

Abstract
The importance of antibodies in activating immune responses against tumors is now better appreciated with the emergence of checkpoint blockade antibodies and with engineered antibody Fc domains featuring enhanced capacity to focus potent effector cells against cancer cells. Antibodies designed with Fc regions of the IgE class can confer natural, potent, long-lived immune surveillance in tissues through tenacious engagement of high-affinity cognate Fc receptors on distinct, often tumor-resident immune effector cells, and through ability to activate these cells under tumor-induced Th2-biased conditions. Here, we review the properties that make IgE a contributor to the allergic response and a critical player in the protection against parasites, which also support IgE as a novel anti-cancer modality. We discuss IgE-based active and passive immunotherapeutic approaches in disparate in vitro and in vivo model systems, collectively suggesting the potential of IgE immunotherapies in oncology. Translation toward clinical application is now in progress.
Keywords: IgE, antibodies, IgE immunotherapy, cancer immunotherapy, antibody effector functions, AllergoOncology
Introduction
Monoclonal antibodies (mAbs) represent a significant addition to the therapeutic armamentarium for a variety of malignancies, with 15 approved antibody-based therapies in current use in oncology.1-4 It is notable, however, that over half of these successful agents are approved for hematological malignancies, i.e., leukemias and lymphomas. Despite the superb specificity and high affinity of mAbs for their target antigens and significant advances in antibody immunotherapy for breast and colorectal cancers, the concept of a “magic bullet” for the treatment of many solid tumors has produced less impressive outcomes. Although many therapeutic mAbs exert their anti-tumor effects through a multitude of mechanisms, Fc-mediated mechanisms of immune system engagement play an important role.5,6 There is now emerging evidence in support of the substantial contributions that Fc-mediated mechanisms make to the observed clinical efficacy of therapeutic antibodies; indeed, Fc-mediated mechanisms of action are attributed to antibodies already in clinical use, such as trastuzumab,7-9 cetuximab,10 and ipilimumab.11 These antibodies lend credence to the notion that effector cell activation is an important, albeit often less well-appreciated contributor to the anti-tumoral properties of many therapeutic mAbs. This knowledge has encouraged the development of several innovative antibody engineering strategies, all aimed at optimizing the antibody-immune system interaction by enhancing Fc-mediated antibody mechanisms of action.12-14
The currently approved mAbs are of the IgG isotype3 due to favorable pharmacokinetic properties and pioneering experiments by Neuberger and colleagues, which demonstrated that IgG, and in particular IgG1, was more effective than other antibody isotypes in activating complement and promoting antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) against tumor cells in vitro.15-17 Because the interaction of a therapeutic antibody with immune effector cells is mediated by its Fc domain, which determines binding to complement and the relevant cellular Fc receptors, it is clear that the class or subclass of an antibody critically determines its effector functions, and ultimately could substantially influence efficacy. In the context of IgG class antibodies, insufficient access of systemically-introduced large molecules like antibodies to solid tumor lesions, the relatively low affinity of antibodies for their cognate Fc receptors, and the presence of inhibitory Fcγ receptors in the tumor microenvironments, together with local immunosuppressive conditions and inefficient recruitment and activation of immune effector cells with sufficient potency to eradicate cancer, may be mechanisms that restrict the effector functions of therapeutic antibodies. Therefore, one strategy to optimize the antibody-immune system interaction has been the use of passive and active immunotherapies that take advantage of the particular properties of antibodies with Fc regions of alternative immunoglobulin classes, such as those of the IgE class. Indeed, work in this area constitutes an important branch of the rapidly growing field of AllergoOncology, which aims to address the potential opportunities posed by IgE-mediated and T helper type 2 (Th2)-biased cellular responses in malignant diseases.18
It has been suggested that the well-documented manifestations of allergic disease and immune surveillance in parasitic infections, namely local immune stimulation, with an ensuing cascade of “allergic” inflammation at the site of antigen provocation, may be harnessed to re-direct potent immune cell populations to induce tumor rejection.19-22 These characteristics, together with the high affinity of IgE for its cognate Fcε receptors and the observation in solid tumors of many critical IgE receptor-expressing immune effector cells, have formed the motivation for several research groups to develop tumor-specific recombinant IgE antibodies, and other immunotherapeutic approaches, aimed at triggering IgE functions to target tumor cells.22-27 These emerging developments are discussed in this review.
The IgE antibody and Its Receptors
IgE is the least abundant circulating antibody class and has a very short serum half-life. Indeed, the serum concentration of IgE in normal individuals is ~50 ng/mL, in stark contrast to IgG, which is present at concentrations in the order of 5–10 mg⁄mL.28,29 In contrast with its short half-life in the circulation (1–2 d, compared with ~3 weeks for IgG), tissue-resident IgE may persist for several days (approximate half-life in the skin of 2 weeks).30-32 This may be a consequence of its extremely high affinity for the IgE Fc receptor, FcεRI, and in particular its slow dissociation from this receptor, resulting in re-binding of dissociated IgE to its receptors, and restricted diffusion away from the tissue within which it resides.32
Both myeloma IgE and normal IgE are heavily glycosylated compared with IgG.33 A glycosyltransferase initiates the addition of carbohydrate chains to Asn at the consensus site, Asn-X-Ser/Thr; one such site in the Fc region is conserved in other immunoglobulin classes including IgG and lies between the two heavy-chains, but there are several other sites, not always fully glycosylated, on the surface of IgE. The particular sugars may be very important for IgE activity and transport. For example, the carbohydrate chains in IgE Fc are of the “high-mannose” type, due to the failure to trim off the branches of the carbohydrate chains that contain this sugar. Mannose is recognized by mannose binding protein, which is present on liver cells that phagocytose IgE. This may contribute to the short half-life of IgE in the circulation, compared with IgG, which in contrast contains “complex-type” carbohydrate chains. Since cells of different types, and also the same types under different conditions, express different glycosyltransferases, the choice of cell lines and growth conditions for the production of recombinant IgEs may be critical for the safety or efficacy of IgE immunotherapy of cancer.34
There are two well-characterized receptors for IgE that are structurally and functionally distinct: the high affinity FcεRI, and the lower affinity CD23 (FcεRII), that bind the Fc epsilon (Fcε) region of IgE with affinities (Ka) of 109‒1011 M−1 and 108–109 M−1, respectively.32,35 In addition, the IgE-binding molecules galectin-3 and galectin-9 recognize oligosaccharides of IgE.36
FcεRI
Human FcεRI exists as both a tetrameric and trimeric structure. Tetrameric FcεRI is composed of an α-chain, a β-chain and a disulfide-linked γ-chain homodimer (αβγ2), whereas for trimeric expression, the α-chain associates with the γ2-dimer in the absence of a β-chain (αγ2). Tetrameric and trimeric FcεRI isoforms have different cellular expression patterns associated with different IgE effector functions. The αβγ2 tetramer is expressed only on basophils and mast cells, where it is expressed in abundance (~200 000 molecules/cell), whereas the αγ2 trimer in humans, is expressed on Langerhans cells, dendritic cells (DCs), monocytes and eosinophils, at 10 – 100 fold lower concentrations.37 The trimer is not expressed in mouse, but is expressed in the rat in the same cell types as in humans.38 This makes rat the more suitable rodent model to study IgE effector functions in allergic disease and cytotoxic killing (ADCC) of tumor cells.22
CD23 (FcεRII)
CD23 (FcεRII) is a type II integral membrane protein that belongs to the calcium-dependent (C-type) lectin superfamily. Its expression can be induced on a broad range of immune cells, such as activated B cells, activated monocytes and macrophages, eosinophils, natural killer T cells, T cells, follicular DCs, and platelets, but also on non-immune cells, such as airway epithelial cells and smooth muscle cells.39-41 Two isoforms of CD23 have been identified, CD23a and CD23b.42 Human CD23a is expressed exclusively on antigen-activated B cells prior to differentiation into antibody-secreting plasma cells, and is involved in IgE antibody-dependent antigen endocytosis, processing and presentation, and the regulation of IgE synthesis and clearance. CD23b is expressed on a variety of effector cells, including B cells and monocytes/macrophages, following interleukin (IL)-4 stimulation.32,42,43 CD23 is a homo-trimer of three chains with C-type lectin domains held together by a coiled-coil stalk in the membrane form. After cleavage at a specific site in the extracellular sequence by metalloprotease, the soluble fragment (sCD23) may be further degraded to leave only the monomeric lectin domain with or without a C-terminal tail region. This soluble CD23 (sCD23) fragment in humans, but not mice, has specificity for complement receptor 2 (CR2) and the binding to CR2 on B cells promotes B cell proliferation and IgE synthesis.44 As IgE production and concentrations increase in the circulation during exposure to allergens in sensitized individuals, this IgE can bind to membrane CD23 to prevent the release of sCD23. This terminates enhancement of IgE synthesis by sCD23 and may induce further inhibitory signals through membrane CD23. The combined activities of membrane CD23 and sCD23 constitute homeostatic mechanism for controlling the concentration of IgE.32
Galectins-3 and -9
In addition to the cell-bound IgE receptors, there are secreted lectins (carbohydrate binding proteins) that recognize particular sugar residues in the carbohydrate chains attached to IgE and other proteins. They may promote (galectin-3) or suppress (galectin-9) allergic inflammation, and undoubtedly affect other functions of IgE and of other glycosylated proteins. Liu and coworkers have performed extensive studies on galectin-3.45 Galectin-9 binds to β-galactoside residues in the carbohydrate chains of IgE and can suppress allergic inflammation.46 It exerts this activity by blocking the binding of antigens to IgE. Galectin-3 is secreted by epithelial cells and immune inflammatory cells and binds to carbohydrate chains containing β-galactoside residues. Galectin-3 binds specifically to oligosaccharide structures with a terminal β-galactose through a lectin–carbohydrate interaction.47 It recognizes IgE and FcεRI, both of which have multiple carbohydrate chains and can activate immune responses.36,48 Due to its ability to form pentameric complexes with oligosaccharides, it can crosslink FcεRI or receptor-bound IgE on the surface of mast cells and basophils.49,50 Despite its potential to enhance mast cell activation and mediate the phagocytotic properties of monocytes, increased expression of galectin-3 has been associated with poor prognosis in thyroid and pancreatic cancers, whereas reduced levels of the protein were detected in colon, ovarian, breast, endometrial and skin cancers, compared with normal tissues.51 Galectin-3 expression in malignancies may have a bearing on the immunogenic nature of the tumor, but this has not yet been fully explored. Although expressed by monocytic cells, in at least one in vitro study, galectin-3 did not appear to mediate tumor cell death.20 To date, there is no evidence linking galectin-3 to IgE antibody-dependent tumor cell killing although further research may shed more light on potential mechanisms that may link galectin-3 to IgE-mediated immune cell activation signals against cancer cells.
The IgE-Mediated Immune Response
Antibodies of the IgE class play a critical pathogenic role in triggering and maintaining allergic inflammation in response to allergens. This allergen-induced immune response is rapidly activated in response to allergens such as the house dust mite or pollen proteins, and results in triggering of mast cell degranulation and eosinophil inflammation at the site of allergen challenge. This allergic inflammatory cascade, however, is thought to have originally evolved from the natural immune defense against parasite infections, which acts to rapidly neutralise invading parasites.52,53
The role of IgE in allergic inflammation
Allergic inflammation is characterized by IgE-dependent activation of mast cells and an infiltration of inflammatory cells to the site of allergen challenge, orchestrated by increased numbers of activated CD4+ Th2 lymphocytes. This occurs in a step-wise manner, beginning with allergen sensitization and the subsequent production of allergen-specific IgE by B cells. Upon re-exposure to the same allergen, a rapid cascade of events is triggered with mast cell and basophil degranulation, followed by early and late phase inflammatory responses.
Allergen sensitization
The immune response in allergy begins with allergen sensitization, a process that culminates in the production and secretion of allergen-specific IgE by B cells that have undergone clonal selection and affinity maturation. First, upon exposure to allergen, antigen presenting cells (APCs) such as DCs, monocytes, or B cells, present the allergen on their cell surface to cognate naive T cells, inducing them to acquire a CD4+ Th2 cell phenotype.30 These Th2 cells then engage cognate B cells through both B cell major histocompatibility complex (MHC) class II and co-stimulatory molecules, such as the CD40 receptor/CD40 ligand, and secrete IL-4 and IL-13, inducing B cells to undergo class-switch recombination (CSR) from IgM or IgD, to IgE.39 Such class switching to IgE may occur, however, after an intermediate switch to IgG, and it is thought that affinity maturation occurs in the IgG-positive cells before switching to IgE.54,55 B cells that have completed this process are irreversibly committed to production of IgE antibodies, expressed as membrane IgE, also known as the B cell receptor (BCR), through which they are stimulated to differentiate into IgE-secreting plasma cells by allergen binding and an appropriate cytokine millieu. CSR can also be induced by IL-4 or IL-13 derived from cells other than Th2 cells, which may include MCs and basophils.56 As a result of allergen sensitization, the B cells that have undergone clonal selection and affinity maturation secrete allergen-specific IgE, which then binds, via its Fc region, to FcεRI receptor-expressing immune effector cells such as mast cells and basophils, leaving its allergen-specific Fab region available for future interaction with allergen.
Early and late phase allergic reactions
The immune response to re-exposure to allergen can be divided into two phases. The first is the classic immediate (type I) hypersensitivity, or early phase reaction, which occurs within minutes of exposure to the allergen. The second, or late phase reaction, occurs 4 – 6 h after the subsidence of the first phase symptoms and can last for days or even weeks.
During the early phase reaction, allergen cross-linking of IgE bound to FcεRI on mast cells (or basophils) causes receptor aggregation and downstream signaling through a Syk-dependent pathway.57 The net result of this is degranulation of these cells and the release of preformed mediators from their cellular granules, including histamine, heparin, serotonin, and mast cell proteases, and de novo synthesized lipid mediators, contributing to the characteristic symptoms of an allergic reaction within minutes.39 Cross-linking of these cells can only be triggered by multivalent antigens because two or more FcεRI receptors need to be engaged for cross-linking and subsequent triggering of the ensuing signaling cascade. As the acute symptoms of immediate hypersensitivity abate, IgE-stimulated mast cells produce chemokines and cytokines that orchestrate the influx and activation of cell types associated with allergy to produce the late phase response.58,59 The recruited inflammatory cells include neutrophils, followed by eosinophils, monocytes and lymphocytes. The resultant chronic inflammation may then lead to tissue remodelling and subsequent exacerbation of the allergic disease.
Crucial cells in this inflammatory infiltrate are monocytes and eosinophils, whose functions include clearance of antigen–IgE complexes, and killing and phagocytosis of pathogens and antigen-expressing tumor cells.20,60 FcεRI and CD23, the expression of which is upregulated by IgE and the cytokines IL-4 and IL-13, respectively, mediate these processes.
The role of IgE in parasitic infections
Besides its critical role in allergy, IgE is generally believed to play a physiological role in immunity against parasites.61 The first in vivo evidence that IgE antibodies could be an essential component of the protective immunity against parasites was provided by passive transfer of monoclonal IgE antibodies directed against schistosomes.62 In this study, a rat monoclonal IgE antibody raised against Schistosoma mansoni afforded a significant level of protection against a challenge infection with S. mansoni when passively transferred into naive recipient rats.62
Furthermore, induction of resistance to infection by adoptive transfer of eosinophils or platelets bearing IgE, indicated that the IgE on these effector cells was crucial.53 Later, support for a role of IgE in parasite immunity was found when it was demonstrated that human eosinophils, platelets and macrophages could harness IgE in vitro to mediate cytotoxicity and phagocytosis, via FcεRI or CD23, respectively, of Schistosoma mansoni or Leishmania major.52,53,63,64 These observations were subsequently established to be relevant to human immunity when epidemiological studies with Schistosoma hematobium provided evidence that host protection against re-infection in S. hematobium-infected populations was associated with high levels of parasite-specific IgE,65 and subsequently IgE antibodies against Schistosoma mansoni were shown to positively predict resistance against re-infection with this blood fluke.66
More recently, studies have demonstrated evidence that IgE antibodies are capable of activating a different cell type, namely mast cells, to induce elimination of parasites through the release of toxic granules.67 Trichinella spiralis infection induces intestinal mastocytosis and heightened IgE responses, and elimination of this parasite requires expulsion of the adult worms from the gut and destruction of the larval cysts deposited in the muscles.68 In IgE-sufficient animals, intense deposition of IgE around the necrotic larval cysts was demonstrated with associated accelerated removal of worms from the intestine and a reduction in the viability of larval parasites in muscle.67 Indeed T. spiralis infection drove a marked splenic mastocytosis and elevated serum levels of mouse mast cell protease-1 (MMCP-1), consistent with a systemic expansion of mast cells driven by the parasite. This mast cell increase was dramatically attenuated in IgE−/− mice, implicating IgE antibodies in this mast cell homeostasis and protection from parasitic infections. Furthermore, protective roles for mast cells during T. spiralis infection have also been observed using mast cell deficient mice and by antibody inhibition of the mast cell marker, c-Kit.68
Based on the evidence, it appears that IgE antibodies play a central physiological role in immunity against parasitic infections, by a number of different mechanisms and through a number of IgE receptor-expressing cell types. Knowledge of these properties, in addition to those that make IgE a crucial contributor in the allergic response, have stimulated researchers to ask whether IgE antibodies may have potential value as a therapeutic agent in cancer. It is hypothesized that the well-documented manifestations of allergic disease and immune surveillance in parasitic infections, namely local immune stimulation, with the ensuing cascade of “allergic” inflammation at the site of antigen provocation may be harnessed to re-direct potent immune cell populations to induce tumor rejection.22 The potential for IgE to induce an “allergic” inflammatory response at the site of a tumor, together with the distinct presence in solid tumors of many critical IgE receptor-expressing immune effector cells, has formed the motivation for several research groups to develop recombinant tumor-specific IgE antibodies and other immunotherapeutic approaches involving triggering IgE functions to target tumor cells.
AllergoOncology: Wielding the Allergic Response against Cancer
The emerging field of AllergoOncology represents a multi-disciplinary effort to determine the relationship between cancer and IgE-mediated immunity, and to exploit this relationship by developing active and passive immunotherapies for the treatment of cancer.18,69
An association between allergic diseases and cancer was first proposed in the 1950s, when experiments were conducted to investigate “allergic responses” toward tumor xenografts.70 The interest of the scientific community with regard to the biological consequences of this so-called “tumor allergy” on cancer progression was further stimulated when a negative correlation between atopy and cancer was first reported over 4 decades ago.71-73 Subsequently, serum IgE levels and allergic reactions in the skin of cancer patients,74,75 provided further evidence of a potential inverse relationship between allergy and cancer, and this seemed to be confirmed by the finding of a decreased prevalence of atopy in cancer patients.76 In the early 1990s, the hypothesis that IgE antibodies possessed a natural surveillance function in malignancies was further fuelled when an immunohistochemical (IHC) study on the distribution of immunoglobulin classes in head and neck cancer revealed that IgE antibodies were the most abundant class.77 Support, however, for a role of IgE in natural tumor immunosurveillance was provided in a subsequent study by Fu et al., who demonstrated significantly elevated levels of serum cancer-specific IgE capable of inducing tumor cytotoxicity in patients with pancreatic cancer patients vs. healthy controls.78 It is possible that these findings are due to either induction of class switching to IgE in Th2-biased milieu within the tumor microenvironment and may be indicative of a redirected, belated or inefficient immune response to cancer growth.79,80 Nonetheless, these findings may suggest that IgE antibodies could also harbor tumor-eradicating functions.
Epidemiological associations of IgE and allergy
A multitude of epidemiological studies have evaluated potential associations between allergy and risk of malignancy. It is clear from these studies that the relationship between allergy and cancer is complex, with a number of hypotheses proposed to explain either the increased or decreased risk of different cancer types reported in the literature.81 Despite the complex relationship between allergy and cancer, strong inverse associations have been consistently reported for a few specific tumor types, including cancers of the brain, pancreas, lymphatic/hematopoietic systems, gastrointestinal tract and gynecological malignancies.82 These epidemiological studies (reviewed by Josephs et al.81), which demonstrate a potential inverse association between a history of allergy and cancer risk, suggest a natural anti-cancer effect of IgE-mediated immunity. In light of these findings, the potential value of IgE, used either as an active or passive immunotherapeutic agent in cancer merited investigation. Indeed, antibodies of the IgE isotype possess several unique properties that make them an attractive option for cancer therapy.
Rationale for the use of antibodies of the IgE class as cancer therapeutics
The particular properties that make IgE a contributor in the allergic response and a crucial player in the immune response against parasites point to its potential value as a therapeutic agent in cancer. These include:
Exceptionally high affinity for IgE receptors
IgE antibodies mediate immune activation by their interaction with two Fcε cell surface receptors: FcεRI and CD23. The affinity of IgE for FcεRI (Ka = 109–1011 M−1) is two to three orders of magnitude higher than that of IgG for FcγRI/CD64 (Ka = 108–109 M−1).35,83 Additionally, the avidity of human IgE for CD23 on the cell surface in its trimeric form is Ka = 108–109 M−1, which is equivalent to the affinity of IgG for FcγRI.84 These high affinities allow IgE antibodies, unlike any other antibody class, to remain bound to immune effector cells even in the absence of antigen.32 This gives IgE the ability to sensitize mast cells for immediate hypersensitivity, the hallmark of the allergic response.
Lack of an inhibitory Fc receptor for IgE
Unlike IgG, which is subject to the inhibitory receptor FcγRIIb, IgE antibodies lack inhibitory Fc receptors. While the Fc receptor-mediated functions of IgG are modulated in the tumor microenvironment by FcγRIIb, lack of IgE inhibitory Fc receptors suggests that IgE antibodies may escape, to some degree, the suppressive effects of the tumor microenvironment.32,39
Tissue residency of IgE antibodies
The differential serum and tissue half-lives of IgE antibodies compared with their IgG counterparts are also likely to confer an advantage to IgE in the treatment of solid tumors. Despite a very short serum half-life of 1.5 d30,83 the half-life of human IgE in tissues is ~2 weeks, which is significantly longer than the 2–3 d tissue half-life of IgG antibodies.31,32 The result is local retention of antibody by IgE receptor-expressing resident immune effector cells, and longer immune surveillance, both of which could be advantageous in the context of cancer. In fact, it is well-established that solid tumors are associated with inflammatory responses that result in infiltration of powerful FcεR-expressing effector cells such as monocytes/macrophages, mast cells, DCs and eosinophils.85 In the absence of tumor antigen-specific IgE antibodies, and as a result of the immunosuppressive tumor microenvironment, these cells may lack the required activity to target tumor cells; however, it is plausible that treatment with a tumor antigen-specific IgE, tightly retained on FcεR-expressing effector cells, would be sufficient to overcome immune suppression and to stimulate these cells against tumors.
Powerful Fc-mediated effector functions of IgE
Potent mechanisms of cell killing are promoted by the binding of IgE to its receptors (FcεRI and CD23) (Fig. 1): (1) degranulation of mast cells and basophils with release of pro-inflammatory mediators and cytokines, (2) ADCC induced by the release of signaling molecules (e.g., nitric oxide), enzymes (e.g., proteases), and cytokines (e.g., tumor necrosis factor) resulting in target cell lysis, and (3) antibody-dependent cell-mediated phagocytosis (ADCP) induced by the activation of macrophages and monocytes, which are normally abundant in tumors. It is suggested, therefore, that these properties, commonly described in allergy and protection from parasitic infections, could be redirected to trigger cytotoxicity and phagocytosis of tumor cells, as well as to initiate IgE antibody-dependent antigen presentation by FcεR-bearing APCs such as DCs, B cells and macrophages. Thus, a combination of passive and active immunity against solid tumors could act in conjunction in tissues naturally populated by IgE effector cells. Furthermore, the combined strength of IgE-mediated immune responses in tissues also carries the expectation of increased potency, as well as longevity of immune surveillance by IgE and effector cells against solid tumors.

Figure 1. Mechanisms of IgE immunotherapy-induced tumor cell killing. (A) Engineered IgE antibodies recognizing tumor-associated antigens can activate IgE receptor-expressing immune effector cells in the tumor microenvironment to mediate tumor cell death by a number of mechanisms (B‒C). (B) Tumor antigen-specific IgE antibodies can mediate effective antibody-dependent tumor cell killing by immune effector cells (e.g., monocytes/macrophages) through activation of FcεR signaling cascades through mechanisms such as cytotoxicity (ADCC) or phagocytosis (ADCP). C) IgE can trigger degranulation upon cross-linking of FcεRI on the surface of effector cells in the presence of antigen-expressing cancer cells. (D) Additionally, similarly to IgG antibodies, tumor antigen-specific IgE antibodies can directly arrest tumor cell proliferation or induce tumor cell death without the aid of effector cells.
Activation of antigen presenting cells to stimulate adaptive immune responses
Antigen presenting cells (APC) such as B lymphocytes, monocytes/macrophages, Langerhans cells and DCs in tumor infiltrates express CD23 or FcεRI. It has been proposed that one of the immune escape mechanisms of tumor cells is the pathway inducing defective differentiation and maturation of APC, thereby reducing adaptive immune responses against tumor antigens. Engagement of IgE with FcεRI on the surface of DCs increases the efficiency of antigen uptake and presentation by 100- to 1,000-fold.86 This may be significant in the context of IgE therapy against tumors, since even small amounts of IgE on the surface of DCs may be sufficient to lead to an efficient activation of autologous T cells and thus provoke powerful anti-tumoral adaptive immunity.25,87-92
When IgE binds to either FcεRI or CD23 on APC, it persists for extended periods. The IgE-FcεRI complex has a half-life of 16 h on cells in suspension (e.g., on circulating APCs), but 2 weeks in tissues.32 This makes the IgE-receptor complex an anticipatory receptor that is particularly effective in surveillance for antigen. When the antigen is internalized and broken into short peptides in the endoplasmic reticulum, several different peptides may be transported to the surface and presented to T cells. Some may contain an epitope different from the one that the capture IgE recognized, which leads to epitope spreading. The production of antibodies that recognize new epitopes on the antigen enhances the effector functions of IgE.39 This is true whether the APC is a macrophage expressing FcεRI or a B cell expressing CD23 and could constitute an additional advantage of IgE therapies.
Therefore, it is proposed, through one or more of their above-mentioned attributes, antibodies of the IgE class directed against tumor cells may provide a novel strategy to increase antibody-mediated tumor cell killing of solid tumors (Fig. 1). While much recent effort has centered on engineering therapeutic antibodies to improve IgG Fc-mediated effector functions, e.g., by increasing affinities to Fc gamma receptors,12-14,93 a few research groups have focused on enhancing antibody-mediated tumor cell killing by exchanging the Fc region of IgG for that of IgE with the aim of exploiting an antibody that binds with exceptionally high affinity for unique Fc receptors to activate monocytes/macrophages, which are known to reside in solid tumors. This concept has also been used in the field of virology, whereby a recombinant CD4-IgE molecule, designed to provide a monoclonal IgE antibody with specificity for the gp120 of human immunodeficiency virus (HIV) has been used to engage type I hypersensitivity effector cells to eliminate HIV-infected cells in vitro.94
IgE receptor expressing immune cells in solid tumor infiltrates
The tumor microenvironment is composed of stromal cells and cells of the immune system, including powerful FcεR-expressing effector cells, which together provide a supportive niche promoting the growth and invasion of tumors. As a result of the immunosuppressive tumor microenvironment, these tumor-resident or infiltrating immune cells are thought to lack the ability to mount an effective anti-tumor immune response. One of the tenets of AllergoOncology, as mentioned above, is that treatment with a tumor antigen-specific IgE antibody, tightly retained on FcεR-expressing effector cells, would overcome immune suppression and stimulate these cells against tumors. The critical infiltrating FcεR-expressing effector cells include:
Mast cells
Mast cells are among the first immune cells to infiltrate the tumor microenvironment, contributing to tumor progression in many aggressive types of cancer such as malignant melanoma and Hodgkin lymphoma.95,96 The early accumulation of mast cells in the tumor microenvironment reflects their role as tissue-resident “sentinel” cells poised to induce a rapid response to pathogens and immunogenic antigens.97 Because the presence of mast cells in many tumors has been associated with poor prognosis,98,99 it has been suggested that they themselves contribute to an immunosuppressive tumor microenvironment, and thereby impede the development of protective anti-tumor immunity. In contrast, in some cancers, e.g., breast cancer, high mast cell density has been associated with favorable prognoses.100 Indeed IHC images of mast cells degranulating near dying tumor cells have suggested a cytotoxic effect of mast cells on breast cancer cells.100
Mast cell infiltration into the tumor microenvironment can thus be correlated with either favorable or unfavorable prognostic significance in human cancers, depending on the type of cancer. Nevertheless, a wealth of evidence from human cancers and mouse models of cancer indicates that mast cells contribute to tumor invasion and angiogenesis, both vital processes for the macroscopic expansion of tumors.101-103 In addition, there is some evidence to suggest that mast cells suppress the development of protective anti-tumor immune responses, in part, by promoting T regulatory cell (T reg) suppression in the tumor microenvironment and also by conditioning DCs to mediate tolerance by controlling DC migration, longevity, and ability to present antigen in an immunogenic fashion.104
There is some evidence, however, that these pro-tumoral activities of mast cells may be subverted by targeting these cells to promote tumor destruction. For example, in a mouse allograft model, triggering of degranulation of mast cells by IgE antibody cross-linking of cell surface FcεRI, resulted in T reg impairment and acute CD4+ and CD8+ T cell-mediated tissue destruction.105 Such acute T cell-mediated destruction is an important goal of immune therapy for cancer, and, therefore, it can be seen that the use of therapeutic anti-tumor antibodies of the IgE class represents one promising approach to elicit mast cell targeting of tumors.
Eosinophils
Another critical tumor-infiltrating FcεR-expressing effector cell type is the eosinophil, which is observed in the peri-tumoral infiltrate of several types of cancer, a phenomenon known as tumor-associated tissue eosinophilia (TATE).106 While eosinophils are traditionally referred to as effector cells in allergic diseases and parasitic infections, the cytotoxic potential of these cells toward tumor cells has only recently been demonstrated in experimental models and in humans. Accordingly, TATE has been suggested to represent a positive prognostic indicator in particular tumors, including colorectal carcinoma, oral and esophageal squamous cell carcinoma, laryngeal carcinoma, pulmonary adenocarcinoma, bladder carcinoma and gastric carcinoma.107-110 As mentioned above, however, TATE has also been associated with poor prognosis in Hodgkin lymphoma.111
Although TATE is often associated with a favorable prognosis, little is known about the exact role of eosinophils in the anti-tumor immune response. Reports of immunotherapeutic approaches and recent in vitro and in vivo studies, however, suggest that eosinophils are involved in tumoricidal activity.112-114 Degranulated eosinophils have been detected in tumors following systemic administration of IL-2, suggesting that they may play an effective role in the anti-tumor response either by inducing direct tumor cell lysis through release of their toxic granules, by antibody-dependent mechanisms of eosinophil activation, or by their immuno-regulation of the tumor microenvironment.112 However, the link between immunotherapeutic anti-tumor efficacy and eosinophilia is mainly based on correlation analyses, and no conclusions can be drawn regarding the actual mechanisms of action of eosinophils in the modulation of tumor growth.
Furthermore, several in vivo studies suggest a link between tumor eradication and eosinophil recruitment. For example, eosinophil infiltration into tumors in wild-type mice has been shown to be an early and persistent response.113 In addition, both IL-4 and IL-5 transgenic mice demonstrated a significant reduction in tumor establishment and growth that correlated with a high level of eosinophil recruitment to the tumor.114,115 However, a subsequent study using the IL-4 transgenic mouse model failed to demonstrate tumoricidal activity of eosinophils, and instead attributed responsibility for tumor suppression, at least in part, to neutrophils.116 Taken together, these results indicate that a Th2-type response involving eosinophils and possibly also neutrophils, is associated with tumor eradication in several animal models. Indeed, it is possible that this response could be harnessed and improved upon in the presence of a therapeutic anti-tumor antibody of the IgE class.
Macrophages
Macrophages, major FcεR-expressing effector cells, have been observed in the tumor infiltrate of almost all tumor types. These so-called tumor-associated macrophages (TAMs) are significant for fostering tumor progression. With the exception of non-small cell lung carcinoma,117 patient prognosis in solid tumors is generally described as correlating inversely with TAM density.118 The pro-tumor properties of TAMs derive from their regulation of angiogenic programming via production of vascular endothelial growth factor A,119 their tissue remodelling abilities, their production of soluble mediators that support proliferation, survival and invasion of malignant cells, and their development of immunosuppressive microenvironments that blunt cytotoxic T cell activities.118 Recent literature recognizes macrophages as major determinants of immune suppression in solid tumors. Although the mechanisms underlying these activities are less well-characterized, TAMs typically express several genes with immunosuppressive potential,120 and are capable of directly limiting T cell proliferation in vitro.121
These different activities of TAMs are dependent on their polarization state. The description of macrophage activation as either classical (M1; interferon (IFN)γ/lipopolysaccharide (LPS)-dependent) or alternative (M2; IL-4/IL-13/IL-10- dependent) has provided a necessary framework for the understanding of TAM polarization.122 However, even though M1/M2 designations represent extreme ends of a scale, the concept is an oversimplification of the diversity of TAM phenotypes. In reality, TAMs receive signals from the particular microenvironment in which they reside, and integration of these signals results in the production of an array of TAM populations/phenotypes within a tumor, each with unique tumor-regulating properties.118
TAMs present attractive therapeutic targets, with therapeutic strategies grouped crudely into four themes: (1) blocking effector function, (2) limiting recruitment, (3) reprogramming from an immunosuppressive to an immunostimulatory phenotype, or (4) preventing polarization to a tumor-promoting subtype. It has recently been established that blocking TAM recruitment or survival in solids tumors (in murine models) improves efficacy of cytotoxic therapies, in a manner dependent upon CD8+ T cells.123 Indeed, emerging therapeutic strategies are now focusing on the repolarization of TAMs as a way of unlocking their anti-tumor potential. For example, agonist antibodies against the co-stimulatory molecule CD40 were able to activate TAMs in human pancreatic ductal adenocarcinoma to become tumoricidal and deplete tumor stroma.124 A potential alternative method to repolarize TAMs would be the use of tumor-specific IgE antibodies with the aim of redirecting the TAM-associated activatory phenotype of these cells to target tumor cells.
Antigen presenting cells
APCs such as DCs, monocytes, Langerhans cells and B cells are often resident in the tumor microenvironment, and express either or both of the IgE Fc receptors. Impaired differentiation and maturation of these cells, with resultant blunting of an anti-tumor adaptive immune response, is suggested to play a role in tumor immune escape. However, the presence of IgE bound to the surface of DCs in the tumor microenvironment may increase the efficacy of antigen uptake and presentation,86 leading to efficient local activation of T cells resulting in an effective anti-tumor adaptive immune response.
IgE, an Antibody Class Less Prone to Tumor-Induced Antibody Blockade
Recent findings provide evidence that IgG4 can impair the cytotoxic properties of IgG1 antibodies in human melanoma, suggesting that therapeutic antibodies of the IgG(1) class may be partly prevented from engaging Fcγ receptors on immune effector cells in tumors and may consequently be less effective in activating these cells to kill cancer cells, while competition for antigen recognition may play less important roles.79 This report is in agreement with evidence for the presence of IgG4 antibody+ B cells in pancreatic cancer, extrahepatic cholangiocarcinoma and squamous cell carcinoma, providing evidence that this phenomenon of IgG4 impairment of the immune response in the tumor microenvironment may not be limited to melanoma.125,126 Increased production of Th2 cytokines such as IL-4 and IL-10, has been described in the tumor microenvironment of melanoma and other cancers;127-129 and IgG4, rather than IgE antibodies have been described to be elevated in individuals chronically exposed to known allergic triggers such as bee venom and animal fur.130 Until recently, however, the combination of these conditions, (i.e., IL-10-driven Th2 inflammatory conditions and chronic exposure to antigens) in relation to their potential to redirect antibody production by B cells had not been investigated in the context of cancer.
Additional findings by our group demonstrate that only a small fraction of patient-derived melanomas tested positive for IgE antibody expression, consistent with chronic exposure to antigens (possibly tumor antigens) and the “alternative” (or IL-10-driven) rather than “classic” (or IL-4-dominant) Th2-driven inflammatory conditions in tumors, which are known to drive immunity away from IgE and in favor of IgG4.79,131 The combination of alternative Th2-biased environments in tumors and chronic antigen exposure are expected to favor class switching to and enhanced production of IgG4 rather than IgE. These reports may uphold the suggestion that perhaps a classic, IgE-biased, rather than an alternative, IgG4-biased, immunity, may tip the balance in favor of tumor clearance by effector cells.
Following allergen immunotherapy, increases in IL-10 and TGFβ-expressing T regs, followed by elevated IgG4 subclass and IgA class antibodies have been reported. These events were proportional to dosing of allergen rather than clinical symptoms. It was found, however, that the small proportion of IgG4 antibodies from treated individuals that recognize the immunizing allergen could also compete with allergen-specific IgE, and that IgG4 antibodies could also restrict IgE-mediated antigen presentation by B cells.132-136 Such mechanisms could also be in operation in cancer where the local cytokine milieu may play crucial roles in promoting IgG4 class switching and production. It is therefore possible that tumor antigen-specific IgG4 antibodies may compete with IgG1 antibodies for recognition of Fc receptors on effector cells, and, although IgE and IgG4 operate through different Fc receptors on effector cells, it is conceivable that IgG4 may also compete with IgE antibodies for target antigen engagement or for tumor antigen presentation.79,131 Further investigations are needed to elucidate the roles of endogenous IgE and IgG4 antibodies in tumors, the anti-tumoral mechanisms of recombinant IgE antibodies in the tumor microenvironment, potential interactions of therapeutic IgE with endogenous IgG4, and the effect of these mechanisms on the use of IgE as novel immunotherapeutics.
Strategies of IgE-Based Immunotherapy
The potential advantages of IgE antibodies in the treatment of solid tumors, and the distinct presence in solid tumors of all of the critical IgE receptor-expressing immune effector cells, formed the basis for several groups to design therapeutic strategies with the aim of exploiting the IgE immune response against cancer. These strategies have encompassed: (1) the utilization of IgE as an adjuvant for cancer immunotherapy,137 (2) the development of IgE-coated cellular vaccines,87,137,138 (3) the development of oral mimotope vaccination programs with the aim of inducing tumor antigen-specific antibodies,139 and (4) the development of recombinant IgE antibodies targeting tumor-associated antigens (Table 1).22-27
Table 1. Summary of therapeutic strategies aimed at harnessing the IgE immune responses against cancer.
| IgE species (route of administration) | IgE specificity | Targeted cancer cells | Mouse Model (immune status) | Reference |
|---|---|---|---|---|
| IgE as an adjuvant | ||||
| Murine (i.p.) | DNP | MC38 murine colon carcinoma cells expressing human CEA (s.c.) * | C57BL/6 (immunocompetent) | 137 |
| IgE-coated cellular vaccines | ||||
|---|---|---|---|---|
| Murine (s.c.) | TNP or DNP | MC38 murine colon carcinoma cells expressing human CEA coated with IgE (s.c.) ** | C57BL/6 (immunocompetent) | 137 |
| Murine (s.c.) | DNP | TS/A-LACK murine mammary carcinoma cells coated with DNP IgE (s.c.) *** | FcεRIα−/− / CD23−/− BALB/c or human FcεRIα transgenic BALB/c (immunocompetent) | 138 |
| Human (truncated) (s.c.) | N/A | TS/A-LACK murine mammary carcinoma cells coated with truncated IgE (s.c.) **** | Human FcεRIα transgenic BALB/c | 87 |
| Recombinant IgE antibodies targeting tumor-associated antigens | ||||
|---|---|---|---|---|
| Murine (i.p.) | Gp36 of MMTV | H2712 (MMTV-positive) murine mammary carcinoma (s.c.) | C3H/HeJ (immunocompetent) | 23 |
| Rat/human chimeric (s.c.) | Murine Ly-2 | E3 murine thymoma (s.c.) | C57BL/6 (immunocompetent) | 26 |
| Rat/human chimeric (i.p.) | Murine Ly-2 | E3 murine thymoma (i.p.) | NOD-SCID (immunocompromised) | 141 |
| Murine and murine/human chimeric (i.v.) | Colorectal cancer antigen | Human COLO 205 (s.c.) | SCID (immunocompromised) | 27 |
| Murine/human chimeric (i.v. or i.p.) | FRα | A. IGROV-1 human ovarian carcinoma cells (s.c.) B. HUA patient-derived ovarian carcinoma (i.p.) |
C.B-17 scid/scid (immunocompromised) nu/nu (immunocompromised) |
20, 21, 60, 140 |
| Human (i.p.) | HER2/neu | D2F2/E2 murine mammary carcinoma cells expressing human HER2/neu (i.p.) | Human FcεRIα transgenic BALB/c (immunocompetent) | 24 |
| Murine/human chimeric (s.c.) | MUC-1 | 4T1 tumor cells expressing MUC-1 (s.c.) | Human FcεRIα transgenic BALB/c (immunocompetent) | 25 |
| Murine/human chimeric (s.c.)***** | PSA | CT26-PSA tumor cells expressing PSA (s.c.) | Human FcεRIα transgenic BALB/c (immunocompetent) | 142 |
Tumour-targeting occurred via a biotinylated anti-CEA IgG followed by streptavidin and then a biotinylated IgE; ** Irradiated tumor cells were coated with IgE in vitro by biotin-avidin bridging; *** Tumour cells were pre-infected with modified Vaccinia virus Ankara (MVA) then conjugated with DNP and coated with anti-DNP IgE; **** Tumour cells expressed truncated human IgE on their surface by pre-infecting with truncated human IgE engineered into MVA (rMVA-tmIgE); ***** Anti-PSA antibody was complexed to PSA antigen prior to injection. Abbreviations: s.c., subcutaneous; i.p., intraperitoneal; MMTV, murine mammary tumor virus; TNP, trinitrophenol; DNP, dinitrophenol; FRα, folate receptor α; PSA, prostate specific antigen.
IgE-Fc-coated tumor cell approaches
One therapeutic strategy designed to harness the IgE immune response against cancer is the utilization of IgE-Fc-coated tumor cells, based on the hypothesis that IgE on the surface of tumor cells could improve tumor immunogenicity by the activation of immune cells within the tumor microenvironment, and that these changes might suppress tumor growth (Fig. 2).137

Figure 2. IgE may be used as an adjuvant/vaccine administered in vivo using different approaches. (A) The three-step biotin-avidin bridge strategy: Tumor cells are introduced, followed by a biotinylated tumor-specific murine IgG antibody (1), then streptavidin (2), and finally by a biotinylated non-specific murine IgE (3). (B‒D) The cellular vaccine approach: (B) Irradiated tumor cells coated with tumor antigen-specific IgE. Immunization was followed by live tumor cell challenge. (C) Modified Vaccinia virus Ankara (MVA)-infected tumor cells (1) conjugated with hapten and then coated with hapten-specific IgE antibody (2). Immunization was followed by live tumor cell challenge. (D) Tumor cells infected with recombinant MVA into which truncated human IgE Fc regions had been engineered (rMVA-tmIgE). Immunization was followed by live tumor cell challenge. All four approaches were designed to trigger an allergic-like inflammatory response through recruitment of FcεR-expressing immune effector cells and with subsequent activation of a memory (CD8+ and CD4+) T cell response.
In the first study, a three-step biotin-avidin bridge strategy was used to target IgE to the tumor (Fig. 2A). Tumor cells were coated with IgE (Fc region facing outwards), leaving IgE Fc free to bind FcεRs on immune cells in the vicinity and initiate an allergic immune response. Immunocompetent mice were injected subcutaneously (s.c.) with syngeneic CEA-2-expressing tumor cells, and two days later, mice were injected intraperitoneally (i.p.) with a biotinylated tumor-specific (anti-CEA-2) murine IgG antibody to target the tumor. The next day, mice were given avidin i.p. (used as a “chase” to clear the unbound, circulating mAb) followed by streptavidin 4 h later to create the avidin-biotin bridge with bound mAb. Finally on day 4, mice were given biotinylated anti-dinitrophenyl (DNP) murine IgE (or biotinylated IgG control antibody) i.p. IgE treatment significantly reduced tumor growth and prolonged survival compared with treatment with the corresponding IgG antibody. Two of 10 mice receiving the IgE treatment completely rejected the tumor. Additionally, these 2 mice also resisted tumor cell growth for up to 60 d after re-challenge with parental (non-CEA-2 expressing) tumor cells, suggesting the induction of a memory response beyond that of the antigen that was initially targeted. The Fc region of the IgE antibodies was shown to play a critical role in their efficacy, since heat inactivation of the biotinylated IgE abrogated the anti-cancer effects of the treatment. Depletion of eosinophils, CD8+, or CD4+ cells also abrogated the anti-tumor effects, demonstrating the requirement for these three cell types in the IgE-mediated growth inhibitory effects, suggesting that IgE-based immunotherapy may trigger innate as well as adaptive mechanisms against tumors. Additionally, these findings were confirmed in a more immunogenic tumor model using the syngeneic murine lymphoma RMA- Thy1.1 cell line under similar conditions.137
IgE-coated cellular vaccines
To further explore the properties of murine IgE as an adjuvant and to determine whether IgE could be used for the purpose of prophylactic immunisation, cellular vaccines were developed using irradiated tumor cells coated with IgE (Fig. 2B).137 Immunocompetent mice were vaccinated twice (2 weeks apart) by s.c. administration of IgE- or IgG-coated CEA-2-expressing irradiated tumor cells. Two weeks after the second immunisation, the animals were challenged with live CEA-2-expressing tumor cells. Cells coated with murine IgG molecules, only showed significant delay in tumor growth at the highest dose of vaccination; however, all doses of cells coated with IgE showed significant anti-cancer properties. These results were confirmed using a similar vaccination schedule and RMA tumor cells.137
The adjuvant effect of IgE-coated tumor cells was later confirmed using a slightly different strategy (Fig. 2C).138 TS/A-LACK murine mammary adenocarcinoma cells were first infected with the highly attenuated modified Vaccinia virus Ankara (MVA) to avoid the need for irradiation of tumor cells prior to vaccination and increasing the immunogenicity of infected tumor cells. Fresh TS/A-LACK-infected cells were then conjugated with the hapten DNP and the haptenised cells coated with murine anti-DNP IgE and used to vaccinate immunocompetent mice. Mice were vaccinated s.c. and then challenged s.c. with live TS/A-LACK tumor cells 15 d after vaccination. A strong anti-cancer effect was observed in these animals. Vaccination with haptenised tumor cells (not bearing IgE) did not induce an anti-tumor response; however, when mice were vaccinated twice with haptenised tumor cells, an anti-tumor effect was observed. This effect was similar to that of animals vaccinated once with IgE-coated tumor cells.
The same experiments were performed in either FcεRI or CD23 knockout mice.138 The anti-cancer effects of vaccination with IgE-coated tumor cells were not observed in FcεRI knockout animals, but remained evident in the CD23 knockout animals, suggesting a central role for FcεRI as a mediator of the anti-cancer effects of the vaccination, and raising the possibility that the IgE adjuvanticity could result from an inflammatory response similar to that observed in an allergic reaction, followed by antigen presentation by well-known APCs such as DCs. Finally, to translate these findings into a human system, the effect of a human IgE-loaded MVA-infected tumor cell vaccine was studied in wild type (WT) and human FcεRIα transgenic mice. Human IgE had no effect in WT mice, but induced a significant protection in the humanised transgenic mice. This result was demonstration of a successful active antitumor vaccination exerted by human IgE.138 Taken together, these studies suggest that the presence of IgE in the tumor microenvironment results in a strong anti-tumor immune activation, and thus provide significant evidence that IgE can act as an adjuvant to activate immunity in cancer therapy.
These results recently led to the development of a novel protocol for IgE-based anti-tumor vaccination that employed membrane IgE, thus eliminating the possibility of toxicities associated with free unbound IgE (Fig. 2D).87 Truncated human IgE lacking its Fab regions (tmIgE) was engineered into a recombinant MVA (rMVA-tmIgE), and used to infect TS/A-LACK tumor cells, with resultant transport of tmIgE to the surface of the infected cells. Transgenic human FcεRIα mice were vaccinated s.c. and then challenged s.c. with live TS/A-LACK tumor cells 14 d after vaccination. Human FcεRIα transgenic mice immunised with the rMVA-tmIgE/TS/A-LACK cellular vaccine showed a significant attenuation of tumor growth compared with mice immunised with the control vaccine (not expressing tmIgE). This anti-tumor protection was completely lost when the cellular vaccine was administered to FcεRIα−/− mice (in which the FcεRIα gene had been knocked out), thereby confirming the central role of FcεRI in IgE active anti-tumor immunotherapy.
Oral mimotope vaccination approaches
In addition to the cellular vaccination protocols described above, an oral mimotope vaccination protocol with the aim of inducing tumor antigen-specific IgE antibodies (Fig. 3) has also been described.139 In this approach, synthetically manufactured epitope mimics, so-called mimotopes, for the epitope recognized by the anti-human epidermal growth factor receptor-2 (HER-2) antibody trastuzumab were developed and used to immunize immunocompetent mice by the oral route under simultaneous neutralisation and suppression of gastric acid, which is a feeding regimen that has been shown to effectively induce Th2 immune responses. These conditions resulted in the formation of serum IgE antibodies toward the HER-2 antigen, and anti-HER-2 IgE-sensitized effector cells were subsequently shown to mediate HER-2-expressing tumor cell lysis in vitro in an antibody-dependent cytotoxicity assay. These experiments indicated that directed and epitope-specific induction of IgE against tumor antigens is feasible with an oral mimotope vaccination regimen, and that these antibodies mediate anti-cancer effects in vitro. It remains to be seen, however, whether this oral mimotope vaccination protocol induces anti-tumor effects in vivo.139
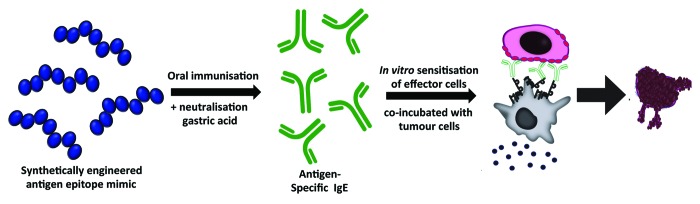
Figure 3. Oral mimotope vaccination strategy triggering tumor-reactive IgE antibody responses. Serum tumor antigen-specific IgE antibodies are induced in vivo by oral administration of a synthetically manufactured tumor antigen epitope mimic (mimotope) under simultaneous neutralisation of gastric acid. Vaccinations resulted in generation of tumor antigen-specific IgE antibodies in vivo which were capable of promoting effector cell-induced tumor cell ADCC in vitro.
Recombinant IgE antibodies targeting tumor-associated antigens
Previous pioneering studies and more recent work have collected in vitro and in vivo evidence that engineered anti-cancer IgE antibodies may be comparable or even superior to their IgG counterparts (Fig. 1).8,20,21,23-27,60,140-142
Murine anti-gp36 IgE
The first study performed with a tumor-targeted IgE antibody was conducted in the early 1990s.23 A murine IgE specific for the major envelope glycoprotein (gp36) of the mouse mammary tumor virus (MMTV) was produced and its anti-cancer efficacy evaluated in immunocompetent mice bearing syngeneic MMTV-positive s.c tumors. Simultaneous with tumor cell inoculation, mice received mouse ascites containing the anti-gp36 IgE or an equivalent amount of normal mouse serum through the i.p. route. Additionally, every 4 d for 8 weeks, the animals were treated i.p. with ascites containing the targeted IgE or normal mouse serum. All animals that received mouse serum died by day 44, but 4 of the 6 animals treated with anti-gp36 IgE were alive at that time. After the cessation of treatment (day 56), 1 mouse treated with IgE started to develop a tumor on day 58 and died on day 72. The remaining 3 animals survived past 175 d post-tumor challenge. This study also evaluated the efficacy of anti-gp36 IgE against tumors growing in the peritoneal cavity with treatment similar to the previously described experiment. All control animals died within 34 d, whereas 2 of the 6 in the IgE treatment group survived until day 139.23 These data demonstrated for the first time the ability of a tumor-targeting murine IgE antibody to improve the survival tumor-bearing mice.
Rat/human chimeric anti-murine Ly-2 IgE
The next tumor-specific IgE antibody to be developed was a rat/human chimeric IgE specific for murine Ly-2, a cell surface marker for murine CD8+ T cells that is also expressed on tumors of T cell origin.26 A strategy was employed in which the anti-Ly-2 IgE was administered together with murine cytotoxic T cells (CTLs) that had been stably co-transfected to express a chimeric “FcεRI-ζ” comprising the human FcεRIα extracellular sequence linked to the human CD3-ζ signaling sequence and human FcγRIIa. Anti-Ly-2 IgE and chimeric FcεRI-ζ CTLs were incubated with Ly-2 expressing murine E3 thymoma cells and then the mixture was injected into immunocompetent syngeneic mice. Control animals received E3 cells alone or E3 cells plus chimeric FcεRI-ζ CTLs (without anti-Ly-2 IgE). Addition of the chimeric FcεRI-ζ CTLs significantly prolonged mouse survival, but 4 out of 5 of these animals developed tumors and succumbed to the disease. The greatest effect was observed when E3 cells plus chimeric FcεRI-ζ CTLs were given with anti-Ly-2 IgE, where survival was prolonged even further and only 1 of 5 animals developed tumor.26 These data suggest that an anti-tumor IgE can be used to redirect engineered CTL to lyse tumor cells in an adoptive transfer therapy setting.
The same strategy was also used in immunodeficient animals utilizing primary human T cells retrovirally transduced with cDNA encoding the extracellular domain of FcεRI linked to the hinge and transmembrane domains of FcεRI and the cytoplasmic domains of the human co-stimulatory molecule CD28, and T cell receptor zeta chain (FcεRI-CD28-zeta) to potentiate the activation signal of the CTL.141 Mice were given Ly-2 expressing thymoma cells i.p., followed by FcεRI-CD28-zeta expressing primary human T lymphocytes (TCL-9 cells) loaded with anti-Ly-2 IgE at 6, 24, 48, and 96 h following tumor challenge. Adoptive transfer of the IgE-coated TCL-9 cells significantly prolonged survival, but complete tumor rejection and long-term survival were not observed, which was explained by the fact that the TCL-9 cells did not persist in the animal and could not be detected in the blood, spleen, or peritoneal cavity 1-week post-administration. This effect was shown to be due to the presence of the chimeric receptor since primary T cells not expressing the receptor showed no anti-tumor protection. Furthermore, tumor targeting by the anti-Ly-2 IgE was also necessary since TCL-9 cells transferred with a non-specific IgE also failed to show a protective effect. These data demonstrated that primary human T cells expressing the chimeric IgE receptor could suppress tumor growth in the presence of a tumor-targeted IgE.
Murine and murine/human chimeric anti-CCA IgE
The same group also developed a murine IgE (30.6) specific for a colorectal cancer antigen (CCA).27 Severe combined immunodeficiency (SCID) mice were injected s.c. with COLO 205 cells and subsequently treated with either the 30.6 murine IgE or the 30.6 mouse/human chimeric IgE given via intravenous (i.v.) injection 5 d after tumor challenge. The chimeric IgE had no effect on tumor growth; however, the murine IgE induced a rapid, but transient (48 h in duration) inhibition of tumor growth.27 No effect was observed against E3 thymoma cells that lack expression of the antigen targeted by 30.6 or with a non-specific murine IgE. These data demonstrated that the anti-cancer effects of murine anti-30.6 IgE were antigen specific and required the murine IgE Fc region. A mouse/human chimeric IgG1 containing the 30.6 variable region had also previously shown anti-cancer activity in this tumor model.143 The murine 30.6 IgE effect, however, was observed at antibody concentrations that were 250-fold lower than those previously reported for the effects seen using 30.6 IgG1, which is consistent with the superior anti-cancer activity of IgE. It is notable that these two antibodies were not tested simultaneously.
Murine/human chimeric anti-human FRα IgE
In 1999, Gould et al. created a murine/human chimeric MOv18 monoclonal IgE antibody specific for the ovarian tumor associated antigen, folate receptor α (FRα).21 The in vivo efficacy of MOv18 IgE was compared with its IgG1 counterpart in two disparate human xenograft models of FRα-expressing ovarian carcinoma grown in immunodeficient mice. In the first model, SCID mice were challenged s.c. with FRα-expressing human ovarian carcinoma (IGROV1) cells. Subsequently, human peripheral blood mononuclear cells (PBMC), added as effector cells, were administered i.v. in the presence of either MOv18 IgE or MOv18 IgG1.21 MOv18 IgE demonstrated a superior and prolonged anti-tumor effect. Since the human Fc region of chimeric IgE does not bind to mouse Fc receptors, MOv18 IgE did not induce an anti-tumor response in the absence of human PBMC.
Further evidence for the anti-tumor efficacy of MOv18 IgE was provided by a second model in which patient-derived FRα-expressing human ovarian carcinoma cells (HUA) were injected i.p. in nude mice in the presence of human PBMC.140 In this model, co-administration of MOv18 IgE with PBMC significantly increased survival to 40 d, whereas the survival of mice treated with PBMC and MOv18 IgG1 was only 22 d. IHC analysis of tumors from MOv18 IgE-treated mice revealed infiltration of human monocytes into tumor lesions, reinforcing the central role played by these effector cells in the anti-tumor efficacy of tumor-specific IgE antibodies.140 Furthermore, the use of monocyte-depleted PBMC in vivo resulted in loss of the survival advantage conferred by MOv18 IgE.60 Flow cytometric analysis revealed two distinct pathways by which human monocytes mediated MOv18 IgE-dependent tumor cell killing: ADCC via FcεRI and ADCP via CD23, both expressed on the surface of IL-4-activated monocytes.20,60 The involvement of human eosinophils as potent effector cells in MOv18 IgE mAb-dependent cytotoxicity in vitro has also been shown.60
Humanized and human anti-HER2/neu IgE
Further evidence for IgE-mediated tumor cell killing by monocytes was demonstrated by Karagiannis et al. who engineered an IgE antibody equivalent of the approved humanized IgG1 mAb trastuzumab, specific for HER2/neu, and evaluated the biological properties of this antibody (“trastuzumab IgE”) using in vitro functional assays.8 Trastuzumab IgE directed monocytic cells to kill HER2/neu-positive tumor cells by ADCC (compared with ADCP by trastuzumab IgG1), while also maintaining the same direct effects on tumor growth arrest reported for trastuzumab IgG1. These studies supported the conclusion that trastuzumab IgE functioned with similar potency, but through different effector cell mechanisms compared with the IgG1 equivalent, supporting a potential role for the development of IgE antibodies to complement or enhance the known mechanisms of existing therapeutic mAbs.
A fully human IgE antibody specific for HER2/neu has also been constructed and administered i.p to HER2/neu-positive i.p. tumor-bearing human FcεRIα transgenic mice 2 and 4 d post tumor inoculation. Anti-HER2/neu IgE significantly prolonged survival of the mice compared with those treated with vehicle control alone.24 In addition, this antibody was the first (and only) therapeutic IgE antibody to be administered to non-human primates (cynomolgus monkeys), albeit at considerably lower doses (up to 0.08 mg/kg). At these doses the anti-HER2/neu IgE was well-tolerated.
Murine/human chimeric anti-human MUC-1 IgE and anti-human CD20
A mouse-human chimeric IgE antibody specific for the epithelial antigen MUC-1 has also been engineered.25 Human FcεRIα transgenic mice were injected s.c. with MUC-1-expressing 4T1 tumor cells, followed by peri-tumoral injection of anti-MUC-1 IgE on days 1, 2, 3, 4 and 5. Modest inhibition of tumor growth was observed, and this was attributed to an inability to deliver adequate amounts of antibody to a poorly vascularized and rapidly growing s.c. tumor, and to a lack of sufficient effector cells in the tumor microenvironment of the transplanted tumors. Therefore, Teo and colleagues created two tumor cell lines that produced anti-hMUC1 mouse IgE or chemoattractant cytokines MCP-1 and IL-5. These cells were mixed and inoculated s.c. in human FcεRIα transgenic mice. Tumors that expressed anti-hMUC1 mouse IgE and both cytokines failed to grow, while tumors expressing the cytokines without the anti-hMUC-1 antibody showed no growth inhibition, thus suggesting that when sufficient amounts of tumor-specific IgE are delivered to an antigen-bearing tumor in the presence of IgE receptor-expressing effector cells, a complete and durable response can be observed.25
The same group also produced a murine/human chimeric IgE antibody specific for the human B cell antigen CD20.25 The anti-hCD20 IgE was capable of inducing ADCC of CD20-expressing lymphoma B cells in vitro, by umbilical cord blood-purified mast cells and basophils. However, the in vivo activity of anti-hCD20 IgE was not evaluated due to concern that significant levels of circulating CD20 antigen in a physiological model of lymphoma would lead to a high risk of anaphylaxis upon IgE-treatment.
Human anti-EGFR IgE
A human monoclonal IgE antibody has also been engineered to target the human epidermal growth factor receptor (EGFR).144 This human anti-EGFR IgE was compared in vitro to two EGFR-targeting IgG antibodies, namely cetuximab and matuximab. Proliferation and cytotoxicity assays demonstrated both signal blocking and tumor cell-killing capabilities of the human anti-EGFR IgE. Interestingly however, while the degree of ADCP of tumor cells was similar between the anti-EGFR IgE and IgG antibodies, ADCC by anti-EGFR IgE increased up to 70%, compared with 30% by its IgG counterpart.144
Mouse/human chimeric anti-PSA IgE
Most recently, a mouse/human chimeric monoclonal IgE antibody raised against prostate-specific antigen (PSA), a prostate cancer-specific tumor associated antigen, has been engineered.142 The murine IgG1 equivalent mAb AR47.47, specific for human PSA, has previously been shown to enhance antigen presentation by human DCs, and induce both CD4+ and CD8+ T cell activation when complexed with PSA.145 In this study, the anti-PSA IgE, complexed with the PSA antigen, was capable of enhancing antigen presentation by human DCs in vitro, inducing CD4+ and CD8+ T cell activation. In vivo, using a prophylactic vaccination strategy in human FcεRIα-transgenic mice, anti-PSA IgE complexed with the PSA antigen was capable of triggering immune activation and significantly prolonging survival of mice challenged with PSA-expressing tumors. This protection, however, was not observed with vaccination with the IgG1 counterpart complexed to PSA, or with PSA alone.142
Safety of IgE Immunotherapy
In spite of the numerous studies and the accumulating evidence that IgE immunotherapy could have a beneficial role in oncology, studies with tumor-specific IgE antibodies have so far not gone beyond preclinical proof-of-concept evaluations. This is partly due to concerns that the systemic use of an IgE molecule targeting a self-protein for therapeutic purposes may potentially induce a systemic type I hypersensitivity (anaphylactic) reaction, through cross-linking of IgE molecules bound to FcεRI on circulating basophils. Notably, polyclonal and monoclonal IgE have previously been administered to man in the context of two early studies investigating the metabolism of IgE.146,147 In these studies, IgE purified from the serum of patients with either hyperimmunoglobulin E-recurrent infection syndrome146 or multiple myeloma147 was radiolabelled with 125I and administered i.v. to healthy volunteers at a maximum dose of 12 µg. No adverse events were observed in this study following administration of IgE. Furthermore, no toxicities were reported during the previously-described in vivo studies of recombinant IgE antibodies targeting tumor-associated antigens, in which the Fc region of the therapeutic antibody was capable of cross-reacting with the animal’s native Fcε receptors, or with co-administered PBMC.8,20,21,23-27,60,140-142
Finally, a thorough in vitro study that examined the propensity of a tumor antigen-specific IgE to induce early events that could lead to Type I hypersensitivity was recently conducted.148 Upon addition of MOv18 IgE to human sera or to whole human blood of patients or healthy volunteers, neither functional degranulation nor significant activation of human basophils were observed. No effector cell activation was recorded even in the presence of detectable levels of shed soluble target antigen FRα, which was shown to be elevated in a larger proportion of ovarian carcinoma patient sera compared with healthy controls.148 This study was replicated using a mouse/human chimeric monoclonal IgE antibody raised against PSA.142 The engineered anti-PSA IgE antibody was capable of triggering effector cell degranulation in vitro and in vivo when artificially cross-linked, but not in the presence of the natural soluble antigen, thereby suggesting that systemic administration of the antibody would be unlikely to trigger anaphylaxis. However, a thorough evaluation of toxicity in vivo is yet be conducted.
Translation of IgE Immunotherapies and Future Perspectives
IgE-based anti-tumor immunotherapies, either active or passive, could constitute potentially advantageous strategies for cancer therapy when given either as monotherapies or in combination with conventional or biological therapeutics. This timely concept merits further study in light of increasing appreciation of the importance of recruiting immune effector cells in tumors, and of triggering the anti-tumoral functions of recruited and tumor-resident immune cells against cancer.149,150 Effective recruitment and activation of immune sentinels may hold the key to a successful immunotherapy in oncology, and IgE immunotherapy may be a strategy to achieve this.
The design of IgE-based anti-tumor immunotherapies, however, is only one facet of the multidisciplinary field of AllergoOncology. This emerging field also includes a wide range of research themes evaluating: (1) evidence of a correlation between IgE/atopy and cancer,81,151 () the critical IgE effector cells in cancer, and their roles in inhibiting or promoting tumor growth,152,153 (3) IgE-mediated antigen uptake and cross-presentation by DCs and the mechanisms by which IgE-antigen complexes trigger activation of adaptive or immunomodulatory responses,154 (4) the role of chemotherapeutics and biological therapeutics in the modulation of IgE responses in cancer,155 (5) evidence linking tumor cell upregulation of ligands for the CD8+ T cell and natural killer cell activating receptor NKG2D, with the induction of a systemic Th2 atopic response,19,156 and (6) redirection of Th2 responses toward inflammatory antibodies such as IgG4.79
The last mentioned theme incorporates another important unknown in the field, whether IgE antibodies against tumor antigens are produced in healthy individuals or in patients, and the nature of the immunomodulatory mechanisms that govern the inefficiency of the immune response from mounting an “allergic” or IgE-mediated response against tumors in patients with cancer. Understanding the nature of the humoral immunity to cancer would provide improved understanding of the mechanisms governing consistently superior efficacy of IgE antibodies using disparate immunotherapy approaches, and may lead to the rational design of targeted treatments, including therapeutic antibodies and potentially combinatorial approaches.
Considerable efforts have so far focused on designing IgE-based anti-tumor immunotherapies that wield the powerful immune effector mechanisms of IgE against cancer, while predicting and minimizing potential toxicities. Specifically, the careful selection of a tumor antigen-specific IgE antibody against (1) single epitopes on tumor antigens, (2) antigenic targets that are not shed as multimeric complexes in the circulation, and (3) antigens highly expressed on tumor cells but with absent or minimal expression and restricted distribution in normal tissues, are factors of critical importance. A critical part of the clinical development of IgE immunotherapies is the production and purification of clinical-grade material at sufficient quantities and of high purity for first-in-human administration. Manufacturing studies should focus on deriving robust medium-to-large scale production and purification to yield highly pure, biologically-active agent, possibly informed by the vast experience with IgG1 class therapeutics, but tailored to the IgE class. These processes should ideally be applicable across the development of many future therapeutics.
Downstream of the pre-clinical evaluations of IgE-based anti-tumor immunotherapies conducted to date and described above, clinical testing of such anti-tumor immunotherapies will represent a crucial milestone for the concept, and will provide valuable scientific insights that stand to advance our understanding of IgE biology, particularly, but not exclusively, in the context of cancer therapeutics. Taking into account the inherent risk of a first-in-class, first-in-human study of these novel and potentially immunomodulatory molecules, a number of measures will be carefully planned to ensure the safety of the patients within these clinical trials. It is expected that clinical trials will be conducted alongside carefully planned metrics and monitoring of patients to inform on safety and efficacy. These could include monitoring for: (1) clinical signs of acute systemic type I hypersensitivity reactions, including evidence of urticaria, (2) serum β-tryptase elevations, (3) serum autoantibodies to the targeted antigen, and (4) serum concentrations of the targeted antigen, in addition to routine monitoring for renal/hepatic/hematological toxicities. The relevance of monitoring for serum autoantibodies and correlation with clinical readouts is that anti-drug antibodies may neutralize and clear the antibody from the circulation, preventing it from reaching tumor sites and reducing efficacy. In the context of a monoclonal IgE antibody, the development of anti-drug antibodies may be less likely because IgE antibodies have a short serum half-life and are therefore rapidly cleared from the circulation.39 In addition, it is likely that any clinical trial protocols will include carefully considered guidelines with respect to the clinical management of emergent systemic reactions upon administration of the investigational IgE-based product in patients.
Increasing evidence is now emerging in support of the potential efficacy of antibodies engineered with human IgE Fc for the treatment of solid tumors that may be superior to that of the corresponding IgG class antibodies. While the mechanisms that drive superior effector cell activation in the context of these therapeutics are being elucidated, numerous IgE-based immunotherapeutic approaches examined using different model systems for a range of tumor indications, support the rationale of translating this concept toward clinical application.
Acknowledgments
The authors acknowledge support from Cancer Research UK (C30122/A11527); KCL Experimental Cancer Medicine Centre, jointly funded by Cancer Research UK, the National Institute for Health Research, Welsh Assembly Government, HSC R&D Office for Northern Ireland and Chief Scientist Office, Scotland; CR UK/EPSRC/MRC/NIHR KCL/UCL Comprehensive Cancer Imaging Centre (C1519/A10331); The research was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Glossary
Abbreviations:
- ADCC
antibody-dependent cell-mediated cytotoxicity
- NO
nitric oxide
- ADCP
antibody-dependent cell-mediated phagocytosis
- APCs
antigen-presenting cells
- CSR
class-switch recombination
- CTL
cytotoxic T cell
- DCs
dendritic cells
- DNP
dinitrophenyl
- EGFR
human epidermal growth factor receptor
- FRα
folate receptor α
- HER2
human epidermal growth factor receptor-2
- IHC
immunohistochemical
- IL-4
interleukin 4
- IFNγ
interferon γ
- Ka
association constant
- LPS
lipopolysaccharide
- mAbs
monoclonal antibodies
- MHC
major histocompatibility complex
- MMCP-1
mouse mast cell protease-1
- PBMC
human peripheral blood mononuclear cells
- PSA
prostate-specific antigen
- SCID
severe combined immunodeficiency
- TAM
tumor-associated macrophage
- TATE
tumor-associated tissue eosinophilia
- Th2
T helper type 2
- T reg
T regulatory cell
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/27029
References
- 1.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–27. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reichert JM. Antibody-based therapeutics to watch in 2011. MAbs. 2011;3:76–99. doi: 10.4161/mabs.3.1.13895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott AM, Allison JP, Wolchok JD. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012;12:14. [PMC free article] [PubMed] [Google Scholar]
- 4.Reichert JM, Dhimolea E. The future of antibodies as cancer drugs. Drug Discov Today. 2012;17:954–63. doi: 10.1016/j.drudis.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Woof JM. Insights from Fc receptor biology: a route to improved antibody reagents. MAbs. 2012;4:291–3. doi: 10.4161/mabs.20100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs. 2010;2:181–9. doi: 10.4161/mabs.2.2.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nahta R, Esteva FJ. Trastuzumab: triumphs and tribulations. Oncogene. 2007;26:3637–43. doi: 10.1038/sj.onc.1210379. [DOI] [PubMed] [Google Scholar]
- 8.Karagiannis P, Singer J, Hunt J, Gan SK, Rudman SM, Mechtcheriakova D, Knittelfelder R, Daniels TR, Hobson PS, Beavil AJ, et al. Characterisation of an engineered trastuzumab IgE antibody and effector cell mechanisms targeting HER2/neu-positive tumour cells. Cancer Immunol Immunother. 2009;58:915–30. doi: 10.1007/s00262-008-0607-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A. 2006;103:4005–10. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 11.Lipson EJ, Drake CG. Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clin Cancer Res. 2011;17:6958–62. doi: 10.1158/1078-0432.CCR-11-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griggs J, Zinkewich-Peotti K. The state of the art: immune-mediated mechanisms of monoclonal antibodies in cancer therapy. Br J Cancer. 2009;101:1807–12. doi: 10.1038/sj.bjc.6605349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chames P, Baty D. Bispecific antibodies for cancer therapy: the light at the end of the tunnel? MAbs. 2009;1:539–47. doi: 10.4161/mabs.1.6.10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov. 2009;8:226–34. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- 15.Brüggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis R, Waldmann H, Neuberger MS. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med. 1987;166:1351–61. doi: 10.1084/jem.166.5.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hale G, Clark M, Waldmann H. Therapeutic potential of rat monoclonal antibodies: isotype specificity of antibody-dependent cell-mediated cytotoxicity with human lymphocytes. J Immunol. 1985;134:3056–61. [PubMed] [Google Scholar]
- 17.Steplewski Z, Sun LK, Shearman CW, Ghrayeb J, Daddona P, Koprowski H. Biological activity of human-mouse IgG1, IgG2, IgG3, and IgG4 chimeric monoclonal antibodies with antitumor specificity. Proc Natl Acad Sci U S A. 1988;85:4852–6. doi: 10.1073/pnas.85.13.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jensen-Jarolim E, Achatz G, Turner MC, Karagiannis S, Legrand F, Capron M, Penichet ML, Rodríguez JA, Siccardi AG, Vangelista L, et al. AllergoOncology: the role of IgE-mediated allergy in cancer. Allergy. 2008;63:1255–66. doi: 10.1111/j.1398-9995.2008.01768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen-Jarolim E, Pawelec G. The nascent field of AllergoOncology. Cancer Immunol Immunother. 2012;61:1355–7. doi: 10.1007/s00262-012-1315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karagiannis SN, Bracher MG, Beavil RL, Beavil AJ, Hunt J, McCloskey N, Thompson RG, East N, Burke F, Sutton BJ, et al. Role of IgE receptors in IgE antibody-dependent cytotoxicity and phagocytosis of ovarian tumor cells by human monocytic cells. Cancer Immunol Immunother. 2008;57:247–63. doi: 10.1007/s00262-007-0371-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gould HJ, Mackay GA, Karagiannis SN, O’Toole CM, Marsh PJ, Daniel BE, Coney LR, Zurawski VR, Jr., Joseph M, Capron M, et al. Comparison of IgE and IgG antibody-dependent cytotoxicity in vitro and in a SCID mouse xenograft model of ovarian carcinoma. Eur J Immunol. 1999;29:3527–37. doi: 10.1002/(SICI)1521-4141(199911)29:11<3527::AID-IMMU3527>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 22.Karagiannis SN, Josephs DH, Karagiannis P, Gilbert AE, Saul L, Rudman SM, Dodev T, Koers A, Blower PJ, Corrigan C, et al. Recombinant IgE antibodies for passive immunotherapy of solid tumours: from concept towards clinical application. Cancer Immunol Immunother. 2012;61:1547–64. doi: 10.1007/s00262-011-1162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagy E, Berczi I, Sehon AH. Growth inhibition of murine mammary carcinoma by monoclonal IgE antibodies specific for the mammary tumor virus. Cancer Immunol Immunother. 1991;34:63–9. doi: 10.1007/BF01741326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daniels TR, Leuchter RK, Quintero R, Helguera G, Rodríguez JA, Martínez-Maza O, Schultes BC, Nicodemus CF, Penichet ML. Targeting HER2/neu with a fully human IgE to harness the allergic reaction against cancer cells. Cancer Immunol Immunother. 2012;61:991–1003. doi: 10.1007/s00262-011-1150-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teo PZ, Utz PJ, Mollick JA. Using the allergic immune system to target cancer: activity of IgE antibodies specific for human CD20 and MUC1. Cancer Immunol Immunother. 2012;61:2295–309. doi: 10.1007/s00262-012-1299-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kershaw MH, Darcy PK, Trapani JA, Smyth MJ. The use of chimeric human Fc(epsilon) receptor I to redirect cytotoxic T lymphocytes to tumors. J Leukoc Biol. 1996;60:721–8. doi: 10.1002/jlb.60.6.721. [DOI] [PubMed] [Google Scholar]
- 27.Kershaw MH, Darcy PK, Trapani JA, MacGregor D, Smyth MJ. Tumor-specific IgE-mediated inhibition of human colorectal carcinoma xenograft growth. Oncol Res. 1998;10:133–42. [PubMed] [Google Scholar]
- 28.Waldmann TA, Strober W. Metabolism of immunoglobulins. Prog Allergy. 1969;13:1–110. doi: 10.1159/000385919. [DOI] [PubMed] [Google Scholar]
- 29.Waldmann TA, Strober W, Blaese RM, Terry WD. Immunoglobulin metabolism in disease. Birth Defects Orig Artic Ser. 1975;11:87–94. [PubMed] [Google Scholar]
- 30.Geha RS, Jabara HH, Brodeur SR. The regulation of immunoglobulin E class-switch recombination. Nat Rev Immunol. 2003;3:721–32. doi: 10.1038/nri1181. [DOI] [PubMed] [Google Scholar]
- 31.Hellman L. Regulation of IgE homeostasis, and the identification of potential targets for therapeutic intervention. Biomed Pharmacother. 2007;61:34–49. doi: 10.1016/j.biopha.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 32.Gould HJ, Sutton BJ, Beavil AJ, Beavil RL, McCloskey N, Coker HA, Fear D, Smurthwaite L. The biology of IGE and the basis of allergic disease. Annu Rev Immunol. 2003;21:579–628. doi: 10.1146/annurev.immunol.21.120601.141103. [DOI] [PubMed] [Google Scholar]
- 33.Arnold JN, Radcliffe CM, Wormald MR, Royle L, Harvey DJ, Crispin M, Dwek RA, Sim RB, Rudd PM. The glycosylation of human serum IgD and IgE and the accessibility of identified oligomannose structures for interaction with mannan-binding lectin. J Immunol. 2004;173:6831–40. doi: 10.4049/jimmunol.173.11.6831. [DOI] [PubMed] [Google Scholar]
- 34.Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, Murphy BA, Satinover SM, Hosen J, Mauro D, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. 2008;358:1109–17. doi: 10.1056/NEJMoa074943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinet JP. The high-affinity IgE receptor (Fc epsilon RI): from physiology to pathology. Annu Rev Immunol. 1999;17:931–72. doi: 10.1146/annurev.immunol.17.1.931. [DOI] [PubMed] [Google Scholar]
- 36.Liu FT, Hsu DK, Zuberi RI, Hill PN, Shenhav A, Kuwabara I, Chen SS. Modulation of functional properties of galectin-3 by monoclonal antibodies binding to the non-lectin domains. Biochemistry. 1996;35:6073–9. doi: 10.1021/bi952716q. [DOI] [PubMed] [Google Scholar]
- 37.Kraft S, Kinet JP. New developments in FcepsilonRI regulation, function and inhibition. Nat Rev Immunol. 2007;7:365–78. doi: 10.1038/nri2072. [DOI] [PubMed] [Google Scholar]
- 38.Dombrowicz D, Quatannens B, Papin JP, Capron A, Capron M. Expression of a functional Fc epsilon RI on rat eosinophils and macrophages. J Immunol. 2000;165:1266–71. doi: 10.4049/jimmunol.165.3.1266. [DOI] [PubMed] [Google Scholar]
- 39.Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol. 2008;8:205–17. doi: 10.1038/nri2273. [DOI] [PubMed] [Google Scholar]
- 40.Dullaers M, De Bruyne R, Ramadani F, Gould HJ, Gevaert P, Lambrecht BN. The who, where, and when of IgE in allergic airway disease. J Allergy Clin Immunol. 2012;129:635–45. doi: 10.1016/j.jaci.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 41.Acharya M, Borland G, Edkins AL, Maclellan LM, Matheson J, Ozanne BW, Cushley W. CD23/FcεRII: molecular multi-tasking. Clin Exp Immunol. 2010;162:12–23. doi: 10.1111/j.1365-2249.2010.04210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yokota A, Yukawa K, Yamamoto A, Sugiyama K, Suemura M, Tashiro Y, Kishimoto T, Kikutani H. Two forms of the low-affinity Fc receptor for IgE differentially mediate endocytosis and phagocytosis: identification of the critical cytoplasmic domains. Proc Natl Acad Sci U S A. 1992;89:5030–4. doi: 10.1073/pnas.89.11.5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong HL, Lotze MT, Wahl LM, Wahl SM. Administration of recombinant IL-4 to humans regulates gene expression, phenotype, and function in circulating monocytes. J Immunol. 1992;148:2118–25. [PubMed] [Google Scholar]
- 44.Cooper AM, Hobson PS, Jutton MR, Kao MW, Drung B, Schmidt B, Fear DJ, Beavil AJ, McDonnell JM, Sutton BJ, et al. Soluble CD23 controls IgE synthesis and homeostasis in human B cells. J Immunol. 2012;188:3199–207. doi: 10.4049/jimmunol.1102689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saegusa J, Hsu DK, Chen HY, Yu L, Fermin A, Fung MA, Liu FT. Galectin-3 is critical for the development of the allergic inflammatory response in a mouse model of atopic dermatitis. Am J Pathol. 2009;174:922–31. doi: 10.2353/ajpath.2009.080500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niki T, Tsutsui S, Hirose S, Aradono S, Sugimoto Y, Takeshita K, Nishi N, Hirashima M. Galectin-9 is a high affinity IgE-binding lectin with anti-allergic effect by blocking IgE-antigen complex formation. J Biol Chem. 2009;284:32344–52. doi: 10.1074/jbc.M109.035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu FT. Regulatory roles of galectins in the immune response. Int Arch Allergy Immunol. 2005;136:385–400. doi: 10.1159/000084545. [DOI] [PubMed] [Google Scholar]
- 48.Frigeri LG, Liu FT. Surface expression of functional IgE binding protein, an endogenous lectin, on mast cells and macrophages. J Immunol. 1992;148:861–7. [PubMed] [Google Scholar]
- 49.Zuberi RI, Hsu DK, Kalayci O, Chen HY, Sheldon HK, Yu L, Apgar JR, Kawakami T, Lilly CM, Liu FT. Critical role for galectin-3 in airway inflammation and bronchial hyperresponsiveness in a murine model of asthma. Am J Pathol. 2004;165:2045–53. doi: 10.1016/S0002-9440(10)63255-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahmad N, Gabius HJ, André S, Kaltner H, Sabesan S, Roy R, Liu B, Macaluso F, Brewer CF. Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J Biol Chem. 2004;279:10841–7. doi: 10.1074/jbc.M312834200. [DOI] [PubMed] [Google Scholar]
- 51.van den Brûle F, Califice S, Castronovo V. Expression of galectins in cancer: a critical review. Glycoconj J. 2004;19:537–42. doi: 10.1023/B:GLYC.0000014083.48508.6a. [DOI] [PubMed] [Google Scholar]
- 52.Gounni AS, Lamkhioued B, Ochiai K, Tanaka Y, Delaporte E, Capron A, Kinet JP, Capron M. High-affinity IgE receptor on eosinophils is involved in defence against parasites. Nature. 1994;367:183–6. doi: 10.1038/367183a0. [DOI] [PubMed] [Google Scholar]
- 53.Capron M, Capron A. Immunoglobulin E and effector cells in schistosomiasis. Science. 1994;264:1876–7. doi: 10.1126/science.8009216. [DOI] [PubMed] [Google Scholar]
- 54.Erazo A, Kutchukhidze N, Leung M, Christ AP, Urban JF, Jr., Curotto de Lafaille MA, Lafaille JJ. Unique maturation program of the IgE response in vivo. Immunity. 2007;26:191–203. doi: 10.1016/j.immuni.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiong H, Dolpady J, Wabl M, Curotto de Lafaille MA, Lafaille JJ. Sequential class switching is required for the generation of high affinity IgE antibodies. J Exp Med. 2012;209:353–64. doi: 10.1084/jem.20111941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med. 2012;18:693–704. doi: 10.1038/nm.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hlavacek WS, Perelson AS, Sulzer B, Bold J, Paar J, Gorman W, Posner RG. Quantifying aggregation of IgE-FcepsilonRI by multivalent antigen. Biophys J. 1999;76:2421–31. doi: 10.1016/S0006-3495(99)77397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burd PR, Thompson WC, Max EE, Mills FC. Activated mast cells produce interleukin 13. J Exp Med. 1995;181:1373–80. doi: 10.1084/jem.181.4.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsu CL, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS One. 2010;5:e11944. doi: 10.1371/journal.pone.0011944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karagiannis SN, Bracher MG, Hunt J, McCloskey N, Beavil RL, Beavil AJ, Fear DJ, Thompson RG, East N, Burke F, et al. IgE-antibody-dependent immunotherapy of solid tumors: cytotoxic and phagocytic mechanisms of eradication of ovarian cancer cells. J Immunol. 2007;179:2832–43. doi: 10.4049/jimmunol.179.5.2832. [DOI] [PubMed] [Google Scholar]
- 61.Finkelman FD, Urban JF., Jr. The other side of the coin: the protective role of the TH2 cytokines. J Allergy Clin Immunol. 2001;107:772–80. doi: 10.1067/mai.2001.114989. [DOI] [PubMed] [Google Scholar]
- 62.Verwaerde C, Joseph M, Capron M, Pierce RJ, Damonneville M, Velge F, Auriault C, Capron A. Functional properties of a rat monoclonal IgE antibody specific for Schistosoma mansoni. J Immunol. 1987;138:4441–6. [PubMed] [Google Scholar]
- 63.Vouldoukis I, Riveros-Moreno V, Dugas B, Ouaaz F, Bécherel P, Debré P, Moncada S, Mossalayi MD. The killing of Leishmania major by human macrophages is mediated by nitric oxide induced after ligation of the Fc epsilon RII/CD23 surface antigen. Proc Natl Acad Sci U S A. 1995;92:7804–8. doi: 10.1073/pnas.92.17.7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vouldoukis I, Mazier D, Moynet D, Thiolat D, Malvy D, Mossalayi MD. IgE mediates killing of intracellular Toxoplasma gondii by human macrophages through CD23-dependent, interleukin-10 sensitive pathway. PLoS One. 2011;6:e18289. doi: 10.1371/journal.pone.0018289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hagan P, Blumenthal UJ, Dunn D, Simpson AJ, Wilkins HA. Human IgE, IgG4 and resistance to reinfection with Schistosoma haematobium. Nature. 1991;349:243–5. doi: 10.1038/349243a0. [DOI] [PubMed] [Google Scholar]
- 66.Dunne DW, Butterworth AE, Fulford AJ, Ouma JH, Sturrock RF. Human IgE responses to Schistosoma mansoni and resistance to reinfection. Mem Inst Oswaldo Cruz. 1992;87(Suppl 4):99–103. doi: 10.1590/S0074-02761992000800014. [DOI] [PubMed] [Google Scholar]
- 67.Gurish MF, Bryce PJ, Tao H, Kisselgof AB, Thornton EM, Miller HR, Friend DS, Oettgen HC. IgE enhances parasite clearance and regulates mast cell responses in mice infected with Trichinella spiralis. J Immunol. 2004;172:1139–45. doi: 10.4049/jimmunol.172.2.1139. [DOI] [PubMed] [Google Scholar]
- 68.Watanabe N, Bruschi F, Korenaga M. IgE: a question of protective immunity in Trichinella spiralis infection. Trends Parasitol. 2005;21:175–8. doi: 10.1016/j.pt.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 69.Penichet ML, Jensen-Jarolim E. Cancer and IgE: Introducing the Concept of AllergoOncology. Springer, 2010. [Google Scholar]
- 70.Molomut N, Spain DM, Kreisler L, Warshaw LJ. The effect of an allergic inflammatory response in the tumor bed on the fate of transplanted tumors in mice. Cancer Res. 1955;15:181–3. [PubMed] [Google Scholar]
- 71.Ure DM. Negative assoication between allergy and cancer. Scott Med J. 1969;14:51–4. doi: 10.1177/003693306901400203. [DOI] [PubMed] [Google Scholar]
- 72.Schlitter HE. [Is there an allergy against malignant tumor tissue and what can it signify in regard to the defense of the body against cancer?] Strahlentherapie. 1961;114:203–24. [PubMed] [Google Scholar]
- 73.McCormick DP, Ammann AJ, Ishizaka K, Miller DG, Hong R. A study of allergy in patients with malignant lymphoma and chronic lymphocytic leukemia. Cancer. 1971;27:93–9. doi: 10.1002/1097-0142(197101)27:1<93::AID-CNCR2820270114>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 74.Augustin R, Chandradasa KD. IgE levels and allergic skin reactions in cancer and non-cancer patients. Int Arch Allergy Appl Immunol. 1971;41:141–3. doi: 10.1159/000230505. [DOI] [PubMed] [Google Scholar]
- 75.Jacobs D, Landon J, Houri M, Merrett TG. Circulating levels of immunoglobulin E in patients with cancer. Lancet. 1972;2:1059–61. doi: 10.1016/S0140-6736(72)92341-0. [DOI] [PubMed] [Google Scholar]
- 76.Allegra J, Lipton A, Harvey H, Luderer J, Brenner D, Mortel R, Demers L, Gillin M, White D, Trautlein J. Decreased prevalence of immediate hypersensitivity (atopy) in a cancer population. Cancer Res. 1976;36:3225–6. [PubMed] [Google Scholar]
- 77.Neuchrist C, Kornfehl J, Grasl M, Lassmann H, Kraft D, Ehrenberger K, Scheiner O. Distribution of immunoglobulins in squamous cell carcinoma of the head and neck. Int Arch Allergy Immunol. 1994;104:97–100. doi: 10.1159/000236714. [DOI] [PubMed] [Google Scholar]
- 78.Fu SL, Pierre J, Smith-Norowitz TA, Hagler M, Bowne W, Pincus MR, Mueller CM, Zenilman ME, Bluth MH. Immunoglobulin E antibodies from pancreatic cancer patients mediate antibody-dependent cell-mediated cytotoxicity against pancreatic cancer cells. Clin Exp Immunol. 2008;153:401–9. doi: 10.1111/j.1365-2249.2008.03726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karagiannis P, Gilbert AE, Josephs DH, Ali N, Dodev T, Saul L, Correa I, Roberts L, Beddowes E, Koers A, et al. IgG4 subclass antibodies impair antitumor immunity in melanoma. J Clin Invest. 2013;123:1457–74. doi: 10.1172/JCI65579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Palm NW, Rosenstein RK, Medzhitov R. Allergic host defences. Nature. 2012;484:465–72. doi: 10.1038/nature11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Josephs DH, Spicer JF, Corrigan CJ, Gould HJ, Karagiannis SN. Epidemiological associations of allergy, IgE and cancer. Clin Exp Allergy. 2013;43:1110–23. doi: 10.1111/cea.12178. [DOI] [PubMed] [Google Scholar]
- 82.Turner MC. Epidemiologicl evidence: IgE and solid tumours. In: Penichet ML, Jensen-Jarolim E, eds. Cancer and IgE: Introducing the concept of AllergoOncology. New York: Springer, 2010:47-77. [Google Scholar]
- 83.Ravetch JV, Kinet JP. Fc receptors. Annu Rev Immunol. 1991;9:457–92. doi: 10.1146/annurev.iy.09.040191.002325. [DOI] [PubMed] [Google Scholar]
- 84.Conrad DH. Fc epsilon RII/CD23: the low affinity receptor for IgE. Annu Rev Immunol. 1990;8:623–45. doi: 10.1146/annurev.iy.08.040190.003203. [DOI] [PubMed] [Google Scholar]
- 85.Pagès F, Galon J, Dieu-Nosjean MC, Tartour E, Sautès-Fridman C, Fridman WH. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. 2010;29:1093–102. doi: 10.1038/onc.2009.416. [DOI] [PubMed] [Google Scholar]
- 86.Bieber T. Fc epsilon RI on human epidermal Langerhans cells: an old receptor with new structure and functions. Int Arch Allergy Immunol. 1997;113:30–4. doi: 10.1159/000237500. [DOI] [PubMed] [Google Scholar]
- 87.Nigro EA, Soprana E, Brini AT, Ambrosi A, Yenagi VA, Dombrowicz D, Siccardi AG, Vangelista L. An antitumor cellular vaccine based on a mini-membrane IgE. J Immunol. 2012;188:103–10. doi: 10.4049/jimmunol.1101842. [DOI] [PubMed] [Google Scholar]
- 88.Luiten RM, Fleuren GJ, Warnaar SO, Litvinov SV. Target-specific activation of mast cells by immunoglobulin E reactive with a renal cell carcinoma-associated antigen. Lab Invest. 1996;74:467–75. [PubMed] [Google Scholar]
- 89.Luiten RM, Warnaar SO, Schuurman J, Pasmans SG, Latour S, Daëron M, Fleuren GJ, Litvinov SV. Chimeric immunoglobulin E reactive with tumor-associated antigen activates human Fc epsilon RI bearing cells. Hum Antibodies. 1997;8:169–80. [PubMed] [Google Scholar]
- 90.Sapino A, Cassoni P, Ferrero E, Bongiovanni M, Righi L, Fortunati N, Crafa P, Chiarle R, Bussolati G. Estrogen receptor alpha is a novel marker expressed by follicular dendritic cells in lymph nodes and tumor-associated lymphoid infiltrates. Am J Pathol. 2003;163:1313–20. doi: 10.1016/S0002-9440(10)63490-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dadabayev AR, Sandel MH, Menon AG, Morreau H, Melief CJ, Offringa R, van der Burg SH, Janssen-van Rhijn C, Ensink NG, Tollenaar RA, et al. Dendritic cells in colorectal cancer correlate with other tumor-infiltrating immune cells. Cancer Immunol Immunother. 2004;53:978–86. doi: 10.1007/s00262-004-0548-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mudde GC, Reischul IG, Corvaïa N, Hren A, Poellabauer EM. Antigen presentation in allergic sensitization. Immunol Cell Biol. 1996;74:167–73. doi: 10.1038/icb.1996.23. [DOI] [PubMed] [Google Scholar]
- 93.Khan KD, Emmanouilides C, Benson DM, Jr., Hurst D, Garcia P, Michelson G, Milan S, Ferketich AK, Piro L, Leonard JP, et al. A phase 2 study of rituximab in combination with recombinant interleukin-2 for rituximab-refractory indolent non-Hodgkin’s lymphoma. Clin Cancer Res. 2006;12:7046–53. doi: 10.1158/1078-0432.CCR-06-1571. [DOI] [PubMed] [Google Scholar]
- 94.Krauss S, Kufer P, Federle C, Tabaszewski P, Weiss E, Rieber EP, Riethmüller G. Recombinant CD4-IgE, a novel hybrid molecule, inducing basophils to respond to human immunodeficiency virus (HIV) and HIV-infected target cells. Eur J Immunol. 1995;25:192–9. doi: 10.1002/eji.1830250132. [DOI] [PubMed] [Google Scholar]
- 95.Ribatti D, Ennas MG, Vacca A, Ferreli F, Nico B, Orru S, Sirigu P. Tumor vascularity and tryptase-positive mast cells correlate with a poor prognosis in melanoma. Eur J Clin Invest. 2003;33:420–5. doi: 10.1046/j.1365-2362.2003.01152.x. [DOI] [PubMed] [Google Scholar]
- 96.Molin D, Edström A, Glimelius I, Glimelius B, Nilsson G, Sundström C, Enblad G. Mast cell infiltration correlates with poor prognosis in Hodgkin’s lymphoma. Br J Haematol. 2002;119:122–4. doi: 10.1046/j.1365-2141.2002.03768.x. [DOI] [PubMed] [Google Scholar]
- 97.Galli SJ, Maurer M, Lantz CS. Mast cells as sentinels of innate immunity. Curr Opin Immunol. 1999;11:53–9. doi: 10.1016/S0952-7915(99)80010-7. [DOI] [PubMed] [Google Scholar]
- 98.Gulubova M, Vlaykova T. Prognostic significance of mast cell number and microvascular density for the survival of patients with primary colorectal cancer. J Gastroenterol Hepatol. 2009;24:1265–75. doi: 10.1111/j.1440-1746.2007.05009.x. [DOI] [PubMed] [Google Scholar]
- 99.Cai SW, Yang SZ, Gao J, Pan K, Chen JY, Wang YL, Wei LX, Dong JH. Prognostic significance of mast cell count following curative resection for pancreatic ductal adenocarcinoma. Surgery. 2011;149:576–84. doi: 10.1016/j.surg.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 100.Rajput AB, Turbin DA, Cheang MC, Voduc DK, Leung S, Gelmon KA, Gilks CB, Huntsman DG. Stromal mast cells in invasive breast cancer are a marker of favourable prognosis: a study of 4,444 cases. Breast Cancer Res Treat. 2008;107:249–57. doi: 10.1007/s10549-007-9546-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–97. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, Evan GI. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat Med. 2007;13:1211–8. doi: 10.1038/nm1649. [DOI] [PubMed] [Google Scholar]
- 103.Gounaris E, Erdman SE, Restaino C, Gurish MF, Friend DS, Gounari F, Lee DM, Zhang G, Glickman JN, Shin K, et al. Mast cells are an essential hematopoietic component for polyp development. Proc Natl Acad Sci U S A. 2007;104:19977–82. doi: 10.1073/pnas.0704620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.de Vries VC, Pino-Lagos K, Nowak EC, Bennett KA, Oliva C, Noelle RJ. Mast cells condition dendritic cells to mediate allograft tolerance. Immunity. 2011;35:550–61. doi: 10.1016/j.immuni.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.de Vries VC, Wasiuk A, Bennett KA, Benson MJ, Elgueta R, Waldschmidt TJ, Noelle RJ. Mast cell degranulation breaks peripheral tolerance. Am J Transplant. 2009;9:2270–80. doi: 10.1111/j.1600-6143.2009.02755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Munitz A, Levi-Schaffer F. Eosinophils: ‘new’ roles for ‘old’ cells. Allergy. 2004;59:268–75. doi: 10.1111/j.1398-9995.2003.00442.x. [DOI] [PubMed] [Google Scholar]
- 107.Fernández-Aceñero MJ, Galindo-Gallego M, Sanz J, Aljama A. Prognostic influence of tumor-associated eosinophilic infiltrate in colorectal carcinoma. Cancer. 2000;88:1544–8. doi: 10.1002/(SICI)1097-0142(20000401)88:7<1544::AID-CNCR7>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 108.Dorta RG, Landman G, Kowalski LP, Lauris JR, Latorre MR, Oliveira DT. Tumour-associated tissue eosinophilia as a prognostic factor in oral squamous cell carcinomas. Histopathology. 2002;41:152–7. doi: 10.1046/j.1365-2559.2002.01437.x. [DOI] [PubMed] [Google Scholar]
- 109.Ishibashi S, Ohashi Y, Suzuki T, Miyazaki S, Moriya T, Satomi S, Sasano H. Tumor-associated tissue eosinophilia in human esophageal squamous cell carcinoma. Anticancer Res. 2006;26(2B):1419–24. [PubMed] [Google Scholar]
- 110.Cuschieri A, Talbot IC, Weeden S, MRC Upper GI Cancer Working Party Influence of pathological tumour variables on long-term survival in resectable gastric cancer. Br J Cancer. 2002;86:674–9. doi: 10.1038/sj.bjc.6600161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.von Wasielewski R, Seth S, Franklin J, Fischer R, Hübner K, Hansmann ML, Diehl V, Georgii A. Tissue eosinophilia correlates strongly with poor prognosis in nodular sclerosing Hodgkin’s disease, allowing for known prognostic factors. Blood. 2000;95:1207–13. [PubMed] [Google Scholar]
- 112.Huland E, Huland H. Tumor-associated eosinophilia in interleukin-2-treated patients: evidence of toxic eosinophil degranulation on bladder cancer cells. J Cancer Res Clin Oncol. 1992;118:463–7. doi: 10.1007/BF01629431. [DOI] [PubMed] [Google Scholar]
- 113.Cormier SA, Taranova AG, Bedient C, Nguyen T, Protheroe C, Pero R, Dimina D, Ochkur SI, O’Neill K, Colbert D, et al. Pivotal Advance: eosinophil infiltration of solid tumors is an early and persistent inflammatory host response. J Leukoc Biol. 2006;79:1131–9. doi: 10.1189/jlb.0106027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Simson L, Ellyard JI, Dent LA, Matthaei KI, Rothenberg ME, Foster PS, Smyth MJ, Parish CR. Regulation of carcinogenesis by IL-5 and CCL11: a potential role for eosinophils in tumor immune surveillance. J Immunol. 2007;178:4222–9. doi: 10.4049/jimmunol.178.7.4222. [DOI] [PubMed] [Google Scholar]
- 115.Tepper RI, Coffman RL, Leder P. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science. 1992;257:548–51. doi: 10.1126/science.1636093. [DOI] [PubMed] [Google Scholar]
- 116.Noffz G, Qin Z, Kopf M, Blankenstein T. Neutrophils but not eosinophils are involved in growth suppression of IL-4-secreting tumors. J Immunol. 1998;160:345–50. [PubMed] [Google Scholar]
- 117.Kim DW, Min HS, Lee KH, Kim YJ, Oh DY, Jeon YK, Lee SH, Im SA, Chung DH, Kim YT, et al. High tumour islet macrophage infiltration correlates with improved patient survival but not with EGFR mutations, gene copy number or protein expression in resected non-small cell lung cancer. Br J Cancer. 2008;98:1118–24. doi: 10.1038/sj.bjc.6604256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lin EY, Li JF, Bricard G, Wang W, Deng Y, Sellers R, Porcelli SA, Pollard JW. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol Oncol. 2007;1:288–302. doi: 10.1016/j.molonc.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ojalvo LS, King W, Cox D, Pollard JW. High-density gene expression analysis of tumor-associated macrophages from mouse mammary tumors. Am J Pathol. 2009;174:1048–64. doi: 10.2353/ajpath.2009.080676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, Coussens LM, Karin M, Goldrath AW, Johnson RS. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70:7465–75. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–96. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 123.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–6. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kawa S, Ito T, Watanabe T, Maruyama M, Hamano H, Maruyama M, Muraki T, Arakura N. The Utility of Serum IgG4 Concentrations as a Biomarker. Int J Rheumatol. 2012;2012:198314. doi: 10.1155/2012/198314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Harada K, Shimoda S, Kimura Y, Sato Y, Ikeda H, Igarashi S, Ren XS, Sato H, Nakanuma Y. Significance of immunoglobulin G4 (IgG4)-positive cells in extrahepatic cholangiocarcinoma: molecular mechanism of IgG4 reaction in cancer tissue. Hepatology. 2012;56:157–64. doi: 10.1002/hep.25627. [DOI] [PubMed] [Google Scholar]
- 127.Nevala WK, Vachon CM, Leontovich AA, Scott CG, Thompson MA, Markovic SN, Melanoma Study Group of the Mayo Clinic Cancer Center Evidence of systemic Th2-driven chronic inflammation in patients with metastatic melanoma. Clin Cancer Res. 2009;15:1931–9. doi: 10.1158/1078-0432.CCR-08-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sheu BC, Lin RH, Lien HC, Ho HN, Hsu SM, Huang SC. Predominant Th2/Tc2 polarity of tumor-infiltrating lymphocytes in human cervical cancer. J Immunol. 2001;167:2972–8. doi: 10.4049/jimmunol.167.5.2972. [DOI] [PubMed] [Google Scholar]
- 129.Agarwal A, Verma S, Burra U, Murthy NS, Mohanty NK, Saxena S. Flow cytometric analysis of Th1 and Th2 cytokines in PBMCs as a parameter of immunological dysfunction in patients of superficial transitional cell carcinoma of bladder. Cancer Immunol Immunother. 2006;55:734–43. doi: 10.1007/s00262-005-0045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.van de Veen W, Stanic B, Yaman G, Wawrzyniak M, Söllner S, Akdis DG, Rückert B, Akdis CA, Akdis M. IgG4 production is confined to human IL-10-producing regulatory B cells that suppress antigen-specific immune responses. J Allergy Clin Immunol. 2013;131:1204–12. doi: 10.1016/j.jaci.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 131.Karagiannis P, Gilbert AE, Nestle FO, Karagiannis SN. IgG4 antibodies and cancer-associated inflammation: Insights into a novel mechanism of immune escape. Oncoimmunology. 2013;2:e24889. doi: 10.4161/onci.24889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Canonica GW, Bousquet J, Casale T, Lockey RF, Baena-Cagnani CE, Pawankar R, Potter PC, Bousquet PJ, Cox LS, Durham SR, et al. Sub-lingual immunotherapy: World Allergy Organization Position Paper 2009. Allergy. 2009;64(Suppl 91):1–59. doi: 10.1111/j.1398-9995.2009.02309.x. [DOI] [PubMed] [Google Scholar]
- 133.Shamji MH, Ljørring C, Francis JN, Calderon MA, Larché M, Kimber I, Frew AJ, Ipsen H, Lund K, Würtzen PA, et al. Functional rather than immunoreactive levels of IgG4 correlate closely with clinical response to grass pollen immunotherapy. Allergy. 2012;67:217–26. doi: 10.1111/j.1398-9995.2011.02745.x. [DOI] [PubMed] [Google Scholar]
- 134.James LK, Bowen H, Calvert RA, Dodev TS, Shamji MH, Beavil AJ, McDonnell JM, Durham SR, Gould HJ. Allergen specificity of IgG(4)-expressing B cells in patients with grass pollen allergy undergoing immunotherapy. J Allergy Clin Immunol. 2012;130:663–70, e3. doi: 10.1016/j.jaci.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 135.Pilette C, Nouri-Aria KT, Jacobson MR, Wilcock LK, Detry B, Walker SM, Francis JN, Durham SR. Grass pollen immunotherapy induces an allergen-specific IgA2 antibody response associated with mucosal TGF-beta expression. J Immunol. 2007;178:4658–66. doi: 10.4049/jimmunol.178.7.4658. [DOI] [PubMed] [Google Scholar]
- 136.Satoguina JS, Weyand E, Larbi J, Hoerauf A. T regulatory-1 cells induce IgG4 production by B cells: role of IL-10. J Immunol. 2005;174:4718–26. doi: 10.4049/jimmunol.174.8.4718. [DOI] [PubMed] [Google Scholar]
- 137.Reali E, Greiner JW, Corti A, Gould HJ, Bottazzoli F, Paganelli G, Schlom J, Siccardi AG. IgEs targeted on tumor cells: therapeutic activity and potential in the design of tumor vaccines. Cancer Res. 2001;61:5517–22. [PubMed] [Google Scholar]
- 138.Nigro EA, Brini AT, Soprana E, Ambrosi A, Dombrowicz D, Siccardi AG, Vangelista L. Antitumor IgE adjuvanticity: key role of Fc epsilon RI. J Immunol. 2009;183:4530–6. doi: 10.4049/jimmunol.0900842. [DOI] [PubMed] [Google Scholar]
- 139.Riemer AB, Untersmayr E, Knittelfelder R, Duschl A, Pehamberger H, Zielinski CC, Scheiner O, Jensen-Jarolim E. Active induction of tumor-specific IgE antibodies by oral mimotope vaccination. Cancer Res. 2007;67:3406–11. doi: 10.1158/0008-5472.CAN-06-3758. [DOI] [PubMed] [Google Scholar]
- 140.Karagiannis SN, Wang Q, East N, Burke F, Riffard S, Bracher MG, Thompson RG, Durham SR, Schwartz LB, Balkwill FR, et al. Activity of human monocytes in IgE antibody-dependent surveillance and killing of ovarian tumor cells. Eur J Immunol. 2003;33:1030–40. doi: 10.1002/eji.200323185. [DOI] [PubMed] [Google Scholar]
- 141.Teng MW, Kershaw MH, Jackson JT, Smyth MJ, Darcy PK. Adoptive transfer of chimeric FcepsilonRI gene-modified human T cells for cancer immunotherapy. Hum Gene Ther. 2006;17:1134–43. doi: 10.1089/hum.2006.17.1134. [DOI] [PubMed] [Google Scholar]
- 142.Daniels-Wells TR, Helguera G, Leuchter RK, Quintero R, Kozman M, Rodríguez JA, Ortiz-Sánchez E, Martínez-Maza O, Schultes BC, Nicodemus CF, et al. A novel IgE antibody targeting the prostate-specific antigen as a potential prostate cancer therapy. BMC Cancer. 2013;13:195. doi: 10.1186/1471-2407-13-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mount PF, Sutton VR, Li W, Burgess J, McKEnzie IF, Pietersz GA, Trapani JA. Chimeric (mouse/human) anti-colon cancer antibody c30.6 inhibits the growth of human colorectal cancer xenografts in scid/scid mice. Cancer Res. 1994;54:6160–6. [PubMed] [Google Scholar]
- 144.Spillner E, Plum M, Blank S, Miehe M, Singer J, Braren I. Recombinant IgE antibody engineering to target EGFR. Cancer Immunol Immunother. 2012;61:1565–73. doi: 10.1007/s00262-012-1287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Berlyn KA, Schultes B, Leveugle B, Noujaim AA, Alexander RB, Mann DL. Generation of CD4(+) and CD8(+) T lymphocyte responses by dendritic cells armed with PSA/anti-PSA (antigen/antibody) complexes. Clin Immunol. 2001;101:276–83. doi: 10.1006/clim.2001.5115. [DOI] [PubMed] [Google Scholar]
- 146.Iio A, Waldmann TA, Strober W. Metabolic study of human IgE: evidence for an extravascular catabolic pathway. J Immunol. 1978;120:1696–701. [PubMed] [Google Scholar]
- 147.Dreskin SC, Goldsmith PK, Strober W, Zech LA, Gallin JI. Metabolism of immunoglobulin E in patients with markedly elevated serum immunoglobulin E levels. J Clin Invest. 1987;79:1764–72. doi: 10.1172/JCI113017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rudman SM, Josephs DH, Cambrook H, Karagiannis P, Gilbert AE, Dodev T, Hunt J, Koers A, Montes A, Taams L, et al. Harnessing engineered antibodies of the IgE class to combat malignancy: initial assessment of FcɛRI-mediated basophil activation by a tumour-specific IgE antibody to evaluate the risk of type I hypersensitivity. Clin Exp Allergy. 2011;41:1400–13. doi: 10.1111/j.1365-2222.2011.03770.x. [DOI] [PubMed] [Google Scholar]
- 149.Marcucci F, Bellone M, Rumio C, Corti A. Approaches to improve tumor accumulation and interactions between monoclonal antibodies and immune cells. MAbs. 2013;5:34–46. doi: 10.4161/mabs.22775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Karagiannis SN, Nestle FO, Gould HJ. IgE interacts with potent effector cells against tumors: ADCC and ADCP. Chapter 8. In: Penichet ML, Jensen-Jarolim E, eds. Cancer and IgE. New York: Humana Press, Springer, 2010:185-213. [Google Scholar]
- 151.Turner MC. Epidemiology: allergy history, IgE, and cancer. Cancer Immunol Immunother. 2012;61:1493–510. doi: 10.1007/s00262-011-1180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dalton DK, Noelle RJ. The roles of mast cells in anticancer immunity. Cancer Immunol Immunother. 2012;61:1511–20. doi: 10.1007/s00262-012-1246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gatault S, Legrand F, Delbeke M, Loiseau S, Capron M. Involvement of eosinophils in the anti-tumor response. Cancer Immunol Immunother. 2012;61:1527–34. doi: 10.1007/s00262-012-1288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Platzer B, Dehlink E, Turley SJ, Fiebiger E. How to connect an IgE-driven response with CTL activity? Cancer Immunol Immunother. 2012;61:1521–5. doi: 10.1007/s00262-011-1127-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bluth MH. IgE and chemotherapy. Cancer Immunol Immunother. 2012;61:1585–90. doi: 10.1007/s00262-011-1170-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Strid J, Sobolev O, Zafirova B, Polic B, Hayday A. The intraepithelial T cell response to NKG2D-ligands links lymphoid stress surveillance to atopy. Science. 2011;334:1293–7. doi: 10.1126/science.1211250. [DOI] [PMC free article] [PubMed] [Google Scholar]