

Abstract
Single B cell technologies, which avoid traditional hybridoma fusion and combinatorial display, provide a means to interrogate the naturally-selected antibody repertoire of immunized animals. Many methods enable the sampling of memory B cell subsets, but few allow for the direct interrogation of the plasma cell repertoire, i.e., the subset of B cells responsible for producing immunoglobulin in serum. Here, we describe the use of a robust and simple fluorescence-based technique, called the fluorescent foci method, for the identification and isolation of antigen-specific IgG-secreting cells, such as plasma cells, from heterogeneous bone marrow preparations. Following micromanipulation of single cells, cognate pairs of heavy and light chain variable region genes were recovered by reverse transcription (RT)-polymerase chain reaction (PCR). During the PCR, variable regions were combined with a promoter fragment and a relevant constant region fragment to produce two separate transcriptionally-active PCR (TAP) fragments that were directly co-transfected into a HEK-293F cell line for recombinant antibody expression. The technique was successfully applied to the generation of a diverse panel of high-affinity, functional recombinant antibodies to human tumor necrosis factor (TNF) receptor 2 and TNF derived from the bone marrow of immunized rabbits and rats, respectively. Progression from a bone marrow sample to a panel of functional recombinant antibodies was possible within a 2-week timeframe.
Keywords: monoclonal, antibody, fluorescent foci, TAP, PCR, plasma cell, bone marrow, IgG
Introduction
Monoclonal antibodies represent an important class of therapeutic molecule for treating serious human diseases. As of mid-2012, 34 monoclonal antibodies have been approved for use in the US or Europe, and an additional ~350 are currently in clinical trials.1 The criteria that therapeutic antibodies must meet in order to be successful are extensive. To ensure that antibodies are developed with the greatest chance of producing a positive outcome in a patient, an efficient platform to facilitate the discovery of lead molecules, with optimal characteristics, is essential.
Although the hybridoma technique described by Köhler and Milstein in 19752 has revolutionized the use of monoclonal antibodies, and it remains a widely used platform throughout industry, the technology is relatively inefficient. Its reliance on fusion of a B cell to a suitable myeloma partner, means that only a very small percentage of splenocytes from an immunized animal are immortalized (5 × 10−6 efficiency with conventional PEG fusion).3 As a result, the vast majority of B cells are not sampled, and it is possible that rare antibodies, with desirable properties, will not be identified and recovered. Hybridoma screening is also mostly restricted to rodent immunizations, limiting the potential diversity.
Display methodologies have also been widely adopted as a technology for producing monoclonal antibodies.4,5 One drawback of most display systems is that libraries are constructed through random combination of antibody variable region genes. Hence, natural cognate pairings that are evolved and selected for in vivo during an immune response are typically lost and unnatural variable region pairs combined unproductively, resulting in reduced specific diversity.6,7 Although naïve antibody libraries provide the opportunity to produce antibodies to targets that are challenging using conventional immunization-based methods, the resulting antibodies usually require in vitro affinity maturation to produce molecules with an acceptable potency profile. This engineering process can sometimes result in the introduction of liabilities into the molecule that affect stability and pharmacokinetics.8,9 The reliance on phage to display antibodies also produces a bias in the repertoire toward those molecules capable of being expressed by E. coli and displayed on a phage particle as an antibody fragment, such as a single-chain variable region fragment (scFv).6,10,11 For this reason some groups have moved to a eukaryotic system, such as yeast, to display the antibody fragments.10,12,13
More recently, there has been an emergence of platforms that allow the direct sampling of the immune repertoire via single B cell analysis, as reviewed by Tiller.14 These technologies avoid the inefficient hybridoma fusion step, thereby allowing a more thorough interrogation of the B cell population, improvement of the likelihood of finding rare antibodies with highly desirable properties, and production of large and diverse panels of antibody lead molecules. Due to the reliance on immunization, these techniques exploit the natural process of affinity, specificity and stability maturation,15,16 and retention of the natural heavy and light chain cognate pairing ensures that beneficial characteristics are preserved in the recombinant molecules.
Several technologies exist that enable monoclonal antibody generation from single B cells. Antigen-specific memory B cells expressing surface IgG have been exploited extensively as a source of monoclonal antibodies. For example, flow cytometry has been used to sort single, antigen-labeled B cells.17-20 B cell panning has also been used to select for antigen-specific memory B cells before recovery of variable region genes by reverse transcription (RT)-PCR.21-23 Alternatively, memory B cell culturing and screening followed by micromanipulation of single antigen-specific B cells24 or single-cell memory B cell cultures25 have also been successfully employed as methods of monoclonal antibody generation.
Flow cytometry has also been applied in the isolation of single plasmablasts. The most common method is to take blood from human donors 7 d following an immunization, vaccination or infection and isolate plasmablasts that appear transiently in the periphery during this small window.6,7,26,27 These plasmablasts are enriched for antigen-specificity and therefore represent a good pool from which to perform single-cell RT-PCR. Although these techniques are moderately efficient, i.e., 50% recovery of cognate VH-VL pairs from sorted B cells with as low as 10% of recombinant antibodies being specific for the target antigen,7 they are limited to larger organisms that allow significant bleed volumes to be taken. The system also relies on the use of a cocktail of antibody reagents specific to a number of cell-surface markers. For these reasons, it is challenging to apply the concept to species other than human.
The terminally-differentiated plasma cell subset of B cells, both the relatively stable population of long-lived plasma cells residing in the bone marrow and the short-lived plasma cells in the spleen and other secondary lymphoid organs, also represent an excellent source of high quality antibodies.28-39 Plasma cells represent <1% lymphoid cells, but are responsible for the production of the vast majority of circulating IgG.31,38 Therefore, following screening of an immune serum for a particular activity, it is an attractive option to “go fishing” for the plasma cells that are directly making the antibodies of interest. Plasma cells also benefit from an increased level of immunoglobulin mRNA compared with memory B cells,31,40,41 thereby facilitating the recovery of variable-region genes from single isolated cells. However, due to the low frequency of antigen-specific plasma cells in the bone marrow and secondary lymphoid organs of immunized animals and the lack of surface-associated IgG and other markers, flow cytometry has not been used extensively to interrogate the plasma cell subset from these important niches.
To exploit the high secretory capacity of plasma cells, a number of techniques have been developed that allow for the identification and isolation of antigen-specific cells. Manz et al.,42 and more recently Carroll and Al-Rubeai,43 described the use of a cell-surface affinity matrix to capture secreted immunoglobulin and allow for phenotypic screening via flow cytometry. The technique, however, has not been widely reported in the literature as a method for plasma cell isolation for the purpose of monoclonal antibody discovery. Babcook et al.44 described a hemolytic plaque assay that allowed the identification of plasma cells producing antibody against a target protein attached to sheep red blood cells. The use of microengraved array chips, designed to harbor and screen single plasma cells or activated B cells, has also been described.45-51 Although the current techniques that avoid flow cytometry provide conditions that favor efficient recovery of variable region genes from single cells,51 the methods described to date are limited to soluble antigen targets. The application of the above methods for the generation of antibodies to cell-surface receptor targets is challenging, and therefore represents a shortcoming in these platforms. Also, in the case of the microengraved array of B cells, the methods rely on specialized fabricated chips that may not be readily available in research laboratories.
Here, we report a robust fluorescence-based system that allows the identification and isolation of antigen-specific plasma cells. The technique, termed the “fluorescent foci method”, was performed on standard microscope slides and employed a micromanipulator device to isolate single antigen-specific IgG-secreting cells from the bone marrow of immunized rabbits and rats. Although the examples in this study were performed with soluble purified forms of antigen, due to the simplicity of the technique, the method is also applicable to the identification of plasma cells producing antibody specific to molecules expressed on the surface of cells. In the rat system, we employed an anti-rat CD138 (syndecan-1) monoclonal reagent to enrich for plasma cells prior to setting the assay up. Having isolated plasma cells from the bone marrow of a number of individual immunized animals, heavy and light chain variable region genes were recovered via single cell reverse-transcription (RT)-PCR in order to retain the natural heavy and light chain cognate pairing. During the PCR process, it was also possible to produce transcriptionally-active PCR (TAP) linear DNA fragments for both the heavy and light chains that encompassed the amplified variable region, a constant region fragment (including a poly-A signal sequence) and a human cytomegalovirus (HCMV) promoter region that could be used directly (without DNA purification) in a mammalian cell transfection to produce recombinant antibody. This high throughput method enabled the functional activity and binding profile of large numbers of recombinant antibody to be determined. The TAP process circumvents the need for traditional cloning methods, and when combined with the fluorescent foci method, enabled the generation of diverse panels of functional recombinant monoclonal antibodies from bone marrow preparations of immunized rats and rabbits within 2 weeks.
Results
Development of the fluorescent foci method for the isolation of antigen-specific plasma cells
The fluorescent foci method is a technique that allows the identification and isolation of antigen-specific IgG-secreting cells, such as plasma cells, from heterogeneous populations of cells. The method is extremely robust and efficient and does not require specialized fabricated microwell devices,45-47,49 but instead relies on standard glass microscope slides. IgG-secreting cells, a source of solid-phase antigen (such as a protein immobilized on a bead or a receptor expressed on a cell) and a FITC-labeled anti-Fcγ -specific secondary reagent are simply mixed together and then plated out as a monolayer on a glass slide. Up to 106 cells can be analyzed on a single slide, but typically 2‒4 × 105 cells are screened. Following a short static incubation at 37°C (30‒60 min), B cells secreting antigen-specific IgG can be visualized through the formation of a halo of fluorescence surrounding those B cells. This concentration of fluorescence forms as antibody accumulates on the solid-phase antigen in the immediate vicinity of the specific B cell (Fig. 1). Single cells can be subsequently isolated using a micromanipulator device and deposited in a PCR tube for variable region gene recovery.

Figure 1. Fluorescent foci method. (A) Schematic of the method. IgG-secreting B cells or plasma cells from a bone marrow sample are mixed with solid-phase immobilised antigen (either streptavidin beads (New England Biolabs) coated with biotinylated antigen, or cell expressing an antigen of interest) and goat anti-species Fcγ fragment-specific FITC conjugate (Jackson Immunoresearch). (B) Schematic showing a monolayer of plasma cells and antigen-coated beads or antigen-bearing cells. After static incubation of a monolayer at 37°C for 1 h, antigen-specific B cells can be identified by the presence of a fluorescent halo, formed by the complex of locally secreted antibody, antibody conjugate and solid-phase immobilised antigen, surrounding that B cell. (C) The left-hand image is a representative fluorescent halo formed against a biotinylated antigen immobilised on a 1 μm streptavidin bead photographed on an Olympus IX70 microscope using a fluorescence filter compatible with FITC. The middle image is the same frame captured under normal illumination conditions showing the presence of a single IgG secreting cell. The right-hand image is an example of a fluorescent halo formed by a B cell producing IgG specific for a receptor expressed on the surface of CHO cells.
We have used the technique extensively for the isolation of specific IgG-secreting cells from 96-well-plate polyclonal B cell cultures that have been pre-screened for activity.24 In this report, we used the fluorescent foci method to identify naturally-occurring, antigen-specific plasma cells that reside in bone marrow of both immunized rabbits and rats. Bone marrow preparations from animals that had generated measurable immune responses were used directly as a source of antigen-specific plasma cells without undertaking a pre-activation culture step. In the case of the rat samples, we used a rabbit anti-rat CD138 (syndecan-1) monoclonal antibody reagent that we had generated previously to enrich for plasma cells prior to assay set-up (see below). Fluorescent foci were successfully generated from all bone marrow preparations tested, hence confirming the presence of antigen-specific plasma cells and other IgG-secreting cells in these samples (details provided in subsequent sections).
Development and optimization of transcriptionally-active PCR (TAP)
We developed transcriptionally-active PCR (TAP) as a rapid method for generating recombinant antibody from amplified heavy and light chain variable region genes. The method provided us with a means of transiently transfecting HEK-293F cells using product directly generated from a PCR reaction. This circumvented the need to clone genes into expression vectors or even to purify fragments from the PCR reaction. Cognate pairs of variable region genes were amplified via 2 rounds of PCR (Fig. 2A). A primary PCR utilized gene-specific primers at both the 5′ and 3′ ends. The 5′ oligonucleotide set bound either in the leader sequence or at the 5′ end of the framework 1 region of the mature variable region sequence. The 3′ reverse primer set annealed to the CH1 or Cκ region. In the secondary PCR, a generic 5‘ forward oligonucleotide that annealed to a “tail” encoded at the 5′ end of the primary PCR product was used with a 3′ primer set that annealed in the J region. Not only did the secondary oligonucleotides introduce restriction sites to facilitate downstream cloning, but they also provided ~25 base-pair overlap regions at the 5′ end with a human cytomegalovirus (HCMV) promoter fragment and at the 3′ end with a heavy or light chain constant region fragment containing a poly-adenylation sequence. Then, in a tertiary PCR, variable region DNA, HCMV promoter fragment and constant region fragment were combined and amplified to produce two separate linear TAP products, one encoding the heavy chain the other the light chain. These were subsequently used directly in a transient transfection of HEK-293F cells to produce recombinant antibody. During optimization of the process, we noted that a 1 in 10 dilution of the secondary PCR product prior to seeding the tertiary PCR significantly increased the percentage of full-length, correctly assembled TAP product. Figure 2B shows the assembly of the three separate DNA fragments into the TAP product for both the rabbit and rat systems. This method was used for cDNA produced from single, antigen-specific plasma cells that had been identified and isolated via the fluorescent foci method.

Figure 2. Transcriptionally-active PCR (TAP). (A) Cognate pairs of variable region genes were amplified via 2 rounds of PCR. A primary PCR utilized gene-specific primers at both the 5′ and 3′ ends. The 5′ oligonucleotide set bound either in the leader sequence (L) or at the 5′ end of the framework 1 region of the mature variable region sequence. The 3′ reverse primer set annealed to the CH1 or Cκ region. In the secondary PCR, a generic 5 ‘ forward oligonucleotide which annealed to a “tail” encoded at the 5′ end of the primary PCR product was used with a 3′ primer set that annealed in the J region. The secondary oligonucleotides provided ~25 base-pair overlap regions; at the 5′ end with a human cytomegalovirus (HCMV) promoter fragment (pink) and at the 3′ end with a heavy or light chain constant region fragment (blue) containing a poly-adenylation sequence (green). Then, in a tertiary PCR, variable region DNA, HCMV promoter fragment and constant region fragment were combined and amplified to produce two separate linear TAP products, one encoding the heavy chain the other the light chain. (B) Agarose gel of rabbit and rat transcriptionally-active PCR (TAP) products. Lane 1, 2-log ladder (New England Biolabs); Lane 2, human CMV fragment; Lane 3, heavy chain variable region; Lane 4, heavy chain constant region fragment containing poly-adenylation sequence; Lane 5, assembled heavy chain TAP fragment; Lane 6, human CMV fragment; Lane 7, light chain variable region; Lane 8, light (kappa) chain constant region fragment containing poly-adenylation sequence; Lane 9, assembled light chain TAP fragment; Lane 10, 2-log ladder (New England Biolabs). Top row indicates the rat system which comprised rat variable regions assembled with mouse constant regions and the bottom row indicates the rabbit system.
Generation of recombinant anti-human tumor necrosis factor receptor 2 monoclonal antibodies from rabbit plasma cells
The rabbit system provides an attractive host from which to generate high quality monoclonal antibodies due to its robust immune response, quantity of immune cells from which to screen from and proteome sequence divergence compared with human and rodent.53-58 We wanted to confirm that it was possible to produce antigen-specific antibodies from plasma cells residing in the bone marrow of immunized rabbits. Four rabbits were immunized with purified human tumor necrosis factor receptor (TNFR)2 extracellular domain fused to rabbit Fc fragment and the sera titers were monitored via ELISA. All four rabbits demonstrated a significant immune response to the target antigen with sera reactivity being detectable at a 1:100 000 dilution (data not shown).
Because the bone marrow is the major niche supporting the survival of long-lived plasma cells, which are considered to be the main cell responsible for generating large amounts of IgG in serum, we harvested bone marrow cells from femurs and used them directly in a fluorescent foci experiment to identify human TNFR2-specific IgG-secreting plasma cells. The numbers of cells screened, the recovery of cognate variable region gene pairs from single cells and the subsequent determination of antigen reactivity of recombinant monoclonal antibodies generated from TAP products transiently transfected into HEK-293F cells are summarized in Table 1.
Table 1. Summary of recovery of recombinant anti-human TNFR2 antibodies from antigen-specific rabbit plasma cells.
| Number of bone marrow cells analyzed | Number of hTNFR2-specific plasma cells isolated | Number of VH and VL cognate gene pairs recovered | Number of hTNFR2-specific recombinant antibodies (via TAP) |
|---|---|---|---|
| 3.8 × 106 | 96 | 72 | 50 |
Bone marrow preparations from 4 immunized rabbits were analyzed via the fluorescent foci method using biotinylated human TNFR2 extracellular domain as the target antigen. Following RT-PCR, recoveries of heavy (VH) and light (VL) chain variable region gene pairs were determined by agarose gel electrophoresis of the secondary PCR products. Binding of recombinant antibody to TNFR2 was determined using supernatants from HEK-293F cells which had been transiently co-transfected with heavy and light chain TAP products. The assay was performed using a fluorescence-based homogeneous format with biotinylated human TNFR2 immobilised on Superavidin beads and an anti-rabbit Fcγ-Cy5 secondary reagent.
Following the screening of 3.8 × 106 bone marrow cells, 96 antigen-specific plasma cells were isolated (0.0025% frequency). Plasma cells were successfully recovered from all four rabbits over a period of 2 d. RT-PCR from the single cells enabled the recovery of heavy and light chain gene pairs from 72 cells (75%), with the subsequent confirmation of binding activity in 50 recombinant antibodies produced from transiently-transfected TAP products in HEK-293F cells.
Expression levels of recombinant rabbit IgG in supernatants of HEK-293F cultures were determined using a sandwich ELISA. Concentrations varied greatly across the panel, but levels as high as 2.5 µg/ml were achieved (Fig. 3). The mean concentration across the data set was 617 ng/ml. At these concentrations, many further assays were possible, including protein-based biochemical screens and cell-based functional assays.

Figure 3. Concentrations of recombinant rabbit IgG generated from TAP products expressed in HEK-293 cells. Histogram plot showing the range of IgG concentrations produced from HEK-293 cells transiently co-transfected with cognate pairs of heavy and light chain transcriptionally active PCR (TAP) products. Concentrations were determined using a sandwich ELISA with a rabbit IgG standard.
Biacore analysis of recombinant anti-human TNFR2 rabbit IgG molecules produced from TAP products
To estimate the affinity of these recombinant antibodies and to assess their ability to block binding of human TNF to TNFR2, we performed a Biacore screen. In this assay, recombinant rabbit IgG was captured from HEK-293F cell culture supernatant before hTNFR2, at a single concentration, was applied to the surface to provide an estimate of KD. Having determined binding kinetics, human TNF was then flowed over the chip in order to identify antibodies that directly blocked binding of TNF to TNFR2 (Table 2). It should be noted that the primary objective of the Biacore experiment was to estimate the affinity of binding. Therefore, antibodies that exhibited fast off-rates and/or low capture levels of TNFR2 could not be reliably identified as blockers or non-blockers of TNF binding. For example, DF119 had an affinity of ~5.7 nM, with relatively fast on and fast off kinetics. Following off-rate measurements, the response units of TNFR2 had dropped from 17 to 2, which made the subsequent TNF association step very challenging because of the low levels of receptor remaining on the surface and the fast off-rate of receptor from the antibody that would continue to feature in the TNF binding step. It is possible that this and similar antibodies were indeed blocking antibodies, but further analysis considered outside the scope of this work would have been required to confirm this conclusion.
Table 2. Biacore analysis of the recombinant rabbit IgG molecules generated from TAP products.
| Sample number | TNFR2 Binding (RU) | TNFR2 level following off-rate step (RU) | TNF Binding (RU) | TNF Blocker? | TNFR2 On Rate (M−1s−1) | TNFR2 Off Rate (s−1) | TNFR2 Affinity (Estimated KD) (pM) |
|---|---|---|---|---|---|---|---|
| DF107 | 11 | 9 | 1 | Yes | 5.30E+05 | 2.10E-04 | 387 |
| DF109 | 4 | 1 | 1 | nd | 2.50E+05 | 5.00E-04 | 2017 |
| DF113 | 25 | 24 | 28 | No | 9.80E+05 | 3.70E-05 | 38 |
| DF116 | 6 | 2 | 1 | nd | 1.20E+05 | 8.20E-04 | 6697 |
| DF119 | 17 | 2 | 1 | nd | 2.00E+06 | 1.10E-02 | 5722 |
| DF120 | 3 | 0 | 1 | nd | Fast-On and Fast-Off | nd | |
| DF121 | 4 | 2 | 1 | nd | Fast-On and Fast-Off | nd | |
| DF122 | 18 | 18 | 31 | No | 1.90E+05 | 4.60E-05 | 240 |
| DF123 | 1 | -3 | 1 | nd | Very Low-Binder | nd | |
| DF128 | 26 | 20 | 23 | No | 4.50E+06 | 2.90E-04 | 65 |
| DF130 | 67 | 6 | -1 | nd | 2.50E+06 | 3.30E-03 | 1320 |
| DF131 | 28 | 23 | 30 | No | 1.90E+06 | 1.40E-04 | 76 |
| DF132 | 8 | -3 | 2 | nd | 8.10E+06 | 2.40E-01 | 29954 |
| DF134 | 2 | -1 | 0 | nd | 3.70E+05 | 3.10E-04 | 835 |
| DF136 | 27 | 21 | 0 | Yes | 1.00E+06 | 2.90E-04 | 279 |
| DF139 | 5 | 1 | 0 | nd | 7.40E+06 | 4.20E-03 | 576 |
| DF143 | 50 | 44 | 45 | No | 1.90E+06 | 1.40E-04 | 73 |
| DF145 | 37 | 12 | 1 | nd | Fast-On and Fast-Off | nd | |
| DF151 | 19 | 9 | 0 | Yes | 1.20E+06 | 7.90E-04 | 661 |
| DF159 | 71 | 11 | 1 | nd | Fast-On and Fast-Off | nd | |
| DF161 | 3 | 2 | 1 | nd | Very Low-Binder | nd | |
| DF163 | 33 | 24 | 1 | Yes | 4.00E+05 | 2.90E-04 | 723 |
| DF164 | 82 | 71 | 72 | No | 1.80E+06 | 1.40E-04 | 79 |
| DF176 | 45 | 22 | 21 | No | 2.50E+06 | 9.00E-04 | 368 |
| DF179 | 59 | 8 | 1 | nd | 3.90E+06 | 8.70E-03 | 2221 |
| DF180 | 55 | 51 | 62 | No | 2.70E+06 | 8.20E-05 | 31 |
| DF185 | 9 | 7 | 7 | No | 1.50E+05 | 4.90E-04 | 3179 |
| DF187 | 78 | 73 | 0 | Yes | 6.70E+06 | 9.80E-05 | 15 |
| DF189 | 65 | 7 | 1 | nd | 4.10E+06 | 8.90E-03 | 2197 |
| DF191 | 37 | 30 | 37 | No | 8.50E+06 | 2.30E-04 | 27 |
| DF192 | 1 | 0 | 4 | nd | Very Low-Binder | nd | |
| DF195 | 24 | 23 | 0 | Yes | 4.60E+05 | 5.40E-05 | 118 |
| DF196 | 1 | 0 | 1 | nd | Very Low-Binder | nd | |
| DF198 | 65 | 57 | 63 | No | 1.40E+07 | 1.50E-04 | 11 |
Data are shown for 34 antibodies that exhibited binding to TNFR2 in the Biacore assay. Recombinant rabbit IgG was captured from HEK-293F cell culture supernatant before hTNFR2 at 20 nM was applied to the surface. Having determined binding kinetics, human TNF at 150 nM was then flowed over the chip in order to identify those antibodies that directly blocked binding of TNF to TNFR2. Response units of capture (RU) is shown for TNFR2, before and after off-rate determinations. Response units of TNF binding is also shown, this was performed after the off-rate determination vs. TNFR2. Estimates of affinity (KD) are quoted in pM and were derived from a 1:1 binding model. In situations where the Biacore software was unable to fit the interaction or when very low levels of binding were observed, affinity values were not determined (marked as nd). Blocking antibodies are in boldface.
Biacore data for 34 recombinant antibodies is shown in Table 2. Six anti-TNFR2 blocking antibodies were identified using this rapid Biacore screen. All six had estimated KD values in the sub-nM range, with DF187 demonstrating the highest affinity binding to TNFR2 measured with an estimated KD of 15 pM. Several very high affinity non-blockers were identified, including DF198, which had an estimated affinity of 11 pM. In the Biacore traces for DF187 (blocker) and DF198 (non-blocker) shown in Figure 4, TNF-binding can be clearly observed when TNFR2 is captured by DF198, whereas no significant association can be detected when DF187 is bound to the receptor.

Figure 4. Biacore traces of a blocking and a non-blocking anti-human TNFR2 rabbit IgG. Recombinant rabbit IgG was captured onto a Biacore CM5 sensor chip from HEK-293F cell culture supernatant using an anti-rabbit Fcγ-specific capture antibody. At time 0 s human TNFR2 was applied to the chip surface. Having determined binding kinetics, after 1200 s, human TNF was then injected to identify those antibodies that directly blocked binding of human TNF to TNFR2. DF187 is an example of a high affinity (15 pM) blocking antibody and DF198 is an example of a high affinity (11 pM) non-blocking antibody.
Diversity assessment of rabbit anti-human TNFR2 antibodies
To determine the diversity of the antibodies that gave a positive result in the Biacore experiment, we generated DNA sequences from 34 cloned variable region genes. Although it is possible to sequence the secondary PCR products directly, we used plasmid DNA to generate sequences for the variable region genes because the quality of the data was better. We performed a Principal Components Analysis (PCA)52 as a means to reduce the dimensionality of the data and generate an easy to interpret 2-dimensional data plot that illustrated the extent of diversity in our recombinant antibody panel (see materials and methods section). The distance between two points is directly proportional to sequence identity based on a concatenated sequence encompassing all six complementarity-determining regions (CDRs). Identical sequences resulted in the co-localization of data points on the 2-D plot. By using color as a measure of blocking activity in the Biacore (green representing blocking) and the size of the data point as an indication of affinity (larger being of higher affinity, i.e., lower KD), information regarding sequence uniqueness and functionality can be visualized on the same plot.
Using this method of visualization, it is clear that there was extensive diversity in the antibody panel likely representing the broad coverage of multiple epitopes on TNFR2 (Fig. 5). Cluster analysis enabled antibodies to be assigned into families. A family encompassed antibodies that demonstrated over 80% sequence identity across the CDRs. This analysis suggested that there were 24 unique families of antibody. Notably, the six blocking antibodies were all different and unique within the data set. These data strongly support the concept that the bone marrow-derived plasma cell repertoire represents a good source of highly diverse and high affinity antibodies in the rabbit.

Figure 5. Diversity assessment of rabbit anti-human TNFR2 antibodies. For each antibody, the 6 CDRs were concatenated, aligned in a pairwise and comprehensive manner to generate a sequence distance value. We performed a Principal Components Analysis (PCA) (Pearson, 1901)52 as a means to reduce the dimensionality of this data and generate an easy to interpret 2-dimensional data plot that illustrated the extent of diversity in our recombinant antibody panel. Data for principle component (PC) 1 and 2 are shown on the X and Y axis respectively. Cluster analysis enabled antibodies to be assigned into families. A family encompassed antibodies that demonstrated over 80% sequence identity across the CDRs. Families containing multiple sequences have been circled (only those data points completely within the circle are considered to be in a family). All other sequences were considered unique. Identical sequences are co-located on the 2-D plot, but indicated with multiple identifier flags. Blocking activity in the Biacore is highlighted by color, with blockers represented in green, non-blockers in red and samples where blocking could not be determined (nd) in blue. The size of the data points are indicative of affinity, with large circles representing high affinity (maximum KD of 11 pM) and smaller circles representing lower affinity (minimum KD of 30 nM). Where affinity values and blocking data are missing, the data points are shown as blue squares.
Generation of recombinant anti-human TNF monoclonal antibodies from rat plasma cells
Despite the rabbit representing an attractive source from which to generate monoclonal antibodies, rodents remain the preferred route to generate monoclonal antibodies destined for clinical development.59 For this reason, we wanted to confirm that the bone marrow of rodents also provided us with an attractive source of antigen-specific plasma cells from which high quality monoclonal antibodies could be produced. We used rats that had been immunized with human TNF, a therapeutically-relevant and well-studied target. Sera titers from five immunized rats confirmed that an effective immune response had been produced in these animals with TNF binding activity observed at a dilution of 1:100 000 (data not shown).
As with the work in the rabbit, we wanted to confirm that the bone marrow-derived plasma cell repertoire harbored cells likely to be responsible for producing antigen-specific IgG that was identified in the serum. Again, we used a combination of the fluorescent foci method, with biotinylated human TNF immobilized on streptavidin beads, and the TAP method to rapidly generate a panel of recombinant monoclonal antibodies.
To improve the efficiency of the fluorescent foci step, we enriched plasma cells from bone marrow preparations using a rabbit anti-rat CD138 reagent that we had generated previously. CD138 (syndecan-1) is a well-known and highly expressed marker of plasma cells in both mouse and human systems.60-62 We generated an antibody specific for the rat CD138 molecule through immunization of rabbits, followed by screening of activated B cell culture supernatants and subsequent variable region cloning from single B cells using a method similar to that described by Lightwood et al.24 (see materials and methods). The rabbit anti-CD138 antibody was then used to purify plasma cells from bone marrow samples via magnetic-activated cell sorting (MACS). Figure 6 shows a flow cytometric analysis of the starting bone marrow material (A), the enriched CD138+ population (B) and the CD138- flow-through (C) stained with the rabbit-anti-rat CD138-PE reagent (UCB) and a mouse anti-rat CD45R FITC antibody (BD biosciences). As discussed, CD138 is thought to be highly expressed on plasma cells whereas CD45R is down-regulated. Highly efficient purification of rat CD138+ cells from a rat bone marrow sample was achieved. Notably, we isolated both CD138+/CD45Rlow and CD138+/CD45Rhigh populations, but we have not separated the two populations and confirmed the location of the antigen-specific plasma cells. Although these CD138+ cells are not likely to be exclusively plasma cells, we have previously shown that this step provides an approximate 4-fold enrichment of IgG-secreting cells (n = 5 rat experiments, data not shown), thereby increasing the frequency of target plasma cells.
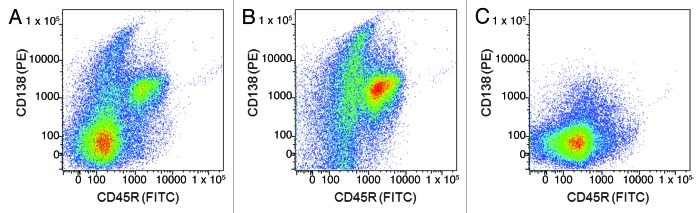
Figure 6. Purification of CD138+ cells from rat bone marrow. Bone marrow cells from immunized rats were labeled with a rabbit anti-rat CD138 monoclonal antibody reagent conjugated to phycoerythrin (PE). CD138+ cells were then enriched using magnetic-activated cell sorting (MACS) (Miltenyi Biotec). Flow cytometry analysis of the starting bone marrow material (A), the enriched CD138+ population (B) and the CD138- flow through (C) stained with the rabbit-anti-rat CD138-PE reagent (in house reagent) and a mouse anti-rat CD45R FITC antibody (BD biosciences).
We enriched CD138+ cells from the bone marrow of five individual rats and used this preparation in a fluorescent foci experiment. The numbers of CD138+ cells screened, the recovery of cognate variable region gene pairs from single cells and the subsequent determination of antigen reactivity of recombinant monoclonal antibodies generated from TAP products transiently transfected into HEK-293F cells are summarized in Table 3. Following the screening of 2.5 × 106 CD138+ bone marrow cells, 287 antigen-specific plasma cells were isolated (0.01% frequency). Plasma cells were successfully recovered from all five rats. RT-PCR from the single cells enabled the recovery of heavy and light chain gene pairs from 226 cells (79%), with the subsequent confirmation of binding activity in 147 recombinant IgG produced from transiently-transfected TAP products in HEK-293F cells.
Table 3. Summary of recovery of recombinant anti-human TNF antibodies from antigen-specific rat plasma cells.
| Number of CD138+ bone marrow cells analyzed | Number of h TNF -specific plasma cells isolated | Number of VH and VL cognate gene pairs recovered | Number of hTNF-specific recombinant antibodies (via TAP) |
|---|---|---|---|
| 2.5 × 106 | 287 | 226 | 147 |
CD138+ bone marrow cells from 5 immunized Sprague Dawley rats were analyzed via the fluorescent foci method using biotinylated human TNF as the target antigen. Following RT-PCR, recoveries of heavy (VH) and light (VL) chain variable region gene pairs were determined by agarose gel electrophoresis of the secondary PCR products. Binding of recombinant IgG to TNF was determined using supernatants from HEK-293F cells which had been transiently co-transfected with heavy and light chain TAP products. The assay was performed using a fluorescence-based homogeneous assay with biotinylated human TNF immobilized on Superavidin beads and a goat anti-rat Fcγ-specific-Cy5 secondary reagent.
Expression levels of recombinant IgG in supernatants of HEK-293F cultures were determined using a sandwich ELISA. Concentrations varied greatly across the panel, but levels as high as 3.5 ug/ml were achieved (Fig. 7). The mean concentration across the data set was 628 ng/ml. This result was very similar to that observed with the recombinant rabbit IgG molecules.

Figure 7. Concentration and blocking activity of recombinant anti-TNF IgG generated from TAP products expressed in HEK-293 cells Scatter plot showing the range of IgG concentrations produced from HEK-293 cells transiently co-transfected with cognate pairs of heavy and light chain transcriptionally active PCR (TAP) products plotted against neutralisation activity. Concentrations were determined using a sandwich ELISA with a mouse IgG standard. TNF neutralisation was determined using a HEK-293-CD40-BLUE cell reporter assay (Invivogen).
Determination of functional activity of recombinant rat-anti- TNF antibodies generated from TAP products
To identify antibodies capable of neutralizing the biological activity of TNF, we performed a cell-based TNF reporter assay. The assay utilized HEK-293-CD40-BLUE cells (Invivogen) engineered to secrete alkaline phosphatase in response to a number of stimuli operating through the NFκB pathway, including human TNF. Antibody-containing supernatants from HEK-293F transfections were used directly in this assay at a single dilution of 1:2.5. As seen in Figure 7, a number of highly potent neutralizing antibodies were identified. Blocking activity was observed with a range of antibodies at various concentrations, i.e., not only in those with very high levels of expression. There were 40 antibodies that exhibited greater than 50% inhibition in this assay. Twenty-three of the highest blockers were selected and successfully cloned into plasmid expression vectors to allow for further characterization of antibodies and DNA sequence analysis of the variable regions.
Blocking activity and affinity data for cloned recombinant rat anti- TNF Fab fragments
Following cloning and re-expression as recombinant Fab molecules (rat variable region-mouse constant regions), antibodies were profiled in the HEK-293-CD40-BLUE reporter assay at a number of concentrations to enable the estimation of IC50 values and determine the maximum percentage inhibition. Antibodies were also analyzed in a Biacore experiment to estimate binding affinities. As shown in Table 4, a large number of antibodies were capable of high affinity binding to human TNF and possessing significant neutralization activity. However, three antibodies were identified as having exceptionally good profiles. TDF0083, TDF0261 and TDF0293, all had an estimated affinity for human TNF below 100 pM, an IC50 value <10 ng/ml and attained > 95% maximum inhibition. With respect to IC50, these Fab molecules were 32-fold, 9-fold and 6-fold more potent in this assay than a positive control, etanercept (human TNF-binding therapeutic), respectively (on a molar basis, correcting for differences in molecular weights). These data demonstrate that bone marrow-derived plasma cells provide a valuable source of high affinity, functional antibodies from immunized rats and that the fluorescent foci method is a viable tool to interrogate this repertoire.
Table 4. Blocking activity and affinity values for anti- TNF antibodies.
| Sample number | Blocking (IC50) (ng/ml) | Max Inhibition (%) | TNF On Rate (M−1s−1) |
TNF Off Rate (s−1) |
Affinity (Estimated KD) (pM) |
|---|---|---|---|---|---|
| TDF0008 | > 2927.2 | 34.30 | 3.00E+06 | 1.82E-03 | 607 |
| TDF0012 | 3.29 | 89.92 | Low binding | nd | |
| TDF0024 | 10791.02 | 60.03 | 1.12E+06 | 6.01E-03 | 5350* |
| TDF0030 | 1177.18 | 67.31 | 8.95E+05 | 2.84E-04 | 317 |
| TDF0047 | 1704.46 | 72.33 | 3.33E+06 | 2.99E-03 | 900 |
| TDF0082 | 35.43 | 94.93 | 3.70E+06 | 6.63E-03 | 1795 |
| TDF0083 | 1.05 | 106.38 | 1.28E+06 | 7.52E-05 | 59 |
| TDF0091 | 981.34 | 106.00 | 5.14E+06 | 2.05E-03 | 399 |
| TDF0164 | > 1419.38 | 48.20 | 1.38E+06 | 7.22E-03 | 5236* |
| TDF0168 | 123.47 | 106.76 | 1.48E+06 | 2.58E-04 | 174 |
| TDF0173 | 519.15 | 87.56 | 1.93E+06 | 1.25E-03 | 649 |
| TDF0174 | 1067.73 | 96.45 | 2.12E+06 | 1.19E-03 | 560 |
| TDF0178 | 501.86 | 96.26 | 3.93E+06 | 1.22E-03 | 310 |
| TDF0180 | > 1287.3 | 17.08 | 6.46E+06 | 8.52E-03 | 1320 |
| TDF0183 | 372.05 | 98.91 | 3.71E+06 | 1.08E-03 | 290 |
| TDF0187 | 132.20 | 90.39 | 2.17E+06 | 2.75E-04 | 127 |
| TDF0245 | > 29.224 | 35.34 | 3.35E+06 | 3.16E-03 | 943 |
| TDF0260 | > 3073.78 | 27.01 | 7.04E+06 | 1.09E-02 | 1543 |
| TDF0261 | 3.67 | 102.88 | 2.39E+06 | 1.50E-04 | 63 |
| TDF0273 | 17.24 | 106.76 | 3.45E+06 | 5.06E-04 | 146 |
| TDF0284 | 40.00 | 93.89 | 4.83E+06 | 1.18E-03 | 245 |
| TDF0289 | > 1526.97 | 47.45 | Low binding | nd | |
| TDF0293 | 6.05 | 95.79 | 2.19E+06 | 1.62E-04 | 74 |
| Etanercept | 102 | 100 | nd | nd | nd |
Data are shown for 23 antibodies (produced from transiently expressed Fab molecules using cloned variable region genes) that originally demonstrated > 50% inhibition of TNF activity as a recombinant antibody derived from TAP products (Fig. 7). Functional activity was determined using the HEK-293-CD40-BLUE system (Invivogen) stimulated with human TNF. Maximum inhibition and estimated IC50 values are presented. A Z’ value of 0.67 was derived for the functional assay. For the Biacore assay, recombinant Fab was captured from cell culture supernatant before h TNF at 10 nM was applied to the surface and binding kinetics measured. Estimates of affinity (KD) are quoted in pM. All estimates of KD were derived from a 1:1 binding model except for those marked with an asterisk. For these, KD was derived from the ka1 and kd1 of a bivalent binding model. In situations where the Biacore software was unable to fit the interaction or when very low levels of binding were observed, affinity values were not determined (marked as nd). Antibodies which demonstrated sub-100 pM binding, >95% maximum inhibition and had an IC50 <10 ng/ml are in boldface. As a comparator, blocking data for etanercept is also shown in ng/ml.
Diversity assessment of rat anti-human TNF antibodies
We determined the diversity of the panel of 23 antibodies that had been selected on the basis of blocking activity through DNA sequencing of the cloned variable region genes. Again, we used principle component analysis52 to assess diversity and used the same 2-D plot method to illustrate sequence variability in the anti-TNF antibody panel. Considerable sequence variability was again observed, with high affinity antibodies being broadly distributed across a highly diverse set of sequences (Fig. 8). Cluster analysis enabled antibodies to be assigned into families. A family encompassed antibodies that demonstrated over 80% sequence identity across the CDRs. This analysis suggested the presence of 15 unique families. The two highly potent blocking antibodies, TDF0261 and TDF0293, had highly homologous sequences and, as expected, were derived from the same animal. However, the other lead molecule, TDF0083, possessed a completely unrelated and unique sequence and was isolated from a separate animal. As with the rabbit, these data suggest that the bone marrow-derived plasma cell repertoire represents a good source of highly diverse antibodies in the rat.

Figure 8. Diversity assessment of rat anti-human TNF antibodies. For each antibody, the 6 CDRs were concatenated, aligned in a pairwise and comprehensive manner to generate a sequence distance value. We performed a Principal Components Analysis (PCA) (Pearson, 1901)52 as a means to reduce the dimensionality of the data and generate an easy to interpret 2-dimensional data plot that illustrated the extent of diversity in our recombinant antibody panel. Data for principle component (PC) 1 and 2 are shown on the X and Y axis respectively. Cluster analysis enabled antibodies to be assigned into families. A family encompassed antibodies that demonstrated over 80% sequence identity across the CDRs. Families containing multiple sequences have been circled (only those data points completely within the circle are considered to be in a family). All other sequences were considered unique. Identical sequences are co-located on the 2-D plot but indicated with multiple identifier flags. TNF-blocking activity in a HEK-293-CD40-BLUE cell reporter assay (Invivogen) is highlighted by intensity of color, with the darkest green coloring indicating the highest IC50 activity. The size of the data points are indicative of affinity, with large circles representing high affinity (maximum KD of 59 pM) and smaller circles representing lower affinity (minimum KD of 5.4 nM).
Discussion
A number of single B cell technologies that allow the efficient sampling of the B cell repertoire have been developed in recent years.14 Despite these advances, most methods do not enable the screening of antigen-specific plasma cells and other IgG-secreting B cell subsets due to the lack of surface IgG32,33,63,64 and the difficulty in culturing these cells.65-67 In this study, we described the use of the fluorescent foci method to isolate naturally-occurring antigen-specific IgG-secreting cells, such as plasma cells, directly from the bone marrow of immunized rabbits and rats. Unlike similar techniques that employ elaborate microwell devices,45-51 the fluorescent foci method uses standard microscope slides facilitating the sampling of up to 106 cells per slide. A skilled scientist can analyze 107 cells, over 10 or more slides, in a single day and isolate several hundred antigen-specific plasma cells. This provides a throughput of plasma cell interrogation and isolation that is not matched by the microarray systems or other techniques for identifying and recovering antigen-specific IgG-secreting cells. An important feature of the fluorescent foci method is that the entire process, including incubation and single cell isolation, is performed in a short period of time (typically <2 h), which favors plasma cell viability over the course of the experiment. This, along with the observation that plasma cells possess very high levels of mRNA41,68 facilitates the efficient recovery of variable region genes from single cells.
The use of a monolayer on a simple glass microscope slide also facilitates the broad application of the fluorescent foci method to multiple target classes, including soluble and membrane-bound targets on cells. For example, an antigen can either be immobilized on a bead or alternatively expressed on the surface of a cell. Both sources of antigen can be used in a fluorescent foci experiment and have been successfully employed for the recovery of antigen-specific IgG secreting cells. This is particularly attractive when the target molecule cannot be readily purified as a soluble molecule and in situations where conformational or functional integrity of a cell-surface receptor is compromised through the extraction of the molecule from the cell membrane environment.
Here, we used the fluorescent foci method to sample the IgG-secreting B cell repertoire from bone marrow of immunized rabbits and rats. The bone marrow is the major niche that supports the stable survival of long-lived plasma cells and these cells are responsible for secreting the vast majority of IgG present in serum.33,38,69,70 Thus, one might expect desirable attributes, revealed in analysis of sera samples, to be directly mirrored in the plasma cell antibody repertoire rather than in the memory B cell repertoire for example. This enables the focused sampling of individual animals that are most likely to harbor cells producing interesting functional antibodies. We have also demonstrated that the bone marrow provides different families of antibodies to those produced from cultured memory B cells derived from the spleen or blood (data not shown). For example, only one of the six high affinity anti-TNFR2 blocking antibodies described in this work (DF195) was identified through screening of over 107 cultured memory B cells from the same immunized rabbits. Although other antibodies with similar properties were isolated from memory B cells and not all memory B cells were sampled, it is tempting to suggest that the bone marrow-derived plasma cell population possesses a unique repertoire of antibodies. Therefore, to maximize the diversity in recombinant antibody panels, it is important to have technologies that sample both the plasma cell and memory B cell repertoires of immunized animals. There is also evidence suggesting that the plasma cell compartment is populated by germinal center B cells that are selected on the basis of high affinity antigen-binding,29,71,72 highlighting their attractiveness as a source of monoclonal antibodies. Although the mechanism by which plasma cells are selected for residence in the bone marrow is poorly understood, it is thought that during an immune response, recently-generated, highly mobile plasmablasts migrate to the bone marrow before they differentiate into plasma cells.29,73 If space becomes limiting in the bone marrow niche (the estimated capacity in mice is 106 plasma cells29), this may result is displacement and subsequent apoptosis of immobile resident long-lived plasma cells. Therefore, following repeated immunizations the bone marrow is likely to become highly polarized in favor of antigen-specificity.30 In mice, it has been estimated that between 2 and 10% of the plasma repertoire can become specific to a single antigen following an immunization regime.37,74 In immunized rats, we have observed up to 30% of the bone marrow-derived plasma cells population exhibiting antigen-specificity (data not shown). These factors suggest that bone marrow represents an extremely attractive compartment from which to isolate antigen-specific B cells and plasma cells and ultimately produce high quality recombinant monoclonal antibodies.
Using this novel method, we successfully identified and isolated human TNFR2-specific IgG-secreting cells, presumably plasma cells, from the bone marrow of immunized rabbits. Following RT-PCR from single cells, we recovered cognate heavy and light chain variable region pairs from 75% of the cells. Using a technique called transcriptionally-active PCR (TAP), we were able to produce recombinant monoclonal Fab fragments from HEK-293F cells transiently-transfected with heavy and light chain linear DNA fragments comprising a human cytomegalovirus (HCMV) promoter fragment, the amplified variable region, a constant region and poly A signal sequence. The DNA products of TAP were used directly in the HEK-293F transfection without the need to perform a purification step. Expression levels of monoclonal antibodies in conditioned media varied considerably, but we were able to demonstrate binding to human TNFR2 in 50 samples (from 96 isolated plasma cells). The reason for the attrition from PCR recovery to binding activity (72 samples down to 50) is unclear, but may be due to poor expression of some recombinant Fab molecules, resulting in the inability to detect binding. Alternatively, in situations where expression was observed (eight negative samples produced > 200 ng/ml; data not shown), negative-binders may have arisen as a result of the isolation of a B cell that was not responsible for generating the observed fluorescent foci or through a random contamination event (although negative controls during PCR did not produce products). Despite this, the efficiency of the platform described here is significantly better than that reported for similar methods based on microwell devices. For example, Ozawa et al.29 isolated 189 hen egg white lysozyme (HEL)-specific rabbit B cells, recovered only 56 VH and VL pairs (30%) and only 24 of those demonstrated binding to HEL as a recombinant antibody.
Following expression of recombinant Fab fragments from TAP products, we then used a Biacore assay to estimate binding affinity and to identify those antibodies that were capable of blocking the interaction of human TNFR2 with human TNF. Although some antibodies could not be examined using this assay due to low expression levels in HEK-293-conditioned media, data were successfully generated for 34 recombinant monoclonal antibodies. Six anti-TNFR2 Fabs exhibited blocking activity, and all had sub-nM KD values. DF187 demonstrated the highest affinity binding to TNFR2 measured at 15 pM. There were also several very high affinity non-blockers identified, including DF198, which had an estimated affinity of 11 pM. In total, 18 Fab fragments exhibited sub-nM affinity binding, 9 of which were sub-100 pM. This suggests that the bone marrow-derived plasma cell repertoire in the rabbit is capable of producing extremely high affinity antibodies and that the fluorescent foci method represents an efficient process by which to sample this population.
We next performed DNA sequencing of the 34 antibodies that had been identified in the Biacore to assess diversity. Principle component analysis using combined CDR sequences suggested that the 34 antibodies could be binned into 24 unique families (based on > 80% identity). This suggests that the plasma cell compartment in rabbits is highly diverse. Notably, the six blocking antibodies were all different and unique; no closely related sibling sequences identified. Although the data set is relatively small, it does imply that high affinity functional antibodies may not always be represented by the most dominant sequences present in a repertoire. This highlights the importance of having a platform capable of identifying rare B cells or plasma cells that may be present at low frequencies. Techniques that fail to identify these cells due to poor sampling power or methods that disrupt the natural cognate VH and VL pairing are unlikely to identify such antibodies. For example, in the ground-breaking work described by Reddy et al.,30 a deep sequencing approach was used to generate separate VH and VL sequence libraries from bone-marrow plasma cells from immunized mice. Antigen-specific recombinant scFv were successfully produced following the synthesis and re-pairing of VH and VL based on relative frequencies of sequences in a library derived from approximately 5000 plasma cells. Despite recovering reactivity in samples where the variable region gene sequences were represented at frequencies between 1 and 10% in a repertoire, it is likely that less abundant antibodies may not be recovered using these techniques.
We also extended the technique to the recovery of anti-human TNF antibodies from immunized rats. To improve the efficiency of the fluorescent foci method, we introduced a plasma cell purification method using a rabbit anti-rat CD138 monoclonal antibody (in house reagent) in combination with magnetic-activated cell sorting (MACS). CD138 (syndecan-1) is an adhesion molecule that is highly expressed on both mouse and human plasma cells.60-62 We reasoned that it would also likely represent a useful marker of rat plasma cells. As expected, using this anti-rat CD138 reagent we were able to enrich a rat bone marrow preparation for plasma cells by ~4-fold. This increased the frequency of antigen-specific plasma cells and improved the efficiency of the process.
We used a combination of the fluorescent foci method and TAP to rapidly generate a panel of recombinant anti-human TNF monoclonal antibodies from rat bone marrow. Isolation of 287 antigen-specific plasma cells followed by RT-PCR from the single cells enabled the recovery of 226 cognate VH and VL gene pairs (79%). Following recombinant IgG expression from HEK-293F cells transiently-transfected with TAP products, TNF-binding activity was observed in 147 samples. Similar to the rabbit experiment, concentrations of recombinant antibody varied greatly across the panel and the attrition from variable region genes to recombinant antibody binders was likely predominantly due to low level expression in some samples. However, we cannot rule out the possibility that some unnatural and unproductive variable regions may have been generated through the annealing of mismatched framework-1-specific oligonucleotides during rat B cell PCR resulting in non-active antibodies. Despite this, it was possible to identify 40 antibodies present in the supernatant of transiently-transfected HEK-293F cells that were capable of blocking the activity of human TNF by more than 50% in a single-point HEK-293-CD40-BLUE reporter assay. Following cloning and re-expression of a selection of recombinant Fab molecules, antibodies were analyzed by Biacore and profiled in the cell-based reporter assay to enable the calculation of IC50 values and determine the maximum percentage inhibition. As seen with rabbit, the rat system also yielded a panel of high affinity antibodies with 16 out of 23 recombinant Fab molecules possessing estimated affinity values for TNF in the sub-nM range. Three antibodies were identified as having exceptionally good profiles. TDF0083, TDF0261 and TDF 0293 all had estimated affinities for human TNF below 100 pM, IC50 values <10 ng/ml and attained >95% maximum inhibition. Based on IC50 values, the three Fab molecules exhibited 32-fold, 9-fold and 6-fold higher activity, respectively, in this assay compared with etanercept, which was a positive control.
In conclusion, this work demonstrates the utility of the fluorescent foci method as an efficient means to identify and isolate antigen-specific plasma cells directly from the bone marrow niche of multiple species. When combined with a rapid process for variable region recovery and the generation of transcriptionally-active, “transfection-ready” PCR (TAP) products, recombinant antibodies can be generated and stringently tested within 1–2 weeks following harvest of bone marrow material. The work here clearly suggests that bone marrow-derived plasma cells provide a valuable source of high affinity, functional antibodies and that the novel method described provides a viable process by which to interrogate this repertoire. Ultimately it is hoped that this approach will facilitate the rapid discovery of therapeutic monoclonal antibodies for treating patients suffering with serious diseases.
Materials and Methods
Immunizations
Four female New Zealand White and two Half-Lop rabbits (>2 kg) were immunized subcutaneously with 500 µg human TNFR2-rabbit-Fc fusion protein and 5 female Sprague Dawley rats (260‒280 g) were immunized subcutaneously with 50 µg human TNF protein. Protein antigens were emulsified in an equal volume of complete Freund’s adjuvant (CFA) by vigorously mixing with a syringe. Booster injections at 21-d intervals using incomplete Freund’s adjuvant (IFA) with bleeds taken from the ear (rabbit) or tail (rat) 14 d post-immunization. Termination occurred 14 d after the final boost with single cell suspensions of spleen, bone marrow and peripheral blood mononuclear cells prepared and frozen in 10% DMSO in FCS at -80 °C.
Purification of CD138+ cells from rat bone marrow
CD138+ cells were enriched from bone marrow preparations using a rabbit anti-rat CD138 reagent that we had previously produced at UCB. We generated a monoclonal antibody specific for the rat CD138 molecule through immunization of rabbits with rat CD138 extracellular domain protein followed by screening of activated B cell culture supernatants and subsequent variable region cloning from single antigen-specific B cells using a method similar to that described by Lightwood et al.23 The purified rabbit anti-CD138 antibody was then labeled with phycoerythrin (PE) using a Lightning-link kit (Innova Biosciences). CD138+ cells were then enriched from bone marrow samples via magnetic-activated cell sorting (MACS) (Miltenyi Biotec) with anti-PE microbeads following the manufacturer’s protocol.
Fluorescent foci method
Streptavidin beads (New England Biolabs) were coated with biotinylated human TNFR2 or human TNF and incubated for 1 h at room temperature. The antigen coated beads were washed five times using a magnetic rack and resuspended in cell culture media (RPMI 1640; Gibco® Life Technologies) containing 8.7% fetal bovine serum (Sigma-Aldrich), 1.7% HEPES buffer (PAA Laboratories Ltd), 1.7% L-glutamine (Gibco® Life Technologies), 0.86% penicillin/streptomycin solution (Gibco® Life Technologies), 0.17% normacin (InvivoGen) and 0.09% 2-mercaptoethanol (Gibco® Life Technologies). Frozen bone marrow samples from immunized rabbits or rats were thawed and washed three times in pre-warmed cell culture media by centrifugation and resuspended at a density of 1.6 × 106 cells/mL in cell culture media. Rat bone marrow cells were enriched for plasma cells through magnetic activated cell sorting (MACS) using a rabbit anti-rat CD138 antibody (described above). Goat F(ab′)2 anti-rabbit Fcγ fragment-specific or goat F(ab′)2 anti-rat Fcγ fragment-specific FITC conjugate (Jackson Immunoresearch) was prepared in a 1:300 dilution in cell culture media. An assay mix was created with antigen-coated beads, IgG-secreting B cells from the bone marrow samples and FITC-conjugated secondary antibody in a 2:1:1 ratio, respectively. The assay mix was spotted onto walled-glass slides that had been pre-coated with Sigmacote (Sigma-Aldrich) and then covered in mineral oil to prevent evaporation. After static incubation at 37 °C for 1 h, slides were examined for fluorescent foci formation on an Olympus IX70 microscope. Antigen-specific antibody-expressing B-cells were isolated using an Eppendorf NK micromanipulator. Individual B cells were placed into 0.2 ml PCR tubes and snap-frozen on dry ice.
RT-PCR and transcriptionally-active PCR (TAP)
cDNA from single B cells was prepared using Superscript III reverse transcriptase (Invitrogen) primed with oligo (dT). Antibody variable-region genes were then recovered via two rounds of PCR (Fig. 2A) using either KOD DNA polymerase (EMD Millipore) or TaqPlus Precision DNA polymerase (Agilent) (rat primary PCR) on an Aviso Onyx liquid handling robot. A primary PCR utilized gene-specific primers at both the 5′ and 3′ ends. Gene-specific primers were designed following analysis of a database of rat and rabbit antibody genes generated using the rapid-amplification of cDNA ends (Smart RACE) method (Clonetech) from spleen and bone marrow cells (data not shown). The 5′ oligonucleotide set bound either at the 5′ end of the leader sequence (for rabbit variable regions) or at the 5′ end of the framework 1 region of the mature variable region sequence (for rat variable regions). The 3′ reverse primer set annealed to CH1 or Cκ region respectively. For the rabbit primary PCR, two forward and two reverse primers were used for amplification of the heavy chain, and two forward and one reverse oligonucleotide were used for the amplification of the light chain. For the rat primary PCR, 34 forward and four reverse oligonucleotides were used for the amplification of the heavy chain, and 42 forward and 2 reverse oligonucleotides were used for the amplification of the light chain. In the secondary PCR, a single 5‘ forward oligonucleotide that annealed to a “tail” encoded at the 5′ end of the primary PCR product was used with a 3′ primer set that annealed in the J region. This secondary reverse set contained two oligonucleotides for rabbit heavy chain variable regions, three oligonucleotides for rabbit light chain variable regions, seven oligonucleotides for rat heavy chain variable regions or five oligonucleotides for rat light chain variable regions. All gene-specific oligonucleotide sets were designed to cover all possible sequences observed in the Smart RACE analysis. Not only did the secondary oligonucleotides introduce restriction sites to facilitate downstream cloning, but they also provided ~25 base-pair overlap regions; at the 5′ end with a human cytomegalovirus (HCMV) promoter fragment (plus a leader sequence for rat-derived fragments that were generated with the framework 1 primer set) and at the 3′ end with a heavy or light chain constant region fragment. Then, in a tertiary PCR, variable region DNA, HCMV promoter fragment and constant region fragment containing a poly-adenylation sequence were combined and amplified to produce two separate linear transcriptionally-active PCR (TAP) products, one encoding the heavy chain and the other the light chain. Rabbit variable regions were recombined with rabbit constant regions in the expression cassette, whereas rat variable regions were assembled with mouse constant regions to produce chimeric recombinant molecules.
Cloning of variable region genes and DNA sequencing
Secondary PCR products were purified from an agarose gel using a Qiagen kit and digested with either Hind III/Xho I (VH) or Hind III/BsiW I (VL). Following purification of the digested DNA (Qiagen), variable region fragments were ligated into an in-house expression vector containing an HCMV promoter, constant region cassette (rabbit (for rabbit V regions) or mouse (for rat V regions)) and poly-adenylation sequence using T4 DNA ligase (Promega). Ligated DNA was then transformed into XL1-Blue E. coli cells (Agilent) and plated out onto agar containing kanamycin (25 µg/ml). Miniprep DNA was then made from 5 ml cultures of single kanamycin-resistant colonies. DNA was sequenced using a forward primer that annealed in the promoter region of the vector.
Recombinant antibody expression from transiently transfected HEK-293F cells
Heavy- and light-chain transcriptionally active PCR fragments or plasmid DNA encoding cloned antibody was transiently co-transfected into HEK293F cells using 293fectin (Invitrogen). Cells were incubated for one week at 37 °C in a 5% CO2 atmosphere before supernatant was harvested by centrifugation and antibody expression determined using a sandwich ELISA.
Homogeneous binding assay
Antibody in HEK-293F supernatant was tested for binding to antigen using Fluorometric Microvolume Assay Technology (FMAT). Briefly, 10 μl of supernatant containing antigen-specific antibody, was transferred into a 96-well black-walled assay plate and mixed with biotinylated human TNFR2 or TNF immobilized on Superavidin™ beads (Bangs laboratories) and a goat anti-species Cy-5 conjugate (Jackson Immunoresearch). Plates were read using an 8200 Cellular Detection System (Applied Biosystems).
Biacore analysis
Surface plasmon resonance was performed using a BIAcore T200 (GE Healthcare). All experiments were performed at 25°C. Goat F(ab’)2 anti-rabbit Fcγ, anti-rabbit Fab, anti-rat Fcγ or anti rat Fab (Jackson Immunoresearch) was immobilised on a CM5 Sensor Chip (GE Healthcare) via amine coupling chemistry to a capture level of ~5000 response units. HBS-EP+ buffer (10 mM HEPES pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.05% (v/v) surfactant P20, (GE Healthcare) was used as the running buffer. A 10‒50 μl injection of each antibody sample (0.1‒4 μg/ml) was used to achieve ~100 RU of capture. A 3 min injection of human TNFR2 (generated in-house) at 20 nM or a 6 min injection of human TNF (generated in house) at 10 nM, was passed over the immobilized antibody. Affinity was estimated following a 15 min dissociation phase. For the rabbit anti-TNFR2 antibodies, the binding of TNFR2 was followed by a 1 min injection of human TNF (generated in house) at 150 nM or HBS-EP+ buffer control, at a flow rate of 30 μl/min. The surface was regenerated at a flow-rate of 10 μl/min by a 60 s injection of 40 mM HCl ×2 and 30 s 5 mM NaOH. Double referenced background subtracted binding curves were analyzed using the T200 Evaluation software (version 1.0) following standard procedures. Kinetic parameters and estimated affinity values for most antibodies were determined from fitting a Biacore algorithm using a 1:1 binding model. Those antibodies fitting a bivalent model are highlighted in the data tables and affinity estimates derived from Ka1 and kd1. It should be noted that the Biacore assay formed part of an antibody screening campaign where only limited amounts of antibody material were available and where the focus was on hit-identification. We have previously shown that these estimated KD values, derived from single concentrations of analyte are a good predictor of true KD, calculated through using multiple analyte concentrations (data not shown).
Identification of TNF blockers using a HEK-293-CD40-Blue reporter assay
To identify antibodies capable of blocking the functional activity of human TNF, we used a reporter assay employing HEK-293-CD40-BLUE cells following the manufacturer’s protocol (Invivogen). These cells have been stably transfected with human CD40 and the secreted embryonic alkaline phosphatase (SEAP) reporter gene under the control of the interferon-β minimal promoter fused to five NFκB binding sites. The cells produce SEAP upon stimulation with a number of cytokines including human TNF. HEK-293 supernatants containing antibody were either tested in a single-point assay (1:2.5 final dilution) to establish a level of inhibition or via a 10-point titration to determine an estimated IC50 value and maximum inhibition. Etanercept (human TNF-binding therapeutic) was used as a positive control.
Sequence diversity assessment
We performed a Principal Components Analysis (PCA)52 as a means to reduce the dimensionality of the data and generate an easy to interpret 2-dimensional data plot that illustrated the extent of diversity in our recombinant antibody panel.
From the N sequences that were analyzed in each set, an N × N sequence similarity distance matrix was generated (using a NeedlemanWunsch pairwise alignment comparison in an all vs. all manner). The next step was then to mean center the data in the distance matrix. This was achieved by subtracting the mean value of each column from each value in that column. The end result of this was to generate a data set with a mean of zero. Using this mean-centered data matrix, a co-variance matrix that showed co-variance across all N dimensions of the data set was generated.
With this co-variance information generated, the matrix was decomposed into a set of eigenvectors and eigenvalues. The eigenvectors describe how sequences correlate together in terms of their sequence distance. Each eigenvector has an eigenvalue associated with it that is used to rank the eigenvectors in terms of how well they describe the variation in the data. The eigenvector with the highest eigenvalue thus will show the most significant relationship between all the dimensions of the data. The eigenvectors are orthogonal to each other, so by taking the 1st and 2nd eigenvectors (also known as principle components), we were able to generate a 2-D plane onto which the relative positions of the sequences in terms of their sequence similarity were plotted. Some information is lost by not including further eigenvectors or dimensions and greater resolution of sequence diversity may have been obtained if these were included. However, for the purpose of illustrating diversity in two dimensions and due to the relative influence compared with principle component (PC) 1 and 2, these values were ignored in this analysis.
A new vector was formed from the two selected eigenvectors. This was then transposed and multiplied against the transposed mean centered data matrix. The operation provided the original data set in terms of the chosen eigenvectors, and allowed the data to be plotted in 2 dimensions rather the original N dimensions.
The relative position of the data points on the 2-D plot can be considered to be directly proportional to sequence identity across the data set based on a concatenated sequence encompassing all six CDRs. Identical sequences resulted in the co-localization of data points on the 2-D plot. By using color as a measure of functional activity and size as an indication of affinity (larger being of higher affinity), information regarding sequence uniqueness and functionality could be visualized on the same plot. Clustering analysis was performed and families of closely related sequences were assigned on the basis of sequence identity in the concatenated CDRs of 80% or higher.
Acknowledgments
We would like to thank Grace Smith for performing DNA sequencing and Fred Vanclef for input on variable region diversity assessment and visualization.
Glossary
Abbreviations:
- RT-PCR
reverse transcription-polymerase chain reaction
- TAP
transcriptionally-active PCR
- ELISA
enzyme-linked immunosorbent assay
- HEK
human embryonic kidney
- VH
heavy chain variable region
- VL
light chain variable region
- Ig
immunoglobulin
- FMAT
Fluorescence microvolume assay technology
- SEAP
secreted embryonic alkaline phosphatase
- HCMV
human cytomegalovirus
- CHO
Chinese hamster ovary
- min
minute
- EDTA
ethylenediaminetetraacetic acid
- NEM
ethylmaleimide
- TNF
tumor necrosis factor
- FACS
fluorescence-activated cell sorting
- MACS
magnetic-activated cell sorting
- FITC
Fluorescein isothiocyanate
- PE
phycoerythrin
- CDR
complementarity-determining region
- ka
on rate
- kd
off rate
- KD
affinity constant
Disclosure of Potential Conflicts of Interest
The work described in this paper was performed at UCB Pharma by UCB Pharma employees. Some of the authors do hold shares in UCB Pharma.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/27044
References
- 1.Reichert JM. Marketed therapeutic antibodies compendium. MAbs. 2012;4:413–5. doi: 10.4161/mabs.19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. 1975. Biotechnology. 1992;24:524–6. [PubMed] [Google Scholar]
- 3.Yu X, McGraw PA, House FS, Crowe JE., Jr. An optimized electrofusion-based protocol for generating virus-specific human monoclonal antibodies. J Immunol Methods. 2008;336:142–51. doi: 10.1016/j.jim.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradbury AR, Sidhu S, Dübel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol. 2011;29:245–54. doi: 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol. 2005;23:1105–16. doi: 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- 6.Meijer PJ, Andersen PS, Haahr Hansen M, Steinaa L, Jensen A, Lantto J, Oleksiewicz MB, Tengbjerg K, Poulsen TR, Coljee VW, et al. Isolation of human antibody repertoires with preservation of the natural heavy and light chain pairing. J Mol Biol. 2006;358:764–72. doi: 10.1016/j.jmb.2006.02.040. [DOI] [PubMed] [Google Scholar]
- 7.Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R, Wilson PC. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc. 2009;4:372–84. doi: 10.1038/nprot.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vugmeyster Y, Guay H, Szklut P, Qian MD, Jin M, Widom A, Spaulding V, Bennett F, Lowe L, Andreyeva T, et al. In vitro potency, pharmacokinetic profiles, and pharmacological activity of optimized anti-IL-21R antibodies in a mouse model of lupus. MAbs. 2010;2:335–46. doi: 10.4161/mabs.2.3.11850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu H, Pfarr DS, Tang Y, An LL, Patel NK, Watkins JD, Huse WD, Kiener PA, Young JF. Ultra-potent antibodies against respiratory syncytial virus: effects of binding kinetics and binding valence on viral neutralization. J Mol Biol. 2005;350:126–44. doi: 10.1016/j.jmb.2005.04.049. [DOI] [PubMed] [Google Scholar]
- 10.Patel CA, Wang J, Wang X, Dong F, Zhong P, Luo PP, Wang KC. Parallel selection of antibody libraries on phage and yeast surfaces via a cross-species display. Protein Eng Des Sel. 2011;24:711–9. doi: 10.1093/protein/gzr034. [DOI] [PubMed] [Google Scholar]
- 11.Saggy I, Wine Y, Shefet-Carasso L, Nahary L, Georgiou G, Benhar I. Antibody isolation from immunized animals: comparison of phage display and antibody discovery via V gene repertoire mining. Protein Eng Des Sel. 2012;25:539–49. doi: 10.1093/protein/gzs060. [DOI] [PubMed] [Google Scholar]
- 12.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–7. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 13.Gai SA, Wittrup KD. Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol. 2007;17:467–73. doi: 10.1016/j.sbi.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tiller T. Single B cell antibody technologies. N Biotechnol. 2011;28:453–7. doi: 10.1016/j.nbt.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–81. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 16.Wang F, Sen S, Zhang Y, Ahmad I, Zhu X, Wilson IA, Smider VV, Magliery TJ, Schultz PG. Somatic hypermutation maintains antibody thermodynamic stability during affinity maturation. Proc Natl Acad Sci U S A. 2013;110:4261–6. doi: 10.1073/pnas.1301810110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Di Niro R, Mesin L, Raki M, Zheng NY, Lund-Johansen F, Lundin KE, Charpilienne A, Poncet D, Wilson PC, Sollid LM. Rapid generation of rotavirus-specific human monoclonal antibodies from small-intestinal mucosa. J Immunol. 2010;185:5377–83. doi: 10.4049/jimmunol.1001587. [DOI] [PubMed] [Google Scholar]
- 18.Dohmen SE, Mulder A, Verhagen OJ, Eijsink C, Franke-van Dijk ME, van der Schoot CE. Production of recombinant Ig molecules from antigen-selected single B cells and restricted usage of Ig-gene segments by anti-D antibodies. J Immunol Methods. 2005;298:9–20. doi: 10.1016/j.jim.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 19.Townsend SE, Goodnow CC, Cornall RJ. Single epitope multiple staining to detect ultralow frequency B cells. J Immunol Methods. 2001;249:137–46. doi: 10.1016/S0022-1759(00)00352-5. [DOI] [PubMed] [Google Scholar]
- 20.Scheid JF, Mouquet H, Feldhahn N, Walker BD, Pereyra F, Cutrell E, Seaman MS, Mascola JR, Wyatt RT, Wardemann H, et al. A method for identification of HIV gp140 binding memory B cells in human blood. J Immunol Methods. 2009;343:65–7. doi: 10.1016/j.jim.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kodituwakku AP, Jessup C, Zola H, Roberton DM. Isolation of antigen-specific B cells. Immunol Cell Biol. 2003;81:163–70. doi: 10.1046/j.1440-1711.2003.01152.x. [DOI] [PubMed] [Google Scholar]
- 22.Lagerkvist AC, Furebring C, Borrebaeck CA. Single, antigen-specific B cells used to generate Fab fragments using CD40-mediated amplification or direct PCR cloning. Biotechniques. 1995;18:862–9. [PubMed] [Google Scholar]
- 23.Lightwood DJ, Carrington B, Henry AJ, McKnight AJ, Crook K, Cromie K, Lawson AD. Antibody generation through B cell panning on antigen followed by in situ culture and direct RT-PCR on cells harvested en masse from antigen-positive wells. J Immunol Methods. 2006;316:133–43. doi: 10.1016/j.jim.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Lightwood D, O’Dowd V, Carrington B, Veverka V, Carr MD, Tservistas M, Henry AJ, Smith B, Tyson K, Lamour S, et al. The discovery, engineering and characterisation of a highly potent anti-human IL-13 fab fragment designed for administration by inhalation. J Mol Biol. 2013;425:577–93. doi: 10.1016/j.jmb.2012.11.036. [DOI] [PubMed] [Google Scholar]
- 25.Kwakkenbos MJ, Diehl SA, Yasuda E, Bakker AQ, van Geelen CM, Lukens MV, van Bleek GM, Widjojoatmodjo MN, Bogers WM, Mei H, et al. Generation of stable monoclonal antibody-producing B cell receptor-positive human memory B cells by genetic programming. Nat Med. 2010;16:123–8. doi: 10.1038/nm.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He XS, Sasaki S, Narvaez CF, Zhang C, Liu H, Woo JC, Kemble GW, Dekker CL, Davis MM, Greenberg HB. Plasmablast-derived polyclonal antibody response after influenza vaccination. J Immunol Methods. 2011;365:67–75. doi: 10.1016/j.jim.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods. 2008;329:112–24. doi: 10.1016/j.jim.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fairfax KA, Kallies A, Nutt SL, Tarlinton DM. Plasma cell development: from B-cell subsets to long-term survival niches. Semin Immunol. 2008;20:49–58. doi: 10.1016/j.smim.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 29.Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dörner T, Hiepe F. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;6:741–50. doi: 10.1038/nri1886. [DOI] [PubMed] [Google Scholar]
- 30.Reddy ST, Ge X, Miklos AE, Hughes RA, Kang SH, Hoi KH, Chrysostomou C, Hunicke-Smith SP, Iverson BL, Tucker PW, et al. Monoclonal antibodies isolated without screening by analyzing the variable-gene repertoire of plasma cells. Nat Biotechnol. 2010;28:965–9. doi: 10.1038/nbt.1673. [DOI] [PubMed] [Google Scholar]
- 31.Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;5:230–42. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- 32.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–72. doi: 10.1016/S1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 33.Slifka MK, Ahmed R. Long-lived plasma cells: a mechanism for maintaining persistent antibody production. Curr Opin Immunol. 1998;10:252–8. doi: 10.1016/S0952-7915(98)80162-3. [DOI] [PubMed] [Google Scholar]
- 34.Smith KG, Light A, Nossal GJ, Tarlinton DM. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 1997;16:2996–3006. doi: 10.1093/emboj/16.11.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tarlinton DM, Smith KG. Apoptosis and the B cell response to antigen. Int Rev Immunol. 1997;15:53–71. doi: 10.3109/08830189709068171. [DOI] [PubMed] [Google Scholar]
- 36.Tarlinton DM, Smith KG. Dissecting affinity maturation: a model explaining selection of antibody-forming cells and memory B cells in the germinal centre. Immunol Today. 2000;21:436–41. doi: 10.1016/S0167-5699(00)01687-X. [DOI] [PubMed] [Google Scholar]
- 37.Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. 1997;388:133–4. doi: 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- 38.Manz RA, Hauser AE, Hiepe F, Radbruch A. Maintenance of serum antibody levels. Annu Rev Immunol. 2005;23:367–86. doi: 10.1146/annurev.immunol.23.021704.115723. [DOI] [PubMed] [Google Scholar]
- 39.Benner R, Hijmans W, Haaijman JJ. The bone marrow: the major source of serum immunoglobulins, but still a neglected site of antibody formation. Clin Exp Immunol. 1981;46:1–8. [PMC free article] [PubMed] [Google Scholar]
- 40.Coronella JA, Telleman P, Truong TD, Ylera F, Junghans RP. Amplification of IgG VH and VL (Fab) from single human plasma cells and B cells. Nucleic Acids Res. 2000;28:E85. doi: 10.1093/nar/28.20.e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen-Bettecken U, Wecker E, Schimpl A. Transcriptional control of mu- and kappa-gene expression in resting and bacterial lipopolysaccharide-activated normal B cells. Immunobiology. 1987;174:162–76. doi: 10.1016/S0171-2985(87)80036-0. [DOI] [PubMed] [Google Scholar]
- 42.Manz R, Assenmacher M, Pflüger E, Miltenyi S, Radbruch A. Analysis and sorting of live cells according to secreted molecules, relocated to a cell-surface affinity matrix. Proc Natl Acad Sci U S A. 1995;92:1921–5. doi: 10.1073/pnas.92.6.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carroll S, Al-Rubeai M. ACSD labelling and magnetic cell separation: a rapid method of separating antibody secreting cells from non-secreting cells. J Immunol Methods. 2005;296:171–8. doi: 10.1016/j.jim.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 44.Babcook JS, Leslie KB, Olsen OA, Salmon RA, Schrader JW. A novel strategy for generating monoclonal antibodies from single, isolated lymphocytes producing antibodies of defined specificities. Proc Natl Acad Sci U S A. 1996;93:7843–8. doi: 10.1073/pnas.93.15.7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin A, Ozawa T, Tajiri K, Obata T, Kondo S, Kinoshita K, Kadowaki S, Takahashi K, Sugiyama T, Kishi H, et al. A rapid and efficient single-cell manipulation method for screening antigen-specific antibody-secreting cells from human peripheral blood. Nat Med. 2009;15:1088–92. doi: 10.1038/nm.1966. [DOI] [PubMed] [Google Scholar]
- 46.Jin A, Ozawa T, Tajiri K, Obata T, Kishi H, Muraguchi A. Rapid isolation of antigen-specific antibody-secreting cells using a chip-based immunospot array. Nat Protoc. 2011;6:668–76. doi: 10.1038/nprot.2011.322. [DOI] [PubMed] [Google Scholar]
- 47.Love JC, Ronan JL, Grotenbreg GM, van der Veen AG, Ploegh HL. A microengraving method for rapid selection of single cells producing antigen-specific antibodies. Nat Biotechnol. 2006;24:703–7. doi: 10.1038/nbt1210. [DOI] [PubMed] [Google Scholar]
- 48.Ogunniyi AO, Story CM, Papa E, Guillen E, Love JC. Screening individual hybridomas by microengraving to discover monoclonal antibodies. Nat Protoc. 2009;4:767–82. doi: 10.1038/nprot.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ozawa T, Piao X, Kobayashi E, Zhou Y, Sakurai H, Andoh T, Jin A, Kishi H, Muraguchi A. A novel rabbit immunospot array assay on a chip allows for the rapid generation of rabbit monoclonal antibodies with high affinity. PLoS One. 2012;7:e52383. doi: 10.1371/journal.pone.0052383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park S, Han J, Kim W, Lee GM, Kim HS. Rapid selection of single cells with high antibody production rates by microwell array. J Biotechnol. 2011;156:197–202. doi: 10.1016/j.jbiotec.2011.08.031. [DOI] [PubMed] [Google Scholar]
- 51.Yoshimoto N, Kida A, Jie X, Kurokawa M, Iijima M, Niimi T, Maturana AD, Nikaido I, Ueda HR, Tatematsu K, et al. An automated system for high-throughput single cell-based breeding. Sci Rep. 2013;3:1191. doi: 10.1038/srep01191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pearson K. On Lines and Planes of Closest Fit to Systems of Points in Space. Philos Mag. 1901;2:559–72. [Google Scholar]
- 53.Bystryn JC, Jacobsen JS, Liu P, Heaney-Kieras J. Comparison of cell-surface human melanoma-associated antigens identified by rabbit and murine antibodies. Hybridoma. 1982;1:465–72. doi: 10.1089/hyb.1.1982.1.465. [DOI] [PubMed] [Google Scholar]
- 54.Feng L, Wang X, Jin H. Rabbit monoclonal antibody: potential application in cancer therapy. Am J Transl Res. 2011;3:269–74. [PMC free article] [PubMed] [Google Scholar]
- 55.Huang Z, Zhu W, Meng Y, Xia H. Development of new rabbit monoclonal antibody to progesterone receptor (Clone SP2): no heat pretreatment but effective for paraffin section immunohistochemistry. Appl Immunohistochem Mol Morphol. 2006;14:229–33. doi: 10.1097/01.pai.0000157906.38495.31. [DOI] [PubMed] [Google Scholar]
- 56.Mage RG, Lanning D, Knight KL. B cell and antibody repertoire development in rabbits: the requirement of gut-associated lymphoid tissues. Dev Comp Immunol. 2006;30:137–53. doi: 10.1016/j.dci.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 57.Rossi S, Laurino L, Furlanetto A, Chinellato S, Orvieto E, Canal F, Facchetti F, Dei Tos AP. Rabbit monoclonal antibodies: a comparative study between a novel category of immunoreagents and the corresponding mouse monoclonal antibodies. Am J Clin Pathol. 2005;124:295–302. doi: 10.1309/NR8HN08GDPVEMU08. [DOI] [PubMed] [Google Scholar]
- 58.Tao GZ, Nakamichi I, Ku NO, Wang J, Frolkis M, Gong X, Zhu W, Pytela R, Omary MB. Bispecific and human disease-related anti-keratin rabbit monoclonal antibodies. Exp Cell Res. 2006;312:411–22. doi: 10.1016/j.yexcr.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 59.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–74. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 60.Luque R, Brieva JA, Moreno A, Manzanal A, Escribano L, Villarrubia J, Velasco JL, López-Jiménez J, Cerveró C, Otero MJ, et al. Normal and clonal B lineage cells can be distinguished by their differential expression of B cell antigens and adhesion molecules in peripheral blood from multiple myeloma (MM) patients--diagnostic and clinical implications. Clin Exp Immunol. 1998;112:410–8. doi: 10.1046/j.1365-2249.1998.00600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Minges Wols HA, Witte PL. Plasma cell purification from murine bone marrow using a two-step isolation approach. J Immunol Methods. 2008;329:219–24. doi: 10.1016/j.jim.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanderson RD, Lalor P, Bernfield M. B lymphocytes express and lose syndecan at specific stages of differentiation. Cell Regul. 1989;1:27–35. doi: 10.1091/mbc.1.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abney ER, Cooper MD, Kearney JF, Lawton AR, Parkhouse RM. Sequential expression of immunoglobulin on developing mouse B lymphocytes: a systematic survey that suggests a model for the generation of immunoglobulin isotype diversity. J Immunol. 1978;120:2041–9. [PubMed] [Google Scholar]
- 64.Halper J, Fu SM, Wang CY, Winchester R, Kunkel HG. Patterns of expression of human “Ia-like” antigens during the terminal stages of B cell development. J Immunol. 1978;120:1480–4. [PubMed] [Google Scholar]
- 65.Cassese G, Arce S, Hauser AE, Lehnert K, Moewes B, Mostarac M, Muehlinghaus G, Szyska M, Radbruch A, Manz RA. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol. 2003;171:1684–90. doi: 10.4049/jimmunol.171.4.1684. [DOI] [PubMed] [Google Scholar]
- 66.Minges Wols HA, Ippolito JA, Yu Z, Palmer JL, White FA, Le PT, Witte PL. The effects of microenvironment and internal programming on plasma cell survival. Int Immunol. 2007;19:837–46. doi: 10.1093/intimm/dxm051. [DOI] [PubMed] [Google Scholar]
- 67.Nossal GJ, Szenberg A, Ada GL, Austin CM. single cell studies on 19s antibody production. J Exp Med. 1964;119:485–502. doi: 10.1084/jem.119.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Calame KL. Plasma cells: finding new light at the end of B cell development. Nat Immunol. 2001;2:1103–8. doi: 10.1038/ni1201-1103. [DOI] [PubMed] [Google Scholar]
- 69.McMillan R, Longmire RL, Yelenosky R, Lang JE, Heath V, Craddock CG. Immunoglobulin synthesis by human lymphoid tissues: normal bone marrow as a major site of IgG production. J Immunol. 1972;109:1386–94. [PubMed] [Google Scholar]
- 70.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–72. doi: 10.1016/S1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 71.Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, Brink R. High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med. 2006;203:2419–24. doi: 10.1084/jem.20061254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith KG, Light A, O’Reilly LA, Ang SM, Strasser A, Tarlinton D. bcl-2 transgene expression inhibits apoptosis in the germinal center and reveals differences in the selection of memory B cells and bone marrow antibody-forming cells. J Exp Med. 2000;191:475–84. doi: 10.1084/jem.191.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, Berek C, Hiepe F, Manz R, Radbruch A, et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. 2005;105:1614–21. doi: 10.1182/blood-2004-07-2507. [DOI] [PubMed] [Google Scholar]
- 74.Pihlgren M, Schallert N, Tougne C, Bozzotti P, Kovarik J, Fulurija A, Kosco-Vilbois M, Lambert PH, Siegrist CA. Delayed and deficient establishment of the long-term bone marrow plasma cell pool during early life. Eur J Immunol. 2001;31:939–46. doi: 10.1002/1521-4141(200103)31:3<939::AID-IMMU939>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]