

Abstract
We set out to gain deeper insight into the potential of antibody light chain variable domains (VLs) as immunotherapeutics. To this end, we generated a naïve human VL phage display library and, by using a method previously shown to select for non-aggregating antibody heavy chain variable domains (VHs), we isolated a diversity of VL domains by panning the library against B cell super-antigen protein L. Eight domains representing different germline origins were shown to be non-aggregating at concentrations as high as 450 µM, indicating VL repertoires are a rich source of non-aggregating domains. In addition, the VLs demonstrated high expression yields in E. coli, protein L binding and high reversibility of thermal unfolding. A side-by-side comparison with a set of non-aggregating human VHs revealed that the VLs had similar overall profiles with respect to melting temperature (Tm), reversibility of thermal unfolding and resistance to gastrointestinal proteases. Successful engineering of a non-canonical disulfide linkage in the core of VLs did not compromise the non-aggregation state or protein L binding properties. Furthermore, the introduced disulfide bond significantly increased their Tms, by 5.5–17.5 °C, and pepsin resistance, although it somewhat reduced expression yields and subtly changed the structure of VLs. Human VLs and engineered versions may make suitable therapeutics due to their desirable biophysical features. The disulfide linkage-engineered VLs may be the preferred therapeutic format because of their higher stability, especially for oral therapy applications that necessitate high resistance to the stomach’s acidic pH and pepsin.
Keywords: VL, single-domain antibody, disulfide linkage, thermal stability, protease resistance
Introduction
As antibody-based therapeutics, full-length monoclonal antibodies have little competition so far.1-3 In fact, most approved monoclonal antibodies and those in regulatory review are canonical IgG antibodies (www.landesbioscience.com/journals/mabs/about/). The disadvantages of these molecules, such as costly and time-consuming production in mammalian cell lines, large (~150 kDa) and complex molecular structures, poor tissue penetration and inability to access cryptic epitopes, and the fact that the Fc portion of the antibody is not needed in many instances or may even be harmful, have resulted in the creation of a niche that can be occupied by antibody fragments.2,4,5 These smaller antibody fragments, including single-domain antibodies (sdAbs), have unique features that may make them the preferred therapeutic format for many applications. Currently, there are numerous antibody fragments in clinical development, with some being sdAbs.2,4
sdAbs, e.g., human VHs, human VLs, camelid VHHs, have become a viable option in the antibody-based therapeutic tool box that also includes IgGs, antigen-binding fragments (Fabs), single chain Fv fragments (scFvs), and their many derivatives. Appealing features of sdAbs include their high affinity (nM - pM equilibrium dissociation constant (KD) range) for cognate antigens,6-29 small size (~15 kDa) and simple structure, single domain nature, modularity, low immunogenicity, high-level expression in microorganisms such as bacteria, high thermal, chemical and protease stabilities, high solubility and aggregation resistance, ability to access cryptic epitopes, and ease of genetic manipulation and display library construction.5,30,31 VHHs are more convenient to obtain due to their better biophysical properties and the existence and accessibility of in vivo naïve and immune VHH repertoire sources, but human VHs and VLs have the perceived advantage of being less immunogenic in human therapy. A number of reports have implied human VLs may be superior therapeutic candidates compared with human VHs because of their lower tendency to aggregate,32-34 which may translate to lower immunogenicity and in turn higher therapeutic efficacy for VLs.
In vivo, human VL constructs are the result of genetic recombination between germline gene segments VL and JL. The first two complementarity-determining regions (CDR1 and CDR2) and a part of the CDR3 up to residue 95 are encoded by VL segment genes; the rest of the CDR3 and the entire framework region (FR) 4 are encoded by JL gene segments.35 Human VLs are classified as either κ or λ subtypes, with seven Vκ gene segment subgroups (Vκ1–7) within the κ class and 11 Vλ gene segment subgroups (Vλ1–11) within the λ class (http://www.imgt.org/).36,37 In general, Vκ domains exhibit higher solubility and stability than Vλ domains, possibly due to a higher packing density in their upper core and a more hydrophilic C-terminus, and among the Vκ subgroups, Vκ3 subgroup members exhibit the best properties in terms of solubility and thermodynamic stability.33,38 A significant proportion of human VLs, predominantly of Vκ class, bind to the B cell super-antigen protein L.39-41
VL domains are similar to VHs in terms of overall structure. They are composed of two β-sheets that are formed by several anti-parallel β-strands and pack face-to-face to form β-sandwich structures.42 Also, similar to VHs, they possess a pair of cysteine residues at spatially equivalent positions (Kabat positions 23 and 88)43 that form a highly conserved disulfide linkage. This linkage, which pins together the two β-sheets in the core of VL domains, plays a critical role in maintaining the structural integrity of VLs.44,45 Previously, it was shown that engineering an additional disulfide linkage in the core of a set of human VHs improved their aggregation resistance and thermostability.46,47 Given the overall structural similarity between VH and VLs, it is hypothesized that the same engineering approach, with similar stability improvements, should be applicable to VLs.
Here, to further explore the merits of human VLs as therapeutic modalities, we set out to perform an extensive biophysical characterization of a set of test VLs. We constructed a naïve human VL phage display library, and from it isolated a diversity of domains with protein L binding property by a phage selection method that was previously shown to be highly selective for non-aggregating human VH domains.48 We then characterized a representative sample of VLs for properties such as expression yield, non-aggregation, thermal stability, reversible thermal unfolding, structural integrity, and protease resistance. Next, we determined if a non-canonical disulfide linkage engineering approach previously shown to improve the thermostability and protease resistance of camelid VHHs and human VHs46,47,49-52 would do the same for the present VLs. Thus, we engineered human VLs with an additional, non-canonical disulfide linkage between Cys48 and Cys64 in β-strands C’ and D. We then performed pair-wise biophysical comparisons between wild-type and their corresponding Cys mutant domains.
Results
Identification and sequence analysis of human VLs
Essentially the same selection method employed to isolate non-aggregating VHs from a human VH phage display library was applied to a human VL library for isolating soluble, monomeric VLs.48 A human VL library with a size of 3 × 106 transformants was constructed. Twenty-four clones (plaques) from the library titer plates were isolated and their VL genes were amplified by PCR and then sequenced. The sequences were diverse in terms of germ-line origin, although 75% of the VLs were of Vλ origin (data not shown). Three rounds of panning against protein L resulted in enrichment for large plaques. Thirty-four of the large plaques were sequenced and 32 unique sequences were identified (Fig. 1). Except for HVLP389, which is from the λ class (subgroup Vλ1, V germline 1b), the remaining 31 VLs belonged to the Vκ class. Of the 31 κ class VLs, 24 fell within the Vκ3 subgroup and 7 fell within the Vκ1 subgroup. Sixteen of the 24 Vκ3 sequences utilized the L6 V germline sequence, while the remaining sequences utilized A27, L2, and L16 V germline sequences. The Vκ1 subgroup VLs originated from the O2/O12 or A30 V germline sequence. Noticeable mutations occurred at position 96. The germline amino acids at this position are aromatic and hydrophobic amino acids Trp, Phe, Tyr, Leu or Ile for κ class VLs, and Tyr, Val or Ala for λ class VLs. In the selected pool of κ class VLs, however, only 5 out of 31 VLs had their germline amino acids at position 96: HVLP325, HVLP349, HVLP388, HVLP3109, and HVLP393; 21 VLs had charged amino acids, of which 20 were positively charged, 2 had Pro, 1 had Gln, 1 had Ser, and one had Thr at positions 96. Moreover, 18 VLs of the κ class had their last three germline residues (105–107) replaced with amino acids Thr, Val, and Leu, which are only found in λ class VLs.

Figure 1. Amino acid sequences of VLs selected from a human VL phage display library by panning against protein L. The dots in the sequence entries indicate amino acid identity with HVLP333. Dashes are included for sequence alignment. See V BASE (http://vbase.mrc-cpe.cam.ac.uk/index.php?&MMN_position=1:1) for sequence numbering and CDR and FR designations. L6, A27, L2, L16, O2/O12, A30, and 1b are V germline gene segment designations. J germline gene segment designations are in brackets. NF, not found.
Expression yield and aggregation status of human VLs
Eight of the selected VLs representing different V germline origins were expressed in E. coli TG1 in 1 L cultures and purified: HVLP324, HVLP325, HVLP335, HVLP342, HVLP351, HVLP364, HVLP389 and HVLP3103 (Fig. 2A; Table 1). All were expressed in good yields ranging from 6.2 mg for HVLP325 to ~75 mg for HVLP335 and HVLP364. The aggregation tendency of the human VLs was assessed by Superdex 75™ size-exclusion chromatography (SEC).47 At a concentration of 0.6 mg/mL (43 µM) all VLs were essentially free of aggregates and gave single, symmetrical peaks (Fig. 3A). HVLP351, HVLP342, HVLP335 and HVLP3103, were still monomers when tested at their highest concentration available, i.e., 0.89 mg/mL (64 µM), 1.0 mg/mL (72 µM), 4.9 mg/mL (352 µM), and 5.9 mg/mL (430 µM), respectively, although slight tailing was observed for the HVLP335 monomeric peak at 5.9 mg/mL, suggesting VL interaction with the column matrix. The apparent molecular masses (Mapps) of VLs, calculated from their elution volumes (Fig. S1), ranged from 6.9 kDa to 24.3 kDa, with a mean Mapp ± SEM of 13.7 ± 2.2 kDa and a Mfor ± SEM of 13.8 ± 0.04 kDa. Variation in Mapps for non-aggregating VLs with similar formula molecular masses (Mfors) has been reported previously.32 Such variation was also observed in the case of highly non-aggregating VHs and may have been the result of weak transient interactions with the column materials or monomer/dimer equilibria.53 The non-aggregating status of VLs was confirmed by a surface plasmon resonance (SPR) assay based on the Ni2+-His6 tag interaction that involved flowing His-tagged VLs over Ni2+-immobilized sensorchip surfaces.46 Similar to previous results obtained with monomeric VHs, for the two dissociation phase windows tested, all the C-terminally His6-tagged VLs gave koffs that are very similar to those of the monomeric VHH control, but drastically faster than those for the dimeric VH control with two C-terminal His6 tags, confirming the monomeric status of the VLs (Fig. 3B). This conclusion was accurately endorsed by the multiangle light scattering (MALS) experiments, which showed that the VLs had MMALSs (molecular masses determined by MALS) that were very similar to their Mfors (Table 1).

Figure 2. Structure of a set of human VLs chosen for detailed biophysical analyses. (A) Amino acid sequences of eight human VLs and their Cys mutant versions. The amino acid positions (48 and 64) where mutations to cysteine residues were made are marked. (B) Homology structures of wild-type (left) and Cys mutant (right) of HVLP324. The native and engineered non-canonical disulfide linkages are shown as green and red spheres, respectively. Protein homology structures were obtained using the Geno3D automatic modeling tool (http://pbil.ibcp.fr/htm/index.php). The figures were drawn with PyMOL (http://www.pymol.org) and Disulfide by Design (version 1.20) freeware.80
Table 1. Biophysical characteristics of VLs.
| VL | Subgroup | Mfor (kDa) | MMALS (kDa)a | Expression yield (mg)b | KD (µM) | VL:protein Lc | Tm (°C) | ΔTm (°C) | TRE (%)d | GI protease resistance (%)e | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 µM | 20 µM | Trypsin | Chymotrypsin | Pepsin | |||||||||
| HVLP324 | Vκ1 | 13.83 | 13.84 ± 0.76 | 2.5, 7 | 0.2, 0.07f | 7:1 | 66.1 | 7.3 | 90 | 78 | 34 | 100 | 22 |
| HVLP324S | 13.87 | 14.68 ± 0.58 | 0.5, 1.1 | 0.1, 0.06f | 7:1 | 73.4 | NDg | NDg | 32 | 36 | 46 | ||
| HVLP325 | Vκ3 | 13.61 | 13.5 ± 0.63 | 0.5, 6.2 | 1 | 3:1 | 68.5 | 14 | 94 | 65 | 67 | 76 | 34 |
| HVLP325S | 13.66 | 13.35 ± 0.64 | 2.2, 3.1 | 1 | 3:1 | 82.5 | NDg | NDg | 81 | 93 | 100 | ||
| HVLP335 | Vκ3 | 13.90 | 15.26 ± 0.50 | 73.5 | 2 | 3:1 | 61.7 | 17.3 | 92 | 95 | 2 | 59 | 0 |
| HVLP335S | 13.94 | 16.8 ± 0.47 | 5.5 | 2 | 3:1 | 79.0 | NDg | NDg | 0 | 32 | 62 | ||
| HVLP342 | Vκ1 | 13.95 | 14.22 ± 0.64 | 1, 7.7 | 0.6, 0.04f | 7:1 | 58.4 | 5.4 | 92 | 70 | 25 | 73 | 3 |
| HVLP342S | 13.99 | 17.02 ± 0.40 | 1.7, 10.8 | 0.2, 0.05f | 7:1 | 63.8 | NDg | NDg | 5 | 40 | 5 | ||
| HVLP351 | Vκ3 | 13.85 | 14.60 ± 0.61 | 1.2, 8.9 | 2 | 2:1 | 62.0 | 9.9 | 87 | 65 | 71 | 100 | 31 |
| HVLP351S | 13.89 | 14.61 ± 0.53 | 1.9, 4.8 | 0.7 | 2:1 | 71.9 | NDg | NDg | 100 | 99 | 94 | ||
| HVLP364 | Vκ3 | 13.95 | 14.20 ± 0.58 | 0.3, 77 | 3 | 3:1 | 57.0 | 15.3 | 100 | 92 | 32 | 58 | 0 |
| HVLP364Sh | 13.97 | 14.98 ± 0.43 | 4.7 | NDg | NDg | 72.3 | NDg | NDg | 0 | 12 | 0 | ||
| HVLP3103 | Vκ3 | 13.85 | 14.29 ± 0.64 | 0.7, 19 | 1 | 3:1 | 65.7 | 10.7 | 94 | 92 | 81 | 90 | 0 |
| HVLP3103S | 13.89 | 14.05 ± 0.68 | 6.5 | 1 | 3:1 | 76.4 | NDg | NDg | 47 | 80 | 89 | ||
| HVLP389 | Vλ1 | 13.69 | 15.12 ± 0.50 | 3, 16.7 | 1 | 1:1 | 51.9 | 14.4 | 91 | 91 | 15 | 74 | 0 |
| HVLP389S | 13.72 | 13.65 ± 0.68 | 6.5 | 1 | 1:1 | 66.3 | NDg | NDg | 64 | 90 | 11 | ||
a Mean ± SEM; bExpression yield values are per liter of bacterial culture. Two expression yield values correspond to two independent expression trials; cStoichiometry of VL-protein L binding; dThermal refolding efficiency at 4 and 20 µM VL concentrations, respectively; ePercentage proteolytic resistance values are at protease concentrations of 10 µg/mL (see also Fig. 6E); fSmaller KD values correspond to the binding of HVLP324, HVLP324S, HVLP342, and HVLP342S to the high affinity sites on protein L; gND, not determined; hHVLP364S additionally gave a significant second peak (Fig. 3A) with a smaller elusion volume and a MMALS of 26.21 ± 0.47 kDa.

Figure 3. Size-exclusion chromatography and SPR analyses of wild-type and mutant VLs. (A) Superdex 75™ size-exclusion chromatograms of VLs with monomeric peaks marked with arrowheads. For HVLP364S, the dimeric VL peak situated to the left of the monomeric peak is visible. (B) SPR analysis of VL binding to a Ni2+-NTA sensorchip. A llama VHH Monomer (A4.249) and a VH Dimer81 were used as controls. Measurements were taken at two dissociation phase windows (315–375 s and 780–840 s) and the mean values obtained from two independent trials performed in duplicates were recorded in the table. SPR experiments were performed with SEC-purified VLs.
Protein L binding of human VLs
As anticipated, all selected VLs bound to protein L in SPR analysis (Fig. S2A; Table 1). The KDs of binding to protein L were in the 0.2–3 µM range, with HVLP324 and HVLP342, which belong to Vκ1 subgroup, showing additional smaller KDs of 0.07 µM and 0.04 µM, respectively, when Biacore analyses were performed at a low concentration range (1–10 nM) (Table 1). The estimated stoichiometry of binding, VL:protein L, were 7 for Vκ1 subgroup members, 3 for all Vκ3 subgroup members except for HVLP351 which was 2, and 1 for HVLP389, a Vλ1 member.
Tms of human VLs
To assess the thermostability of the VLs, Tms of VLs were determined based on ellipticity data assuming a two-state system, which is in agreement with the observed denaturation curves corresponding to a sharp transition into denaturation (Fig. 4A). Tms, taken at midpoint of the sigmoidal denaturation curves of molar ellipticity change (Δθ) vs. temperature (°C), were in the range of 51.9–68.5 °C (median Tm = 61.9°C) (Table 1). The lowest Tm was that of HVLP389, which is the only VL of λ family among the eight VLs, and the highest Tm was that of HVLP325, which showed slight aggregation (Fig. 3A; Table 1). The Tms of eight non-aggregating VHs (HVHP428, HVHP413, HVHP414, HVHP421, HVHP429, HVHP44, HVHP420, and HVHP419) isolated previously in the same manner as VLs48 were also determined for comparison. The Tm range was 54.2–64.2 °C and the median Tm was 57.6°C; the Tm differences between VLs and VHs were not significant (Mann-Whitney test, two-tailed; P = 0.2345) (Fig. 4B and D).

Figure 4. Thermostability analysis of antibody domains. Thermal unfolding curves of (A) wild-type VLs, (B) VHs, and (C) mutant VLs. Tms were calculated and incorporated into Figure 4D and Table 1. The non-aggregating VHs are described in ref. 48. (D) Graph comparing the Tms of wild-type VLs to those of VHs and mutant VLs.
Thermal refolding efficiency of human VLs
The ability of the human VLs to refold following thermal denaturation was assessed by determining their thermal refolding efficiencies (TREs), which is calculated as the ratio of the KDs for the binding of the native VLs to protein L (KDn) to the KDs for the binding of the heat-denatured/cooled (refolded) VLs to protein L (KDref).48,54 KDs were determined by SPR (Fig. 5; Fig. S3; Table 1). Figures 5A and B compare the protein L binding sensorgram profiles for HVLP335 and HVLP325, respectively, in native and refolded states at 20 µM VL concentrations. It can be seen that for HVLP335, with near perfect refolding (95%) (Table 1), the sensorgrams for the native and refolded species are almost superimposable. In contrast, for HVLP325 with 65% TRE (Table 1), a significant drop in binding is observed for refolded species compared with the native species when sensorgrams at the same concentrations are compared. At 4 µM VL concentrations, TREs are in the range of 90–100%, except for HVLP351, which is slightly lower at 87%, indicating a near perfect refolding for VLs (median = 92%) (Fig. 5C). At 20 µM VL concentrations, the TREs significantly decrease, with VLs having a TRE range of 65–95% and a TRE median dropping to 84.5% (Wilcoxon matched-pairs signed rank test, two-tailed; P = 0.0469) (Fig. 5C). However, for four VLs (HVLP335, HVLP364, HVLP389, HVLP3103), TRE values still remains above 90%, and for three of the four, TREs do not change when VL concentrations are increased from 4 µM to 20 µM. For comparison, the TREs of the eight non-aggregating VHs at 4 µM and 20 µM VH concentrations are also included (Fig. 5C; Fig. S4). Two of the eight VHs (HVHP428 and HVHP413) showed low TREs of 48% and 7% at 4 µM, with the remaining 6 VHs (HVHP414, HVHP421, HVHP429, HVHM44, HVHP420, HVHP419) having TREs of 90–98% (median = 92%). As with VLs, the TREs of VHs at 20 µM concentrations decreased significantly (median = 78.5%, Wilcoxon matched-pairs signed rank test, two-tailed; P = 0.0078), with HVHP428 and HVHP413 VHs failing to refold (TRE = 2% and 1%, respectively). Statistical analysis of TREs revealed that VLs were comparable to VHs in terms of reversibility of thermal unfolding (Mann-Whitney test, two-tailed; P = 0.8785 [4 µM], P = 0.5054 [20 µM]). Moreover, the drops in TRE values as a function of increased concentrations of VHs and VLs are significant and expected as previously reported.55 No significant correlation between TRE and Tm was found.
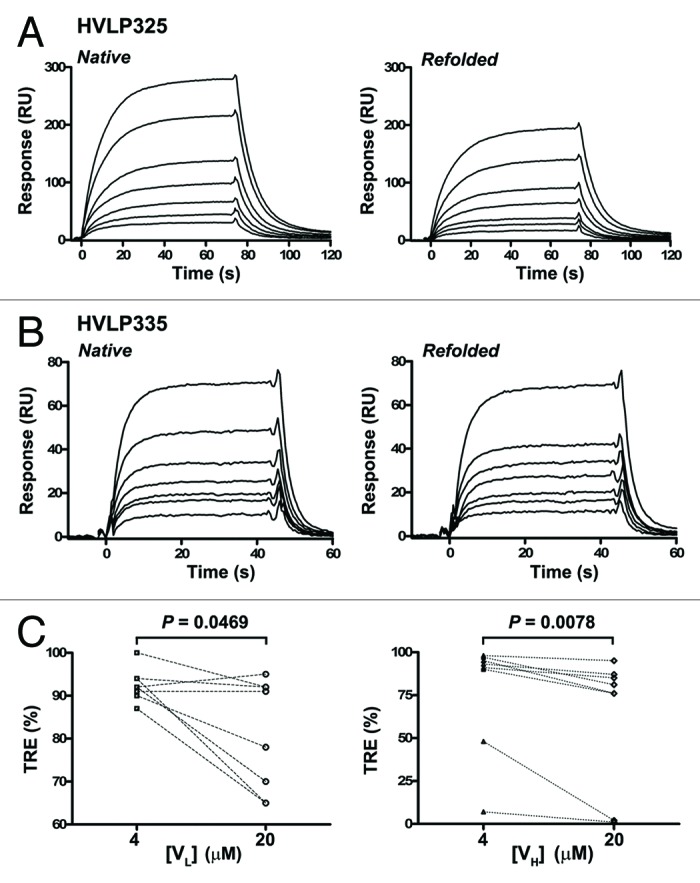
Figure 5. Thermal refolding efficiency determination of human VLs and VHs by SPR. (A), (B) Representative SPR sensorgrams for the binding of native and heat-denatured/cooled (refolded) human VLs to immobilized protein L. Data are from TRE experiments performed at 20 µM VL concentrations. VL concentrations used to construct each sensorgram set was 12.5, 18.8, 25, 37.5, 50, 75, and 100 nM for HVLP325 and 12.5, 20, 25, 30, 38, 50, and 75 nM for HVLP335. See Figure S3 and S4 for SPR data for all VLs and VHs. The KDns and KDrefs determined from “Native” and “Refolded” sensorgram pairs were used to determine TREs in (C). (C) TREs of wild-type VLs and VHs obtained under 4 µM and 20 µM domain concentration conditions in refolding experiments. Lines connect TREs for the same clones.
Protease resistance of human VLs
In addition to thermostability, e.g., Tm, protease resistance is also a measure of protein stability. To determine protease resistance, VLs were treated with major gastrointestinal (GI) proteases, trypsin, chymotrypsin, and pepsin, at various protease concentrations (Fig. 6; Table 1). As expected, a gradual decrease in protease resistance of VLs was observed as a function of protease concentration for all three proteases (Fig. 6A, B, and C). At the highest trypsin concentration (20 µg/mL), the protease resistance of VLs was in the range of 0–75% with a median resistance of 16% (Fig. 6A). The VLs, however, demonstrate higher resistance to chymotrypsin (Fig. 6B). For example, the VLs demonstrated a median resistance of 75% at a chymotrypsin concentration of 10 µg/mL compared with 33% for trypsin at the same concentration; at a higher chymotrypsin concentration of 20 µg/mL, VLs had a protease resistance range and median of 34–100% and 67.5%, respectively. VLs showed the least resistance to pepsin (Fig. 6C). At 1 µg/mL pepsin concentration, two of the VLs (HVLP364 and HVLP389) were digested almost completely with 8% and 2% pepsin resistance, respectively, and at 10 µg/mL pepsin concentration, the resistance of all VLs decreased to below 34% (median resistance = 1.5%) (Table 1). At 20 µg/mL pepsin concentration, the pepsin resistance range and median were 0–23% and 0%, respectively.

Figure 6. GI protease resistance profiles for human VLs and VHs. (A), (B), (C) Trypsin, chymotrypsin, and pepsin resistance (res.) of human VLs (open circles) and their corresponding Cys mutants (closed circles). (D) Graph comparing VLs to VHs in terms of resistance to trypsin, chymotrypsin, and pepsin at 10 µg/mL protease concentrations. Horizontal lines in graphs (A)–(D) represent medians. (E) Graph showing trypsin, chymotrypsin, and pepsin resistance of human VLs (open circles) and their corresponding Cys mutants (closed circles) in pair-wise manner at 10 µg/mL protease concentrations (see also Table 1). Lines connect protease resistances value for each VL to that for its corresponding mutant version. The P values in graphs (D) and (E) were obtained by the Mann-Whitney test (two-tailed) and Wilcoxon matched-pairs signed rank test (two-tailed), respectively, using GraphPad Prism (GraphPad Software). (F) A correlation graph of pepsin resistance vs. Tm (Pearson’s correlation, 0.7807; P = 0.0004, r2 = 0.6095). Data are from digestion experiments performed at 10 µg/mL pepsin concentrations. Open circles, wild-type VLs; closed circles, mutant VLs.
We compared VLs to the eight non-aggregating VHs in terms of resistance to the three proteases at 10 µg/mL protease concentration (Fig. 6D). We found that there was no significant difference between VLs and VHs with respect to resistance to trypsin (Mann-Whitney test, two-tailed; P = 0.1605). VLs were significantly more resistant to chymotrypsin than VHs (Mann-Whitney test, two-tailed; P = 0.0148), while the opposite was true with respect to resistance to pepsin (Mann-Whitney test, two-tailed; P = 0.0011). Theoretical numbers of protease cleavage sites were determined by a protease digestion prediction webware (http://web.expasy.org/peptide_cutter). It was found that VHs had significantly higher number of protease cleavage sites than VLs for all three proteases (trypsin sites medians: 8.5 and 10.5 for VL and VH, respectively, [Mann-Whitney test, two-tailed; P = 0.0207]; chymotrypsin sites medians: 10 and 13.5 for VL and VH, respectively, [Mann-Whitney test, two-tailed; P = 0.0070]; pepsin sites medians: 30 and 39.5 for VL and VH, respectively [Mann-Whitney test, two-tailed; P = 0.0002]).
Disulfide linkage engineering of human VLs
To improve the stability of human VLs, we created 8 VL mutants (HVLP324S, HVLP325S, HVLP335S, HVLP342S, HVLP351S, HVLP364S, HVLP389S, and HVLP3103S) with a pair of Cys substitutions at amino acid positions 48 and 64 (Fig. 2A). All were expressed well in E. coli, albeit with lower yields compared with wild-type VLs, with expression yields ranging from 1.1 mg/L of bacterial culture in shaker flasks for HVLP324S to ~11 mg/L for HVLP342S (Table 1).
To determine if the engineered Cys pairs formed the desired disulfide linkages in the mutants, mass spectrometry (MS) was performed by analyzing tryptic digests of mutant VLs. The MS analyses revealed that the disulfide linkage was formed as intended in all VL mutants (Fig. S5; Table 2). The disulfide-linked peptide ions appeared prominent in the survey of LC-MS chromatograms with tryptic peptides of the mutant VLs. The expected disulfide-linked peptide sequences corresponding to each mutant VLs were confirmed by manual de novo sequencing. When there was only one disulfide linkage between two peptides, the exact disulfide linkage position was confirmed by an almost complete disulfide-linked y fragment ion series from one peptide with the other peptide attached as a modification via a disulfide bond that remains intact under collision-induced dissociation (CID),47,49,56 e.g., the disulfide linked peptides ATLSCR (P1) and GSGTLFTLTISSLEPEDSAVYFCQQR (P2) of HVLP325S (Table 2; MS2 data not shown). When there were three peptides linked by two disulfide bonds, the y fragment ion containing the linkage close to the N-terminal of two peptides was difficult to observe. Nevertheless, an almost complete y ion series of each peptide was observed. For example, a prominent ion at m/z 1042.66 (6+) from HVLP324S tryptic disulfide-linked peptide LLCFAASTLQSGVPSR (P1), FSCSGSGTDFTLTISNLQPEDFATYYCQQSYSTPR (P2) and VTITCR (P3) was observed from the survey of LC-MS chromatogram (Fig. S5B (top panel);Table 2). Informative y fragment ions were observed from P2 with P3 as a modification via a disulfide bond, and an almost complete y ion series of P1 was observed as well (Fig. S5B, top panel). To further confirm the above disulfide bond formation in the mutant HVLP324S, the electron transfer dissociation (ETD)-MS2 spectrum of the peptide ion [M + 5H]5+ at m/z 1250.99 (5+) from the same disulfide-linked peptide of HVLP324S was acquired (Fig. S5B, middle panel). The most abundant charge-reduced ETD fragment ion [M + 5H]4+• at m/z 1563.49 (4+) was selected for CID to obtain the ETD-CID-MS3 spectrum of the m/z 1250.99 (5+) ion (Fig. S5B, bottom panel). The intact P1, P2, and P3 ions at m/z 691.47 (1+), 1648.86 (1+), and 1956.85 (2+), respectively, were all observed at relatively high abundances upon dissociation of the disulfide linkages of the three linked peptides by ETD. Tryptic peptides linked by the engineered disulfide bonds were positively identified for all mutant VLs. These fragments are recorded in Table 2.
Table 2. Disulfide linkage determination of VLs by MS analyses.
| VLs | Tryptic peptidesa |
Mfor (Da) |
Mexp (Da) |
ΔMc (Da) |
|---|---|---|---|---|
| HVLP324S | VTITCR...LLCFAASTLQSGVPSR...FSCSGSGTDFTLTISNLQPEDFATYYCQQSYSTPR | 6249.91b | 6249.96b | -0.05b |
| HVLP325S | ATLSCR...GSGTLFTLTISSLEPEDSAVYFCQQR | 3495.66 | 3495.56 | 0.10 |
| LLCFDTSNR...FSCR | 1576.71b | 1576.68b | 0.03b | |
| HVLP335S | LLCYGTSNR...FSCSGSGTHFTLTINR | 2750.29b | 2750.32b | -0.03b |
| ATLSCR...LEPGDFAVYYCQQYGSSPR | 2826.27 | 2826.20 | 0.07 | |
| HVLP342S | VTITCR...LCYGASSLQGGVPSR...FSCSGSGTEFTLTI SGLQPEDFATYYCLQHHTYPR | 6136.85b | 6136.80b | 0.05b |
| HVLP351S | ATLSCR...LLCYDASNR...FSCSGSGTDFTLTISSLEPE DFAVYYCQQR | 5049.26b | 5049.00b | 0.26b |
| HVLP364S | LLCYGASSR...FSCSGSGTDFTLTISR | 2644.23b | 2644.32b | -0.09b |
| ATFSCR...LEPEDFAVYYCQQYDTSPR | 3004.29 | 3004.35 | -0.06 | |
| HVLP389S | LLCYGNDK...FSCSK | 1492.65b | 1492.59b | 0.06b |
| VTISCSGSSYNIGENSVSWYQQLPGTAPK…SGTSAT LGITGLQTGDEADYYCGTWDSNLR | 6221.84 | 6221.97 | -0.13 | |
| HVLP3103S | ATLSCR…LLCYGASTR…FSCSGSGTDFTLTISSLQV EDVAVYYCQQYYTTPK | 5528.56b | 5528.52b | 0.04b |
a Major tryptic peptides containing disulfide linkages are shown, with connecting cysteine residues single or double underlined (native or engineered Cys (C), respectively) and boldfaced (see Fig. S5 for experimental details). The triple dots between peptides denote sequence discontinuity, which was caused by the loss of VL sequences after trypsin digestion; the discontinuing peptides, however, are held together by disulfide linkage(s); bThe very close match between Mfor (formula molecular mass) and Mexp (experimental molecular mass) indicates the presence of the Cys48-Cys64 disulfide linkage. cΔM = Mfor- Mexp. In addition, the disulfide linkages were confirmed by de novo sequencing the CID or ETD spectra of the disulfide linked peptides (e.g., see Fig. S5B).
Aggregation status of disulfide linkage-engineered human VLs
Next, we aimed to determine if the engineered disulfide linkage compromised the non-aggregation status of VLs. To this end, we assessed the mutant VLs by Superdex 75™ SEC. Except for HVLP364S, which seemingly formed dimeric aggregates at ~17%, the remaining seven VL mutants were monomeric (Fig. 3A), indicating that similar to wild-type VLs, mutant VLs with the extra disulfide linkage were aggregation-resistant. Moreover, the slight aggregation observed in HVLP325 disappeared in the mutant version, HVLP325S. The Mapps of mutant VLs, calculated from their elution volumes, were similar to those of wild-type versions, ranging from 4.9 kDa to 23.5 kDa with a mean Mapp ± SEM of 11.4 ± 2.3 kDa compared with 13.7 ± 2.2 kDa for the wild-type VLs. The SEC results were further confirmed by the MALS data, which showed that the VLs were indeed monomeric as their experimental MMALSs were very close to their theoretical Mfors (Table 1). Furthermore, the minor HVLP364S peak identified as corresponding to a VL dimer in SEC had indeed the MMALS for a dimeric VL: 26.21 ± 0.47 kDa (vs the expected Mfor of 27.94 kDa).
Probing conformational changes in disulfide linkage-engineered human VLs by protein L binding
To probe any possible structural changes brought about by the non-canonical disulfide linkage in VL mutants, the binding of VL mutants to protein L was quantified by SPR. We found that all VL mutants, with the exception of HVLP324S, HVLP342S, and HVLP351S, bound to protein L with almost the same KDs (and kons and koffs) as their wild-type counterparts (Fig. S2; Table 1), indicating that, for the majority of VLs, there were no structural changes due to the engineered disulfide linkage, or if there were any, they were too subtle to be sensed by protein L. HVLP324S, HVLP342S and HVLP351S showed 2- to 3-fold affinity increase toward protein L low affinity binding sites compared with wild-type counterparts. In contrast, in all cases the stoichiometry of VL:protein L binding remained unchanged between the wild-type and corresponding mutant VLs. In the case of HVLP364S, the KD seemed to be in the low micromolar range, but a reliable KD, and consequently a reliable stoichiometry, could not be determined by SPR due to aggregate contamination in the VL sample. A homology structure of HVLP324S suggests that the non-canonical disulfide linkage and protein L binding site occupy distinct locations in mutant VLs (Fig. 7). In contrast, for a VH with a similar disulfide linkage mutation, the non-canonical disulfide linkage is very intimate with and imbedded within the protein A binding site (Fig. 7).
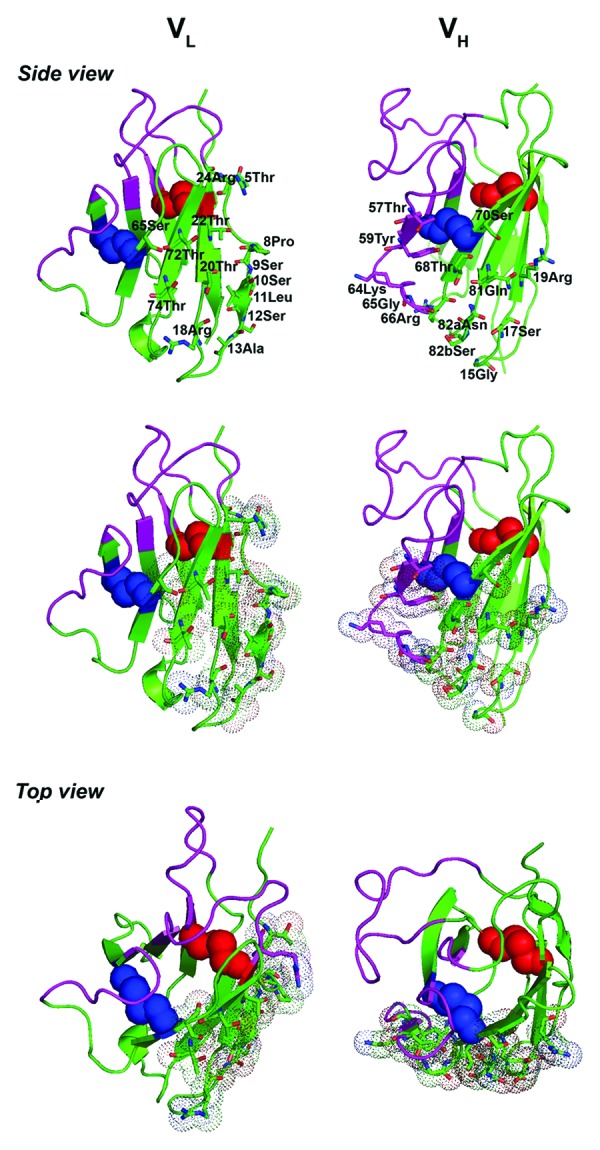
Figure 7. Homology structures of HVLP324S VL and huVHAm302S VH, comparing the positioning of protein L binding site to protein A binding site relative to the engineered non-canonical disulfide linkages (blue spheres) for the VL and VH, respectively. huVHAm302S is a previously described human VH that lost its protein A binding activity by 3.5-fold upon the introduction of a non-canonical disulfide linkage at positions 49 and 69.46 Red spheres represent the native, canonical disulfide linkage, pink- and green-colored regions CDRs and FRs, respectively. Amino acid residues forming protein L binding sites 1 and 2 (top, left panel) and protein A binding site (top, right panel) are shown. The binding site residues were identified based on the published crystal structures of the complex between a human antibody Fab fragment (2A2, κ1 subtype, VH3 family) and a single Peptostreptococcus magnus protein L domain64 (VL) and between Fab 2A2 and domain D of Staphylococcus aureus protein A82 (VH). Middle and bottom panels show side and top views of the protein L and protein A binding sites in dots and sticks presentations. Protein homology structures were obtained as described in the Figure 2 legend. The figures were drawn with PyMOL (http://www.pymol.org) and manipulated using Adobe Photoshop CS2 software.
Thermal stability of disulfide linkage-engineered human VLs
To determine the effect of the non-canonical disulfide linkage on the thermal stability of VLs, the Tms of mutant VLs were determined and compared with those for wild-type VLs. The Tms were in the range of 63.8–82.5 °C (median Tm = 72.9°C) compared with 51.9–68.5 °C (median Tm = 61.9°C) for wild-type VLs (Fig. 4C and D; Table 1). The thermostability improvements were significant, reflecting a Tm increase (ΔTm) range and median of 5.4–17.3 °C and 12.4°C, respectively (Fig. 4D; Table 1) (Wilcoxon matched-pairs signed rank test, two-tailed; p = 0.0078). Although the result indicated that the engineered disulfide linkage stabilized the VLs regardless of their germline subtypes (κ or λ), the thermostability improvements were more pronounced for the κ3 and λ1 subgroup members compared with the κ1 subgroup members.
GI protease resistance of disulfide linkage-engineered human VLs
Previously, VHHs were shown to have acquired protease resistance with the addition of a similar non-canonical disulfide linkage.49 We therefore investigated the effect of the non-canonical disulfide linkage on the resistance of VLs to trypsin, chymotrypsin, and pepsin. Mutant VLs were digested with varying concentrations of proteases under the same digestion conditions as for wild-type VLs (Fig. 6). We observed that there was no significant overall difference between wild-type and mutant VLs with respect to resistance to trypsin or chymotrypsin (Fig. 6A, 6B, and 6E; Wilcoxon matched-pairs signed rank test, two-tailed; P = 0.2969, 0.8438, 0.8438, and 1.0625 for trypsin at 1 µg/mL, 5 µg/mL, 10 µg/mL, and 20 µg/mL, respectively; P = 0.0625, 0.1563, 0.1484, and 0.0781 for chymotrypsin at 1 µg/mL, 5 µg/mL, 10 µg/mL, and 20 µg/mL, respectively).
Mutant VLs, however, showed improved resistance to pepsin. The pepsin resistance medians of wild-type VLs were 73% and 1.5% at enzyme concentrations of 1 µg/mL and 10 µg/mL, respectively, and decreased to 0% at the concentration of 20 µg/mL (Fig. 6C). In contrast, mutant VLs had pepsin resistance medians of 100%, 54%, 39.5%, and 25.9% at enzyme concentrations of 1, 10, 20, and 50 µg/mL, respectively. The pepsin resistance improvements were significant at 1 and 10 µg/mL enzyme concentrations (Fig. 6E; Wilcoxon matched-pairs signed rank test, two-tailed; P = 0.0078 and 0.0156, respectively), but not so at 20 and 50 µg/mL enzyme concentrations (p = 0.0625 for both concentration conditions). Moreover, the correlation between protease resistance and Tm was explored (Fig. 6F). It was found that in general, VLs with higher Tms had higher resistance to pepsin (Pearson’s correlation, 0.6077; P = 0.0125, r2 = 0.3693, at 1 µg/mL enzyme concentration; Pearson’s correlation, 0.7807; P = 0.0004, r2 = 0.6095, at 10 µg/mL enzyme concentration; Pearson’s correlation, 0.7438; P = 0.0010, r2 = 0.5532, at 20 µg/mL enzyme concentration; Pearson’s correlation, 0.6837; P = 0.0035, r2 = 0.4674, at 50 µg/mL enzyme concentration). No significant correlation was observed in the case of trypsin or chymotrypsin (data not shown).
Discussion
Numerous publications in the past two decades have firmly established the suitability of sdAbs as therapeutic and diagnostic agents. Human VH and VL sdAbs, in particular, have been pursued as therapeutics due to their expected lower immunogenicity in patients compared with other classes of sdAbs such as camelid VHHs and shark VNARs.4,5,32 A number of studies have highlighted VLs as bona fide affinity reagents, and a few, in particular, have pointed to an inherent property of VLs as being more aggregation resistant than VHs.23,32-34,57-61 Thus, from the aggregation point of view, human VLs may be preferable over human VHs as immunotherapeutics. In this study, we set out to obtain a deeper understanding of VLs with respect to a number of biophysical properties, including their aggregation tendencies.
Human VH domains are known for their general tendency to aggregate. Previously, a phage selection method was used to obtain exclusively non-aggregating human VH domains from a naïve human VH phage display library that was propagated as plaques.48 The library, with a size of 6 × 108 transformants, was panned against the B cell super-antigen protein A, and sequencing of more than 110 clones with complete VH open reading frames yielded a total of 15 non-aggregating VHs. By applying essentially the same selection approach, we obtained a total of 32 unique sequences out of 34 screened clones from a 200-fold smaller-sized VL library. Given that the selection method has been shown to select only for non-aggregating domains, this high yield isolation of VLs implies that human VLs are more frequently aggregation resistant than human VHs, which is consistent with previous findings.32,33 This is further supported by the fact that our subset of randomly selected protein L binding VLs representing different germline origins were indeed non-aggregating as shown by size-exclusion chromatography, hexa-histidine capture SPR and multiangle light scattering experiments. This also confirms the power of the aforementioned selection approach for the isolation of non-aggregating proteins. However, an intrinsic ability of protein L to screen out structurally compromised, aggregating domains in favor of non-aggregating protein L binders during panning experiments is a possibility that may have contributed to the strong selective power of the approach.
It is not surprising that from a library consisting mostly of Vλ class VL domains, the vast majority of the protein L binding VLs isolated (31 out of 32 unique sequences) were of Vκ type (Vκ3 and Vκ1 subgroups) with only one being of a Vλ type (HVLP389). Previous studies have shown that protein L predominantly binds to the Vκ type domains, specifically to those in the subgroups Vκ1, Vκ3, and Vκ4, with a paucity of binding to λ class VLs.40,41,62-66 Given the selective nature of our approach for stable (non-aggregating) domains, the predominance of Vκ3 subgroup type VLs (24/31) followed next by Vκ1 subgroup type VLs (7/31), and the absence of any Vκ4 subgroup type VLs, in the pool of selected binders may be a reflection of the relative stability of VLs. Previously, it was shown that of the four VL domains representing the consensus sequences of human subgroups Vκ1–4, the Vκ3 VL was the most thermodynamically stable followed by the Vκ1 VL, with the Vκ4 VL appearing to be the least stable of the three.33 Also, unlike the first two VLs, which were monomeric, the Vκ4 VL formed dimers, an indication of its aggregation tendency.33 In fact, a bleak selection for the Vλ class VLs in this study may not have been just the result of their general lack of binding to protein L, but also the result of their lower stability compared with Vκ class VLs.33 Consistent with this is the fact that our lone λ class binder belongs to the Vλ1 subgroup, a subgroup whose one representative was shown to be more thermodynamically stable than two other VLs representing subgroups Vλ2 and Vλ3. It is also possible, however, that the relative proportion of Vκ3 and Vκ1 binders may simply reflect their relative proportions in the original, unselected VL library.
Mutations with respect to VL germline sequences were observed for the pool of selected VLs, but, in the absence of mutational studies, it is very difficult to assign solubility roles to mutation positions. Significant mutations at position 96, e.g., mutations from a hydrophobic germline amino acid to a positively-charged amino acid in the vast majority of VLs, including 6 of the 8 non-aggregating, representative VLs, suggest a solubility role for position 96. Consistent with this, previous studies with immunoglobulin κ1 light chains have suggested that while an aromatic or hydrophobic residue at position 96 enhances dimerization, a charged amino acid (Arg) at the same position results in stable light chain monomers.67 It was explained that Arg-Arg charge repulsion at positions 96 of monomers would interfere with dimer formation.67 Further mutational studies are required to determine if the presence of a positively-charged amino acid at position 96 leads to VLs that are more aggregation resistant, and if so, whether a negatively-charged amino acid would have the same effect. Other studies with immunoglobulin VH domains showed that substitution with negatively-charged amino acids, Asp substitution in particular, were more effective than positively-charged substitutions in increasing the aggregation resistance of VHs.68
We also observed that at FR4 positions 105, 106, and 107, instead of the typical Asp/Glu, Ile, and Lys, the majority of the κ class VLs had Thr, Val, and Leu, amino acids, respectively, which are characteristic of λ class VLs. Whether the substitutions have a role in improving the biophysical properties of the κ class VLs remains to be seen. The importance of FR4 residues in improving the aggregation resistance of immunoglobulin variable domains (VHHs and VHs) has been suggested.69,70 For example, VHHs, which are known for their high aggregation resistance, have a highly conserved 105Q mutation compared with the aggregation prone VHs. Similarly, VHs with the 105Q mutation have been shown to have improved aggregation resistance compared with a corresponding wild-type VH. Also, the role of J segment, which codes for the FR4 amino acids, in increasing the thermal stability and non-aggregation of VHHs has been shown by others.69
SPR binding experiments on the eight representative non-aggregating VLs confirmed their protein L binding activity. Of interest were the two Vκ1 type binders, which unlike the Vκ3 and Vλ type VLs that bound to protein L with similar micromolar affinities (1–3 µM), bound to protein L with low (0.2 µM and 0.6 µM) and high (0.04 µM and 0.07 µM) affinities. High and low affinity binding of human Fab and light chain of Vκ1-subgroup type to 2 distinct sites on single Ig binding domains of protein L have been reported previously.63,64 Here a differential VL:protein L stoichiometry was also observed, with the λ type VL having a 1:1 binding stoichiometry, the Vκ3 VLs having a 3:1 binding stoichiometry in the majority of cases, and Vκ1 having a 7:1 binding stoichiometry. Previous SPR binding studies with 5 Ig-binding domains of a protein L showed that while all 5 domains bound to a human Vκ light chain of Vκ1 subgroup, only 3 bound to a human Vκ light chain of Vκ3 subgroup, supporting the higher observed stoichiometry for Vκ1 binders compared with Vκ3 binders and the 3:1 stoichiometry found for the Vκ3 binders. A binding stoichiometry of 7:1 in the case Vκ1 subgroup binders is plausible, given that our protein L consisted of 4 Ig-binding domains with each Ig-binding domain having up to 2 binding sites that can simultaneously engage with 2 sites on Vκ1 subgroup binders.63,64,71 A higher affinity and stoichiometry (higher avidity) in the case of Vκ1 type antibodies should translate to their more sensitive detection by protein L. The low affinity and lack of avidity as a result of a 1:1 stoichiometry, as shown here for the Vλ type HVLP389, may be the reason for the reported weak interactions between human Ig λ-light chains and protein L.41,65,66 Thus, failing to detect a Vλ-protein L interaction should not be interpreted as the lack of protein L binding activity on the part of a Vλ type antibody. It should, however, be mentioned that the stoichiometry values are estimates. If the surface is not fully active the theoretical Rmax for 1:1 binding cannot be attained, resulting in underestimation of the binding stoichiometry. Isothermal titration calorimetry may provide a more accurate means of determining the binding stoichiometries.
Consistent with being highly non-aggregating at concentrations as high as ≈450 μM, the VLs showed high reversibility of thermal unfolding at relatively high VL concentrations. In this respect, and with respect to Tm, the VLs performed as well as our set of non-aggregating VHs. The observed lack of correlation between Tm and aggregation resistance, expressed in terms of TREs, is consistent with previous findings that showed that aggregation resistant VHs may not necessarily have high Tm or thermodynamic stability.72 Consistent with this finding is the fact that the VL with the highest Tm from among the eight VLs in this study (HVLP325) was also the only one that showed some degree of aggregation.
The VLs were different from VHs with respect to GI protease resistance patterns. That is, while VLs were more resistant to chymotrypsin, the VHs were more so to pepsin, with both VLs and VHs being comparable in terms of resistance to trypsin. The observed protease resistance data for only chymotrypsin could be explained in terms of the number of potential cleavage sites, i.e., VLs had a significantly lower number of potential chymotrypsin cleavage sites than VHs. Even when only the more protease accessible CDR sequences were considered in the calculation of theoretical number of cleavage sites, the obtained numbers did not fully explain the observed differences between protease resistance profiles of VLs and VHs (data not shown). Previously for VHHs, increases in pepsin resistance were correlated with increases in Tms, but this correlation cannot explain the better pepsin resistance of VHs here because both VHs and VLs had very similar Tms.49 As discussed previously, protease sensitivity is a function of a number of variables including the theoretical number of proteolytic sites, the location of proteolytic sites, and protein compactness and thermodynamic stability.49,73,74
In an effort to further improve the biophysical properties of VLs, we introduced a pair of cysteine residues at amino acid positions 48 and 64, hypothesizing that this would lead to the formation of a disulfide linkage in the sdAb core. Previously, it was shown that the substitution for a pair of cysteine at spatially equivalent positions in VHHs and VHs led to the formation of disulfide linkages in all domains tested, with subsequent improvements in thermostability and protease resistance.46,49-52 We too find here that all the VLs with the added Cys pair have the intended disulfide linkage, as well as improved thermostability (Tm) and protease resistance. We find that the increases in Tms (ΔTm) are relatively high (5.4–17.3 °C, median ΔTm: 12.4°C) compared with those obtained for a previously reported set of VHHs with similar engineered disulfide linkages (ΔTm: 4–12°C, median ΔTm: 7°C).49 This may, at least partly, be due to the fact that Tms were obtained under different assay conditions and instrument settings. It cannot be excluded, however, that a non-canonical disulfide linkage may have been a better fit to the overall fold of VLs, leading to their overall higher ΔTm gains.49 This may also explain the differential Tm gains observed among the mutant VLs that were characterized under identical conditions. We also find that the thermostability gain due to the engineered disulfide linkage is more pronounced for the κ3 and λ1 members, compared with the κ1 members; however, general conclusions should await further experiments involving a statistically appropriate sample size.
In terms of protease resistance, the non-canonical disulfide linkage led to increases in pepsin resistance of VLs, without compromising their trypsin or chymotrypsin resistance. This is similar to the results obtained with a set of VHHs with similar non-canonical disulfide linkages.49 As with the VHHs, positive correlations between pepsin resistance and Tm were also observed. The higher pepsin resistance of the mutant VLs may be due to the fact that they may have a more compact and thermodynamically stable structure—equating to higher resistance to acid-induced unfolding under pepsin digestion conditions (pH = 2; pepsin is more effective on denatured proteins)—compared with the wild-type VLs without the non-canonical disulfide linkage.49 Higher Tms of mutants, suggesting their higher thermodynamic stabilities, and the fact that VHHs with similar engineered disulfide linkage became resistant to unfolding at pH 2 and 37°C support this speculation.49 Importantly, the introduction of the non-canonical disulfide linkage into VLs does not appear to compromise their aggregation resistance. This was shown to be the case for VHHs and VHs with similar non-canonical disulfide linkages as well.46,49 In the case of VHs, this led to improvements in the aggregation resistance of mutants compared with the wild-type counterparts without the non-canonical disulfide linkage.
The biophysical improvements gained through the introduction of the non-canonical disulfide linkage do come at the expense of expression yield as mutants demonstrated significantly lower expression yields than wild-type VLs in E. coli. This was also reported in the case of VHHs, which like the VLs here, were expressed in E. coli.49 Thus, the relatively lower expression yields of Cys mutant domains compared with wild-type ones may have to do with the limited capacity of E. coli in folding proteins with higher disulfide linkages such as the Cys mutant VLs in this study. This should be resolved by expressing the mutant VLs in eukaryotic microorganisms, e.g., yeast or mammalian cells, with the capacity to fold complex proteins such as those with multiple disulfide linkages.
The biophysical improvements also come at the expense of undesirable conformational changes for mutants, which were also reported in the case of VHHs and VHs.46,49 The observed differential protease resistance profiles between wild-type and corresponding mutant VLs, as well as changes to protein L binding for some of the mutants, support this conclusion. However, for the majority of VLs, the conformational changes, as determined by binding measurements of wild-type and Cys mutant VLs against protein L, are too subtle to be sensed by protein L, which binds to VLs in a conformation-dependent manner.41 This is in sharp contrast to the results obtained with our Cys mutant VHs,46 where structural changes as a result of the introduction of non-canonical disulfide linkages were more easily probed with protein A and led to up to 10-fold reductions in protein A binding of mutant VHs. Such discrepancy could be due to the fact that, while for VLs the protein L binding site is too far from the engineered disulfide linkage to be affected by it, for VHs the protein A binding site is in the influencing range of the non-canonical disulfide linkage. This is clearly supported by our homology structure data of a VL and a VH with similar non-canonical disulfide linkages (see Fig. 7). The conformational changes in VL mutants were not reflected in SPR stoichiometry data either, as all wild-type/mutant pairs had the same stoichiometry of binding to protein L.
In conclusion, we demonstrated the suitability of VL sdAbs as affinity reagents, in particular, as immunotherapeutics, and provided insights into the biophysical characteristics of VLs. We identified a diversity of non-aggregating VL domains that could form the basis of therapeutics when incorporated as library scaffolds (e.g., into sdAb phage display libraries) as has already been demonstrated.32 As, irrespective of the degree of the stability of the original library scaffold, loop randomization always leads to a proportion of the library consisting of unstable domains, a coupling of affinity selection to stability selection during the panning experiments to filter out unstable binders from the pool of binders is advisable.53,55,72,75 Moreover, we presented a general strategy based on disulfide linkage engineering for stabilizing VLs in terms of thermostability and pepsin resistance. The disulfide linkage engineered VLs would be the preferred scaffolds for constructing VL phage display libraries over versions without. Such libraries, especially if affinity selected under high temperature conditions, should yield non-aggregating immunotherapeutics that are also thermodynamically stable, like those VH domains that were selected by panning a phage display library under the acidic conditions.72 Due to the positive correlation between pepsin resistance and Tm, affinity selection under heat should yield binders which are also pepsin resistant, hence suitable as oral therapeutics for GI tract applications. The disulfide linkage engineering approach should thus be viewed as an efficacy engineering one for therapeutic VLs. The fact that the affinity reagents obtained from the aforementioned VL libraries would have protein L binding properties offers biotechnological advantages similar to those offered by VHs that bind to B cell super-antigen protein A.48
Beyond single domains, the disulfide linkage engineering approach can be applied to domains in the context of scFvs, Fabs, and IgGs. Previously, when a similar disulfide linkage engineering approach was applied to a VH in the context of a Fab, it resulted in a significant increase in the thermostability of the Fab.76 However, the introduction of the disulfide linkage results in conformational changes that may further lead to undesirable affinity and specificity compromises as has been shown in the case of VHHs with similar disulfide linkage engineering.49-51 This should not be a concern with domains that are selected from libraries already containing the non-canonical disulfide linkage.
Materials and Methods
Human VL library construction and panning
A VL phage display library in a multivalent display format and with plaque formation as the selectable marker was constructed in a similar fashion as a previously described VH phage display library.48 It was anticipated that, as in the case of the VH phage display library, phages displaying non-aggregating domains (VLs in the present study) would form larger plaques on bacterial lawns than phages displaying aggregation prone domains, and, as a result, panning the library against super-antigen protein L for several cycles should eventually enrich the library for phage-displayed, aggregation-resistant VLs that bind protein L. Such VLs can subsequently be retrieved and expressed as soluble, autonomous domains. Briefly, cDNA was synthesized from human spleen mRNA as previously described.48 The cDNA was used as a template for PCR to amplify VL genes in 50 µL reaction volumes using six Vκ and 11 Vλ back primers77 and four Vκ and two Vλ forward primers.78 The back and forward primers were modified to have flanking ApaLI and NotI restriction sites, respectively, for subsequent cloning purposes. Forward primers were pooled together in ratios that reflected their degree of degeneracy. Vλ genes were subjected to PCR in 11 separate reactions using the pooled Vλ forward primers with 11 individual Vλ back primers. Similarly, Vκ genes were amplified in six separate reactions using the pooled Vκ forward primers with six individual Vκ back primers. The PCR products were gel-purified and digested with ApaLI and NotI restriction endonucleases, and the library was constructed as previously described.48 PCR was performed on individual library colonies, and the amplified VL genes were sequenced as described before.79 Panning (against protein L; Pierce) and the germline assignment of VLs were performed as previously described.48
Cloning, expression, purification, SEC, and MALS analysis
Introduction of cysteine residues at Kabat positions 48 and 6443 of VLs was performed using a splice overlap extension method.46 VL expression, purification, concentration determination, and SEC were performed as described for VHs.47,55 Size-exclusion chromatograms were normalized as described.46,47 The elution volume values obtained from chromatograms were used to calculate Mapps of VLs using the Log M vs. Ve standard curve described in Figure S1. UPLC-SEC-MALS was performed with a Waters BioAcquity system equipped with a Waters PDA detector (Waters). The column temperature was maintained at 30°C with a CM-A column compartment. The samples (10–20 µg) were injected onto a Waters BEH125 SEC column (4.6 mm × 150 mm, 1.7 µm particles) at 0.4 mL/min with PBS (–Ca –Mg, Hyclone Thermo Scientific) as the solvent. MALS data was measured on a Wyatt HELEOS 8+ detector (Wyatt Technology Corporation) and weight average molecular masses (MMALSs) were calculated using a protein concentration determined using the A280 from the PDA detector with extinction coefficients calculated from the amino acid sequences. Experiments were performed in duplicates. MALS data processing was performed with ASTRA 6.1 software (Wyatt Technology Corporation).
Disulfide linkage determination
Disulfide linkage determination for Cys mutant VLs was performed as described elsewhere.46,49 Briefly, tryptic fragments for subsequent MS analysis were prepared as described.47 Purified VLs at ~15 pmol/µL in 50% (v/v) acetonitrile + 0.1% (v/v) formic acid were infused at 1 µL/min for electrospray ionization (ESI) mass spectrometric mass measurements of the VLs using a Q-TOF 2TM mass spectrometer (Waters). The mass spectra of the VLs were de-convoluted using the MaxEnt 1 program in the MassLynx software package (Waters). Aliquots of VL proteolytic digests were re-suspended in 0.1% (v/v) formic acid (aq) and subsequently analyzed by nano-flow reversed-phase HPLC MS (nanoRPLC-ESI-MS) with data-dependent analysis (DDA) using CID on a nanoAcquity UPLC system coupled to a Q-TOF UltimaTM hybrid quadrupole/TOF mass spectrometer (Waters). The peptides were first loaded onto a 180 µm I.D. × 20 mm 5 µm symmetry®C18 trap (Waters) and then eluted into a 100 µm I.D. × 10 cm 1.7 µm BEH130C18 column (Waters) using a linear gradient from 0% to 36% solvent B (acetonitrile + 0.1% formic acid) over 36 min followed by 36–90% solvent B for 2 min. Solvent A was 0.1% formic acid in water. The peptide MS2 spectra were compared with mutant VL protein sequences using the Mascot™ database searching algorithm (Matrix Science). The MS2 spectra of the disulfide-linked peptides were de-convoluted using the MaxEnt 3 program (Waters) for de novo sequencing to confirm and determine the exact disulfide linkage positions. NanoRPLC-ESI-MS analyses with DDA using a combination of ETD and CID were performed on an LTQXL mass spectrometer fitted with an ETD source (Thermo Fisher Scientific) and coupled to a nanoAcquity UPLC system (Waters) using the LC conditions described above. Briefly, for the DDA experiments, the most abundant peptide ion from the survey scan was used for the automatic acquisition of ETD-MS2 spectra using 30% normalized collision energy followed by the acquisition of ETD-CID-MS3 spectra of the most abundant charge-reduced ions produced by ETD.
SPR binding studies
VLs were subjected to Superdex 75™ (GE Healthcare) SEC in HBS-EP buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA and 0.005% P20 surfactant) at 0.5 mL/min prior to BIACORE analysis and purified monomer peaks were collected even in the absence of any evidence of aggregated material. Binding kinetics for the interaction of VLs to protein L were determined by SPR using a BIACORE 3000 biosensor system (GE Healthcare). For wild-type VLs, ~600 resonance units (RUs) of protein L or 800 RUs of a Fab reference were immobilized onto research grade CM5 sensorchips (GE Healthcare). For mutant VLs, ~650 RUs of protein L (for HVLP324S, HVLP325S, and HVLP342S) or 400 RUs of protein L (for HVLP335S, HVLP351S, HVLP389S, and HVLP3103S) or 400 RUs of ovalbumin (Sigma-Aldrich) as a reference protein were immobilized. For HVLP324S and HVLP342S, ~1,700 RUs of protein L or 2,600 RUs of a ovalbumin reference were immobilized onto research grade CM5 sensorchips to determine binding data for the low affinity binding sites on protein L. Immobilization was performed at protein concentrations of 20 or 50 µg/mL in 10 mM acetate buffer, pH 4.5, using an amine coupling kit supplied by the manufacturer (GE Healthcare). All measurements were performed at 25°C in HBS-EP buffer at 40 or 50 µL/min. The surfaces were regenerated through washing with the running buffer. Data were evaluated using BIAevaluation 4.1 software (GE Healthcare). The stoichiometry of binding (VL:protein L) was estimated by comparing the observed Rmaxs and theoretical Rmaxs for 1:1 binding.
SPR analysis for the binding of VLs, a control llama VHH monomer (A4.2), and a control VH homodimer to a Ni2+-NTA sensorchip was performed as described previously.46 All VL/VH/VHH domains had His6 tags at their C-termini and the protein preparations lacked contaminating species without the His6 tag as determined by Western blot and SDS-PAGE analyses. The VH homodimer is formed by the non-covalent association of two VH monomer units, and, as a result, has two C-terminal His6 tags. SPR experiments were performed with protein fractions corresponding to the dimeric peak in the case of the VH dimer control and monomeric peaks for the remaining VL/VHH domains. RUs from duplicate data sets were averaged and then normalized to obtain %RU. The SPR binding of antibody domains to an activated NTA sensorchip was determined at 25°C using a BIACORE 3000. In each cycle, NiCl2 was injected, followed by an injection of VL/VH/VHH. Sensorgrams were run in duplicates. The NTA chip was regenerated with EDTA before the next cycle. Dissociation rate constants (koffs) were obtained over 60 s periods between 315–375 s and 780–840 s and data were analyzed with BIAevaluation 4.1 software.
Protease digestion experiments
Digestion experiments were performed in a total volume of 30 µL with 6 µg of VL in phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4) at 37°C for 1 h with an enzyme to VL ratio of 1:200, 1:40, 1:20, and 1:10 (trypsin/chymotrypsin), corresponding to 1 µg/mL, 5 µg/mL, 10 µg/mL, and 20 µg/mL protease concentrations, respectively, or 1:200, 1:20, 1:10, and 1:4 (pepsin), which corresponds to 1 µg/mL, 10 µg/mL, 20 µg/mL, and 50 µg/mL protease concentrations, respectively. Tryptic and chymotryptic digestion experiments were performed using sequencing grade enzymes (Roche Diagnostics) according to the manufacturer’s instructions. Peptic digestion experiments were performed at pH 2.0 using pepsin from Sigma-Aldrich. In control experiments, enzymes were replaced with equal volumes of reaction buffers. Reactions were stopped by adding an equal volume of SDS-PAGE sample buffer containing 0.2 M dithiothreitol and boiling mixtures at 95°C for 5 min. The samples were then subjected to SDS-PAGE analysis and the percentages of intact sdAbs after protease digestions were determined by spot density analysis as described.49 For VHs, digestion experiments were performed as described for VLs with an enzyme to VH ratio of 1:20.
TRE experiments
TREs were determined by SPR as previously described.48 Briefly, VLs or VHs were incubated at 85°C for 20 min at concentrations of 4 or 20 µM, slowly cooled to room temperature for 30 min, centrifuged at 16,000 X g for 5 min at 22°C, and the supernatants, termed “refolded,” were kept for subsequent SPR analysis. Following this, binding analysis against protein L and protein A for VLs and VHs, respectively, was performed. Native and refolded domains were analyzed under identical conditions. For VLs at 4 µM, 600 RUs of protein L and 700 RUs of a Fab reference were immobilized at 25 or 50 µg/mL in 10 mM acetate buffer, pH 4.5. For VHs at 4 µM, 600 RUs of protein A (Sigma-Aldrich) or ovalbumin reference were immobilized at 50 µg/mL in 10 mM acetate buffer, pH 4.5. For VLs at 20 µM, 2,100 RUs of protein L and 1,200 RUs of ovalbumin reference were immobilized at 100 µg/mL in acetate buffer, pH 4.0, and 50 µg/mL in 10 mM acetate buffer, pH 4.5, respectively. For VHs at 20 µM, 650–1,100 RUs of protein A and 600–1,200 RUs of ovalbumin were immobilized at 50 µg/mL in 10 mM acetate buffer, pH 4.5. In all instances, the analyses were performed at 25°C in HBS-EP buffer at 40 µL/min. The surfaces were washed thoroughly with the running buffer for regeneration. Data were analyzed with BIAevaluation 4.1 software and KDs were analyzed with 1:1 binding models. Refolding efficiencies were expressed in terms of the ratio of KD (refolded)/KD (native).48
Tm measurements
Circular dichroism spectra and Tms of VLs and VHs were obtained as described previously47,49 using a Jasco J-815 spectropolarimeter (Jasco) equipped with a Peltier thermoelectric type temperature control system. The circular dichroism spectra were recorded over a temperature range of 30°C to 96°C in 100 mM sodium phosphate buffer, pH 7.4, and with a protein concentration of 50 µg/mL. Ellipticity changes at 203 nm for HVLP325 and HVLP389, at 218 nm for HVLP324, HVLP335, HVLP342, HVLP351, HVLP364, and HVLP3103, at 210 nm for Cys mutant VLs, at 205 nm for HVHP421, HVHP428, and HVHP429, at 200 nm for HVHP413, HVHP419, and HVHP420, and at 202 nm for HVHP44 and HVHP414 were used for constructing thermal unfolding curves and subsequent calculations of Tms. At these wavelengths, wide differences in ellipticity values between the folded (at 30°C) and fully denatured (at 90°C) domains allowed reliable determination of Tms (Fig. S6).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
We thank Sonia Leclerc for DNA sequencing.
Supplemental Materials
Supplemental material can be found at www.landesbioscience.com/journals/mabs/article/26844
Glossary
Abbreviations:
- CDR
complementarity-determining region
- CID
collision induced dissociation
- DDA
data dependent analysis
- ETD
electron transfer dissociation
- Fab
fragment antigen-binding
- FR
framework region
- HBS-EP buffer
10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA and 0.005% P20 surfactant
- GI
gastrointestinal
- KD
equilibrium dissociation constant
- MALS
multiangle light scattering
- Mapp
apparent molecular mass
- Mfor
formula molecular mass
- MMALS
molecular mass determined by MALS
- MS
mass spectrometry
- RU
resonance unit
- sdAb
single-domain antibody
- SEC
size-exclusion chromatography
- scFv
single chain Fv fragment of an antibody
- SPR
surface plasmon resonance
- Tm
melting temperature
- TRE
thermal refolding efficiency
- VH
antibody heavy chain variable domain
- VHH
camelid heavy chain antibody variable domain
- VL
antibody light chain variable domain
- VNAR
shark IgNAR (Ig New Antigen Receptor) variable domain
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/26844
References
- 1.Fox JL. Anthrax drug first antibacterial mAb to win approval. Nat Biotechnol. 2013;31:8. doi: 10.1038/nbt0113-8. [DOI] [PubMed] [Google Scholar]
- 2.Nelson AL. Antibody fragments: hope and hype. MAbs. 2010;2:77–83. doi: 10.4161/mabs.2.1.10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson AL, Reichert JM. Development trends for therapeutic antibody fragments. Nat Biotechnol. 2009;27:331–7. doi: 10.1038/nbt0409-331. [DOI] [PubMed] [Google Scholar]
- 4.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23:1126–36. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 5.Holt LJ, Herring C, Jespers LS, Woolven BP, Tomlinson IM. Domain antibodies: proteins for therapy. Trends Biotechnol. 2003;21:484–90. doi: 10.1016/j.tibtech.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Ma X, Barthelemy PA, Rouge L, Wiesmann C, Sidhu SS. Design of synthetic autonomous VH domain libraries and structural analysis of a VH domain bound to vascular endothelial growth factor. J Mol Biol. 2013;425:2247–59. doi: 10.1016/j.jmb.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 7.Matz J, Kessler P, Bouchet J, Combes O, Ramos OH, Barin F, Baty D, Martin L, Benichou S, Chames P. Straightforward selection of broadly neutralizing single-domain antibodies targeting the conserved CD4 and coreceptor binding sites of HIV-1 gp120. J Virol. 2013;87:1137–49. doi: 10.1128/JVI.00461-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukherjee J, Tremblay JM, Leysath CE, Ofori K, Baldwin K, Feng X, Bedenice D, Webb RP, Wright PM, Smith LA, et al. A novel strategy for development of recombinant antitoxin therapeutics tested in a mouse botulism model. PLoS One. 2012;7:e29941. doi: 10.1371/journal.pone.0029941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jamnani FR, Rahbarizadeh F, Shokrgozar MA, Ahmadvand D, Mahboudi F, Sharifzadeh Z. Targeting high affinity and epitope-distinct oligoclonal nanobodies to HER2 over-expressing tumor cells. Exp Cell Res. 2012;318:1112–24. doi: 10.1016/j.yexcr.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Wei G, Meng W, Guo H, Pan W, Liu J, Peng T, Chen L, Chen CY. Potent neutralization of influenza A virus by a single-domain antibody blocking M2 ion channel protein. PLoS One. 2011;6:e28309. doi: 10.1371/journal.pone.0028309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trilling AK, de Ronde H, Noteboom L, van Houwelingen A, Roelse M, Srivastava SK, Haasnoot W, Jongsma MA, Kolk A, Zuilhof H, et al. A broad set of different llama antibodies specific for a 16 kDa heat shock protein of Mycobacterium tuberculosis. PLoS One. 2011;6:e26754. doi: 10.1371/journal.pone.0026754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skottrup PD, Leonard P, Kaczmarek JZ, Veillard F, Enghild JJ, O’Kennedy R, Sroka A, Clausen RP, Potempa J, Riise E. Diagnostic evaluation of a nanobody with picomolar affinity toward the protease RgpB from Porphyromonas gingivalis. Anal Biochem. 2011;415:158–67. doi: 10.1016/j.ab.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldman ER, Anderson GP, Zabetakis D, Walper S, Liu JL, Bernstein R, Calm A, Carney JP, O’Brien TW, Walker JL, et al. Llama-derived single domain antibodies specific for Abrus agglutinin. Toxins (Basel) 2011;3:1405–19. doi: 10.3390/toxins3111405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emmerson CD, van der Vlist EJ, Braam MR, Vanlandschoot P, Merchiers P, de Haard HJW, Verrips CT, van Bergen en Henegouwen PM, Dolk E. Enhancement of polymeric immunoglobulin receptor transcytosis by biparatopic VHH. PLoS One. 2011;6:e26299. doi: 10.1371/journal.pone.0026299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hmila I, Saerens D, Ben Abderrazek R, Vincke C, Abidi N, Benlasfar Z, Govaert J, El Ayeb M, Bouhaouala-Zahar B, Muyldermans S. A bispecific nanobody to provide full protection against lethal scorpion envenoming. FASEB J. 2010;24:3479–89. doi: 10.1096/fj.09-148213. [DOI] [PubMed] [Google Scholar]
- 16.Monegal A, Ami D, Martinelli C, Huang H, Aliprandi M, Capasso P, Francavilla C, Ossolengo G, de Marco A. Immunological applications of single-domain llama recombinant antibodies isolated from a naïve library. Protein Eng Des Sel. 2009;22:273–80. doi: 10.1093/protein/gzp002. [DOI] [PubMed] [Google Scholar]
- 17.Li S, Zheng W, Kuolee R, Hirama T, Henry M, Makvandi-Nejad S, Fjällman T, Chen W, Zhang J. Pentabody-mediated antigen delivery induces antigen-specific mucosal immune response. Mol Immunol. 2009;46:1718–26. doi: 10.1016/j.molimm.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 18.Deckers N, Saerens D, Kanobana K, Conrath K, Victor B, Wernery U, Vercruysse J, Muyldermans S, Dorny P. Nanobodies, a promising tool for species-specific diagnosis of Taenia solium cysticercosis. Int J Parasitol. 2009;39:625–33. doi: 10.1016/j.ijpara.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Abderrazek RB, Hmila I, Vincke C, Benlasfar Z, Pellis M, Dabbek H, Saerens D, El Ayeb M, Muyldermans S, Bouhaouala-Zahar B. Identification of potent nanobodies to neutralize the most poisonous polypeptide from scorpion venom. Biochem J. 2009;424:263–72. doi: 10.1042/BJ20090697. [DOI] [PubMed] [Google Scholar]
- 20.Koide A, Tereshko V, Uysal S, Margalef K, Kossiakoff AA, Koide S. Exploring the capacity of minimalist protein interfaces: interface energetics and affinity maturation to picomolar D of a single-domain antibody with a flat paratope. J Mol Biol. 2007;373:941–53. doi: 10.1016/j.jmb.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopsidas G, Roberts AS, Coia G, Streltsov VA, Nuttall SD. In vitro improvement of a shark IgNAR antibody by Qß replicase mutation and ribosome display mimics in vivo affinity maturation. Immunol Lett. 2006;107:163–8. doi: 10.1016/j.imlet.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 22.Shen J, Vil MD, Jimenez X, Iacolina M, Zhang H, Zhu Z. Single variable domain-IgG fusion. A novel recombinant approach to Fc domain-containing bispecific antibodies. J Biol Chem. 2006;281:10706–14. doi: 10.1074/jbc.M513415200. [DOI] [PubMed] [Google Scholar]
- 23.Colby DW, Chu Y, Cassady JP, Duennwald M, Zazulak H, Webster JM, Messer A, Lindquist S, Ingram VM, Wittrup KD. Potent inhibition of huntingtin aggregation and cytotoxicity by a disulfide bond-free single-domain intracellular antibody. Proc Natl Acad Sci U S A. 2004;101:17616–21. doi: 10.1073/pnas.0408134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cortez-Retamozo V, Backmann N, Senter PD, Wernery U, De Baetselier P, Muyldermans S, Revets H. Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 2004;64:2853–7. doi: 10.1158/0008-5472.CAN-03-3935. [DOI] [PubMed] [Google Scholar]
- 25.Nuttall SD, Humberstone KS, Krishnan UV, Carmichael JA, Doughty L, Hattarki M, Coley AM, Casey JL, Anders RF, Foley M, et al. Selection and affinity maturation of IgNAR variable domains targeting Plasmodium falciparum AMA1. Proteins. 2004;55:187–97. doi: 10.1002/prot.20005. [DOI] [PubMed] [Google Scholar]
- 26.Saerens D, Kinne J, Bosmans E, Wernery U, Muyldermans S, Conrath K. Single domain antibodies derived from dromedary lymph node and peripheral blood lymphocytes sensing conformational variants of prostate-specific antigen. J Biol Chem. 2004;279:51965–72. doi: 10.1074/jbc.M409292200. [DOI] [PubMed] [Google Scholar]
- 27.Nuttall SD, Krishnan UV, Doughty L, Pearson K, Ryan MT, Hoogenraad NJ, Hattarki M, Carmichael JA, Irving RA, Hudson PJ. Isolation and characterization of an IgNAR variable domain specific for the human mitochondrial translocase receptor Tom70. Eur J Biochem. 2003;270:3543–54. doi: 10.1046/j.1432-1033.2003.03737.x. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka T, Lobato MN, Rabbitts TH. Single domain intracellular antibodies: a minimal fragment for direct in vivo selection of antigen-specific intrabodies. J Mol Biol. 2003;331:1109–20. doi: 10.1016/S0022-2836(03)00836-2. [DOI] [PubMed] [Google Scholar]
- 29.Davies J, Riechmann L. Affinity improvement of single antibody VH domains: residues in all three hypervariable regions affect antigen binding. Immunotechnology. 1996;2:169–79. doi: 10.1016/S1380-2933(96)00045-0. [DOI] [PubMed] [Google Scholar]
- 30.Arbabi-Ghahroudi M, Tanha J, MacKenzie R. Prokaryotic expression of antibodies. Cancer Metastasis Rev. 2005;24:501–19. doi: 10.1007/s10555-005-6193-1. [DOI] [PubMed] [Google Scholar]
- 31.Muyldermans S. Single domain camel antibodies: current status. J Biotechnol. 2001;74:277–302. doi: 10.1016/s1389-0352(01)00021-6. [DOI] [PubMed] [Google Scholar]
- 32.Hussack G, Keklikian A, Alsughayyir J, Hanifi-Moghaddam P, Arbabi-Ghahroudi M, van Faassen H, Hou ST, Sad S, MacKenzie R, Tanha J. A VL single-domain antibody library shows a high-propensity to yield non-aggregating binders. Protein Eng Des Sel. 2012;25:313–8. doi: 10.1093/protein/gzs014. [DOI] [PubMed] [Google Scholar]
- 33.Ewert S, Huber T, Honegger A, Plückthun A. Biophysical properties of human antibody variable domains. J Mol Biol. 2003;325:531–53. doi: 10.1016/S0022-2836(02)01237-8. [DOI] [PubMed] [Google Scholar]
- 34.Dubnovitsky AP, Kravchuk ZI, Chumanevich AA, Cozzi A, Arosio P, Martsev SP. Expression, refolding, and ferritin-binding activity of the isolated VL-domain of monoclonal antibody F11. Biochemistry (Mosc) 2000;65:1011–8. [PubMed] [Google Scholar]
- 35.Tomlinson IM, Cox JP, Gherardi E, Lesk AM, Chothia C. The structural repertoire of the human Vκ domain. EMBO J. 1995;14:4628–38. doi: 10.1002/j.1460-2075.1995.tb00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lefranc MP. Nomenclature of the human immunoglobulin lambda (IGL) genes. Exp Clin Immunogenet. 2001;18:242–54. doi: 10.1159/000049203. [DOI] [PubMed] [Google Scholar]
- 37.Lefranc MP. Nomenclature of the human immunoglobulin kappa (IGK) genes. Exp Clin Immunogenet. 2001;18:161–74. doi: 10.1159/000049195. [DOI] [PubMed] [Google Scholar]
- 38.Knappik A, Ge L, Honegger A, Pack P, Fischer M, Wellnhofer G, Hoess A, Wölle J, Plückthun A, Virnekäs B. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Biol. 2000;296:57–86. doi: 10.1006/jmbi.1999.3444. [DOI] [PubMed] [Google Scholar]
- 39.Viau M, Longo NS, Lipsky PE, Björck L, Zouali M. Specific in vivo deletion of B-cell subpopulations expressing human immunoglobulins by the B-cell superantigen protein L. Infect Immun. 2004;72:3515–23. doi: 10.1128/IAI.72.6.3515-3523.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Åkerström B, Nilson BH, Hoogenboom HR, Björck L. On the interaction between single chain Fv antibodies and bacterial immunoglobulin-binding proteins. J Immunol Methods. 1994;177:151–63. doi: 10.1016/0022-1759(94)90152-X. [DOI] [PubMed] [Google Scholar]
- 41.Nilson BH, Solomon A, Björck L, Åkerström B. Protein L from Peptostreptococcus magnus binds to the κ light chain variable domain. J Biol Chem. 1992;267:2234–9. [PubMed] [Google Scholar]
- 42.Bork P, Holm L, Sander C. The immunoglobulin fold. Structural classification, sequence patterns and common core. J Mol Biol. 1994;242:309–20. doi: 10.1016/S0022-2836(84)71582-8. [DOI] [PubMed] [Google Scholar]
- 43.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of Proteins of Immunological Interest, 5th ed., National Institutes of Health Publication 91-3242, National Institutes of Health, Bethesda, MD 1991. [Google Scholar]
- 44.Lesk AM, Chothia C. Evolution of proteins formed by β-sheets. II. The core of the immunoglobulin domains. J Mol Biol. 1982;160:325–42. doi: 10.1016/0022-2836(82)90179-6. [DOI] [PubMed] [Google Scholar]
- 45.Amzel LM, Poljak RJ. Three-dimensional structure of immunoglobulins. Annu Rev Biochem. 1979;48:961–97. doi: 10.1146/annurev.bi.48.070179.004525. [DOI] [PubMed] [Google Scholar]
- 46.Kim DY, Kandalaft H, Ding W, Ryan S, van Faassen H, Hirama T, Foote SJ, MacKenzie R, Tanha J. Disulfide linkage engineering for improving biophysical properties of human VH domains. Protein Eng Des Sel. 2012;25:581–9. doi: 10.1093/protein/gzs055. [DOI] [PubMed] [Google Scholar]
- 47.Kim DY, Ding W, Tanha J. Solubility and stability engineering of human VH domains. Methods Mol Biol. 2012;911:355–72. doi: 10.1007/978-1-61779-968-6_21. [DOI] [PubMed] [Google Scholar]
- 48.To R, Hirama T, Arbabi-Ghahroudi M, MacKenzie R, Wang P, Xu P, Ni F, Tanha J. Isolation of monomeric human VHs by a phage selection. J Biol Chem. 2005;280:41395–403. doi: 10.1074/jbc.M509900200. [DOI] [PubMed] [Google Scholar]
- 49.Hussack G, Hirama T, Ding W, Mackenzie R, Tanha J. Engineered single-domain antibodies with high protease resistance and thermal stability. PLoS One. 2011;6:e28218. doi: 10.1371/journal.pone.0028218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saerens D, Conrath K, Govaert J, Muyldermans S. Disulfide bond introduction for general stabilization of immunoglobulin heavy-chain variable domains. J Mol Biol. 2008;377:478–88. doi: 10.1016/j.jmb.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 51.Chan PH, Pardon E, Menzer L, De Genst E, Kumita JR, Christodoulou J, Saerens D, Brans A, Bouillenne F, Archer DB, et al. Engineering a camelid antibody fragment that binds to the active site of human lysozyme and inhibits its conversion into amyloid fibrils. Biochemistry. 2008;47:11041–54. doi: 10.1021/bi8005797. [DOI] [PubMed] [Google Scholar]
- 52.Hagihara Y, Mine S, Uegaki K. Stabilization of an immunoglobulin fold domain by an engineered disulfide bond at the buried hydrophobic region. J Biol Chem. 2007;282:36489–95. doi: 10.1074/jbc.M707078200. [DOI] [PubMed] [Google Scholar]
- 53.Jespers L, Schon O, Famm K, Winter G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat Biotechnol. 2004;22:1161–5. doi: 10.1038/nbt1000. [DOI] [PubMed] [Google Scholar]
- 54.Arbabi-Ghahroudi M, Mackenzie R, Tanha J. Site-directed mutagenesis for improving biophysical properties of VH domains. Methods Mol Biol. 2010;634:309–30. doi: 10.1007/978-1-60761-652-8_22. [DOI] [PubMed] [Google Scholar]
- 55.Arbabi-Ghahroudi M, To R, Gaudette N, Hirama T, Ding W, MacKenzie R, Tanha J. Aggregation-resistant VHs selected by in vitro evolution tend to have disulfide-bonded loops and acidic isoelectric points. Protein Eng Des Sel. 2009;22:59–66. doi: 10.1093/protein/gzn071. [DOI] [PubMed] [Google Scholar]
- 56.Wu SL, Jiang H, Lu Q, Dai S, Hancock WS, Karger BL. Mass spectrometric determination of disulfide linkages in recombinant therapeutic proteins using online LC-MS with electron-transfer dissociation. Anal Chem. 2009;81:112–22. doi: 10.1021/ac801560k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schiefner A, Chatwell L, Körner J, Neumaier I, Colby DW, Volkmer R, Wittrup KD, Skerra A. A disulfide-free single-domain VL intrabody with blocking activity towards huntingtin reveals a novel mode of epitope recognition. J Mol Biol. 2011;414:337–55. doi: 10.1016/j.jmb.2011.09.034. [DOI] [PubMed] [Google Scholar]
- 58.Lee WR, Jang JY, Kim JS, Kwon MH, Kim YS. Gene silencing by cell-penetrating, sequence-selective and nucleic-acid hydrolyzing antibodies. Nucleic Acids Res. 2010;38:1596–609. doi: 10.1093/nar/gkp1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paz K, Brennan LA, Iacolina M, Doody J, Hadari YR, Zhu Z. Human single-domain neutralizing intrabodies directed against Etk kinase: a novel approach to impair cellular transformation. Mol Cancer Ther. 2005;4:1801–9. doi: 10.1158/1535-7163.MCT-05-0174. [DOI] [PubMed] [Google Scholar]
- 60.Colby DW, Garg P, Holden T, Chao G, Webster JM, Messer A, Ingram VM, Wittrup KD. Development of a human light chain variable domain (VL) intracellular antibody specific for the amino terminus of huntingtin via yeast surface display. J Mol Biol. 2004;342:901–12. doi: 10.1016/j.jmb.2004.07.054. [DOI] [PubMed] [Google Scholar]
- 61.van den Beucken T, van Neer N, Sablon E, Desmet J, Celis L, Hoogenboom HR, Hufton SE. Building novel binding ligands to B7.1 and B7.2 based on human antibody single variable light chain domains. J Mol Biol. 2001;310:591–601. doi: 10.1006/jmbi.2001.4703. [DOI] [PubMed] [Google Scholar]
- 62.Enever C, Tomlinson IM, Lund J, Levens M, Holliger P. Engineering high affinity superantigens by phage display. J Mol Biol. 2005;347:107–20. doi: 10.1016/j.jmb.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 63.Housden NG, Harrison S, Housden HR, Thomas KA, Beckingham JA, Roberts SE, Bottomley SP, Graille M, Stura E, Gore MG. Observation and characterization of the interaction between a single immunoglobulin binding domain of protein L and two equivalents of human κ light chains. J Biol Chem. 2004;279:9370–8. doi: 10.1074/jbc.M312938200. [DOI] [PubMed] [Google Scholar]
- 64.Graille M, Stura EA, Housden NG, Beckingham JA, Bottomley SP, Beale D, Taussig MJ, Sutton BJ, Gore MG, Charbonnier JB. Complex between Peptostreptococcus magnus protein L and a human antibody reveals structural convergence in the interaction modes of Fab binding proteins. Structure. 2001;9:679–87. doi: 10.1016/S0969-2126(01)00630-X. [DOI] [PubMed] [Google Scholar]
- 65.Åkerström B, Björck L. Protein L: an immunoglobulin light chain-binding bacterial protein. Characterization of binding and physicochemical properties. J Biol Chem. 1989;264:19740–6. [PubMed] [Google Scholar]
- 66.Björck L. Protein L. A novel bacterial cell wall protein with affinity for Ig L chains. J Immunol. 1988;140:1194–7. [PubMed] [Google Scholar]
- 67.Stevens FJ, Westholm FA, Solomon A, Schiffer M. Self-association of human immunoglobulin κI light chains: role of the third hypervariable region. Proc Natl Acad Sci U S A. 1980;77:1144–8. doi: 10.1073/pnas.77.2.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dudgeon K, Rouet R, Kokmeijer I, Schofield P, Stolp J, Langley D, Stock D, Christ D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci U S A. 2012;109:10879–84. doi: 10.1073/pnas.1202866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gorlani A, Hulsik DL, Adams H, Vriend G, Hermans P, Verrips T. Antibody engineering reveals the important role of J segments in the production efficiency of llama single-domain antibodies in Saccharomyces cerevisiae. Protein Eng Des Sel. 2012;25:39–46. doi: 10.1093/protein/gzr057. [DOI] [PubMed] [Google Scholar]
- 70.Tanha J, Nguyen TD, Ng A, Ryan S, Ni F, Mackenzie R. Improving solubility and refolding efficiency of human VHs by a novel mutational approach. Protein Eng Des Sel. 2006;19:503–9. doi: 10.1093/protein/gzl037. [DOI] [PubMed] [Google Scholar]
- 71.Svensson HG, Wedemeyer WJ, Ekstrom JL, Callender DR, Kortemme T, Kim DE, Sjöbring U, Baker D. Contributions of amino acid side chains to the kinetics and thermodynamics of the bivalent binding of protein L to Ig κ light chain. Biochemistry. 2004;43:2445–57. doi: 10.1021/bi034873s. [DOI] [PubMed] [Google Scholar]
- 72.Famm K, Hansen L, Christ D, Winter G. Thermodynamically stable aggregation-resistant antibody domains through directed evolution. J Mol Biol. 2008;376:926–31. doi: 10.1016/j.jmb.2007.10.075. [DOI] [PubMed] [Google Scholar]
- 73.Hubbard SJ, Beynon RJ, Thornton JM. Assessment of conformational parameters as predictors of limited proteolytic sites in native protein structures. Protein Eng. 1998;11:349–59. doi: 10.1093/protein/11.5.349. [DOI] [PubMed] [Google Scholar]
- 74.Frenken LG, Egmond MR, Batenburg AM, Verrips CT. Pseudomonas glumae lipase: increased proteolytic stability by protein engineering. Protein Eng. 1993;6:637–42. doi: 10.1093/protein/6.6.637. [DOI] [PubMed] [Google Scholar]
- 75.Christ D, Famm K, Winter G. Repertoires of aggregation-resistant human antibody domains. Protein Eng Des Sel. 2007;20:413–6. doi: 10.1093/protein/gzm037. [DOI] [PubMed] [Google Scholar]
- 76.McConnell AD, Spasojevich V, Macomber JL, Krapf IP, Chen A, Sheffer JC, Berkebile A, Horlick RA, Neben S, King DJ, et al. An integrated approach to extreme thermostabilization and affinity maturation of an antibody. Protein Eng Des Sel. 2013;26:151–64. doi: 10.1093/protein/gzs090. [DOI] [PubMed] [Google Scholar]
- 77.de Haard HJ, van Neer N, Reurs A, Hufton SE, Roovers RC, Henderikx P, de Bruïne AP, Arends JW, Hoogenboom HR. A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J Biol Chem. 1999;274:18218–30. doi: 10.1074/jbc.274.26.18218. [DOI] [PubMed] [Google Scholar]
- 78.Sblattero D, Bradbury A. A definitive set of oligonucleotide primers for amplifying human V regions. Immunotechnology. 1998;3:271–8. doi: 10.1016/S1380-2933(97)10004-5. [DOI] [PubMed] [Google Scholar]
- 79.Tanha J, Muruganandam A, Stanimirovic D. Phage display technology for identifying specific antigens on brain endothelial cells. Methods Mol Med. 2003;89:435–49. doi: 10.1385/1-59259-419-0:435. [DOI] [PubMed] [Google Scholar]
- 80.Dombkowski AA. Disulfide by DesignTM: a computational method for the rational design of disulfide bonds in proteins. Bioinformatics. 2003;19:1852–3. doi: 10.1093/bioinformatics/btg231. [DOI] [PubMed] [Google Scholar]
- 81.Baral TN, Chao SY, Li S, Tanha J, Arbabi-Ghahroudi M, Zhang J, Wang S. Crystal structure of a human single domain antibody dimer formed through VH-VH non-covalent interactions. PLoS One. 2012;7:e30149. doi: 10.1371/journal.pone.0030149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Graille M, Stura EA, Corper AL, Sutton BJ, Taussig MJ, Charbonnier JB, Silverman GJ. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc Natl Acad Sci U S A. 2000;97:5399–404. doi: 10.1073/pnas.97.10.5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.