

Abstract
In vitro selection technologies are an important means of affinity maturing antibodies to generate the optimal therapeutic profile for a particular disease target. Here, we describe the isolation of a parent antibody, KENB061 using phage display and solution phase selections with soluble biotinylated human IL-1R1. KENB061 was affinity matured using phage display and targeted mutagenesis of VH and VL CDR3 using NNS randomization. Affinity matured VHCDR3 and VLCDR3 library blocks were recombined and selected using phage and ribosome display protocol. A direct comparison of the phage and ribosome display antibodies generated was made to determine their functional characteristics.
Keywords: antibody, IL-1R1, ribosome display, phage display, mutagenesis, recombination
In our analyses, we observed distinct differences in the pattern of beneficial mutations in antibodies derived from phage and ribosome display selections, and discovered the lead antibody Jedi067 had a ~3700-fold improvement in KD over the parent KENB061. We constructed a homology model of the Fv region of Jedi067 to map the specific positions where mutations occurred in the CDR3 loops. For VL CDR3, positions 94 to 97 carry greater diversity in the ribosome display variants compared with the phage display. The positions 95a, 95b and 96 of VLCDR3 form part of the interface with VH in this model.
The model shows that positions 96, 98, 100e, 100f, 100 g, 100h, 100i and 101 of the VHCDR3 include residues at the VH and VL interface. Importantly, Leu96 and Tyr98 are conserved at the interface positions in both phage and ribosome display indicating their importance in maintaining the VH-VL interface.
For antibodies derived from ribosome display, there is significant diversity at residues 100a to 100f of the VH CDR3 compared with phage display. A unique deletion of isoleucine at position 102 of the lead candidate, Jedi067, also occurs in the VHCDR3.
As anticipated, recombining the mutations via ribosome display led to a greater structural diversity, particularly in the heavy chain CDR3, which in turn led to antibodies with improved potencies. For this particular analysis, we also found that VH-VL interface positions provided a source of structural diversity for those derived from the ribosome display selections. This greater diversity is a likely consequence of the presence of a larger pool of recombinants in the ribosome-display system, or the evolutionary capacity of ribosome display, but may also reflect differential selection of antibodies in the two systems.
Introduction
Antibodies are routinely engineered for higher affinity using a variety of different molecular techniques that typically incorporate mutagenesis leading to changes in the primary amino acid sequence, alongside methodologies for selecting the improved variant.1 Display technologies, be they bacteriophage, yeast or other cell surface display, or completely cell-free systems, such as ribosome and mRNA display, are widely established as platforms for selecting and screening large populations of protein variants.1-4 In the case of antibody affinity maturation, examples of affinity gains of greater than > 10,000-fold and final dissociation constants (KD) in the low picomolar range have been described using different display technologies.5-7 Multiple mutations acting in concert are typically required to achieve such high final affinities.
Recombination of advantageous individual mutations using display technologies are limited by the size of the libraries that can be technically achieved. For phage and cell-based display systems, sizes are limited by the volume of cells that can be comfortably grown for transformation at the bench. It is feasible to generate library sizes in the order of ~1010, thus restricting recombination, for example, to 105 different variants derived from individual VH and VL chain repertoires. Cell-free display systems, such as ribosome display, which do not require a cellular transformation step, can theoretically reach populations of over 1012 in size.8 Indeed, functional library sizes have been reported as being as high as 1011 antibodies.9 Previously, we reported on the successful affinity maturation of phage display selection output antibody populations by up to 40-fold using ribosome display.10
IL-1RI is an 80 kDa membrane-associated protein and a member of the IL-1R/Toll-like receptor (TLR) superfamily.11 IL-1RI has three ligands, interleukin-1alpha (IL-1α), interleukin-1beta (IL-1β), and an inhibitory ligand, interleukin-1 receptor antagonist (IL-1Ra). IL-1Ra binds to IL-1RI, but cannot initiate signaling and plays an important role limiting the extent of IL-1 pathway activation in vivo. IL-1RI-dependent signaling, instigated by either IL-1α or IL-1β binding, is critical to the initiation and maintenance of inflammatory responses and has been described as a likely driving factor in a broad spectrum of diseases.12 As such, IL-1RI is a potential target for therapeutic blockade.
Here, we describe the affinity maturation of an anti-IL-1RI antibody using targeted mutagenesis of the VH and VL CDR3 loops, followed by recombination and further selection of advantageous combinations using phage display and cell-free ribosome display. We undertook this comparative strategy to understand the affect that VH and VL recombined library size, expression system and PCR-based mutagenesis would have on the antibody variants isolated from the two approaches. Improved antibody variants from the two different platforms were compared at a structural level, as well as for affinity/potency improvements. Distinct differences in the pattern of beneficial mutations derived from the same initial pools were observed, and from our experiments we also identified variations at the VH-VL interface positions as a source of structural diversity, particularly among the ribosome display antibodies. A larger panel of improved variants was generated from ribosome display, indicating that the choice of display technology can play a profound role during in vitro protein evolution.
Results
Isolation of IL-1RI antibody KENB061
IL-1RI binding antibodies were isolated from a large phage library displaying human single-chain Fv (scFv) by carrying out solution-phase selections using recombinant human IL-1RI (Fig. 1A). Individual scFvs from selection outputs were assessed for their ability to functionally inhibit human IL-1β binding to IL-1RI in a single-point, 96-well based homogeneous assay. Individual scFvs that showed inhibition of binding in the assay were re-expressed, purified using Ni-NTA chromatography and retested as a serial titration, giving, IC50 values ranging from 11 nM to > 83 nM (data not shown). One of the most potent scFvs, KENB061, had a measured IC50 value of 31 nM for inhibition of binding of IL-1β to IL-1RI. KENB061 was reformatted to a full-length IgG2 molecule and tested for the ability to neutralize recombinant human IL-1β-induced IL-8 release from HeLa cells, giving a measured IC50 of 473 nM (n = 3, Table 1) in this assay. Additionally, the apparent affinity of KENB061 for recombinant IL-1RI was measured at ~48 nM (Table 1). To improve the affinity and cellular potency of KENB061, we therefore initiated in vitro affinity maturation.
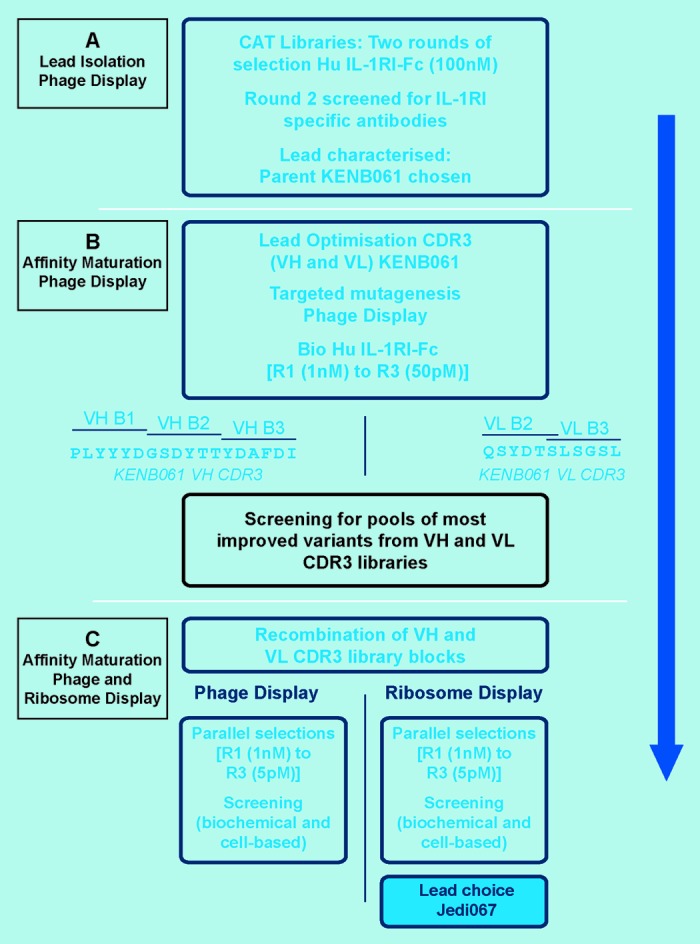
Figure 1. The lead generation strategy. (A) Lead isolation phage display. (B) Affinity maturation phage display. (C) Affinity maturation phage and ribosome display.
Table 1. Optimized anti-IL-1RI antibodies derived from VH and VL CDR3 recombination by phage and ribosome display.
| Text | Text |
| Text | Text |
Italicized font represents the final antibody lead. P, phage display; R, ribosome display; NT, not tested; ND, not determined; *Predicted VH-VL interface residues; ƒKD of KENB061 and Jedi067 IgG2 binding to monomeric sIL-1RI ECD calculated by SPR Jedi067 KD also measured in a Kinetic Exclusion Assay. KD – 2.47pM. See Figure S1. +Purified scFv and IgG were tested for inhibition of IL-1b binding to IL-1RI-Fc in a HTRF® assay.
Targeted mutagenesis of KENB061 CDR3 loops
We focused on alterations to the VH and VL CDR3 loops to increase the affinity and biological potency of KENB061. Separate scFv libraries of KENB061 were generated by targeted mutagenesis in the VH and VL CDR3 regions (Fig. 1B). For the VH CDR3, three individual libraries were constructed, designed to span the 18 residue loop in blocks of 6 amino acids using an NNS library format. For the VL CDR3, two libraries were constructed in blocks of 6 amino acids with a central 1 amino acid overlap for this 11 amino acid loop. Improved scFv were selected using affinity-based phage display. After the completion of three rounds of selection, individual scFvs from each library were screened in a homogeneous assay for inhibition of binding of FLAG-tagged human IL-1β to human IL-1RFc as undiluted crude periplasmic extracts.
Over 1300 scFvs were screened from the phage display selection outputs to establish which randomized CDR blocks yielded the greatest affinity and potency gains (Table 2). A second homogenous screening assay, based upon inhibition of binding of the parental KENB061 antibody to IL-1RI, was used to identify further improved variants. In this assay, a reduction in assay signal by 66% was defined as a hit. The heavy chain CDR3 block 2 (amino acid positions 100a to 100f) showed an increase in hit rate from round 2 to round 3 (6.8 to 16.5%, respectively). Heavy chain block 1 (amino acid positions 95 to 100) showed a reduction in hit rate from rounds 2 to 3 while VH CDR3 block 3 (amino acid positions 100 g to 102) did not identify any hits at round 2 and few at round 3. Light chain block 2 (amino acids 94 to 97) showed a significant improvement in % hit rate from round 2 to 3 (14.8 to 36.4%), whereas in block 1 (amino acids 89 to 94) no improved clones were identified at round 2 and few at round 3.
Table 2. A Summary of HTRF® screening (inhibition of binding of IL-1β binding to human IL-1RFc). Improved scFv variants from VH and VL CDR3 libraries (rounds 2 - 3) were screened as crude periplasmic extracts from E. coli.
| (A) Heavy Chain CDR3 Libraries | |||
|---|---|---|---|
| Block | Selection antigen | Round | % hit1 |
| VH block 1 | 500nM | 2 | 12.5 |
| VH block 1 | 50nM | 3 | 5.7 |
| VH block 2 | 500nM | 2 | 6.8 |
| VH block 2 | 50nM | 3 | 16.5 |
| VH block 3 | 500nM | 2 | 0 |
| VH block 3 | 50nM | 3 | 6.8 |
| (B) Light Chain CDR3 Libraries | |||
|---|---|---|---|
| Block | Selection antigen | round | % hit1 |
| VL block 1 | 500nM | 2 | 0 |
| VL block 1 | 50nM | 3 | 3.4 |
| VL block 2 | 500nM | 2 | 14.8 |
| VL block 2 | 50nM | 3 | 36.4 |
1 Hit defined as reduction in assay signal of 66%.
From this analysis, it was apparent that the third round of selection using a 50 pM antigen concentration with the VH3 block 2 and VL3 block 2 libraries generated the highest total numbers of improved variants, while maintaining a high diversity (88/88 scFvs had unique patterns of mutations in the randomized area).
Recombination of the VH and VL CDR3 libraries and affinity-based selection using phage and ribosome display
We adopted a strategy of recombination and affinity-based selection using both technologies in parallel (Fig. 1C) and compared the data sets.
In the first instance, we compared diversity of the randomized VH and VL CDR3 sequences and efficiency of recombination (by sequencing 88 randomly picked scFvs), as well as library size. For the phage display library, we observed that parental KENB061 VH CDR3 sequence made up 25% of the total library. Overall, 73% of the sequenced recombination library was diverse as defined by changes to the VH and VL CDR3 sequence, with a library size of 5.8 × 108 based on total number of transformants. For the ribosome-displayed recombination library, the diversity of the VH and VL CDR3 sequences was 98%. The recombined output sizes were in the order of 106 for VH and VL, giving a theoretical library size of 1012 for the ribosome display approach.8 The functional diversity of a ribosome display library can be considered as being the number of ribosomal complexes that display a functional protein. An estimate of the active complexes with folded protein can be obtained from the number of mRNA molecules that can be isolated after one round of ribosome display. Previous ribosome display experiments have estimated library size at as high as ~1011 per milliliter of reaction.9
A number of different selection conditions were explored to understand each technology’s capacity to tolerate increased stringency, i.e., reduction in antigen concentration. Figure 2A illustrates the cDNA outputs generated from the round 2 ribosome display selections with antigen concentrations at 1 nM, 500 pM and 50 pM. It is apparent that the cDNA signal decreased with reduced antigen concentration, with the 50 pM cDNA signal just being visible. For both technologies, a minimum antigen concentration of 1 nM was found to yield an effective selection at round 1, and in subsequent rounds 2 and 3, equivalent antigen concentration of 50 pM and 5 pM were found to be the most stringent antigen concentrations that still enabled successful selection (Fig. 2B).

Figure 2. Selection strategy used for phage and ribosome display to generate potent anti-IL-1RI Antibodies. (A) Gel photograph of cDNA outputs from Round 2 of Ribosome Display Selections with antigen concentrations at 1 nM, 500 pM and 50 pM. (B) Selection pathway (high-lighted in light blue) followed to generate high affinity IL-1RI variants using phage and ribosome display.
The relative success of the two approaches was assessed in terms of the numbers of improved variants in the parental antibody competition assay. For this post-CDR3 recombination screen, a more stringent definition of an improved variant, i.e., a reduction in assay signal by 85%, was employed. As a comparison, parental KENB061 reduced the assay signal by ~30% in this assay. From the ribosome display-based library, a total of 1584 scFvs were screened and yielded 34 hits. From the phage display-based library, 1056 scFvs were screened and 6 hits identified. The improved variants from both approaches were sequenced, giving 25 unique scFvs that were expressed and purified for further testing.
Potency of lead antibody variants from phage and ribosome display CDR3 recombinations
From the 25 unique improved variants identified, the 4 from the phage display recombination selections and the remaining 21 from the ribosome displayed selections were screened in a cell-based assay (IL-1β induced IL-8 release from HeLa cells) and the 3 most improved variants from each approach were converted to full-length IgG2 and compared in the assay (Table 1). The most potent lead from the ribosome display CDR3 recombination strategy, Jedi067, had an 8-fold improved potency over the best phage display lead (Jedi066) as scFv and ~2-fold as IgG. Despite the clear success of the optimization, the potential differences in library size between ribosome (~1011) and phage display (~108) and the multiple structural solutions available, it is perhaps surprising that these potency gains are not more significant.
It is also interesting that we did not see improvements in IC50 values on conversion of scFv to IgG for our lead panel. We hypothesize that, during interaction of the IgG with the IL-1RI, there is a steric constraint on the basis of the epitope that prevents two binding events with dimeric IgGs and a single IL-1R1-Fc.
The top three leads, Jedi067, Jedi069, and Jedi072, completely inhibited the binding of a recombinant form of IL-1Ra, the antagonistic ligand, to the receptor (Fig. 3), suggesting at least a partially shared epitope/binding site on IL-1RI. The most potent variant achieved an inhibitory activity equivalent to IL-1Ra (mean IC50 of 6 donors Jedi067 = 229 pM; IL-1Ra = 279 pM) in a physiologically-relevant whole blood assay.
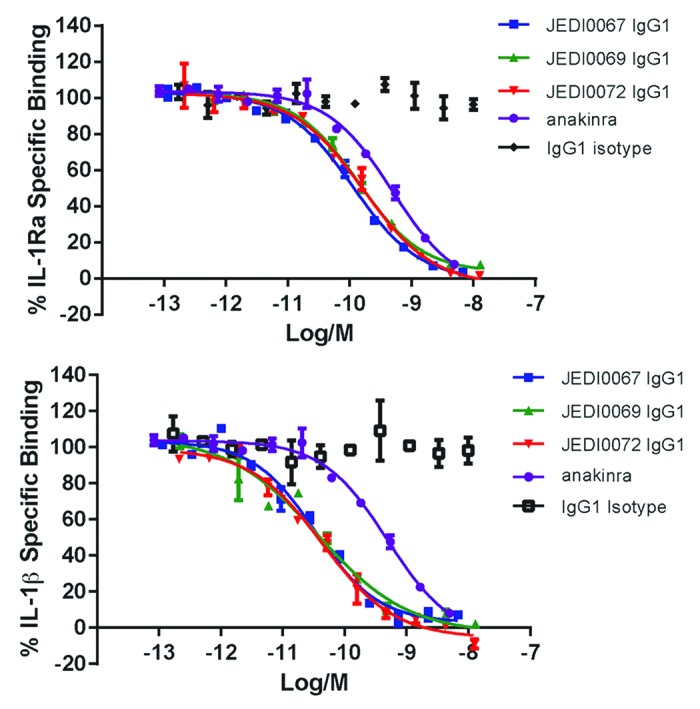
Figure 3. Inhibition of IL-1 receptor ligand binding to IL-1 Receptor. Anti-IL-1 receptor lead panel antibodies from ribosome display completely inhibit both IL-1Ra* binding and IL-1β binding to IL-1R1Fc in biochemical binding HTRF assay. Data representative of n = 2 independent experiments. KENB061 inhibited IL-1β binding to IL-1RI with an IC50 of 473 nM (n = 3, Table 1). *Anakinra is a drug used to treat rheumatoid arthritis. It is an interleukin-1 (IL-1) receptor antagonist. Interleukin-1 receptor antagonist IL-1Ra binds to IL-1RI, but cannot initiate signaling and plays an important role limiting the extent of IL-1 pathway activation in vivo
Sequence analysis of affinity matured antibody variants
The VH and VL CDR3 sequences of antibodies with increased potency from phage and ribosome display selections were analyzed (Fig. 4). It is immediately apparent from the phage display VH CDR3 analysis that there are limited amino acid substitutions within the leads (Fig. 4A), and that these substitutions were contained within the block of residues that were randomized. For ribosome display, recombination mutations occur from position 100a through position 102 in the VHCDR3. There were greater numbers of mutations (particularly for the 100b and the 100c positions within that block) and more extensive coverage of the VH CDR3 compared with phage display. Mutations have occurred at least once per amino acid with one antibody, Jedi067, sustaining a deletion of the final residue in the block (I102). For VL CDR3 analysis (Fig. 4B), two of the four improved antibodies derived from the phage display recombination were improved due to changes in the light chain alone. Multiple substitutions were selected in the VL CDR3 block for ribosome display-derived antibodies, with no mutations occurring outside of this targeted region.

Figure 4. (A) Amino acid usage within the VH CDR3 loops of the improved potency anti-IL-1R1 variants from phage and ribosome display. (B) Amino acid usage within the VL CDR3 loops of the improved potency anti-IL-1R1 variants from phage and ribosome display
Affinity determination of parent KENB061 and affinity matured variant Jedi67
The apparent affinity (dissociation constant, KD) and binding rate constants for the interaction of KENB061 and Jedi67 IgGs for human sIL-1RI were measured on a Biacore T100 surface plasmon resonance (SPR) instrument. The KD of KENB061 was measured at 48 nM (Table 1; Fig. S1; Table S1). The KD of the affinity matured variant Jedi67 was measured at 13 pM, which is considerably higher than that reported for IL-1Ra for IL-1RI (150 pM).13 The extremely slow dissociation rate for Jedi67, however, was considered beyond the practical limits of SPR and prevented confident assessment of the affinity, and so a kinetic exclusion assay (KinExA) 14 was used to further investigate the KD of this variant. KinExA is a flow spectrofluorometric-based methodology that can be used to quantify high affinity interactions, including those in the sub-picomolar range.15
The KinExA analysis of Jedi067 monomeric Fab fragment for human IL-1RI-Fc was measured at 2.5 pM, with calculated 95% confidence interval between 0.96 and 4.4 pM (Fig. S2), suggesting that the SPR methodology did indeed underestimate the monovalent affinity of Jedi67 for IL-1RI.
Structural analysis of affinity matured variants
A homology model of the Fv region of Jedi067 was constructed for structural analysis of specific positions where mutations occurred in the CDR3 loops (Fig. 5). Positions 96, 98, 100e, 100f, 100 g, 100h, 100i, and 101 of VHCDR3 include residues at the VH-VL interface. In the ribosome display variants, mutations occur in positions 100e to 102 of the C-terminus of VHCDR3. The unique deletion of isoleucine at position 102 also occurred in the VHCDR3 C-terminus. The side chains of residues at positions 96, 98, 100e, 100f, and 100h points toward the interface with VL. The model showed that the side chain of the aspartate at position 100h, which is almost completely conserved among the ribosome display variants, forms a hydrogen bond with the backbone nitrogen of VL His34. The backbone nitrogen of residue at position 100i, which almost always had a small side chain (Ala or Gly) among the ribosome display variants, formed a hydrogen bond with the side chain hydroxyl of VL Tyr36. The model suggests that these two hydrogen bonds may be critical for stabilizing the VH-VL interaction. Asp100h and A100i are completely conserved in the phage display variants. Similarly, Leu96 and Tyr98 are conserved at the interface positions in both phage display and ribosome display variants. Such conservation of the interface residues indicated their importance in maintaining the VH-VL interface. On the other hand, in this example, more variations occurred at interface positions 100e and 100f of the VHCDR3 C-terminal half, i.e., end of block 2 residues, for the ribosome display variants.

Figure 5. A structural model of Jedi067 Fv. A view of the structural model of Jedi067 Fv looking down the CDRs is shown. The VH-VL interface residues discussed in the text are shown as sticks and labeled. Color code: cyan-VH; pink-VL; blue-VHCDRs; magenta-VLCDRs; yellow dotted lines-hydrogen bonds. The conserved aspartate at position 101 in VH domain is substituted by phenylalanine.
For the VL CDR3, greater numbers of mutations were seen from position 94 through to 97 for the ribosome display leads. The C-terminal half positions 95a, 95b, and 96 of VLCDR3 form part of the interface with VH in this model. Notably, position 97 at the end of VLCDR3, with a side chain faces the upper core of the VL domain, tolerated significant side chain substitutions among the variants.
Discussion
Combination of CDR mutations is a technique that has been shown to be very successful in the in vitro evolution of antibodies.6,7 We previously employed this technique to recombine whole populations of phage-displayed antibody fragment variants with targeted mutations in the VH and VL CDR3 loops. Mutations that, taken alone, confer only modest potency gains are allowed to recombine in large populations, leading to synergistic affinity and potency gains. Antibodies with affinities in the femtomolar range that exhibited high potencies in biologically-relevant assays have been described using this technique.7
For any combinatorial approach, it is assumed that increasing the practical size of the population of possible recombinants should allow a greater chance of selecting those with improved binding. For protein display methodologies such as phage display, bacterial transformation efficiency provides a practical limit to library size (109–1010) because of the constraints that come from working with large volumes of cells. For a single recombination of two equally-sized populations of mutants, the maximum size of each that will allow every permutation to be theoretically covered will be around 105. Cell-free technologies such as ribosome display are not limited by cellular transformation efficiency, and should theoretically be limited only by the number of ribosomes and transcription/translation components. Yields can also vary according to the mechanical losses of material upon selection and mRNA isolation, incomplete folding of the antibody fragment and the presence of complexes with incompletely translated protein chain. Functional library size can be assessed by quantifying mRNA retrieved from the library after one round of selection, and have been estimated at as high as ~1011 per milliliter of reaction.8,9
In this study, we compared phage and ribosome displayed recombination of populations of heavy and light chain CDR3 advantageous mutations to determine the effects of the disparity between the library sizes, library diversity, and the effect of error-prone mutagenesis on the structural diversity of the antibodies generated by ribosome display.
As anticipated, recombining the mutations via ribosome display led to a greater structural diversity, particularly in the heavy chain CDR3, among the antibodies with significantly improved potencies. This greater diversity was likely to be a direct consequence of the presence of a greater pool of recombinants in the ribosome-display system, but may also reflect differential selection of antibodies in the two systems. The cell-free nature of ribosome display potentially represents a very different milieu for protein expression and folding, compared with the bacterial periplasm, in the case for phage display, which may also be playing a role in the greater diversity of improved leads. Previously, we reported the benefits that evolution during selection can bring in maturation approaches using ribosome display, and this greater diversity is certainly also a direct consequence of error introduced by this PCR-based methodology.10
Interestingly though, the potency of the recombined improved leads was not substantially higher for the variants derived using ribosome display, the most potent variant from the phage selections, Jedi066, having an IC50 of 116 pM in the cellular assay compared with 20 pM for the most potent variant from the ribosome display selection, Jedi067. There were a number of possible explanations for this modest difference in potencies, compared with the potentially enormous difference in diversity of the starting pools, one of which is that, as both sets of selections started from a pool of VH and VL CDR3 mutants derived via phage display selections, there may already be a pre-selected bias in the structural diversity, e.g., for bacterial expression and folding in the periplasm. The greater potential diversity of the ribosome displayed VH/VL recombinations may not therefore represent as high a functional diversity as, say, if the initial selections on the individual VH and VL CDR3 libraries had been performed in the ribosome display format. We are currently investigating whether such an approach would lead to further improvements in diversity and final affinity and potency of selected antibodies. Alternatively, there may be a potency ceiling for the biological system, (IL-1 β binding to IL-1RI) being investigated here. It is intriguing that the natural antagonist for IL-1RI, IL-1Ra, has a very similar potency in the biological assays, particularly as the lead antibodies generated here appear to be binding in a similar location as the natural antagonist, as demonstrated by competitive binding (Fig. 3), even with a substantially higher affinity for IL-1R1 (Jedi067: monovalent KD = 2.5 pM; Fig. S2), compared with that of the reported antagonist (150 pM). Previous reports of antibodies generated against the IL-1RI, with a similar competitive mechanism of action also exhibited very similar potencies to the IL-1Ra, suggesting a limit to achievable potency when targeting this epitope.
Taken together, these hypotheses may also explain why, physiologically, there are multiple strategies to inhibit the potent activity of IL-1, which include IL-1Ra but also release of soluble receptors (solIL-1RI, solIL-1RII), as well as a non-signaling decoy receptor (IL-1RII).16,17
The final affinity-matured antibody, Jedi067, showed high potency in an IL-1 β-dependent HeLa cell assay, inhibiting IL-8 production with an IC50 of 50 pM; an improvement over the parental KENB061 antibody of ~10,000-fold. This lead also showed high potency in an assay measuring IL-1 β-induced IL-6 production in human whole blood. It thus represents a potential antibody lead for therapeutic development for the treatment of inflammatory diseases. The presence of a deletion mutation at the end of the VH CDR3 loop of Jedi067 is of note (I102) because this type of mutation is likely to have resulted from an amplification error during one of the PCR steps of the ribosome display, and is highly unlikely to be generated during the primer-based construction of the phage display mutagenesis library. A loss of the parental aspartate at position 101, which has been widely observed to form a salt bridge with a positively charged amino acid at position 94 was also apparent.10, 18
The loss of this salt bridge may increase the flexibility of orientation of the VH CDR3 loop, and in this case resulted in the generation of the most potent lead molecule, which was produced solely through use of this ribosome display-based recombination method, thus demonstrating its utility for in vitro affinity maturation.
Sequence and structural analysis of the phage display and ribosome display variants revealed that variations occur at the VH-VL interface positions. VH-VL domain association can affect antigen binding by contributing to the structural aspects of antigen combining site formation.19,20 Conservation of the hydrogen bonds formed by residues at VH interface positions 100h (Asp) and 100i (Ala/Gly) indicates that certain structural constraints must be maintained for VH-VL association for this panel of antibodies. On the other hand, the variations observed at these positions, and more so at other VH and VL interface positions, suggests that structural plasticity at the VH-VL interface has been tolerated to generate the variants. It is interesting to note that, for our study, the ribosome display variants show more VH-VL interface variations than the phage display variants. In addition, the deletion mutant at position 102 also was found in the most potent antibody Jedi067. This suggests the potential for ribosome display to offer more structural routes to achieving hits compared with phage display
Materials and Methods
Heat inactivated fetal bovine serum was obtained from Sigma-Aldrich (UK). Streptomycin, penicillin, GlutaMAX I, sodium pyruvate, Dulbecco’s modified Eagle’s medium and RPMI 1640 were purchased from Invitrogen. Cell culture flasks and 96-well tissue culture plates were obtained from Fisher Scientific. Oligonucleotides were obtained from Eurogentec. Azlactone beads were obtained from Fisher Scientific (Loughborough, UK), Streptococcal Protein G and bovine serum albumin were obtained from Sigma-Aldrich. Cy5-labeled mouse anti-human heavy and light chain specific antibody was supplied by Jackson Immunoresearch. Recombinant human soluble IL-1R1 extracellular domain (ECD) was obtained from R&D Systems. All other chemicals were purchased from Sigma-Aldrich.
Expression and purification of recombinant human IL-1R1-Fc
A cDNA encoding the sequence of human IL-1RI extracellular domain (amino acid residues 1 to 336 NP_000868) was amplified from human liver cDNA via PCR using primers based on the human IL-1RI cDNA sequence (Ref Seq NM_00877). The resulting cDNAs were subcloned following the manufacturer’s instructions into pENTR/D-TOPO (Invitrogen). The cDNA fragments coding the IL-1RI extracellular domains were then transferred to mammalian expression vector pDEST12.2 (Invitrogen) using LR Gateway® reaction (Invitrogen). The pDEST12.2 vector had been modified to contain the human IgG1 Fc coding region in-frame with the inserted gene of interest, and also by insertion of the oriP origin of replication from the pCEP4 vector (Invitrogen) allowing episomal plasmid replication upon transfection into cell lines expressing the EBNA-1 gene product. After transient transfection into HEK293-EBNA cells, the soluble secreted IL-1R1-Fc fusion protein was purified from the conditioned media using Protein G chromatography followed by size exclusion chromatography.
To enable detection in biochemical assays and recovery in soluble selections, amino groups on human IL-1RI were biotinylated using standard NHS chemistry techniques (Perbio/Pierce EZ-link NHS-LC-Biotin reagent).
Isolation of anti-IL-1R1 antibodies
Large scFv phage-display human antibody scFv (CAT) libraries cloned into a phagemid vector based on the filamentous phage M13 were used for selections (Fig. 1A).21,22 Anti-IL-1R1 scFv antibodies were isolated from the phage display libraries using a series of selection cycles on recombinant human biotinylated IL-1R1.21,23 IL-1RI neutralizing antibodies were identified from the selections by screening individual scFvs expressed from E. coli for inhibition in an IL-1β/IL-1R1 homogeneous binding assay, as described below. Neutralizing scFvs with unique sequences were then expressed in E. coli and purified by affinity chromatography. The potency of the purified scFvs was then determined in the IL-1β/IL-1R1 assay and the HeLa IL-8 release assay in response to IL-1β, as described below.
FLAG IL-1β and IL-1 receptor homogeneous binding assay
ScFv and IgG at various stages were screened in an HTRF® assay binding assay for inhibition of the binding of FLAG-IL-1β to IL-1RI-Fc. These were tested as undiluted crude periplasmic extracts containing scFv prepared in assay buffer [50 nM 4-morpholinepropanesulfonic acid buffer (pH 7.4), 0.5 mM EDTA, and 0.5 M sorbitol] or as purified scFv or IgG diluted in assay buffer (phosphate buffered saline (PBS) containing 0.4 M potassium fluoride and 0.1% bovine serum albumin). Inhibitors were added to black Costar low volume non-binding microtiter plates and preincubated by the addition of IL-1RFc (0.5 nM) for 1 h at room temperature. FLAG IL-1β (1 nM) was then added along with anti-FLAG IgG labeled with XL and anti-Fc IgG labeled with cryptate. The assay plates were centrifuged and incubated in the dark for 3 h at room temperature prior to reading of time-resolved fluorescence at 620 nm excitation wavelength and 665 nm emission wavelength using an EnVision plate reader (Perkin Elmer). Data were analyzed by calculating percent ΔF values for each sample. ΔF was determined according to the methodology recommended by the manufacturer. Data was expressed as percentage of specific binding. The assay was adjusted and optimized to enable identification of increased potency clones as required during the affinity maturation process, for example by increasing the amount of FLAG IL-1β per reaction to 10nM, and using scFv periplasmic extracts diluted to 0.2% v/v in assay buffer.
Reformatting of scFv to IgG2
Clones were converted from scFv into IgG format by subcloning the VH and VL domains into plasmids expressing whole-antibody heavy (pEU9.4) and light (pEU3.4 for κ light chain or pEU4.4 for λ light chain) chains, respectively. The plasmids are based on those originally described,24 with an additional oriP element engineered into each. To obtain IgGs, we transfected the heavy chain and light chain IgG-expressing vectors into HEK-EBNA cells. IgGs were expressed and secreted into the medium. Harvests were pooled and filtered prior to purification. Individual IgGs were purified using standard Protein A chromatography. The eluted material was buffer exchanged into PBS. The concentration of the IgG was determined at A280 using an extinction coefficient based on the amino acid sequence of IgG.25
IL-1Ra/IL-1 receptor homogeneous binding assay
Purified IgG derived from lead isolation/optimization were tested for inhibition of IL-1Ra binding to IL-1RI-Fc in an HTRF® assay in a similar fashion. Purified IgG to be tested were diluted in assay buffer (PBS containing 0.4 M potassium fluoride and 0.1% bovine serum albumin) and added to black Costar low volume non-binding microtiter plates. Directly cryptate-labeled IL-1RI-Fc (0.1 nM) was pre-incubated with inhibitors for 1 h at room temperature. FLAG IL-1Ra (0.15 nM) was then added along with anti-FLAG IgG labeled with XL. The assay plates were centrifuged and incubated in dark for 3 h at room temperature prior to reading of time-resolved fluorescence at 620 nm excitation wavelength and 665 nm emission wavelength using an EnVision plate reader (Perkin Elmer). Data were analyzed as for the above described HTRF® assay.
HeLa IL-1β-induced IL-8 release assay
HeLa cells (European Collection of Cell Cultures, ECACC catalog number 93021013) were maintained as recommended by the suppliers in MEM plus 10% fetal bovine serum plus 1% non-essential amino acids. For inhibition assays, cells were seeded at 1.5x104 cells/well in 96-well flat-bottomed tissue culture assay plates and cultured overnight. To test inhibitors, a titration was prepared in culture media and this dilution series was added to the HeLa cells without removing overnight culture medium and pre-incubated for 30–60 min at 37° C. This was followed by addition of recombinant human IL-1β (R&D Systems) and incubation for 4–5 h at 37° C/ 5% CO2. Following this incubation period, culture supernatants were harvested and IL-8 levels determined by ELISA using human IL-8 Duoset (R&D Systems).
Affinity maturation: Generation of targeted mutagenesis libraries (VH and VL CDR3)
Large scFv phage libraries derived from KENB061 were created by oligonucleotide-directed mutagenesis of the VH and VL CDR3 using degenerate oligonucleotides to randomize blocks of six amino acids using Kunkel mutagenesis (Fig. 1B).26 For each CDR3, separate scFv libraries were made from overlapping blocks of six fully randomized codons. The scFv libraries were cloned into phagemid vectors, followed by expression of libraries phage particles. The libraries were subjected to affinity-based selections to select variants with a higher affinity for IL-1RI. The phage libraries were incubated with biotinylated IL-1RI-Fc in solution. ScFv phage bound to antigen were then captured on streptavidin-coated magnetic beads (Dynabeads® M 280) following the manufacturer’s recommendations. The selected scFv phage particles were then rescued with addition of helper phage, and the selection process was repeated in the presence of decreasing concentrations of bio-human IL-1RI-Fc (1 nM to 50 pM for 3 rounds).
Affinity maturation: Generation of recombined VH and VL CDR3 libraries and selections using ribosome and phage display
For phage display, recombined VH and VL libraries were made using Kunkel Mutagenesis (Fig. 1C).20 The DNA products of mutagenesis reactions were electroporated into TG1 cells. For ribosome display, VH and VL sequences were amplified from selected libraries using PCR and full-length scFv was created using recombinatorial PCR.27 This recombined scFv library was purified and further recombined with the geneIII tether, which allows the scFv to be displayed out of the ribosome tunnel for ribosome display selections.27,28 This product was purified and amplified by PCR to generate the final ribosome display construct used for transcription, translation and selection.
For phage display, anti-IL-1RI scFv antibodies were isolated from the phage display recombined libraries using a series of selection cycles on recombinant human biotinylated IL-1RI fused to an Fc domain. For selections, the antigen concentration was reduced from 1 nM in round 1 down to a final concentration of 5 pM in R3. For ribosome display, the recombined libraries were converted into ribosome display format and used in affinity-based ribosome display selections essentially as described previously.27,29,30 The selection process was repeated on the obtained population for further rounds of selections with decreasing concentrations of biotinylated human IL-1RI (as per phage display) to enrich, and therefore select, clones with a higher affinity for IL-1RI. The outputs from selection rounds 2 and 3 were subcloned into pCANTAB6 phagemid vector for bacterial expression as scFvs, and improved clones were identified as periplasmic extracts in the IL-1β/IL-1RI competition assay.
IL-1β mediated IL-6 release from whole blood
Whole blood was collected from normal human volunteers (supplied by the National Blood Service, UK), and incubated with Jedi067, IL-1Ra (anakinra, supplied commercially) or an isotype control antibody (MAB005), and then stimulated with IL-1β at a final concentration of 30 pM, a concentration predetermined to give 50% maximal release of IL-6. IL-6 levels in the supernatant were determined 18 h later by enzyme-linked immunosorbent assay (ELISA) (R&D Systems IL-6 Duoset).
Affinity determination by surface plasmon resonance (SPR)
Apparent affinity determination measurements for KENB061 and Jedi67 IgG were performed on a Biacore T100 instrument running appropriate control and evaluation software (- version 2.0.1). C1 biosensor chips, amine coupling kits, HEPES buffered saline-based buffers (HBS-EP) and other buffers were from GE Healthcare (Little Chalfont, UK) and were used according to the manufacturer’s instructions. For the Protein G’ capture surfaces, the Sigma recombinant Protein G′ Tris lyophilisate was reconstituted in water and buffer exchanged into Dulbecco’s PBS via a PD-10 column (GE Healthcare). This Protein G’ was further diluted into 10 mM sodium acetate pH 3.6 and amine coupled to chips using an amine coupling kit. IgG were captured on the Protein G surface and monomeric recombinant soluble IL-1RI was flowed over the chip. For the IgG binding studies, the final level of active immobilized IgG was usually low enough to ensure that only 20 RUs or less of the antigen was bound at saturation (Rmax). This level of antigen binding is below the level considered likely to induce mass-transport artifacts, especially when combined with the relatively high, 50 μL min−1 assay flow-rates used during the kinetic measurement steps.31
Affinity determination by kinetic exclusion assay
The affinity of Jedi067 to IL-1RI was also measured using kinetic exclusion assay (KinExA™) technology. KinExA™ is a flow spectrofluorometric-based technology that can be used to accurately quantify high-affinity interactions, including those in the subpicomolar range. The assay was devised to minimize avidity effects by only allowing one moiety to be presented in a dimeric format. The monomerized Fab of Jedi067 was allowed to come to equilibrium with IL-1RI-Fc and analyzed using KinExA 3200 technology. By performing a titration range of different concentrations of antibody and IL-1RI-Fc, and measuring free antibody after each of these conditions, a KD was estimated for the antibody to the receptor (least squares fitting, using a 1:1 reversible bimolecular interaction model within the supplied KinExA Pro software). Sample buffers was composed of Dulbecco’s PBS (D-PBS) supplemented with 1 mg/mL bovine serum albumin and 0.02% sodium azide. Flow buffer was the same buffer prepared without the albumin. All buffers used in the KinExA experiments were 0.2 μm filter sterilized. The fluorescent secondary detection reagent was Cy5-labeled mouse anti-human heavy and light chain specific antibody. For the sampling bead column, 100 mg of UltraLink Biosupport azlactone beads were mixed with 100 μg human IL-1R1 in 3 mL 50 mM sodium hydrogen carbonate pH 8.4 at room temperature with constant agitation for 90 min. Rinsing and blocking was achieved with 10 mg/mL BSA in 1 M Tris pH 8.7. Before use the re-suspended beads were diluted into D-PBS + 0.02% sodium azide.
Structural modeling and analysis
A model of Jedi067 Fv region was generated using software tools from Accelrys Software Inc. For the structural analysis, residues whose surface area changes with the upon VH-VL association are considered as the VH-VL interface residues. Interface residues are defined here as residues whose surface area changes upon VH-VL domain association. A probe radius of 0.6 Å was used in surface area calculation. Structure figures were generated using The PyMol Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.
Supplementary Material
Acknowledgments
We thank Jon Large for assistance with the figures for this publication.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/27261
References
- 1.Wilkinson T, Lowe D, Vaughan TJ. Affinity maturation approaches for antibody lead optimization. In Antibody drug discovery 2013; (Wood, C. R., ed.): pp. 85, IMPERIAL COLLEGE PRESS. [Google Scholar]
- 2.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006;6:343–57. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 3.Gai SA, Wittrup KD. Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol. 2007;17:467–73. doi: 10.1016/j.sbi.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Löfblom J. Bacterial display in combinatorial protein engineering. Biotechnol J. 2011;6:1115–29. doi: 10.1002/biot.201100129. [DOI] [PubMed] [Google Scholar]
- 5.Boder ET, Midelfort KS, Wittrup KD. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc Natl Acad Sci U S A. 2000;97:10701–5. doi: 10.1073/pnas.170297297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steidl S, Ratsch O, Brocks B, Dürr M, Thomassen-Wolf E. In vitro affinity maturation of human GM-CSF antibodies by targeted CDR-diversification. Mol Immunol. 2008;46:135–44. doi: 10.1016/j.molimm.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Finch DK, Sleeman MA, Moisan J, Ferraro F, Botterell S, Campbell J, Cochrane D, Cruwys S, England E, Lane S, et al. Whole-molecule antibody engineering: generation of a high-affinity anti-IL-6 antibody with extended pharmacokinetics. J Mol Biol. 2011;411:791–807. doi: 10.1016/j.jmb.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 8.Plückthun A. Ribosome display: a perspective. Methods Mol Biol. 2012;805:3–28. doi: 10.1007/978-1-61779-379-0_1. [DOI] [PubMed] [Google Scholar]
- 9.Zahnd C, Amstutz P, Plückthun A. Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat Methods. 2007;4:269–79. doi: 10.1038/nmeth1003. [DOI] [PubMed] [Google Scholar]
- 10.Groves M, Lane S, Douthwaite J, Lowne D, Rees DG, Edwards B, Jackson RH. Affinity maturation of phage display antibody populations using ribosome display. J Immunol Methods. 2006;313:129–39. doi: 10.1016/j.jim.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 11.Sims JE, March CJ, Cosman D, Widmer MB, MacDonald HR, McMahan CJ, Grubin CE, Wignall JM, Jackson JL, Call SM, et al. cDNA expression cloning of the IL-1 receptor, a member of the immunoglobulin superfamily. Science. 1988;241:585–9. doi: 10.1126/science.2969618. [DOI] [PubMed] [Google Scholar]
- 12.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–52. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dripps DJ, Brandhuber BJ, Thompson RC, Eisenberg SP. Interleukin-1 (IL-1) receptor antagonist binds to the 80-kDa IL-1 receptor but does not initiate IL-1 signal transduction. J Biol Chem. 1991;266:10331–6. [PubMed] [Google Scholar]
- 14.Darling RJ, Brault PA. Kinetic exclusion assay technology: characterization of molecular interactions. Assay Drug Dev Technol. 2004;2:647–57. doi: 10.1089/adt.2004.2.647. [DOI] [PubMed] [Google Scholar]
- 15.Rathanaswami P, Roalstad S, Roskos L, Su QJ, Lackie S, Babcook J. Demonstration of an in vivo generated sub-picomolar affinity fully human monoclonal antibody to interleukin-8. Biochem Biophys Res Commun. 2005;334:1004–13. doi: 10.1016/j.bbrc.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Fredericks ZL, Forte C, Capuano IV, Zhou H, Vanden Bos T, Carter P. Identification of potent human anti-IL-1RI antagonist antibodies. Protein Eng Des Sel. 2004;17:95–106. doi: 10.1093/protein/gzh012. [DOI] [PubMed] [Google Scholar]
- 17.Shirai H, Kidera A, Nakamura H. H3-rules: identification of CDR-H3 structures in antibodies. FEBS Lett. 1999;455:188–97. doi: 10.1016/S0014-5793(99)00821-2. [DOI] [PubMed] [Google Scholar]
- 18.Shirai H., Kidera A. & Nakamura H. (1999) H3-rules: identification of CDR-H3 structures in antibodies. FEBS Lett.455, 188-197 [DOI] [PubMed]
- 19.Nakanishi T, Tsumoto K, Yokota A, Kondo H, Kumagai I. Critical contribution of VH-VL interaction to reshaping of an antibody: the case of humanization of anti-lysozyme antibody, HyHEL-10. Protein Sci. 2008;17:261–70. doi: 10.1110/ps.073156708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abhinandan KR, Martin ACR. Analysis and prediction of VH/VL packing in antibodies. Protein Eng Des Sel. 2010;23:689–97. doi: 10.1093/protein/gzq043. [DOI] [PubMed] [Google Scholar]
- 21.Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 1996;14:309–14. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- 22.Lloyd C, Lowe D, Edwards B, Welsh F, Dilks T, Hardman C, Vaughan T. Modelling the human immune response: performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng Des Sel. 2009;22:159–68. doi: 10.1093/protein/gzn058. [DOI] [PubMed] [Google Scholar]
- 23.Hawkins RE, Russell SJ, Winter G. Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J Mol Biol. 1992;226:889–96. doi: 10.1016/0022-2836(92)90639-2. [DOI] [PubMed] [Google Scholar]
- 24.Persic L, Roberts A, Wilton J, Cattaneo A, Bradbury A, Hoogenboom HR. An integrated vector system for the eukaryotic expression of antibodies or their fragments after selection from phage display libraries. Gene. 1997;187:9–18. doi: 10.1016/S0378-1119(96)00628-2. [DOI] [PubMed] [Google Scholar]
- 25.Mach H, Middaugh CR, Lewis RV. Statistical determination of the average values of the extinction coefficients of tryptophan and tyrosine in native proteins. Anal Biochem. 1992;200:74–80. doi: 10.1016/0003-2697(92)90279-G. [DOI] [PubMed] [Google Scholar]
- 26.Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–82. doi: 10.1016/0076-6879(87)54085-X. [DOI] [PubMed] [Google Scholar]
- 27.Lewis L, Lloyd C. Optimisation of antibody affinity by ribosome display using error-prone or site-directed mutagenesis. Methods Mol Biol. 2012;805:139–61. doi: 10.1007/978-1-61779-379-0_9. [DOI] [PubMed] [Google Scholar]
- 28.Groves MA, Nickson AA. Affinity maturation of phage display antibody populations using ribosome display. Methods Mol Biol. 2012;805:163–90. doi: 10.1007/978-1-61779-379-0_10. [DOI] [PubMed] [Google Scholar]
- 29.Plückthun A, Schaffitzel C, Hanes J, Jermutus L. In vitro selection and evolution of proteins. Adv Protein Chem. 2000;55:367–403. doi: 10.1016/S0065-3233(01)55009-3. [DOI] [PubMed] [Google Scholar]
- 30.Schaffitzel C, Hanes J, Jermutus L, Plückthun A. Ribosome display: an in vitro method for selection and evolution of antibodies from libraries. J Immunol Methods. 1999;231:119–35. doi: 10.1016/S0022-1759(99)00149-0. [DOI] [PubMed] [Google Scholar]
- 31.Myszka DG. Improving biosensor analysis. J Mol Recognit. 1999;12:279–84. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<279::AID-JMR473>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.