

Abstract
A growing body of evidence suggests the non-protein coding human genome is of vital importance for human cell function. Besides small RNAs, the diverse class of long non-coding RNAs (lncRNAs) recently came into focus. However, their relevance for infection, a major evolutionary driving force, remains elusive. Using two commercially available microarray systems, namely NCode™ and Sureprint™ G3, we identified differential expression of 42 ncRNAs during influenza A virus (IAV) infection in human lung epithelial cells. This included several classes of lncRNAs, including large intergenic ncRNAs (lincRNAs). As analyzed by qRT-PCR, expression of one lincRNA, which we termed virus inducible lincRNA (VIN), is induced by several IAV strains (H1N1, H3N2, H7N7) as well as vesicular stomatitis virus. However, we did not observe an induction of VIN by influenza B virus, treatment with RNA mimics, or IFNβ. Thus, VIN expression seems to be a specific response to certain viral infections. RNA fractionation and RNA-FISH experiments revealed that VIN is localized to the host cell nucleus. Most importantly, we show that abolition of VIN by RNA interference restricts IAV replication and viral protein synthesis, highlighting the relevance of this lincRNA for productive IAV infection. Our observations suggest that viral pathogens interfere with the non-coding portion of the human genome, thereby guaranteeing their successful propagation, and that the expression of VIN correlates with their virulence. Consequently, our study provides a novel approach for understanding virus pathogenesis in greater detail, which will enable future design of new antiviral strategies targeting the host’s non-protein coding genome.
Keywords: lincRNA, non-protein coding genome, host factor, IAV, VIN
Introduction
The surprising observation that only 1.5% of the mammalian genome is protein coding and the identification of vast numbers of non-protein coding transcripts in large-scale transcriptomic studies initiated a debate about the role of ncRNAs in cell biology.1 It has been speculated that in contrast to protein coding genes, the non-coding portion of a genome correlates with organism complexity and functions in crucial regulatory processes.2,3 With a length of ~22 nucleotides, microRNAs (miRNAs) constitute the best-studied class of small regulatory RNAs and have assigned functions in several cellular processes, including development, proliferation, and disease.4,5 However, recent identification and characterization of lncRNAs indicate a more complex cell regulatory network than previously anticipated. So far, lncRNAs are categorized as transcripts longer than 200 nucleotides that do not contain a significant open reading frame but are often polyadenylated and spliced.6 The genomic origin of lncRNAs is manifold, including intergenic, intronic, gene-overlapping, or antisense transcription. Although only a few have been characterized in detail, their functional repertoire is expanding. Several studies have revealed that lncRNAs are important for gene regulation,7,8 such as XIST, which regulates dosage compensation of the mammalian X chromosome.9 Mechanisms of lncRNA action are diverse and they are involved in widespread biological processes like imprinting, pluripotency, cell differentiation, development, apoptosis, and development of disease.5 However, although infection is considered a major driving force of human genome evolution, little is known about potential roles of lncRNAs in this context. Recently, transcriptome-wide deep sequencing revealed the differential expression of more than a thousand potential lncRNAs during severe acute respiratory syndrome coronavirus (SARS-CoV) infection in mice, indicative of a function in infection.10 Several of these mouse lncRNAs were shown to be similarly regulated upon infection with IAV, a highly contagious virus causing annual epidemics and frequent pandemic outbreaks.11 To understand the process of IAV infection in humans and develop new antiviral strategies, genome-wide studies have aimed to identify host cell proteins necessary for productive IAV replication.12,13 Host cell miRNAs are known to be differentially regulated by IAV and implicated in control of infection outcome,14,15 whereas the functional significance of lncRNA expression during IAV infection remains elusive.
We addressed this question based on differential expression analysis of human lncRNAs during influenza A/WSN/33 infection, using NCode™ and Sureprint™ G3 microarrays. We show that expression of virus inducible lincRNA (VIN), a highly regulated lncRNA during A/WSN/33 infection, is specifically induced during infection with different IAV strains and vesicular stomatitis virus (VSV) but not with influenza B virus (IBV). We further demonstrate a confined nuclear expression of VIN. Analysis of A/WSN/33 replication in VIN-knockdown cells revealed a significant decrease of viral titers, thus highlighting the role of the non-protein coding human genome during infectious disease.
Results
Host cell lncRNAs are differentially expressed during influenza A/WSN/33 (H1N1) infection
Commercially available microarray systems such as NCode™ and Sureprint™ G3 contain probe sequences that incorporate ncRNAs from a variety of classes. In addition to protein-coding genes, NCode™ contains probe sequences of uncharacterized transcripts identified in cDNA and EST sequencing projects in addition to predicted functional ncRNA genes. Besides other non-coding RNA classes, the Sureprint™ G3 annotation is mainly based on human lincRNAs identified during ChIP-seq projects. Due to differences in nomenclature and annotation of both microarrays, NCode™ and Sureprint™ G3 ncRNA probe sequences were re-annotated using the current Ensembl Genome Annotation (Release 68). According to the latest sequence information, microarrays were uniformly annotated to enable cross-platform comparison. Re-annotation revealed the relative contributions of different ncRNA classes to ncRNA probes represented on these microarrays (Fig. 1A), and demonstrated that on NCode™ and Sureprint™ G3 microarrays, respectively, small RNA, uncharacterized miscellaneous RNA, and gene-overlapping RNA made up relatively minor proportions of probes, whereas pseudogene, antisense RNA, and lincRNA probes made up higher proportions. From the 3415 (NCode™) and 3201 (Sureprint™ G3) probes representing non-coding transcripts, 1342 (~40%) were common to both microarray systems (Fig. 1A).

Figure 1. Host cell lncRNAs are differentially expressed during influenza A/WSN/33 (H1N1) infection. (A) The contributions of different ncRNA classes on NCode™ and Sureprint™ G3 microarrays alone and common to both are shown. Re-annotation of microarrays was performed according to Ensembl human genome annotation (Release 68). (B) A549 cells were infected with influenza A/WSN/33 (H1N1) virus (MOI 1) for 8 or 24 h. Columns 1–4: log2-fold expression change of 17 lincRNAs (labeled by Ensembl transcript ID) that were differentially regulated at least 2-fold between uninfected and infected samples at both time points using NCode™ and Sureprint™ G3 microarrays. Columns 5–6: regulation of lincRNA expression by UV-treated, infected cell supernatants.
We used the re-annotated arrays to investigate potential infection-induced changes to lncRNA expression patterns at 8 h and 24 h post-infection (p.i.) with the influenza A/WSN/33 (H1N1) virus at a multiplicity of infection (MOI) of 1 in A549 lung epithelial cells. To avoid platform-specific effects, we focused our analysis on ncRNAs represented on both platforms. The expression of the known antiviral genes myxovirus resistance 1 (MX1) and 2’-5′-oligoadenylate synthetase-like (OASL), as well as several other interferon (IFN)-inducible genes, was upregulated on microarrays, which confirmed successful infection in this system (Fig. S1). In addition to protein-coding transcripts, the transcription of 42 ncRNAs common to both microarrays was up- or downregulated at least 2-fold upon infection, at both time points (Fig. S2). As expected, the distribution of the differentially regulated non-coding RNA genes in both microarray systems mirrored the distribution of the represented ncRNA classes. Thus, the most abundantly represented class was lincRNAs, which are listed in Figure 1B. Following preliminary microarray re-annotation and data analysis, the differential expression of some lncRNA transcripts was confirmed by quantitative reverse transcription (qRT) PCR (Fig. S3).
To further investigate the nature of the infection-induced regulation of ncRNA expression, we exposed infected cell supernatants to ultraviolet light to inactivate virions (Fig. S4A and B). Non-infected cells were then treated with supernatants and extracted RNA applied to Sureprint™ G3 microarrays. IFN-inducible genes were differentially regulated following incubation with UV-treated supernatants (Fig. S4C). However, lncRNA expression, including the subset of 17 lincRNAs induced 8 h and 24 h p.i., was not affected (Fig. 1B). Thus, several lncRNA classes were differentially regulated during infection in response to intact viruses, not as the indirect result of soluble factors released by infected cells.
Infection-induced expression of a novel lincRNA is not specific to A/WSN/33 (H1N1) virus
The non-coding transcript ENST00000412690 was among the most highly induced lincRNAs in the microarray analyses, a finding confirmed by qRT-PCR (Fig. 1B; Fig. S3). We focused on this annotated transcript, hereafter called VIN, for further functional analyses. To check if other viruses can also alter its expression, two additional IAV strains were selected, one IBV strain and vesicular stomatitis virus (VSV)—a non-segmented negative-sense single-stranded RNA virus. The levels of VIN expression in cells infected with the IAV strains A/Panama/2007/99 (H3N2) and A/FPV/Bratislava/79 (H7N7) increased approximately 30–60-fold, compared with 10-fold increased levels in A/WSN/33 (H1N1) virus-infected cells at 6 h p.i. (Fig. 2A). Interestingly, a similar level of expression was induced following infection with VSV but not with the IBV strain B/Brisbane/60/08. Infection with all tested viruses yielded a cell infection rate between 50–70%, excluding the possibility that different infectivities adversely affected VIN expression (Fig. S5). qRT-PCR time course investigations over 8 h suggested that infection-induced expression of VIN increased during late infection [VSV and A/WSN/33 (H1N1); Fig. S6]. In line with initial investigations at 6 h p.i. (Fig. 2A), IBV infection failed to induce VIN expression at all time points investigated (Fig. S6).
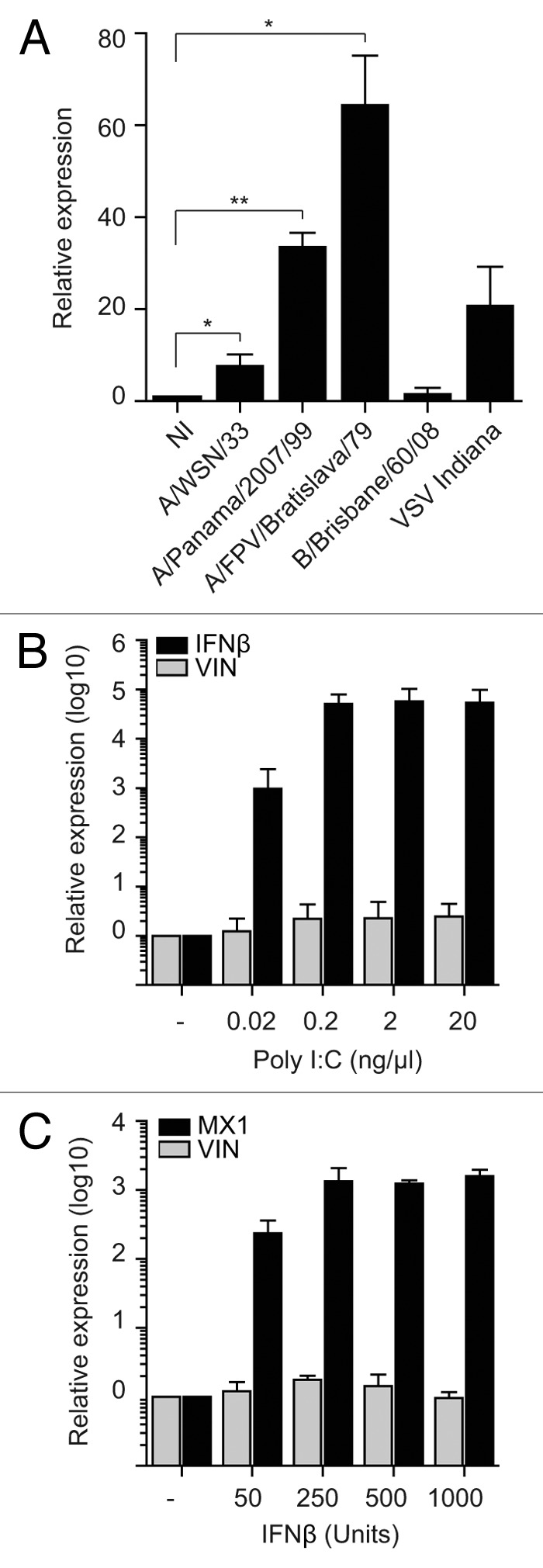
Figure 2. Infection-induced expression of a novel lincRNA is not specific to A/WSN/33 (H1N1) virus. (A) A549 cells were infected with three IAV strains, IBV, and VSV with MOIs 1 for 6 h. qRT-PCR data are presented as mean fold-changes in VIN expression (+/− SD) compared with non-infected (NI) reference. Data from three independent experiments were analyzed using one-sample t test (* P < 0.01; ** P < 0.005). (B) A549 cells were transfected with poly I:C and RNA was isolated 24 h post-transfection. qRT-PCR was performed for VIN and IFNβ. (C) A549 cells were treated with IFNβ and RNA isolated 8 h later. qRT-PCR was performed for VIN and MX1. Data in (B and C) are presented as fold-changes of expression compared with mock control (means of three independent experiments (+/− SD).
To investigate whether the factor responsible for induction of VIN was viral RNA, we treated A549 cells with polyinosinic:polycytidylic acid (poly I:C)—a synthetic analog of double-stranded RNA commonly used to mimic viral RNA intermediates that are present during virus infection. A wide range of poly I:C concentrations failed to induce VIN expression, whereas expression of IFNβ increased in a concentration-dependent manner (Fig. 2B). In line with experiments using UV-inactivated supernatants, treatment with IFNβ at a range of concentrations did not induce VIN expression, although upregulation of the known IFNβ target gene, MX1, was successfully demonstrated in this system (Fig. 2C).
To summarize, VIN expression was induced upon infection with a number of IAV viruses, and also after infection with VSV, suggesting that this lincRNA may have broader functionality during virus infection. However, since IBV, viral RNA mimics, or IFNβ are not able to induce VIN, this induction is likely to be a specific response and not due the presence of viral RNA itself.
In silico characterization of VIN
LncRNAs have only recently been identified and recognized for their pivotal roles in biology, and accordingly, the characterization of these ncRNAs is a developing field.16,17 Bioinformatic analyses of predicted lncRNAs can provide valuable information to help functionally characterize predicted lncRNAs.18,19 The VIN gene is located ~90 kbp downstream of the ACTR3 protein coding gene on the forward strand of chromosome 2 and the transcript is annotated as an intergenic 844 base pair non-coding RNA [Ensembl ENST00000412690 (Release 68); RefSeq LOC440900] (Fig. 3A). UCSC genome browser analysis revealed the presence of high methylation levels of lysine 4 of histone H3 (H3K4me) and acetylation of lysine 27 of histone H3 (H3K27ac) upstream of VIN, markers for transcriptional activation20 (Fig. 3A). Together with the clustering of several transcription factor binding sites in this region and the presence of a CpG island, this supports the notion that VIN is actively transcribed (Fig. 3A). Several other databases, including the recently released LNCipedia compendium, also list VIN as non-coding RNA.21 This database classifies long non-coding transcripts according to Coding Potential Calculator (CPC) analysis. This algorithm takes multiple features such as peptide length, amino acid composition, secondary structure, and protein homology into consideration.22 Like the majority of identified lncRNAs,6,23 VIN is encoded by two exons and contains a 3′-polyadenylation signal (AAUAAA).

Figure 3. In silico characterization of VIN. (A) Genomic context of VIN. Shown is the annotation by UCSC genome browser [http://genome.ucsc.edu, Human Feb. 2009 (GRCh37/hg19) Assembly], depicting the position of LOC440900 (VIN, red arrow), an uncharacterized transcript LOC100499194, H3K27ac, and H3K4me1 marks, location of a CpG island and transcription factor binding site clusters. (B) RNA secondary structure prediction of VIN (RNAfold web server, University of Vienna). Shown is a minimal free energy structure (MFE = -396.90 kcal/mol). Base pairing probabilities have been color coded using a scale from 0 (blue) to 1 (red). (C) RNase A stability of VIN. Nuclear RNA extracts of A549 cells were treated with RNase A followed by purification of RNA. qRT-PCR was performed for GAPDH mRNA and VIN. Data from three independent experiments (mean +/− SD) are depicted as VIN transcripts per GAPDH transcript (1/2^delta CT).
In silico prediction of lncRNA secondary structure is another useful method to assign putative functions to non-coding transcripts, based upon the widely held assumption that highly folded structures impart functionality through binding interactions with proteins/nucleotides.24-26 Characterization of VIN using RNAfold minimum free energy estimations predicted a highly folded secondary structure with several hairpin loops (Fig. 3B).27 Interestingly, unlike GAPDH mRNA, VIN was largely insensitive to endonuclease A (RNase A) digestion (Fig. 3C). Since RNase A preferentially cleaves single-stranded RNA, this supported the idea that VIN adopts stable secondary structures, and thus, has a functional role in cells, perhaps in complex with other cellular components.
VIN is localized to host cell nuclei during A/WSN/33 (H1N1) infection
In addition to sequence and structural information, defining subcellular distributions of lncRNAs can also help assign function. RNA fractionation experiments revealed that VIN was more abundant in the nuclear RNA fraction of A549 cells compared with the cytoplasmic fraction (Fig. 4A). The enrichment of VIN in the nuclear fraction was similar to that for the RNU1-1 nuclear RNA, whereas GAPDH mRNA was distributed approximately equally in both nuclear and cytoplasmic fractions. Western blotting with antibodies specific for the cytoplasmic 14-3-3 proteins and the nuclear protein Lamin A/C confirmed that fractionation was successful (Fig. S7). RNA fluorescence in situ hybridization (RNA-FISH) was performed to further investigate the subcellular localization of VIN. Mock- and A/WSN/33 (H1N1) virus-infected A549 cells were probed with a VIN-specific digoxigenin (DIG)-labeled RNA oligonucleotide. In uninfected A549 cells, a faint diffuse but nuclear DIG signal was observed (Fig. 4B, panel 1), which increased upon infection with A/WSN/33 (H1N1) virus (Fig. 4B, panel 2). The specificity of RNA-FISH was determined via hybridization of probes targeting nuclear RNU1-1 and the ubiquitously distributed GAPDH mRNA (Fig. S8A, panels 1 and 2). A control condition without applying any probe showed that signals detected in the Cy2 channel were not influenced by simultaneous staining of viral nucleoprotein (NP) with Cy3 (Fig. S8A, panel 3). In addition, knockdown of VIN by siRNAs decreased the nuclear DIG signal (Fig. S8B), confirming the specificity of RNA-FISH probes. The observation that nuclear NP levels were decreased in cells depleted of VIN prompted us to hypothesize that VIN expression affected virus protein expression, potentially supporting viral infectivity.
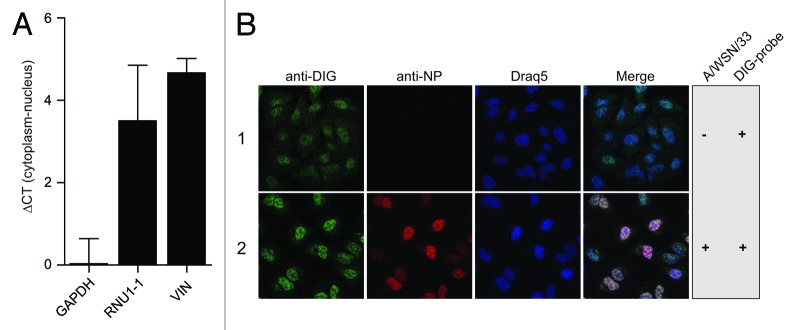
Figure 4. VIN is localized to host cell nuclei. (A) Nuclear and cytoplasmic RNA fractions were prepared from A/WSN/33 (H1N1) virus-infected A549 cells (MOI 5, 8 h). qRT-PCR was performed for GAPDH, RNU1-1 and VIN. Data are presented as mean ∆CT values (cytoplasm-nucleus) +/− SD of three independent experiments. (B) DIG-labeled probes were hybridized to A549 cells mock-infected or infected with A/WSN/33 (H1N1) virus (MOI 5, 6 h p.i.) followed by immunofluorescence staining of DIG (green) and viral NP protein (red) (RNA-FISH). Nuclei were visualized using Draq5 (blue). Images shown are representatives from four independent experiments.
VIN is essential for productive A/WSN/33 (H1N1) virus infection in human lung epithelial cells
To determine its role during productive A/WSN/33 infection, we analyzed viral replication upon knockdown of VIN. A549 cells were transfected with VIN-targeted siRNAs, followed by A/WSN/33 (H1N1) virus infection. Virus replication was allowed to occur for 48 h, thereafter virus-containing supernatants were titrated onto MDCK cells. After an additional 6 h of infection, immunofluorescence analysis was performed and influenza A NP-positive MDCK cells were counted to quantify infection. Viral titers of the titrated cell supernatants were calculated according to infectivity rates. Notably, knockdown with three different siRNAs individually and in conjunction (approx. 60% knockdown efficiency, Fig. S9A), reduced viral titers more than 10-fold compared with Allstars siRNA-treated control cells (Fig. 5A and B), highlighting the importance of VIN for productive IAV infection. Furthermore, the expression of key viral proteins was reduced in VIN-knockdown cells compared with Allstars siRNA-treated control cells, which confirms the observation that it is required for the H1N1 viral lifecycle (Fig. 5C). Cell viability (Fig. S9B) and type I IFN response (Fig. S9C) were unchanged in knockdown cells, and thus, could not have caused decreased virus titers indirectly. These data demonstrate that VIN expression supports completion of the IAV lifecycle, and thus, viral propagation.

Figure 5. VIN is essential for productive A/WSN/33 (H1N1) virus infection in human lung epithelial cells. (A) Three unique siRNAs designed to target VIN were used individually (1, 2, and 3) and collectively (P) to transfect A549 cells. Images show NP immunofluorescent staining in MDCK cells infected with supernatants from VIN knockdown cells (1, 2, 3, and P) compared with Allstars control. Immunofluorescence images shown are representatives from at least three independent experiments. (B) NP-positive MDCK cells from infection experiments were quantified using ScanR software and viral titers calculated. Data are presented as mean A/WSN/33 (H1N1) viral titers (plaque forming units (PFU)/ml) +/− SD from at least three independent experiments). Mann Whitney U tests were used for statistical analysis, * P < 0.01; ** P < 0.005; *** P < 0.001. (C) Western blot analysis of IAV protein expression 48 h p.i. in VIN A549 siRNA knockdown cells (1, 2, and 3, and P) compared with siRNA Allstars control. β-Actin expression is shown as a loading control. Blot is a representative of two independent experiments (HA, Hemagglutinin; NP, Nucleoprotein; NS1, non-structural protein 1; M2, Matrix protein 2).
Discussion
Recent advances in genome and transcriptome sequencing revealed the expression of a high proportion of uncharacterized and mainly non protein-coding RNAs.1 The tightly controlled transcription of lncRNAs and the fact that they appear to play a role in many tissues,28 highlights their potential in regulating important aspects of the cellular machinery. Research into host-pathogen interactions has so far focused mostly on host cell proteins12,13,29and not yet considered the role of lncRNAs in detail. Given that small ncRNAs such as miRNAs14,15,30,31have already been shown to have important functions during the host cell response to infections, it is likely that lncRNAs also play important roles.
We identified the lincRNA VIN as an essential player during IAV replication, showing a more than 10-fold decrease of IAV titer upon VIN knockdown. Viruses are known to hijack the host cellular machinery for their own replication and suppress antiviral responses by a variety of mechanisms. The relevance of host cell factors during IAV replication has been previously identified, revealing the involvement of several cellular networks in IAV replication.12,29,32,33 Since these processes need tight control to ensure successful virus propagation, we propose a role for VIN in gene expression control during IAV infection.
We identified VIN as differentially expressed during influenza A/WSN/33 virus infection using NCode™ and Sureprint™ G3 microarrays. Interestingly, dynamic changes in expression levels of lncRNAs have been identified upon lipopolysaccharide stimulation of macrophages, demonstrating the importance of lncRNAs during immune surveillance.34 The first report about lncRNAs in the context of viral infections showed induction of a lncRNA upon Japanese encephalitis and rabies virus infection in mice.35 More recently, a high-throughput sequencing approach described the differential lncRNA transcriptome upon SARS-CoV infection in mice and regulation of some of these lncRNAs was confirmed in IAV infection and upon IFNβ treatment.10 In contrast, regulation of lncRNAs was mainly dependent on the presence of infectious virus in our system, comparing infection of live and inactivated IAV viruses in human cells.
Strikingly, we show that VIN expression is not affected by viruses in general, viral RNA, or the type I IFN response. Instead, it is induced only by specific viruses, which may be related to differences in virulence. Notably, the induction levels of VIN observed here mirror the different pandemic potential of IAV strains,11 with H7N7 being more pathogenic than H1N1 and H3N2. Differences in the protein repertoire encoded by VSV, IAV, and IBV, or differential host cell signaling during infection, could also be responsible for distinct VIN regulation profiles.36,37 However, the contribution of virus-specific factors on VIN expression, and the function of VIN during VSV and IBV infection, will need further investigation.
Similarly to many other characterized lncRNAs,38,39 our data suggest nuclear localization of VIN. Nuclear lncRNAs have been implicated in the maintenance of sub-nuclear architecture,40 direct transcriptional regulation,41 post-transcriptional control,42 and chromatin remodeling.8 However, it remains to be demonstrated which function VIN conducts in the nucleus of IAV-infected cells. In silico secondary structure analysis and RNase A sensitivity analyses suggest that VIN folds into highly stable structures that may reflect its function, perhaps in complex with other cellular components. Gene regulatory functions of lncRNAs have already been proposed during immune responses.43 The Th1-selective lincRNA NeST, by acting as an enhancer RNA, contributes to IFNγ expression, thereby controlling susceptibility to bacterial and viral pathogens.44,45 In contrast, lncRNAs repressing antiviral or enhancing proviral gene expression might aid in supporting viral replication, rendering them interesting targets for the development of new host-directed antiviral strategies.
Here, we identified the first human lncRNA, which functions in IAV propagation. Induction of VIN expression was observed in different virus infections, and VIN loss-of-function analysis revealed its importance during productive IAV replication. Nuclear expression of VIN suggests an involvement in gene-regulatory processes and our observation that VIN is functionally relevant during pathogenesis of IAV infection strengthens the view that lncRNAs are major players in diverse biological processes. Elucidating the mechanism of VIN action in more detail will broaden our understanding of lncRNAs in general and their implications in fighting infectious disease.
Materials and Methods
Cells and viruses
The A549 human lung epithelial cell line (CCL-185, ATCC-LGC) was grown in DMEM media (Invitrogen) supplemented with 4 mM l-glutamine, 4 mM sodium pyruvate, and 10% fetal calf serum (FCS, Biochrom) at 37 °C and 5% CO2. The Madin-Darby canine kidney cells (MDCK, CCL-34, ATCC-LGC) were grown in DMEM supplemented with 4 mM l-glutamine and 10% FCS. A549-ISRE luciferase reporter cells were generated via lentiviral transduction of pCignal Lenti-TRE-Reporter Gene (Qiagen, CLS-008L) of A549 human lung epithelial cells. The influenza virus strains A/WSN/33 (H1N1), A/Panama/2007/99 (H3N2), A/FPV/Bratislava/79 (H7N7), and B/Brisbane/60/2008 were grown in the allantoic cavities of 11-d-old embryonated chicken eggs. The Vesicular Stomatitis virus strain VSV Indiana was propagated in MDCK cells. Virus stocks were titrated by standard plaque assay on MDCK cells using an agar overlay medium.
Virus infection
Cells were washed with PBS and then infected with viruses at the indicated MOIs in infection buffer (PBS supplemented with 0.2% bovine serum albumin) for 1 h at room temperature. Cells were incubated for the indicated time periods at 37 °C in DMEM supplemented with 0.2% bovine serum albumin, 4 mM l-glutamine, and 100U/ml penicillin-streptomycin.
Microarray analysis
Microarray experiments were performed with dual-color hybridizations and independent dye-reversal color swap was applied to compensate for dye-specific effects. Quality control and quantification of total RNA was assessed using a NanoDrop 1000 UV-Vis spectrophotometer (Kisker) and an Agilent 2100 Bioanalyzer (Agilent Technologies). RNA labeling was performed with the Low RNA Input Linear Amplification Kit PLUS (Agilent Technologies). In brief, mRNA was reverse transcribed and amplified using an oligo-dT-T7 promoter primer, and cRNA was subsequently labeled with Cyanine 3-CTP or Cyanine 5-CTP. After precipitation and purification, 1.25 μg of each labeled cRNA was fragmented and hybridized to NCode™ Human Non-coding RNA Microarray (NCRAH, Invitrogen) or SurePrint™ G3 Human GE 8x60K Microarray (G4851B, Agilent) according to the supplier’s protocol. Scanning of microarrays was performed with 5 μm resolution using a G2565CA high-resolution laser microarray scanner (Agilent Technologies) with XDR extended range. Raw microarray image data were analyzed with the Image Analysis/Feature Extraction software G2567AA v. A.10.5.1 (Agilent Technologies) using default settings. The extracted MAGE-ML files were analyzed further with the Rosetta Resolver Biosoftware, Build 7.2.2 SP1.31 (Rosetta Biosoftware). Ratio profiles comprising single hybridizations were error-weighted and combined to create ratio experiments. The microarray data from this publication have been submitted to the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) and assigned the accession number GSE45399.
Re-annotation of microarrays was performed by BLAST analysis of 60mer oligonucleotide probe sequences from NCode™ and Sureprint™ G3 microarrays against the Ensembl human genome annotation (Release 68). Criterion for annotation was an unambiguous match to a transcript (minimum 95% identity). Omitting protein-coding genes and redundantly represented genes, probe sequences covered 3415 and 3201 unique ncRNAs on NCode™ and Sureprint™ G3, respectively.
RNA isolation and qRT-PCR
Total RNA was isolated by the TRIzol (Invitrogen) method or RNeasy mini kit (Qiagen) following the manufacturer’s protocol. Quantitative RT-PCR analysis was performed with the one-step SYBR-green method using the RNA-to-Ct assay in accordance with the manufacturer’s protocol (Applied Biosystems). One hundred nanograms of RNA were used for each reaction. Relative expression levels were determined by applying the ΔΔCt method using GAPDH as endogenous control and normalization to mock-treated cells. Primer sequences are given in Table 2.
RNA fractionation
6 × 105 cells were washed in 3 ml RSB (10 mM Tris, pH 7.4; 10 mM NaCl; 3 mM MgCl2) and lysed in 500 µl RSBG40 [10 mM Tris, pH 7.4; 10 mM NaCl, 3 mM MgCl2; 10% glycerol, 0.5% Nonident P-40; 0.5 mM dithiothreitol (DTT), and 100 U/ml rRNAsin (Promega)]. After 10 min incubation on ice, nuclei were pelleted at 7000 rpm for 3 min at 4 °C. The supernatant was recovered as cytoplasmic fraction. Nuclear pellets were resuspended two times in 200 µl RSBG40, incubated on ice for 5 min, pelleted, and cytoplasmic fractions pooled. Trizol was added to nuclei and cytoplasmic fractions and RNA and protein extracted following the Trizol procedure.
Immunoblotting
For immunoblotting, cells were washed with PBS and lysed in 1 × SDS sample buffer containing 75 mM TRIS-HCl (pH 6.8), 25% glycerol, 0.6% SDS, 7.5% β-mercaptoethanol, and 0.001% bromophenol blue. Protein lysates were loaded and separated on 10% SDS-polyacrylamide gels. Separated proteins were transferred to a PVDF membrane and detected using antibodies depicted in Table 1. Staining was performed with ECL western blotting detection reagent (Amersham).
Table 1. Antibodies used.
| Antibodies | ||
|---|---|---|
| Primary antibodies | Company | Dilution |
| mouse monoclonal β-Actin | Sigma-Aldrich | 1:3000 |
| mouse monoclonal viral ion channel protein | Santa Cruz | 1:1000 |
| rabbit polyclonal 14–3-3-epsilon (YWHAE) p(Ser58) (ABIN318720) | antibodies-online | 1:3000 |
| rabbit polyclonal Lamin A/C (2032) | Cell Signaling | 1:3000 |
| rabbit anti-H1N1 Hemagglutinin (ABIN399002) | antibodies-online | 1:1000 |
| mouse anti Influenza A Nucleoprotein (MCA0400) | AbD Serotec | 1:2500 |
| mouse anti Influenza B Nucleoprotein (BM3149) | Acris Antibody | 1:1000 |
| goat polyclonal anti Influenza Non-Structural Protein 1 | Santa Cruz | 1:1000 |
| mouse monoclonal anti-VSV Glycoprotein (P5D4) | Sigma-Aldrich | 1:500 |
| mouse anti-Digoxigenin IgG (11333062910) | Roche | 1:200 |
| Secondary antibodies | Company | Dilution |
| donkey anti-goat IgG HRP (sc2020) | Santa Cruz | 1:3000 |
| sheep anti-mouse IgG HRP (NA931) | Amersham | 1:3000 |
| donkey anti-rabbit IgG HRP (NA934) | Amersham | 1:3000 |
| Cy3-goat-anti-mouse IgG (115–165–146) | Dianova | 1:100 |
| Cy2-rabbit-anti-mouse IgG (315–225–003) | Dianova | 1:100 |
RNA-FISH
Cells were fixed in 3% paraformaldehyde for 15 min, rinsed in PBS, and permeabilized with 0.5% Triton X-100/ 5mM Vanadyl ribonucleoside complex (VRC, NEBiolabs) on ice for 10 min. Following 3x washes in PBS and 2 × SSC for 10 min, cells on coverslips were pre-hybridized for 1 h at RT (20% formamide, 10% dextran sulfate, 10% 20×SSC, 120 µg yeast tRNA, 0.5 µl rRNasin). In vitro-transcribed (Ambion MEGAscript T7) Digoxigenin-11-UTP (Roche)-labeled RNA hybridization probes were generated via cloning of GAPDH, RNU1-1, and VIN-specific sequences into pGEMT-Easy (Promega) (primer sequences Table 2). Cells were covered with 2× SSC, heated for 4 min at 95 °C and incubated with denatured probes in hybridization buffer (20% formamide, 10% dextran sulfate, 10% 20x SSC, 100 g yeast tRNA, 0.5 µl rRNasin) in a humidified chamber at 37 °C overnight. Stringency washes were performed three times with 2× SSC/ 50% formamide at 37 °C, three times with 2× SSC at 42 °C, three times with 1× SSC at 42 °C, and one time with 4× SSC at room temperature. For immunofluorescence labeling, cells were permeabilized with 4× SSC/0.1% Triton, blocked, and incubated with mouse anti-digoxigenin IgG (Roche, 1:100) for 1 h. After washing, secondary antibody staining was performed with Cy2-rabbit-anti-mouse IgG (315-225-003, Dianova) and Draq5 (Thermo Scientific, 62252) for 1 h. Influenza Nucleoprotein (NP-Cy3 conjugate) staining was subsequently applied. After washing, coverslips were mounted using Mowiol (Sigma, 324590). Confocal immunofluorescence images were acquired using a Leica TCS SP-E microspcope.
Table 2. Primer sequences.
| Primer sequences | ||
|---|---|---|
| Gene | primer forward 5′-3′ | primer reverse 5′-3′ |
| GAPDH | GGTATCGTGGAAGGACTCATGAC | ATGCCAGTGAGCTTCCCGTTCAG |
| RNU1–1 | ATACTTACCTGGCAGGGGAG | CAGGGGAAAGCGCGAACGCA |
| MX1 | GTTTCCGAAGTGGACATCGCA | GAAGGGCAACTCCTGACAGT |
| VIN | CTAGGAGACACCCGGACAGT | GCCCTGTGAGATGGGTTTAG |
| IFNβ | CAGCTCTTTCCATGAGCTAC | CAGCCAGTGCTAGATGAATC |
| ENST00000511543 | AACCACCCCATCTACCATCA | TGGCTCAGCTGTACGATTTG |
| ENST00000499418 | TGGAGCTTGCCTTCAACTTT | TTATTCTGCCACCAGGGAAG |
| ENST00000512341 | ACTCAGTGATTTGCCCAAGG | CCAACAGGAAGATGGGACTC |
Transfection and treatments
SiRNA transfections were performed with HiPerFect according to the fast-forward protocol (Qiagen). 50 000 A549 cells were transfected with 20 nM siRNA (Table 3) or unspecific Allstars control (1027281,Qiagen) for 48 h.
Table 3. siRNA sequences.
| siRNA sequences | |
|---|---|
| siRNA | sequence 5′-3′ |
| IAV Nucleoprotein | AAGGAUCUUAUUUCUUCGGAG |
| VIN siRNA 1 | CTGTGACATGTAGATTGCTAA |
| VIN siRNA 2 | CCGGAGCCGTTTACAGTTTGA |
| VIN siRNA 3 | CGCGCCCTGTCCCGCCATATA |
Polyinosinic:polycytidylic acid (Poly I:C, InvivoGen tlrl-picw-250) transfection was performed one day after cell seeding, using HiPerFect.
IFNβ treatment (I4151, Sigma) was performed 24 h after cell seeding. Endoribonuclease A (100 µg, Fermentas) treatments were performed on nuclear A549 cell extracts for 30 min at 37 °C followed by RNA isolation.
Replication assay
To quantify infectious virus particles in infected cell culture supernatants, 12 000 MDCK cells were seeded in 96-well plates. Twenty-four hours later, cells were washed, infected with a dilution series of cell culture supernatants, and incubated at room temperature for 1 h. Cells were incubated in DMEM supplemented with 0.2% bovine serum albumin, 4 mM l-glutamine, and antibiotics at 37 °C, 5% CO2 for 6 h, followed by fixation with 3.7% formaldehyde, antibody staining and automatic image processing, as described in “Indirect immunofluorescence labeling.”
Indirect immunofluorescence labeling and image analysis
Cells were fixed with 3.7% formaldehyde and permeabilized with 0.3% Triton X-100, 10% FCS in PBS. Samples were sequentially incubated with a primary antibody against viral nucleoprotein (Table 1) in PBS with 10% FCS, 0.1% Tween 20 for 1 h at room temperature, followed by an incubation with the secondary Cy3-conjugated antibody in PBS with 10% FCS, 0.1% Tween 20, and 0.1% Hoechst dye. Numbers of infected vs. non-infected cells were determined using automated microscopy (Olympus, Soft Imaging Solutions). Images were taken with DAPI and Cy3 filter sets (AHF-Analysetechnik). ScanR Analysis Software (Olympus Soft Imaging Solutions) was used to automatically identify and quantify influenza nuclear protein (NP) and cell nuclei.
WST-1 cell proliferation assay
Determination of host cell viability upon siRNA transfection was performed using cell proliferation assay WST-1 (Roche). WST-1 reagent was diluted 1:10 in the cell culture medium 48 h after siRNA transfection and incubated at 37 °C for 2 h. Absorbance was measured at 460 nm and at the reference wavelength 590 nm. Non-targeting siRNA Allstars and siPLK1 were used as a positive and negative control, respectively.
Luciferase assay
Induction of type I IFN by VIN siRNAs was analyzed using A549-ISRE luciferase reporter cells (see Cells and Viruses). One day after transfection with 20 nM siRNAs or treatment with 1 pg Poly I:C (InvivoGen) Beetle-Lysis-Juice (102512, p.j.k.) was added to the cells and luminescence was measured by Envision reader (PerkinElmer).
Bioinformatic tools
Secondary structure analysis was performed with RNAfold (Vienna package, http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Ethics Statement
All animal work was conducted in accordance with European regulations and approved by the Berlin state authorities, Landesamt für Gesundheit und Soziales (Reg No.: 0321/08).
Author Contributions
Winterling C and Karlas A designed the experiments, Winterling C, Koeppel M, and Koch M performed experiments, Garcia-Alcalde F and Winterling C analyzed the data, Winterling C wrote the manuscript. Meyer TF supervised the project and revised the manuscript.
Acknowledgments
The authors would like to thank Dr Hans Mollenkopf for assistance with the microarray analysis and Drs Kate Holden-Dye and Rike Zietlow for editing the manuscript. Winterling C was supported by the International Max-Planck Research School (IMPRS-IDI).
Glossary
Abbreviations:
- CPC
coding potential calculator
- DIG
digoxigenin
- IAV
influenza A virus
- IBV
influenza B virus
- IFN
interferon
- lincRNA
large intergenic ncRNA
- miRNA
microRNA
- MOI
multiplicity of infection
- MX1
myxovirus resistance 1
- OASL
2’-5′-oligoadenylate synthetase-like
- p.i.
post-infection
- poly I:C
polyinosinic:polycytidylic acid
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- SARS-CoV
acute respiratory syndrome coronavirus
- VIN
virus inducible lincRNA
- VSV
vesicular stomatitis virus
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/27504
References
- 1.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489:101–8. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amaral PP, Dinger ME, Mercer TR, Mattick JS. The eukaryotic genome as an RNA machine. Science. 2008;319:1787–9. doi: 10.1126/science.1155472. [DOI] [PubMed] [Google Scholar]
- 3.Mattick JS. RNA regulation: a new genetics? Nat Rev Genet. 2004;5:316–23. doi: 10.1038/nrg1321. [DOI] [PubMed] [Google Scholar]
- 4.Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–55. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Non-coding RNAs: regulators of disease. J Pathol. 2010;220:126–39. doi: 10.1002/path.2638. [DOI] [PubMed] [Google Scholar]
- 6.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–23. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JT. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009;23:1831–42. doi: 10.1101/gad.1811209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng X, Gralinski L, Armour CD, Ferris MT, Thomas MJ, Proll S, Bradel-Tretheway BG, Korth MJ, Castle JC, Biery MC, et al. Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. MBio. 2010;1:e00206–10. doi: 10.1128/mBio.00206-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medina RA, García-Sastre A. Influenza A viruses: new research developments. Nat Rev Microbiol. 2011;9:590–603. doi: 10.1038/nrmicro2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463:818–22. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe T, Watanabe S, Kawaoka Y. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe. 2010;7:427–39. doi: 10.1016/j.chom.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Chan EY, Li J, Ni C, Peng X, Rosenzweig E, Tumpey TM, Katze MG. MicroRNA expression and virulence in pandemic influenza virus-infected mice. J Virol. 2010;84:3023–32. doi: 10.1128/JVI.02203-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meliopoulos VA, Andersen LE, Brooks P, Yan X, Bakre A, Coleman JK, Tompkins SM, Tripp RA. MicroRNA regulation of human protease genes essential for influenza virus replication. PLoS One. 2012;7:e37169. doi: 10.1371/journal.pone.0037169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–9. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 17.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–14. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dinger ME, Pang KC, Mercer TR, Mattick JS. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput Biol. 2008;4:e1000176. doi: 10.1371/journal.pcbi.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Washietl S, Will S, Hendrix DA, Goff LA, Rinn JL, Berger B, Kellis M. Computational analysis of noncoding RNAs. Wiley Interdiscip Rev RNA. 2012;3:759–78. doi: 10.1002/wrna.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Volders PJ, Helsens K, Wang X, Menten B, Martens L, Gevaert K, Vandesompele J, Mestdagh P. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013;41:D246–51. doi: 10.1093/nar/gks915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–9. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–27. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bovolenta M, Erriquez D, Valli E, Brioschi S, Scotton C, Neri M, Falzarano MS, Gherardi S, Fabris M, Rimessi P, et al. The DMD locus harbours multiple long non-coding RNAs which orchestrate and control transcription of muscle dystrophin mRNA isoforms. PLoS One. 2012;7:e45328. doi: 10.1371/journal.pone.0045328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15:R17–29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 26.Washietl S, Hofacker IL, Lukasser M, Hüttenhofer A, Stadler PF. Mapping of conserved RNA secondary structures predicts thousands of functional noncoding RNAs in the human genome. Nat Biotechnol. 2005;23:1383–90. doi: 10.1038/nbt1144. [DOI] [PubMed] [Google Scholar]
- 27.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–31. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mattick JS. The genetic signatures of noncoding RNAs. PLoS Genet. 2009;5:e1000459. doi: 10.1371/journal.pgen.1000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hao L, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, Ahlquist P, Kawaoka Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008;454:890–3. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma YJ, Yang J, Fan XL, Zhao HB, Hu W, Li ZP, Yu GC, Ding XR, Wang JZ, Bo XC, et al. Cellular microRNA let-7c inhibits M1 protein expression of the H1N1 influenza A virus in infected human lung epithelial cells. J Cell Mol Med. 2012;16:2539–46. doi: 10.1111/j.1582-4934.2012.01572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song L, Liu H, Gao S, Jiang W, Huang W. Cellular microRNAs inhibit replication of the H1N1 influenza A virus in infected cells. J Virol. 2010;84:8849–60. doi: 10.1128/JVI.00456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–54. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.König R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, et al. Human host factors required for influenza virus replication. Nature. 2010;463:813–7. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ravasi T, Suzuki H, Pang KC, Katayama S, Furuno M, Okunishi R, Fukuda S, Ru K, Frith MC, Gongora MM, et al. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res. 2006;16:11–9. doi: 10.1101/gr.4200206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saha S, Murthy S, Rangarajan PN. Identification and characterization of a virus-inducible non-coding RNA in mouse brain. J Gen Virol. 2006;87:1991–5. doi: 10.1099/vir.0.81768-0. [DOI] [PubMed] [Google Scholar]
- 36.Ehrhardt C, Wolff T, Ludwig S. Activation of phosphatidylinositol 3-kinase signaling by the nonstructural NS1 protein is not conserved among type A and B influenza viruses. J Virol. 2007;81:12097–100. doi: 10.1128/JVI.01216-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, et al. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science. 2012;337:199–204. doi: 10.1126/science.1222213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen LL, Carmichael GG. Decoding the function of nuclear long non-coding RNAs. Curr Opin Cell Biol. 2010;22:357–64. doi: 10.1016/j.ceb.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–72. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clemson CM, Hutchinson JN, Sara SA, Ensminger AW, Fox AH, Chess A, Lawrence JB. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell. 2009;33:717–26. doi: 10.1016/j.molcel.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–30. doi: 10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147:358–69. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pang KC, Dinger ME, Mercer TR, Malquori L, Grimmond SM, Chen W, Mattick JS. Genome-wide identification of long noncoding RNAs in CD8+ T cells. J Immunol. 2009;182:7738–48. doi: 10.4049/jimmunol.0900603. [DOI] [PubMed] [Google Scholar]
- 44.Collier SP, Collins PL, Williams CL, Boothby MR, Aune TM. Cutting edge: influence of Tmevpg1, a long intergenic noncoding RNA, on the expression of Ifng by Th1 cells. J Immunol. 2012;189:2084–8. doi: 10.4049/jimmunol.1200774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomez JA, Wapinski OL, Yang YW, Bureau JF, Gopinath S, Monack DM, Chang HY, Brahic M, Kirkegaard K. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell. 2013;152:743–54. doi: 10.1016/j.cell.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.