

Abstract
Novel therapies are needed for the treatment of hypoglycemia resulting from both endogenous and exogenous hyperinsulinema. To provide a potential new treatment option, we identified XMetD, an allosteric monoclonal antibody to the insulin receptor (INSR) that was isolated from a human antibody phage display library. To selectively obtain antibodies directed at allosteric sites, panning of the phage display library was conducted using the insulin-INSR complex. Studies indicated that XMetD bound to the INSR with nanomolar affinity. Addition of insulin reduced the affinity of XMetD to the INSR by 3-fold, and XMetD reduced the affinity of the INSR for insulin 3-fold. In addition to inhibiting INSR binding, XMetD also inhibited insulin-induced INSR signaling by 20- to 100-fold. These signaling functions included INSR autophosphorylation, Akt activation and glucose transport. These data indicated that XMetD was an allosteric antagonist of the INSR because, in addition to inhibiting the INSR via modulation of binding affinity, it also inhibited the INSR via modulation of signaling efficacy. Intraperitoneal injection of XMetD at 10 mg/kg twice weekly into normal mice induced insulin resistance. When sustained-release insulin implants were placed into normal mice, they developed fasting hypoglycemia in the range of 50 mg/dl. This hypoglycemia was reversed by XMetD treatment. These studies demonstrate that allosteric monoclonal antibodies, such as XMetD, can antagonize INSR signaling both in vitro and in vivo. They also suggest that this class of allosteric monoclonal antibodies has the potential to treat hyperinsulinemic hypoglycemia resulting from conditions such as insulinoma, congenital hyperinsulinism and insulin overdose.
Keywords: hypoglycemia, negative allosteric modulation, monoclonal antibody, insulin receptor, insulin, antagonist
Introduction
Insulin initiates the regulation of cellular glucose metabolism by binding to the insulin receptor (INSR) on the cell surface, a process that activates the receptor’s intrinsic kinase activity.1 When activated, the INSR undergoes autophosphorylation, followed by the recruitment and phosphorylation of INSR signaling molecules, including the IRS proteins and members of the phosphotidylinositol 3-kinase (PI3K)-Akt pathway.2 In cells, activation of this pathway by insulin results in the translocation of glucose transporters to the cell surface with subsequent uptake of glucose.3,4
It has been suggested that monoclonal antibodies (mAbs) to the INSR may be useful in various disorders of glucose metabolism.5 Hypoglycemia due to insulin excess from both exogenous and endogenous sources is not an infrequent clinical condition.6-8 In some instances, the current treatments for insulin-induced hypoglycemia do not adequately restore normoglycemia, resulting in prolonged hospitalization or neurological damage.6 Although not currently available, therapies that attenuate insulin signaling via inhibition of the INSR, such as antagonist mAbs, could prove to be effective for the treatment of sustained and life threatening hyperinsulinemic hypoglycemia.
Studies of antibodies that inhibit insulin activation of the INSR, including both spontaneously occurring human autoantibodies and mouse mAbs, have been reported.9-11 In humans, autoantibodies to the INSR typically bind at the insulin binding site (the orthosteric site), and directly compete with insulin for binding. In most cases, these antibodies cause severe insulin resistance and diabetes despite compensatory hyperinsulinemia.12-15 Orthosteric INSR autoantibodies isolated from humans have been studied in rats, and have been shown to be weak agonists that cause hypoglycemia at low concentrations and hyperglycemia at high concentrations.16 Thus, these types of orthosteric antibodies are not likely clinical candidates for the treatment of hyperinsulinemic hypoglycemia.
Allosteric antibodies, antibodies that do not bind at the ligand binding site of receptors, can regulate cell signaling.17,18 In theory, these allosteric antibodies have the potential to bind and regulate receptors more selectively than orthosteric antibodies due to lower sequence and structural homology at allosteric sites relative to orthosteric sites.19 Allosteric regulation of the INSR by glucose and peptides has been previously described.20-23 We recently generated an allosteric, human mAb that activated the INSR both in vitro and in vivo, and normalized fasting glucose levels in diabetic mice.24,25 These types of mAbs have been classified as selective insulin receptor modulators.26 It is possible, therefore, that allosteric antibodies to the INSR that inhibit its activation could also be generated, and be useful for the treatment of hyperinsulinemic hypoglycemia. To date, such antibodies have not been reported.
In the study reported here, we identified allosteric antibodies that antagonized INSR signaling and selected one, XMetD, for further characterization. This allosteric antibody antagonized insulin action both in vitro and in vivo.
Results
XMetD discovery
Allosteric modulating antibodies targeting the human INSR (hINSR) were identified by panning naïve human antibody phage display libraries using the recombinant extracellular domain of the hINSR complexed to insulin. Antibodies binding the hINSR-insulin complex were identified by FACS screening of bacterial periplasmic extracts. Six that had high affinity to both the human and the mouse INSRs were reformatted to fully human IgG2 antibodies. The most efficacious of these antibodies, XMetD, was employed in all subsequent studies described herein.
Binding of XMetD to the hINSR
Binding of XMetD to Chinese hamster ovary (CHO) cells expressing either isoform of the hINSR (hINSR-A or hINSR-B) was demonstrated using FACS (Fig. 1). XMetD bound similarly to both isoforms of the hINSR. In contrast, XMetD bound minimally to parental CHO cells (Fig. 1) and to CHO cells expressing the insulin-like growth factor-1 receptor (IGF-1R) (data not shown). Most likely this minimal binding was due to the interaction with the low concentration of endogenous hamster INSR.
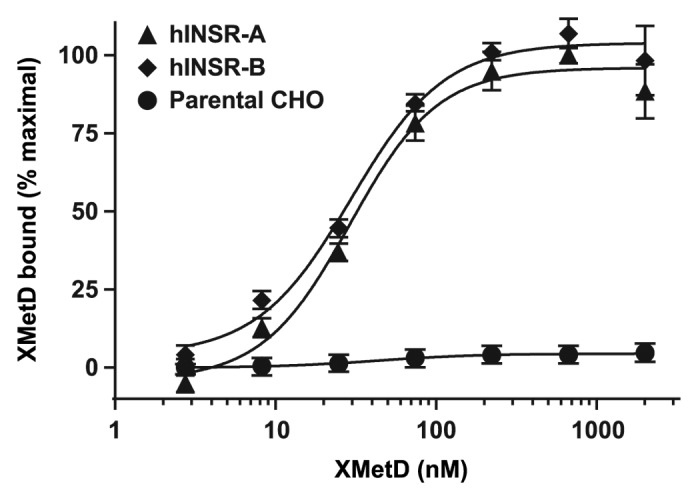
Figure 1. Binding of XMetD to the A and B isoforms of the INSR by FACS. Intact CHO-hINSR cells expressing either the A or B isoform of the INSR were incubated for 120 min at 15 °C with increasing concentrations of XMetD. XMetD binding to the INSR was measured by flow cytometry (FACS). Mean ± SD of triplicate determinations are shown.
To quantify the binding affinity of XMetD to the hINSR and to measure the effect of insulin on the kinetics of XMetD binding to the hINSR, we employed surface plasmon resonance (SPR) analysis using solubilized hINSR (B isoform) that had been captured on the sensor surface (Fig. 2). In the absence of insulin, the on-rate for XMetD binding to the hINSR was 3.8 × 104 M−1sec−1 and the off-rate was 3.1 × 10−4 sec−1 (KD = 8 nM). In the presence of insulin, the affinity of XMetD was ~3-fold lower (KD = 26 nM). With insulin, the on-rate for XMetD binding to the hINSR was 4.4 × 104 M−1sec−1 and the off-rate was 1.6 × 10−3 sec−1.

Figure 2. Quantitative kinetic analysis of XMetD binding to the hINSR. (A) Kinetic analysis of XMetD binding to the INSR solubilized from CHO-hINSR cells in the absence of insulin by SPR. (B) Kinetic analysis of XMetD binding to the INSR solubilized from CHO-hINSR cells in the presence of 1 μg/ml insulin (in running buffer) by SPR. For (A) and (B), the INSR was solubilized from CHO cells expressing the B isoform of the receptor and captured on the sensor surface via an immobilized monoclonal anti-INSR β subunit antibody (clone CT-3). XMetD concentrations ranging from 1.64‒133 nM were injected over the captured receptor to obtain association and dissociation kinetics. Residuals from the curve fit are shown adjacent to each SPR sensorgram.
The effect of XMetD on the binding of insulin to the hINSR
We next studied whether XMetD inhibited the binding of insulin to the hINSR. Employing biotinylated human insulin in a FACS-based assay, we observed that 1.0 μM XMetD only partially inhibited insulin binding to the hINSR, with a ~3-fold reduction in the observed affinity of biotinylated insulin (Fig. 3). No additional inhibitory effects on insulin binding to the hINSR were observed at higher XMetD concentrations. The EC50 for insulin binding in the absence of XMetD was 47 nM (95% CI: 31 to 71 nM) and increased to 125 nM (95% CI: 99 to 166 nM) in the presence of 1.0 μM XMetD. The observations that insulin only partially inhibits XMetD binding to the hINSR and, reciprocally, that XMetD only partially inhibits insulin binding to the hINSR, are characteristic of negative allosteric modulation rather than competitive orthosteric inhibition.19,27
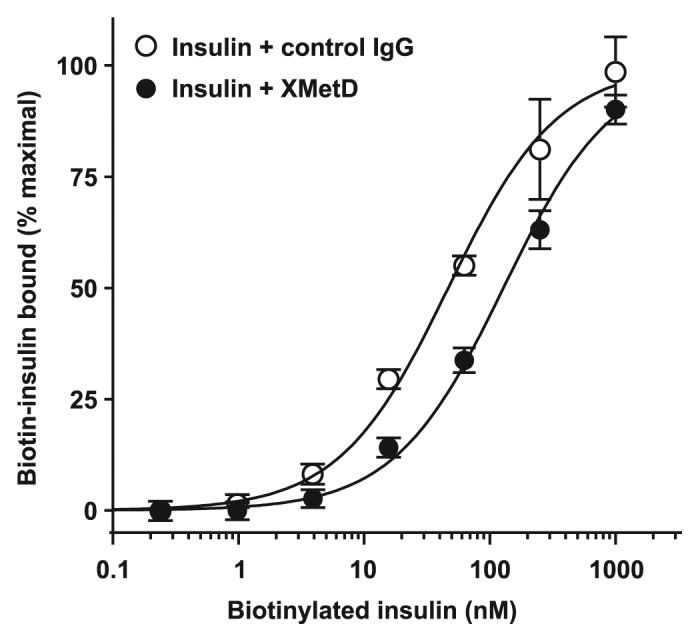
Figure 3. Insulin binding to the hINSR by FACS. CHO-hINSR cells (B isoform) were preincubated for 10 min at 4 ° with 1 μM of either XMetD or control IgG followed by a 30 min incubation with increasing concentrations of biotinylated insulin. Biotinylated insulin binding to the INSR was measured by flow cytometry (FACS). Mean ± SD of triplicate determinations are shown.
Monoclonal antibodies can induce desensitization of the INSR.28,29 We therefore studied the effect of XMetD on internalization (Fig. S1A) and downregulation (Fig. S1B). Insulin in the presence of a negative control IgG had little or no effect on these functions. XMetD as well did not induce either INSR internalization or downregulation in the presence or absence of insulin. In contrast a positive control IgG, in the presence of insulin, did induce both INSR internalization and downregulation by greater than 50%.
The effects of XMetD on insulin-induced hINSR signaling
We then assessed the effect of XMetD at 333 nM (the maximal inhibitory concentration for this function) on insulin activation of the hINSR. The EC50 for insulin-stimulated hINSR autophosphorylation in the absence of XMetD was 7.3 nM (95% CI: 6.0 to 8.9 nM). XMetD treatment resulted in a greater than 40-fold decrease in the sensitivity of insulin-stimulated hINSR autophosphorylation with an EC50 of 310 nM, (95% CI: 230 to 416 nM) (Fig. 4). As a control, IGF-1R autophosphorylation by IGF-1 was studied. XMetD did not influence this autophosphorylation (data not shown). The much greater inhibition by XMetD of insulin-mediated hINSR autophosphorylation, relative to its inhibitory effect on insulin binding to the hINSR, indicated that the antagonistic effects of the antibody were mediated only in part by its negative modulation of insulin binding affinity. Thus, XMetD was also a negative modulator of hINSR activation.19,27
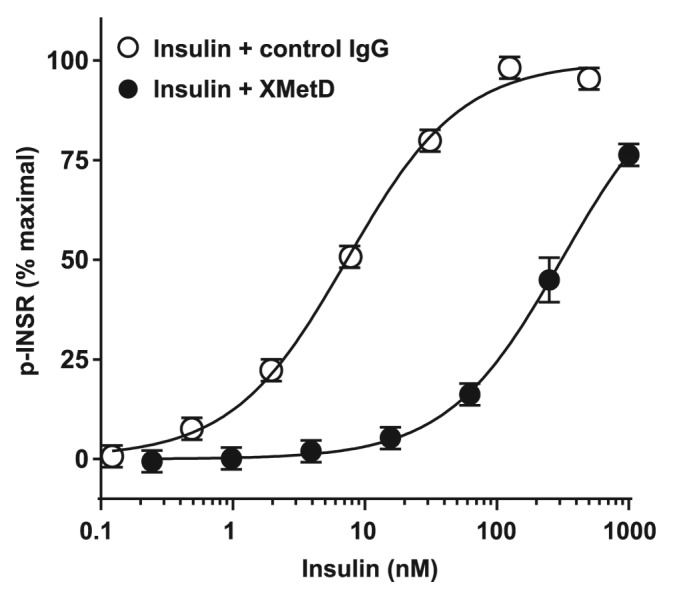
Figure 4. XMetD is an antagonist of the INSR autophosphorylation. CHO-hINSR cells (B isoform) were preincubated for 30 min at 37 °C with 333 nM of either XMetD or control IgG followed by a 10 min incubation with increasing concentrations of insulin. INSR autophosphorylation was measured by ELISA. Mean ± SD of triplicate determinations are shown.
Accordingly, the negative modulating effects of XMetD on hINSR activation were further characterized by investigating how XMetD influenced insulin-dependent activation of intracellular signaling pathways. The serine phosphorylation of Akt is a critical downstream mediator of the metabolic effects of the activated hINSR.1 To characterize the effect of XMetD on insulin-dependent Akt phosphorylation, we employed CHO-hINSR cells exposed to XMetD concentrations ranging from 0 to 3333 nM (Fig. 5A). XMetD, at a maximal concentration for this function (3333 nM), induced a greater than 100-fold decrease in the sensitivity of insulin-dependent Akt phosphorylation. The EC50 for insulin-stimulated pAkt in the absence of XMetD was less than 1 nM (95% CI: 0.04 to 0.34 nM) and increased to 197 nM (95% CI: 174 to 223 nM) in the presence of 3333 nM XMetD. XMetD alone had no effect on Akt phosphorylation (Fig. 5A). The effects of XMetD on the A isoform of the hINSR were similar to those observed with the B isoform of the hINSR (data not shown). When the EC50 of insulin-induced Akt phosphorylation was plotted as a function of XMetD concentration, a sigmoidal curve was observed (Fig. 5B). The half-maximal XMetD concentration was determined to be 110 nM (95% CI: 76 to 159 nM). Thus, these data indicated that, in addition to moderately inhibiting insulin binding, XMetD profoundly inhibited hINSR signaling efficacy via an allosteric mechanism.27

Figure 5. XMetD is an antagonist of insulin dependent Akt and Erk phosphorylation. (A) CHO-hINSR cells (B isoform) were preincubated for 30 min at 37 °C with a wide range of XMetD concentrations from 0 to 3333 nM followed by a 10 min incubation with increasing concentrations of insulin. Phosphorylation of Akt was measured using an electrochemiluminescence based assay. Mean of duplicate determinations are shown. (B) Insulin EC50 values (from Fig. 5A) were determined for each concentration of XMetD and plotted as a function of XMetD concentration. Mean of duplicate determinations are shown. (C) CHO-hINSR cells (B isoform) were preincubated for 30 min at 37 °C with 333 nM of either XMetD or control IgG followed by a 10 min incubation with increasing concentrations of insulin. Phosphorylation of Erk was measured using an electrochemiluminescence based assay. Mean ± SD of triplicate determinations are shown.
Erk, when activated by tyrosine phosphorylation, is another critical downstream mediator INSR activity.1 We therefore evaluated the effect of XMetD on insulin-dependent Erk phosphorylation. As with Akt phosphorylation, XMetD induced a similar decrease in the sensitivity of insulin-dependent Erk phosphorylation in CHO-hINSR cells. In the presence of control IgG, the EC50 for insulin-induced Erk phosphorylation was 1.8 nM (95% CI: 1.4 to 2.3 nM); whereas, in the presence of XMetD, the EC50 for insulin-induced Erk phosphorylation was 107 nM (95% CI: 97 to 117 nM), (Fig. 5C).
The receptor for IGF-1 has structural and sequence similarity to the INSR.30 We therefore assessed whether XMetD affected IGF-1-activativation of Akt in CHO cells expressing the human IGF-1R (CHO-hIGF-1R). IGF-1 treatment induced Akt phosphorylation in these cells, but unlike the prior studies with hINSR, XMetD exhibited little or no antagonism of IGF-1-mediated activation of Akt in CHO-hIGF-1R cells (Fig. 6). These data, in concert with the aforementioned lack of both XMetD binding to the IGF-1R, and XMetD inhibition of IGF-1R phosphorylation, strongly suggested that the inhibitory effect of XMetD was specific for the INSR.
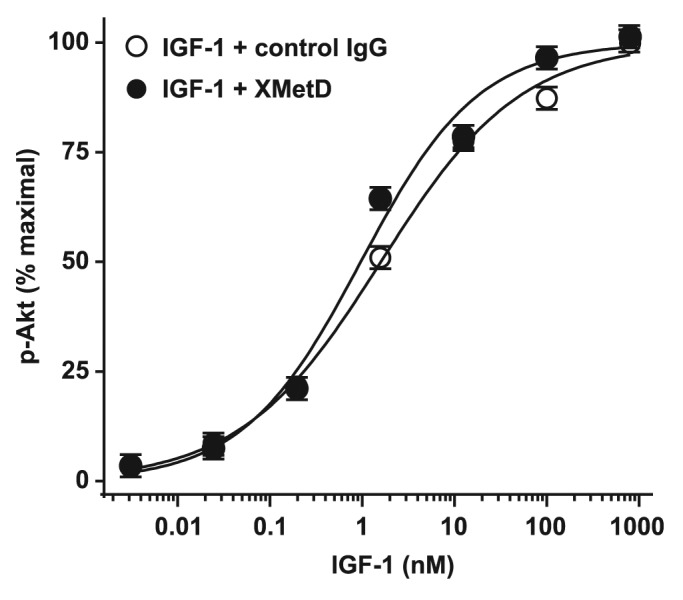
Figure 6. XMetD is not an antagonist of IGF-1 dependent Akt phosphorylation. CHO-hIGF-1R cells were preincubated for 30 min at 37 °C with 333 nM of either XMetD or control IgG followed by a 10 min incubation with increasing concentrations of IGF-1. Phosphorylation of Akt was measured using an electrochemiluminescence based assay. Mean ± SD of triplicate determinations are shown.
To study whether XMetD inhibited glucose transport in muscle, a major target tissue for insulin, we employed cultured L6 muscle cells. These cells express both the hINSR and human GLUT-4, providing robust insulin sensitivity and responsiveness.31 In these cells, XMetD antagonized insulin stimulation of glucose uptake (Fig. 7) by 70-fold. The EC50 for insulin-stimulated glucose uptake in the absence of XMetD was 0.25 nM (95% CI: 0.17 to 0.35 nM) and increased to 18 nM (95% CI: 12 to 26 nM) in the presence of 333 nM XMetD. Thus, the above studies indicated that inhibition of insulin-stimulated INSR activation by XMetD resulted in greatly diminished metabolic signal activation. In these studies, XMetD had no effect on INSR signaling in the absence of insulin (data not shown).
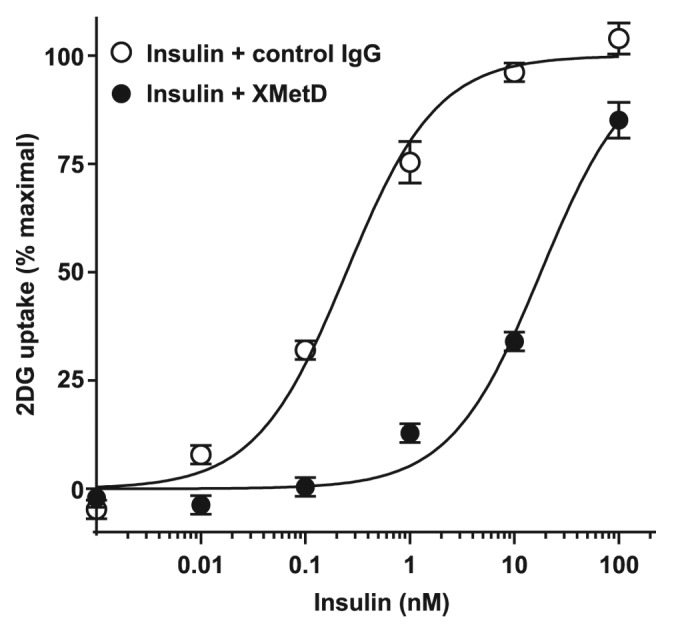
Figure 7. XMetD inhibits insulin-mediated 2-deoxy-D-glucose uptake in L6 muscle cells. L6 cells expressing both isoform B of the hINSR and GLUT-4 were preincubated with 333 nM of either XMetD or control IgG for 60 min at 37 °C followed by a 10 min incubation with increasing concentrations of insulin. [3H]-2-deoxy-D-glucose (2DG) was added and uptake was measured after 20 min. Mean ± SD of triplicate determinations are shown.
In addition to its effects on metabolic functions such as glucose transport, insulin can also stimulate the growth and proliferation of cancer cells.32-34 In COLO-205 human colon cancer cells, insulin stimulated growth with a half maximal effect at 0.5 nM. In contrast to the observed impact of XMetD on insulin-stimulated glucose transport, XMetD had little or no effect on insulin-stimulated proliferation (Fig. S2). Similar data were observed with other cancer cell lines including Saos-2 osteosarcoma cells (data not shown).
Binding of XMetD to mINSR and effects on mINSR-cell signaling
To determine if the mouse would be a suitable species for the characterization of XMetD in vivo, we evaluated the ability of XMetD to act as a negative modulator of the mouse INSR (mINSR) in vitro. XMetD bound to the mINSR in a manner similar to that of hINSR (data not shown). As with the hINSR, XMetD inhibited insulin-dependent Akt activation by the mINSR (Fig. 8). The EC50 for insulin-stimulated Akt activation in the absence of XMetD was 0.97 nM (95% CI: 0.72 to 1.30 nM) and increased to 103 nM (95% CI: 88 to 119 nM) in the presence of 333 nM XMetD.
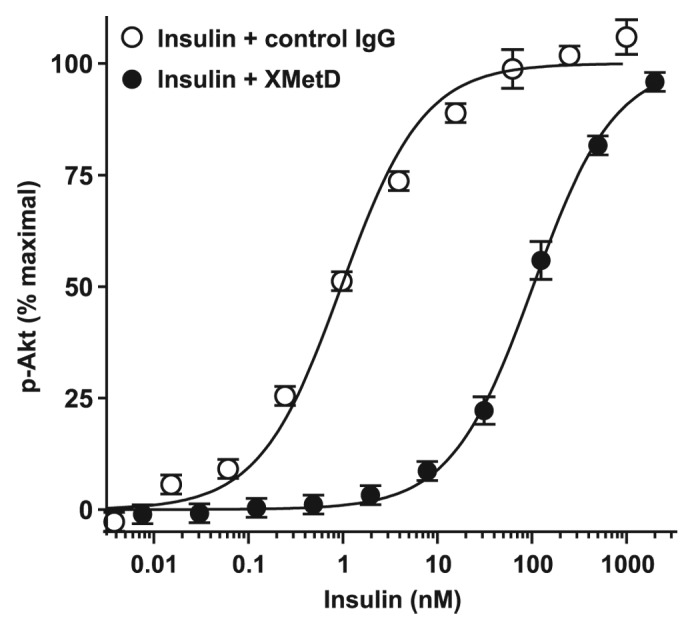
Figure 8. XMetD inhibition of mINSR signaling. CHO-mINSR (B isoform) cells were preincubated for 30 min at 37 °C with 333 nM of either XMetD or control IgG followed by a 10 min incubation with increasing concentrations of insulin. Phosphorylation of Akt was measured using an electrochemiluminescence based assay. Mean ± SD of triplicate determinations are shown.
Effect of XMetD in normal mice
To assess whether the inhibitory effects of XMetD on the INSR observed in vitro were also observed in vivo, studies were performed in normal C57BL/6 mice. In fasted animals treated with control IgG, blood glucose levels were under 100 mg/dL (Fig. 9A). Treatment with XMetD resulted in slightly elevated glucose levels that were not significantly higher than controls. In mice treated with control IgG, plasma insulin levels were 0.73 ± 0.13 ng/ml (mean ± SEM) (Fig. 9B). In fasted mice treated with XMetD, plasma insulin levels were much higher at 12.6 ± 0.2 ng/ml.

Figure 9. XMetD induces insulin resistance in fasted mice and non-fasted mice. Normal male C57BL/6 mice were treated either with either 10 mg/kg XMetD or control IgG. Following a 14 h fast and 24 h after XMetD treatment, glucose (A) and insulin (B) concentrations were then measured. In addition, normal male C57BL/6 mice were treated either with either 10 mg/kg XMetD or control IgG. With ad libitum feeding and 24 h after XMetD treatment, glucose (C) and insulin (D) concentrations were then measured. For all groups n = 6. *P < 0.05 vs control IgG. Values are the mean ± SEM.
We also studied whether XMetD inhibited the INSR activity in non-fasted mice. In non-fasted animals treated with control IgG for 24 h, blood glucose levels were under 200 mg/dL (Fig. 9C). In contrast, non-fasted animals treated with XMetD had significantly higher glucose levels. In mice treated with control IgG, plasma insulin levels were 1.1 ± 0.4 ng/ml (mean ± SEM) (Fig. 9D). In fasted mice treated with XMetD, plasma insulin levels were much higher at 66.0 ± 14.1 ng/ml. These studies indicated that both in fasted and non-fasted mice, XMetD induced insulin resistance with concomitant hyperinsulinemia.
Effect of XMetD in mice with insulin implants
To explore whether XMetD would antagonize hypoglycemia induced by insulin excess, we created a mouse model of hyperinsulinemic hypoglycemia via the insertion of slow-release insulin implants into normal mice. In this model, the implants continually released insulin for over 2 weeks, providing sustained exposure to high concentrations of the hormone. Three days post-implantation, and after a 6 h fast, mice became hypoglycemic with blood glucose levels in the range of 50 mg/dL (Fig. 10). Following a one week treatment with control IgG, blood glucose levels remained in the range of 50 mg/dL in fasted mice with implants. In contrast, following a one week treatment with XMetD, fasted mice with implants were no longer hypoglycemic, having blood glucose levels of approximately 100 mg/dL; these levels were not significantly different from control mice (Fig. 10).
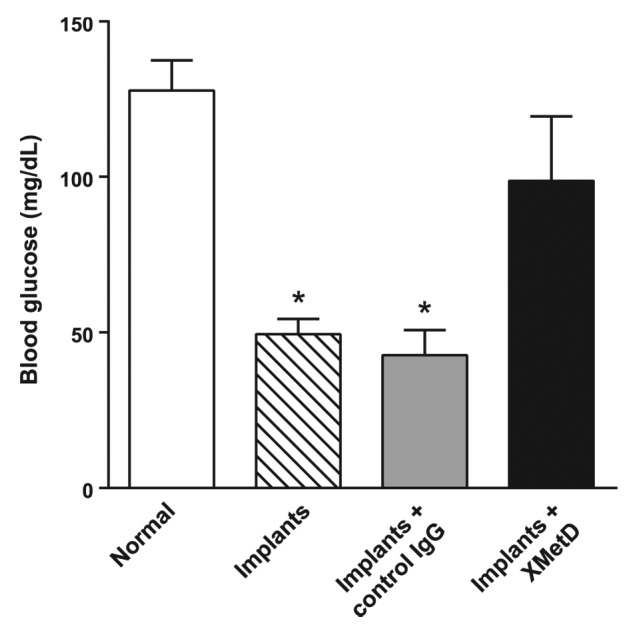
Figure 10. XMetD reverses insulin-induced hypoglycemia in mice. Normal male C57BL/6 mice were given insulin implants and fasting glucose levels were measured after no treatment or following treatment with either 10 mg/kg XMetD or control IgG. Open bar, untreated normal mice. Striped bar, untreated mice with insulin implants. Grey bar = implanted mice treated with control IgG. Solid bar, implanted mice treated with XMetD. For all groups (n = 6), *P < 0.05 vs normal mice. Values are the mean ± SEM.
Discussion
In this study, we identified and characterized a fully human mAb, XMetD, and found it to be a novel antagonist of insulin activation of the INSR. XMetD demonstrated the properties of an allosteric antibody. To identify this type of antibody, we employed the insulin-INSR complex rather than the INSR alone to screen phage libraries. This approach reduced the potential for obtaining antibodies that bound to the orthosteric site of the INSR, because the latter site was occupied by its ligand, insulin. XMetD bound directly to the INSR, and its binding was only partially reduced by a saturating concentration of insulin. Conversely, XMetD only partially inhibited insulin binding to its receptor. These observations are characteristic of a reciprocal relationship between a negative allosteric modulator (XMetD) and an orthosteric ligand (insulin).19,35
After insulin binds to the INSR, it stimulates receptor autophosphorylation and intrinsic kinase domain activation. The metabolic effect of insulin signaling through the INSR on both glucose transport and other aspects of metabolism occurs in large part through the PI3K-Akt pathway.21,36 In cultured cells, XMetD markedly antagonized insulin-dependent INSR autophosphorylation and downstream metabolic effects, including Akt phosphorylation and glucose transport. Therefore, these studies indicated that XMetD was a highly effective inhibitor of insulin-mediated INSR signaling in vitro. It was notable that XMetD did not inhibit insulin-stimulated cancer cell growth in vitro. The reason for the lack of inhibition of this function has not been determined but may reflect an intrinsic aspect of malignant transformation. Further studies will be needed to understand this process.
The inhibitory effects of XMetD on insulin-mediated Akt activation with cells expressing the mINSR were similar to cells expressing the hINSR. Therefore, we undertook studies in mice to determine if this antibody would induce insulin resistance in vivo. In normal non-fasted mice, XMetD elevated blood glucose to levels within the diabetic range. Extreme insulin resistance was induced as reflected by the marked elevation of plasma insulin levels. In normal fasted mice treated with XMetD, hyperglycemia was not observed, but there was strong evidence of insulin resistance, as insulin levels in the fasted state were also markedly elevated.
Thus, the results of these studies indicated that XMetD is a highly effective INSR antagonist, causing insulin resistance both in vitro and in vivo. These findings suggested that this type of antibody would be effective at blocking hypoglycemia induced by insulin excess. Since XMetD was active on the mINSR, we tested XMetD in mice that were made hyperinsulinemic by the insertion of sustained-release insulin implants. As a result of this procedure, the animals became hypoglycemic, but glucose levels returned to the normal range when these hyperinsulinemic mice were treated with XMetD.
Not infrequently, diabetic patients treated with either insulin or insulin-releasing agents develop symptomatic hypoglycemia. This hypoglycemia can be severe and sustained, requiring visits to the emergency department and or prolonged hospitalization.7,8 Our results indicate that allosteric antagonist antibodies to the INSR, such as XMetD, can inhibit INSR signaling in vivo. This observation raises the possibility that this class of antibodies may be useful for the short-term treatment of hypoglycemia caused by exogenous insulin.
In addition, rare conditions of sustained endogenous hyperinsulinemia where current therapies are not always adequate can occur. These conditions include insulinoma,6,8,37 excess secretion of IGF-II38 and congenital hyperinsulinemia (CHI).39-41 In the latter condition, particularly in those patients with defects of the ATP-regulated potassium channel, control of hypoglycemia can be extremely difficult.41,42 Preliminary studies, in the SUR-1 knockout mouse (an animal model of CHI), indicate that XMetD treatment blunts the effect of exogenous insulin.43 Moreover, this antibody prevents fasting hypoglycemia in this animal model.43 It is therefore possible that antagonist allosteric INSR antibodies could be a potential new treatment for patients with hypoglycemia due to both exogenous and endogenous hyperinsulinism. Additional long-term studies with this class of antibodies will be needed to understand their use and clinical utility.
Materials and Methods
XMetD discovery
The extracellular domain of the human insulin receptor (hINSR; R&D Systems, #1544-IR) was biotinylated with Sulfo-NHS-LC-Biotin (Pierce, #21327) according to the manufacturer’s protocol. To obtain allosteric antibodies to the INSR, panning and subsequent screening were performed with the biotinylated extracellular domain of the hINSR maintained in the presence of a saturating insulin concentration (10 micromolar; Sigma-Aldrich, #19278) to block the orthosteric binding site, and prevent the selection of orthosteric antibodies. This biotinylated receptor-ligand complex was immobilized on streptavidin-coated magnetic Dynabeads® M-280 (Invitrogen Dynal AS, #11205D) and panned against antibody phage display libraries (XOMA Corporation).44,45 Following each round of panning, phage were deselected against streptavidin-coated magnetic Dynabeads® M-280 to remove non-specific phage antibodies. After three rounds of panning and deselection, bead-bound phage were eluted and used to infect TG1 bacterial cells (Stratagene, #200123). Phage were then rescued with helper phage M13KO7 (New England Biolabs, #N13KO7). Individual colonies were picked and grown in 96-well plates to generate bacterial periplasmic extracts according to standard methods.45 The lysate supernatants were assayed for INSR binding to suspension-adapted CHO cells (vide infra) by flow cytometry. The CHO cells employed were transfected with either the human or mouse INSR. Antibodies with ortholog cross-reactivity were reformatted into fully human IgG2 mAbs and tested for antagonist activity. The IgG2 with the greatest INSR inhibitory activity was XMetD.45
Engineered cells used to study the activity of XMetD
CHO cells were engineered to express either human INSR isotype A (CHO-hINSR-A), human INSR isotype B (CHO-hINSR-B), or mouse INSR isotype B (CHO-mINSR-B). INSR transfected cell lines had ~250,000 receptors per cell compared with the untransfected cells, which had less than 5,000 INSR per cell as determined by flow cytometry.46 For the majority of the subsequent functional studies shown herein, we employed CHO cells transfected with the B isoform of either the hINSR (CHO-hINSR) or mINSR (CHO-mINSR). The B isoform of the INSR was employed because it is the predominant isoform of the INSR in adult metabolic insulin responsive tissues.47-49 CHO cells were also engineered to express human IGF-1R (CHO-hIGF-1R) at 200,000 receptors per cell. Rat L6 myocytes were engineered to express both human GLUT-4 and the hINSR (100,000 receptors per cell).24
Binding of XMetD to the INSR by FACS
For flow cytometry (FACS), CHO-hINSR-A, CHO-hINSR-B and CHO-mINSR-B cells (2 × 106 per ml) were washed and resuspended in phosphate buffered saline with 0.5% fatty-acid-free bovine serum albumin (BSA) and 0.1% sodium azide (FACS buffer). XMetD was then added and cells were incubated for 120 min at 15 °C. Cells were washed and resuspended in Alexa Fluor® 647-conjugated goat anti-human IgG (1:200; Invitrogen, #A-21445) followed by a 30 min incubation at 4 °C. Subsequently, the cells were washed and analyzed on a FACScanTM flow cytometer (Becton Dickinson). All FACS data were analyzed using Prism software (GraphPad, Inc) and multiplied by a common factor so that the Bmax value for the curve fit with the greatest plateau (CHO-hINSR-B cells) was normalized to 100%.
Quantitative kinetic analysis of XMetD binding to the hINSR by SPR
XMetD kinetics and binding affinity were determined by SPR performed on the ProteOn XPR36 instrument (Bio-Rad) at 25 °C using the one-shot kinetics method.50,51 SPR measures biomolecular interactions in real-time in a label-free environment.52 CHO-hINSR-B cells were solubilized in Tris lysis buffer (TLB) consisting of 150 mM NaCl, 20 mM Tris pH 7.5, 1 mM EDTA, 1 mM EGTA, and 1% TritonTM X-100 supplemented with a protease inhibitor cocktail (Roche, #05892791001). Briefly, 1 ml of TBL was added to ~2 × 106 cells, and the cell suspensions were agitated on a rocker at 4 °C. After 2 h, this solution was centrifuged at 4 °C for 20 min at 14 000 rpm using a tabletop centrifuge. The supernatant containing solubilized INSR was filtered and kept frozen at -80 °C until it was thawed just prior to analysis. To prepare the SPR detection surface for INSR capture, CT-3 mAb (Fisher, #MS-636-PABX), which recognizes the carboxy-teminal moiety of INSR, was covalently immobilized on the sensor chip (Bio-Rad GLM, #176–5012) using standard amine-coupling chemistry. Briefly, the GLM sensor chip was preconditioned with successive injections of 10 mM SDS, 50 mM NaOH, 100 mM Tris pH 9.5, and running buffer consisting of 10 mM HEPES, 150 mM sodium chloride, 3 mM EDTA, 0.05% Polysorbate 20 (Teknova, #H8022) at a flow rate of 100 μL per min. The chip surface was then activated with a five min injection of a freshly prepared 1:1 solution of 0.1M N-hydroxysuccinimide (NHS) and 0.4 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide at a flow rate of 25 µL per min. Following activation, 10 µg/mL of CT-3 antibody in pH 4.5 acetate buffer was injected at a flow rate of 30 μL per min until ~10,000 RU of the capture antibody was immobilized on the sensor surface. To block the surface, 1 M ethanolamine hydrochloride-NaOH pH 8.5 was injected for five min.
Solubilized INSR diluted 1:1 in running buffer supplemented with 1 mg/ml BSA, with or without 1 µg/ml human insulin (Sigma, #A30590), was captured at a density of 1000-1200 RU on a vertical flow channel coated with the CT-3 antibody. After switching to a horizontal orientation and following 10 min of baseline stabilization, XMetD was injected over the captured solubilized INSR at concentrations of 133, 44, 14.7, 4.9, and 1.64 nM at a flow rate of 30 μl per min. Association was monitored for 5 min, and dissociation was monitored for 10 min. Surfaces were regenerated with 100 mM HCl following the XMetD injections. Double-referenced data were curve fit with a simple 1:1 binding model using ProteOn™ software to yield kinetic parameters for on-rate (ka) and off-rate (kd). Equilibrium binding constant (KD) values were calculated from a ratio of the kinetic parameters (kd/ka).
The effect of XMetD on insulin binding affinity for the INSR
The effect of XMetD on insulin binding to the INSR was assessed using a FACS-based assay. CHO-hINSR-B cells were preincubated for 10 min at 4 °C with a saturating concentration of XMetD or an anti-keyhole limpet hemocyanin (KLH) IgG2 isotype control IgG (XOMA Corporation) followed by a 30 min incubation with increasing concentrations of biotinylated insulin (R&D Systems, custom reagent). Cells were then washed and resuspended in allophycocyanin-conjugated streptavidin (1:200 Invitrogen, #SA1005) followed by a 30 min incubation at 4 °C. Subsequently, the cells were washed and analyzed as above.
The effect of XMetD on INSR internalization and downregulation
To study the effect of XMetD on INSR internalization, we employed IM-9 human lymphoblasts.53 These cells were maintained in RPMI media (Gibco, #22400-089) containing 10% fetal bovine serum (FBS) (Hyclone, #SH3007.03). To assess INSR internalization, the cells were incubated for 5 h in RPMI media without serum supplemented with 0.25% BSA. Next, IM-9 cells were treated for 2 h at 37 °C with antibodies plus a range of insulin concentrations. In addition to XMetD, we included both a negative control IgG (anti-KLH) and a downregulating positive control IgG (anti-INSR mAb, XOMA Corporation) all at 66 nM. After 2 h the cells were washed and then stained with the anti-INSR mAb MA-20 (Santa Cruz Biotechnology, #SC-57344)53 for 30 min at 4 °C. MA-20 does not compete with either XMetD or the positive control mAb for binding to the INSR. Following this incubation, cells were washed twice and resuspended in APC-conjugated goat anti-human IgG (1:200; Jackson ImmunoResearch, #115-136-146) followed by a 30 min incubation at 4 °C. Subsequently, the cells were washed and analyzed on an AccuriTM C6 flow cytometer (Becton Dickinson). Fluorescence data reflecting surface INSR were analyzed using the instrument software (CFlow Plus).
To study the effect of XMetD on INSR downregulation, we employed 3T3-L1 mouse preadipocyte cells (ATCC, #CL-173). These cells were grown on collagen coated plates, differentiated and cultured as described,54 except that cells were incubated without insulin following the removal of the differentiation media. Differentiated adipocytes were treated for 2 h at 37 °C in DMEM + 0.25% BSA media with antibodies at plus a range of insulin concentrations. In addition to XMetD, we employed the aforementioned negative and positive control IgGs, all at 66 nM. After 2 h the cells were lysed with Tris-lysis buffer (TLB) and analyzed for total INSR content using the Millipore, STAR ELISA Assay Kit (#17-483).
INSR signaling in cultured cells
To evaluate the effect of XMetD on hINSR autophosphorylation, CHO-hINSR-B cells were preincubated at 37 °C with either 333 nM XMetD or control IgG for 30 min, followed by incubation with increasing concentrations of insulin for 10 min. The phosphotyrosine content of the INSR was measured by ELISA (Millipore, #CBA038). To evaluate the effect of XMetD on insulin mediated Akt phosphorylation, CHO-hINSR-A, CHO-hINSR-B or CHO-mINSR-B cells were preincubated at 37 °C with either XMetD or control IgG for 30 min followed by an incubation with increasing concentrations of insulin in for 10 min. Both total Akt and Akt phosphorylated at Ser473 were measured using an electrochemiluminescent assay (Meso Scale Discovery, #K150MND).24 The same basic protocol was employed for investigating the effect of XMetD on IGF-1 mediated IGF-1R signaling. Insulin-dependent Erk phosphorylation was performed as previously described using an electrochemiluminescent assay (Meso Scale Discovery, #K151DWD).24
Glucose transport assay
To measure 2-deoxy-glucose uptake, rat L6 muscle cells expressing both hINSR and human GLUT-424 were preincubated at 37 °C with either 333 nM XMetD or control IgG for 60 min followed by incubation with increasing concentrations of insulin for 10 min. [3H]-2-deoxy-D-glucose was then added for 20 min and its uptake measured.55
Proliferation assay
COLO-205 colorectal carcinoma cells (ATCC, #CCL-222), were cultured in RPMI 1640 media supplemented with 10% FBS. For proliferation assays, cells were seeded in microtiter plates (Costar, #3917) at a density of 20,000 cells per well. The media was then changed to RPMI (Gibco, #12676) supplemented with 0.5% FBS that had been both charcoal-dextran stripped and heat inactivated. After 24 h, increasing concentrations of insulin were added in the presence and absence of XMetD. Cells were then incubated for 48 h at 37 °C. Proliferation was measured using the CellTiter-Glo® luminescent cell viability assay (Promega, #G7571).
In all the aforementioned in vitro studies, the viability of the cells was approximately 90% as assessed by both microscopic observation and by trypan blue exclusion. Incubation with XMetD did not influence cell viability. All in vitro studies were performed at least 3 times and representative studies are shown. The in vitro signaling, downregulation and proliferation data were analyzed using Prism software (GraphPad, Inc) and normalized so that the curve fit maximal values equaled 100%.
Effects of XMetD in normal fasted and non-fasted mice
All animal experiments were approved by the XOMA Institutional Animal Care and Use Committee (IACUC) and performed in accordance with IACUC guidelines. All animals were maintained in a pathogen-free environment and allowed free access to food and water prior to study. In the first series of experiments, male C57BL/6J mice 6 to 8-weeks of age (The Jackson Laboratory, #000664) were employed and acclimated to a 12 h light-dark cycle. Twenty-four animals were randomly divided into two cohorts (n = 12); one cohort was treated with XMetD and the other with the aforementioned anti-KLH isotype control IgG (both antibodies were administered intraperitoneally at 10 mg/kg). All mice were then allowed free access to food for 10 h. Subsequently, each cohort was divided into two groups (n = 6). One group from each cohort was fasted for 14 h, while the other group was allowed free access to food during the dark cycle. At 24 h after antibody administration, blood from the tail vein was obtained and glucose was measured using a glucometer. Plasma was also obtained at this time by cardiac puncture, and insulin levels measured by ELISA (Alpco Diagnostics, 80-INSMSU-E01). XMetD did not cross-react with either capture or detection antibodies used in these systems (data not shown).
Effects of XMetD in mice with insulin implants
Male C57BL/6J mice, 6- to 8-weeks of age (The Jackson Laboratory, #000664) were randomly divided into two groups. The first group (n = 6) went untreated. In the second group (n = 12), two insulin implants (LinBits, Linshin Inc, #Re-1-T) were placed subcutaneously under the mid-dorsal skin as recommended by the manufacturer. According to the manufacturer, a single implant continually releases 0.1 units of insulin over 24 h in vivo for over 2 weeks. Insulin levels in control mice were less than 500 pg/mL compared with greater than 3000 pg/mL in mice with implants. After 3 d, both untreated and implanted mice were fasted 6 h and blood glucose was measured. On the next day, half of the implant group (n = 6) was treated intraperitoneally (10 mg/kg) with XMetD and the other half of the group (n = 6) was treated with the control IgG: this injection was repeated 5 d later. One week after initiation of antibody treatment, mice were fasted for 6 h and blood glucose was subsequently measured.
Supplementary Material
Disclosure of Potential Conflicts of Interest
Corbin JA, Bhaskar V, Goldfine ID, Issafras H, Bedinger DH, Lau A, Michelson K, Gross LM, Kuan HF, Tran C, Lao L, Handa M, Watson SR, Narasimha AJ, Zhu S, Levy R, Webster L, Wijesuriya SD, Liu N, Wu X, Chemla-Vogel D, Lee SR, Wong S, Wilcock D, Rubin P, and White ML have been or are employees of XOMA Corporation.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/mabs/article/26871
Acknowledgments
Corbin JA led the research team, designed the in vitro experiments and wrote the manuscript. Bhaskar V designed the in vivo experiments, researched the data and edited the manuscript. Goldfine ID wrote and edited the manuscript and contributed to the discussion. Wilcock D, Rubin P, and White ML edited the manuscript and contributed to the discussion. Issafras H, Bedinger DH, Michelson K, Maddux BA, Kuan HF, Tran C, Lao L, Handa M, Watson S, Zhu S, Levy R, Webster L, Wijesuriya SD, Liu N, Wu X, and Chemla-Vogel D researched the in vitro data. Lau A, Gross LM, Narasimha AJ, Lee SR, and Wong S researched the in vivo data. Corbin JA takes responsibility for the content and originality of this manuscript. The authors wish to thank Dr Patrick J Scannon and Dr Marina K Roell, XOMA Corporation, for critical reading of the manuscript and helpful comments.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/26871
References
- 1.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 2.Asano T, Fujishiro M, Kushiyama A, Nakatsu Y, Yoneda M, Kamata H, Sakoda H. Role of phosphatidylinositol 3-kinase activation on insulin action and its alteration in diabetic conditions. Biol Pharm Bull. 2007;30:1610–6. doi: 10.1248/bpb.30.1610. [DOI] [PubMed] [Google Scholar]
- 3.Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med. 2004;10:65–71. doi: 10.2119/2005-00029.Saltiel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simpson IA, Cushman SW. Hormonal regulation of mammalian glucose transport. Annu Rev Biochem. 1986;55:1059–89. doi: 10.1146/annurev.bi.55.070186.005211. [DOI] [PubMed] [Google Scholar]
- 5.Ussar S, Vienberg SG, Kahn CR. Receptor antibodies as novel therapeutics for diabetes. Sci Transl Med. 2011;3:13ps47. doi: 10.1126/scitranslmed.3003447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cryer PE, Axelrod L, Grossman AB, Heller SR, Montori VM, Seaquist ER, Service FJ, Endocrine Society Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2009;94:709–28. doi: 10.1210/jc.2008-1410. [DOI] [PubMed] [Google Scholar]
- 7.Budnitz DS, Lovegrove MC, Shehab N, Richards CL. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med. 2011;365:2002–12. doi: 10.1056/NEJMsa1103053. [DOI] [PubMed] [Google Scholar]
- 8.Service FJ. Hypoglycemic disorders. N Engl J Med. 1995;332:1144–52. doi: 10.1056/NEJM199504273321707. [DOI] [PubMed] [Google Scholar]
- 9.Goldfine ID, Roth RA. Monoclonal antibodies to the insulin receptor as probes of insulin receptor structure and function. Horiz Biochem Biophys. 1986;8:471–502. [PubMed] [Google Scholar]
- 10.Soos MA, Siddle K, Baron MD, Heward JM, Luzio JP, Bellatin J, Lennox ES. Monoclonal antibodies reacting with multiple epitopes on the human insulin receptor. Biochem J. 1986;235:199–208. doi: 10.1042/bj2350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siddle K, Soos MA, O’Brien RM, Ganderton RH, Taylor R. Monoclonal antibodies as probes of the structure and function of insulin receptors. Biochem Soc Trans. 1987;15:47–51. doi: 10.1042/bst0150047. [DOI] [PubMed] [Google Scholar]
- 12.Lupsa BC, Chong AY, Cochran EK, Soos MA, Semple RK, Gorden P. Autoimmune forms of hypoglycemia. Medicine (Baltimore) 2009;88:141–53. doi: 10.1097/MD.0b013e3181a5b42e. [DOI] [PubMed] [Google Scholar]
- 13.Le Marchand-Brustel Y, Gorden P, Flier JS, Kahn CR, Freychet P. Anti-insulin receptor antibodies inhibit insulin binding and stimulate glucose metabolism in skeletal muscle. Diabetologia. 1978;14:311–7. doi: 10.1007/BF01223022. [DOI] [PubMed] [Google Scholar]
- 14.De Pirro R, Roth RA, Rossetti L, Goldfine ID. Characterization of the serum from a patient with insulin resistance and hypoglycemia. Evidence for multiple populations of insulin receptor antibodies with different receptor binding and insulin-mimicking activities. Diabetes. 1984;33:301–4. doi: 10.2337/diab.33.3.301. [DOI] [PubMed] [Google Scholar]
- 15.Arioglu E, Andewelt A, Diabo C, Bell M, Taylor SI, Gorden P. Clinical course of the syndrome of autoantibodies to the insulin receptor (type B insulin resistance): a 28-year perspective. Medicine (Baltimore) 2002;81:87–100. doi: 10.1097/00005792-200203000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Dons RF, Havlik R, Taylor SI, Baird KL, Chernick SS, Gorden P. Clinical disorders associated with autoantibodies to the insulin receptor. Simulation by passive transfer of immunoglobulins to rats. J Clin Invest. 1983;72:1072–80. doi: 10.1172/JCI111032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cazorla M, Arrang JM, Prémont J. Pharmacological characterization of six trkB antibodies reveals a novel class of functional agents for the study of the BDNF receptor. Br J Pharmacol. 2011;162:947–60. doi: 10.1111/j.1476-5381.2010.01094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rizk SS, Paduch M, Heithaus JH, Duguid EM, Sandstrom A, Kossiakoff AA. Allosteric control of ligand-binding affinity using engineered conformation-specific effector proteins. Nat Struct Mol Biol. 2011;18:437–42. doi: 10.1038/nsmb.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 20.Root-Bernstein R, Vonck J. Glucose binds to the insulin receptor affecting the mutual affinity of insulin and its receptor. Cell Mol Life Sci. 2009;66:2721–32. doi: 10.1007/s00018-009-0065-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Meyts P, Gauguin L, Svendsen AM, Sarhan M, Knudsen L, Nøhr J, Kiselyov VV. Structural basis of allosteric ligand-receptor interactions in the insulin/relaxin peptide family: implications for other receptor tyrosine kinases and G-protein-coupled receptors. Ann N Y Acad Sci. 2009;1160:45–53. doi: 10.1111/j.1749-6632.2009.03837.x. [DOI] [PubMed] [Google Scholar]
- 22.De Meyts P. The insulin receptor: a prototype for dimeric, allosteric membrane receptors? Trends Biochem Sci. 2008;33:376–84. doi: 10.1016/j.tibs.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 23.Jensen M, Hansen B, De Meyts P, Schäffer L, Ursø B. Activation of the insulin receptor by insulin and a synthetic peptide leads to divergent metabolic and mitogenic signaling and responses. J Biol Chem. 2007;282:35179–86. doi: 10.1074/jbc.M704599200. [DOI] [PubMed] [Google Scholar]
- 24.Bhaskar V, Goldfine ID, Bedinger DH, Lau A, Kuan HF, Gross LM, Handa M, Maddux BA, Watson SR, Zhu S, et al. A fully human, allosteric monoclonal antibody that activates the insulin receptor and improves glycemic control. Diabetes. 2012;61:1263–71. doi: 10.2337/db11-1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhaskar V, Lau A, Goldfine ID, Narasimha AJ, Gross LM, Wong S, Cheung B, White ML, Corbin JA. XMetA, an allosteric monoclonal antibody to the insulin receptor, improves glycaemic control in mice with diet-induced obesity. Diabetes Obes Metab. 2013;15:272–5. doi: 10.1111/dom.12019. [DOI] [PubMed] [Google Scholar]
- 26.Vigneri R, Squatrito S, Frittitta L. Selective insulin receptor modulators (SIRM): a new class of antidiabetes drugs? Diabetes. 2012;61:984–5. doi: 10.2337/db12-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–74. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- 28.Grunfeld C, Van Obberghen E, Karlsson FA, Kahn CR. Antibody-induced desensitization of the insulin receptor. Studies of the mechanism of desensitization in 3T3-L1 fatty fibroblasts. J Clin Invest. 1980;66:1124–34. doi: 10.1172/JCI109942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grunfeld C, Jones DS, Shigenaga JK. Autoantibodies against the insulin receptor. Dissociation of the acute effects of the antibodies from the desensitization seen with prolonged exposure. Diabetes. 1985;34:205–11. doi: 10.2337/diab.34.3.205. [DOI] [PubMed] [Google Scholar]
- 30.Sacco A, Morcavallo A, Pandini G, Vigneri R, Belfiore A. Differential signaling activation by insulin and insulin-like growth factors I and II upon binding to insulin receptor isoform A. Endocrinology. 2009;150:3594–602. doi: 10.1210/en.2009-0377. [DOI] [PubMed] [Google Scholar]
- 31.Maddux BA, See W, Lawrence JC, Jr., Goldfine AL, Goldfine ID, Evans JL. Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by mircomolar concentrations of alpha-lipoic acid. Diabetes. 2001;50:404–10. doi: 10.2337/diabetes.50.2.404. [DOI] [PubMed] [Google Scholar]
- 32.Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30:586–623. doi: 10.1210/er.2008-0047. [DOI] [PubMed] [Google Scholar]
- 33.Sciacca L, Le Moli R, Vigneri R. Insulin analogs and cancer. Front Endocrinol (Lausanne) 2012;3:21. doi: 10.3389/fendo.2012.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurtzhals P, Schäffer L, Sørensen A, Kristensen C, Jonassen I, Schmid C, Trüb T. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes. 2000;49:999–1005. doi: 10.2337/diabetes.49.6.999. [DOI] [PubMed] [Google Scholar]
- 35.Roell MK, Issafras H, Bauer RJ, Michelson KS, Mendoza N, Vanegas SI, Gross LM, Larsen PD, Bedinger DH, Bohmann DJ, et al. Kinetic approach to pathway attenuation using XOMA 052, a regulatory therapeutic antibody that modulates interleukin-1beta activity. J Biol Chem. 2010;285:20607–14. doi: 10.1074/jbc.M110.115790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saini V. Molecular mechanisms of insulin resistance in type 2 diabetes mellitus. World J Diabetes. 2010;1:68–75. doi: 10.4239/wjd.v1.i3.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Starke A, Saddig C, Mansfeld L, Koester R, Tschahargane C, Czygan P, Goretzki P. Malignant metastatic insulinoma-postoperative treatment and follow-up. World J Surg. 2005;29:789–93. doi: 10.1007/s00268-005-7743-y. [DOI] [PubMed] [Google Scholar]
- 38.Dynkevich Y, Rother KI, Whitford I, Qureshi S, Galiveeti S, Szulc AL, Danoff A, Breen TL, Kaviani N, Shanik MH, et al. Tumors, IGF-2 and Hypoglycemia: Insights from the clinic, the laboratory and the historical archive. Endocr Rev. 2013 doi: 10.1210/er.2012-1033. [DOI] [PubMed] [Google Scholar]
- 39.Hussain K. Congenital hyperinsulinism. Semin Fetal Neonatal Med. 2005;10:369–76. doi: 10.1016/j.siny.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 40.Arnoux JB, de Lonlay P, Ribeiro MJ, Hussain K, Blankenstein O, Mohnike K, Valayannopoulos V, Robert JJ, Rahier J, Sempoux C, et al. Congenital hyperinsulinism. Early Hum Dev. 2010;86:287–94. doi: 10.1016/j.earlhumdev.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Palladino AA, Bennett MJ, Stanley CA. Hyperinsulinism in infancy and childhood: when an insulin level is not always enough. Clin Chem. 2008;54:256–63. doi: 10.1373/clinchem.2007.098988. [DOI] [PubMed] [Google Scholar]
- 42.Henwood MJ, Kelly A, Macmullen C, Bhatia P, Ganguly A, Thornton PS, Stanley CA. Genotype-phenotype correlations in children with congenital hyperinsulinism due to recessive mutations of the adenosine triphosphate-sensitive potassium channel genes. J Clin Endocrinol Metab. 2005;90:789–94. doi: 10.1210/jc.2004-1604. [DOI] [PubMed] [Google Scholar]
- 43.Patel P, Corbin JA, Goldfine ID, Rubin P, De Leon DD. A unique allosteric insulin receptor monoclonal antibody that prevents hypoglycemia in the SUR-1−/− mouse model of KATP hyperinsulinism. Diabetes. 2013;62(suppl1):A-93. doi: 10.1080/19420862.2018.1457599. [abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barbas CF, Burton DR, Scott JK, Silverman GJ. Phage Display: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Laboratory Press, 2001. [Google Scholar]
- 45.Aitken R. Methods in Molecular Biology. In: O'Brien PM, ed. Antibody phage display: methods and protocols. Totowa, NJ: Humana Press, 2001. [Google Scholar]
- 46.Zloza A, Sullivan YB, Connick E, Landay AL, Al-Harthi L. CD8+ T cells that express CD4 on their surface (CD4dimCD8bright T cells) recognize an antigen-specific target, are detected in vivo, and can be productively infected by T-tropic HIV. Blood. 2003;102:2156–64. doi: 10.1182/blood-2002-07-1972. [DOI] [PubMed] [Google Scholar]
- 47.Seino S, Bell GI. Alternative splicing of human insulin receptor messenger RNA. Biochem Biophys Res Commun. 1989;159:312–6. doi: 10.1016/0006-291X(89)92439-X. [DOI] [PubMed] [Google Scholar]
- 48.Moller DE, Yokota A, Caro JF, Flier JS. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol Endocrinol. 1989;3:1263–9. doi: 10.1210/mend-3-8-1263. [DOI] [PubMed] [Google Scholar]
- 49.Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, Goldfine ID, Belfiore A, Vigneri R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. 1999;19:3278–88. doi: 10.1128/mcb.19.5.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bravman T, Bronner V, Lavie K, Notcovich A, Papalia GA, Myszka DG. Exploring “one-shot” kinetics and small molecule analysis using the ProteOn XPR36 array biosensor. Anal Biochem. 2006;358:281–8. doi: 10.1016/j.ab.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 51.Abdiche YN, Lindquist KC, Pinkerton A, Pons J, Rajpal A. Expanding the ProteOn XPR36 biosensor into a 36-ligand array expedites protein interaction analysis. Anal Biochem. 2011;411:139–51. doi: 10.1016/j.ab.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 52.Piliarik M, Vaisocherová H, Homola J. Surface plasmon resonance biosensing. Methods Mol Biol. 2009;503:65–88. doi: 10.1007/978-1-60327-567-5_5. [DOI] [PubMed] [Google Scholar]
- 53.Forsayeth JR, Montemurro A, Maddux BA, DePirro R, Goldfine ID. Effect of monoclonal antibodies on human insulin receptor autophosphorylation, negative cooperativity, and down-regulation. J Biol Chem. 1987;262:4134–40. [PubMed] [Google Scholar]
- 54.Ronnett GV, Knutson VP, Lane MD. Insulin-induced down-regulation of insulin receptors in 3T3-L1 adipocytes. Altered rate of receptor inactivation. J Biol Chem. 1982;257:4285–91. [PubMed] [Google Scholar]
- 55.Brunetti A, Maddux BA, Wong KY, Hofmann C, Whittaker J, Sung C, Goldfine ID. Monoclonal antibodies to the human insulin receptor mimic a spectrum of biological effects in transfected 3T3/HIR fibroblasts without activating receptor kinase. Biochem Biophys Res Commun. 1989;165:212–8. doi: 10.1016/0006-291X(89)91056-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.