi: 10.1038/cr.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013;41:4336–43. doi: 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–9. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–12. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 20.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–4. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–56. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran NQ, Rezende LF, Qimron U, Richardson CC, Tabor S. Gene 1.7 of bacteriophage T7 confers sensitivity of phage growth to dideoxythymidine. Proc Natl Acad Sci U S A. 2008;105:9373–8. doi: 10.1073/pnas.0804164105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.