

Abstract
Alzheimer's disease (AD) is a devastating neurodegenerative disorder currently affecting over 35 million people worldwide. Pathological hallmarks of AD are massive amyloidosis, extracellular senile plaques, and intracellular neurofibrillary tangles accompanied by an excessive loss of synapses. Major constituents of senile plaques are 40–42 amino acid long peptides termed β-amyloid (Aβ). Aβ is produced by sequential proteolytic processing of the amyloid precursor protein (APP). APP processing and Aβ production have been one of the central scopes in AD research in the past. In the last years, lipids and lipid-related issues are more frequently discussed to contribute to the AD pathogenesis. This review summarizes lipid alterations found in AD postmortem brains, AD transgenic mouse models, and the current understanding of how lipids influence the molecular mechanisms leading to AD and Aβ generation, focusing especially on cholesterol, docosahexaenoic acid (DHA), and sphingolipids/glycosphingolipids.
1. APP Processing
Amyloid plaques are composed of aggregated amyloid-β peptides, derived from sequential proteolytic processing of the amyloid-precursor protein (APP), a type-I transmembrane protein with a large extracellular N-terminal domain and a short intracellular C-terminal tail [1]. APP and its gene family members, the APP-like proteins APLP1 and APLP2, are highly conserved proteins expressed in numerous species and tissues pointing out their physiological importance [2]. Indeed, triple knockout of APP/APLP1/APLP2 in mice results in postnatal lethality involving brain development abnormalities and cortical dysplasia [3]. In contrast, single knockout of APP has a minor phenotype consisting of reduced body weight [2], commissure defects [4], and hypersensitivity to epileptic seizures [5] demonstrating the mutual functional compensation of the gene family members and additionally illustrates the physiological role of APP. Furthermore, a potential contribution to the formation of dendritic and synaptic structures as well as in long-term potentiation (LTP) [6–8] suggests a possible impact on cognitive function.
APP can be cleaved in two distinct pathways, the amyloidogenic and nonamyloidogenic pathways. The non-amyloidogenic processing of APP by α-secretases avoids the formation of Aβ peptides by cleaving inside the Aβ domain [9]. Thereby, the large N-terminal ectodomain α-secreted APP (sAPPα) is released into the extracellular matrix, whereas the short C-terminal part (α-CTF) remains within the membrane for further processing. Notably, sAPPα has neuroprotective and memory enhancing properties [10, 11], hypothesizing that sAPPα might mediate a major physiological function of APP [12]. The α-secretases belonging to the ADAM family (a disintegrin and metalloprotease), in particular ADAM10 and ADAM17 (tumor necrosis factor α converting enzyme/TACE), have emerged as predominant α-secretases [13–15]. Like APP itself, these proteins are type I integral membrane proteins.
The amyloidogenic pathway, on the other hand, is initiated by the β-secretase BACE1 (beta-site APP cleaving enzyme) that generates soluble β-secreted APP (sAPPβ) and the membrane-tethered fragment β-CTF. BACE1 is a membrane-bound aspartyl protease belonging to the pepsin family, expressed in neurons [16]. The main β-secretase activity is found in the secretory pathway including the Golgi compartments, secretory vesicles, and endosomes [17, 18]. Apart from its amyloidogenic effect, less is known about the physiological function of BACE1; however, in BACE1 knock-out mice, myelination is affected [19, 20]. Initial cleavage of APP by α-secretase in the non-amyloidogenic pathway, or by β-secretase in the amyloidogenic pathway, is typically followed by γ-secretase processing, a multimeric complex consisting of at least four subunits—presenilin 1 (PS1) or presenilin 2 (PS2), anterior pharynx defective 1 homologue (APH1), presenilin enhancer 2 (PEN2), and nicastrin [21] with PS1/PS2 being the catalytic centre [22, 23]. Importantly, mutations inside the PS genes are responsible for early onset AD [24]. PS1 and PS2 are multitransmembrane spanning aspartyl proteases cleaving the C-terminal stubs α- and β-CTF within the centre of their transmembrane domain [25], generating p3 and Aβ peptides of varying length (e.g., Aβ38, Aβ40, and Aβ42). Among the Aβ species generated, hydrophobic Aβ42 peptides self-aggregate to small oligomers before being manifested as senile plaques composed of a dense core of amyloid fibrils [26, 27]. Simultaneously, the APP intracellular domain (AICD), which is discussed to regulate gene transcription, is released into the cytosol [28]. The substrate APP and the secretases involved in APP cleavage are all transmembrane proteins, suggesting that the surrounding lipid microenvironment may play a pivotal role in the pathogenesis of the disease.
2. Cholesterol and AD
The human brain has a very high cholesterol content, mainly associated with myelin. Due to the limited transport over the blood-brain barrier (BBB), the brain cholesterol level is largely independent of the serum cholesterol concentrations. The vast majority of brain cholesterol is provided by glial de novo synthesis. Noteworthy, cholesterol transport between neurons and glia cells is mostly provided by clusterin/apolipoprotein J (ApoJ) and apolipoprotein E (ApoE) containing lipoproteins. Intriguingly, the ApoEε4 allele genotype is the predominant genetic risk factor for AD, whereas the ε2 allele seems to be protective and ε3 being the most common form. A possible explanation for this might be reduced Aβ-clearance and/or increased formation of amyloid in the presence of the ε4 genotype, but many other mechanisms have also been suggested [29–31]. Recent genome-wide association studies (GWASs) have greatly extended our knowledge on AD risk genes. Interestingly, two main functional risk clusters were identified. Most GWAS-identified risk genes are either linked to inflammation or to lipid/membrane processes. Besides ApoE polymorphism, single-nucleotide polymorphisms for clusterin (CLU), ABCA7, and PICALM were discovered to raise the individual risk for developing AD [32–34]. On one hand, clusterin serves as a major lipid binding protein [35]. Interestingly, clustrin mRNA and protein levels are increased in AD, thereby, correlating with disease severity [36, 37]. Furthermore, clusterin may be involved in modulation of apoptosis, inflammation, and Aβ aggregation [38–40]. ABCA7, belonging to the ATP-binding cassette transporters, is expressed throughout the brain and especially in hippocampal neurons [41, 42]. ABCA7 is involved in cholesterol efflux from cells to ApoE and affects APP processing [43]. These genetic links between AD risk and cholesterol are supported by strong epidemiological evidence linking hypercholesterolemia with dementia [44, 45]. Like many other life-style influenced AD risk factors, elevated blood cholesterol levels, even if only moderately increased, are most relevant if present at midlife [46]. Additionally, elevated 24OH-cholesterol levels were observed in the serum of AD patients [47].
Cholesterol is an essential component of mammalian cell membranes. Based on its extraordinary structure, consisting of a fused rigid ring system, a polar hydroxyl group, and a hydrocarbon tail, cholesterol is essential for bilayer's function and organisation. Due to the impact of the rigid ring system, cholesterol can increase the order within the membrane and thereby affects membrane fluidity. Especially in lipid microdomains, envisioned as so-called “lipid rafts,” primarily found in the plasma membrane, the trans-Golgi, and endosomal membranes, this feature is extremely important. Lipid rafts are strongly enriched in sphingomyelin, glycosphingolipids, and cholesterol. Cholesterol provides tight packing of the lipids in these microdomains. Some membrane proteins are preferentially sited in these ordered microdomains. Mechanistically important is the transient colocalization of APP with the amyloidogenic secretases, β-secretase, and γ-secretase, in the lipid rafts, pointing out that within the amyloidogenic pathway the close colocalization of these proteins is, at least partly, mediated by cholesterol [48, 49]. Nonamyloidogenic processing and the α-secretases, however, are localized outside the lipid rafts [50]. This characteristic implies the possible regulation of the nonamyloidogenic and amyloidogenic pathways by altered membrane cholesterol amounts. Indeed, evidence suggests that cholesterol tends to form complexes with β-CTF and potentially with full-length APP. Thereby, the translocation of β-CTF/APP is promoted to the cholesterol-enriched microdomains and thus providing them to the amyloidogenic processing [51, 52]. Additionally, it is well established that cholesterol directly stimulates β- and γ-secretases; vice versa cholesterol depletion by cyclodextrin or statins leads to a decreased activity [53–55]. Recently, it was described that statins influence APP maturation and phosphorylation not by cholesterol lowering but by the loss of cholesterol precursors [56]. Remarkably, cholesterol further affects the thickness of the lipid bilayer which directly influences the γ-secretase cleavage activity and specificity. With increasing membrane thickness, smaller Aβ species (e.g., Aβ38 and Aβ40) were preferentially produced, whereas Aβ42/43 only occurred in smaller amounts [57]. Additionally, α-secretase cleavage is increased by lowering cholesterol levels. This effect was attributed to increased membrane fluidity and impaired APP internalisation. After treatment with lovastatin, ADAM10 protein level was elevated [50]. Aside from its impact on the Aβ generation, cholesterol might even modify Aβ aggregation and the subsequent neurotoxicity. Under physiological conditions, mainly monomeric Aβ peptides develop from APP processing [58]. Though, in AD these monomers are prone to form oligomers, protofibrils, and fibrils, whereby the oligomers are considered to be the most toxic subspecies [59]. It was highlighted that cholesterol-enriched microdomains promote amyloid aggregation, while depletion of cholesterol leads to reduced aggregation [60]. Recent findings implicated that cholesterol facilitates β-sheet formation by direct interaction with Phe19 amino acid of the Aβ peptide [61]. In line with these findings, some studies showed a relationship between elevated cholesterol level and increased toxicity of Aβ oligomers [62, 63]. Contradictory to this, other investigations reported cholesterol to be protective [64, 65]. Hence, although cholesterol is essential for human brain development and function, it can as well be attributed to a potent role in mediating aggregation and neurotoxicity.
In line with the strong impact of cholesterol on APP processing, there is evidence that the cleavage products of APP themselves affect cholesterol metabolism. In mouse embryonic fibroblasts deficient in APP/APLP2−/− or PS1/2−/− and therefore unable to produce Aβ, cholesterol levels were highly elevated. Interestingly, these cholesterol aberrations were abrogated by treatment with Aβ1-40 displaying a negative feed-back cycle of Aβ1-40 by inhibiting the 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR), the rate-limiting step in cholesterol de novo synthesis [66, 67]. Additionally, decreased membrane fluidity and increased cholesterol content in lipid rafts were observed in cells devoid of Aβ [68]. Within the APP family of proteins, APP apparently has an especially important role for cholesterol homeostasis. In APP knock-out mice, cholesterol is strongly increased [66, 69], and APP knockout has a number of additional cholesterol- and lipid-related consequences, including reduced diet-dependent atherosclerosis [70] and increased Niemann-Pick cholesterol phenotype [71]. To some part, this appears to be caused by a lack of AICD, resulting in altered LRP1 levels [72]. LRP1 belongs to a lipoprotein-receptor family mainly involved in the cholesterol uptake of neurons, whereby cholesterol is provided by ApoE-containing lipoproteins [72]. Changes in membrane cholesterol content will trigger homeostatic actions affecting the regulation and membrane content of many other lipids. Accordingly, many other lipids affect the cholesterol regulation and several of these lipids are targeted by APP/Aβ or themselves change Aβ production.
Taking all of these findings into consideration (Figure 1; See Table S1 in Supplementary Material available online at http://dx.doi.org/10.1155/2013/814390), pharmacological intervention in cholesterol metabolism might be a possible target in AD treatment. It seems likely that cholesterol lowering by administration of HMGCR inhibitors (e.g., statins) might refer to reduced Aβ levels accompanied by slower cognitive decline and improved mental status.
Figure 1.
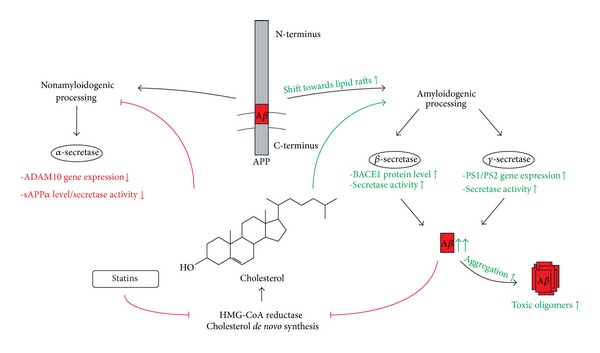
Schematic representation of the proposed mechanisms of cholesterol on APP processing and Aβ aggregation.
Animal model studies associated hypercholesteremia with elevated Aβ levels [80–82]. On the opposite, reduced Aβ accumulation was achieved after administration of cholesterol lowering drugs (e.g., statins), underlining their potential role in the disease treatment [83–86]. However, one study reported unaffected Aβ levels, whereas another one even observed increased Aβ deposition [87, 88]. Interestingly, the latter alteration was only detected in female mice. These deviations might be contributed to differences in animal models, used drugs, and period of drug administration. A potential beneficial effect of cholesterol lowering drugs was further investigated in observational studies and randomized controlled trials, whereas many observational studies with high number of participants associated statin intake with reduced development of AD [89–91], others exhibited no differences [92, 93]. However, recent randomized, double-blind, placebo-controlled studies reported no benefit of statin use in individuals suffering from mild to moderate AD [94, 95]. Since in these trials patients were already affected by AD, statins might be ascribed to a more preventive than therapeutic function. Thus, further studies have to evaluate a protective effect in earlier clinical stages.
3. Docosahexaenoic Acid and Alzheimer's Disease
Docosahexaenoic acid (DHA) is an essential ω-3 polyunsaturated fatty acid (PUFA) mainly found in marine food, especially in oily fish. Only a small amount of DHA can be produced endogenously by synthesis out of α-linoleic acid through elongation and desaturation [96], whereas the main DHA is provided by dietary intake. Approximately 60% of the unsaturated fatty acids in neuronal membranes consist of DHA, thus, representing the most common ω-3 fatty acid in the human brain. DHA rapidly incorporates into phospholipids of cellular membranes and changes the membrane fluidity by the formation of highly disordered domains concentrated in PUFA-containing phospholipids but depleted in cholesterol (reviewed in [97]). Especially in synapses, these alterations in membrane fluidity play an important role in neurotransmission, ion channel formation, and synaptic plasticity. This suggests a potential role of DHA in memory, learning, and cognitive processes. In young rats, the administration of DHA leads to an improved learning ability [98]. Additionally, DHA is involved in neuronal differentiation [99], neurogenesis [100], and protection against synaptic loss [101]. Therefore, DHA is discussed to be involved in pathological processes of AD.
DHA is decreased in certain regions of AD postmortem brains, like pons, white matter and, in particular, frontal grey matter, and hippocampus [102]. Additionally, peroxidation products of DHA, which is very susceptible to lipid peroxidation due to its six double bounds, are elevated in AD brains hypothesizing that DHA loss can be ascribed to the increased oxidative stress [103]. These lipid alterations seem to occur not only in advanced AD patients but also in early stages of the disease [104].
Several epidemiological studies tried to correlate DHA plasma levels or the amount of dietary intaken fish with cognitive decline. The Rotterdam study presented the first findings about an inverse correlation between increased fish intake and all-cause dementia [105]. However, reexamination with a longer follow-up period found no association between dietary intake of ω-3 fatty acids and dementia [106]. Nevertheless, further studies like the PAQUID study [107] or the CHAP study [108] supported the protective effect of fish or DHA consumption initially found in the Rotterdam study. These effects were even more pronounced in individuals not carrying the ApoE ε4 allele [109, 110].
Beside the epidemiological studies, findings from animal models and in vitro experiments suggest that increased dietary intake of DHA is associated with a reduced risk of AD. In a 3xTg-AD mouse model, that exhibits both Aβ and tau pathologies [111], DHA supplementation caused a significant reduction in soluble and intraneuronal Aβ levels as well as tau phosphorylation [112]. Similar results were obtained with APPSwedish transgenic mice, revealing significant plaque reduction in the hippocampus and parietal cortex accompanied by alterations of APP cleavage products [113]. In AD-model rats, produced by infusion of Aβ1-40 into the cerebral ventricle, DHA also improved learning ability [114]. In vitro, Aβ fibrillation was found to be decreased in the presence of DHA [73].
Recently, we and others elucidated the molecular mechanisms involved in the reduced Aβ burden in the presence of DHA (summarized in Table 1). In the neuroblastoma cell line SH-SY5Y, DHA directly inhibited amyloidogenic β- and γ-secretase activities, resulting in a dose-dependent reduction of Aβ levels. On the other hand, non-amyloidogenic APP processing was increased in DHA-treated cells, as we observed elevated sAPPα levels caused by an enhanced ADAM17 protein stability [74]. Interestingly, DHA exhibited various interactions with cholesterol homeostasis. Beside a direct inhibition of the HMGCR, DHA induced a cholesterol shift from lipid raft to the nonraft fractions, illustrating an alternative secretase activity modulating pathway [74]. In line with these findings the DHA-mediator NPD1, derived from DHA processing by cytosolic phospholipase A2 and 15-lipooxygenase, is known to directly affect APP processing resulting in elevated sAPPα and lower sAPPβ and accordingly Aβ levels [75, 76]. Interestingly, phospholipase A2 and 15-lipooxygenase and as a consequence NPD1 were found to be reduced in the hippocampus of AD patients and in AD mouse models [75, 76]. Beside the observed shift of APP processing from the amyloidogenic pathway to the non-amyloidogenic pathway, NPD1 also acts neuroprotective by downregulating inflammatory signalling, apoptosis, and Aβ-induced neurotoxicity [76, 115].
Table 1.
Summary of mechanisms of DHA and DHA derivates on APP processing. DHA both affects amyloidogenic and non-amyloidogenic pathways via multiple mechanisms resulting in a decrease in Aβ production. In opposite to cholesterol it has been reported that DHA decreases Aβ aggregation and toxicity. Direct effects of DHA on APP processing are further enhanced by a DHA-mediated decrease in cholesterol de novo synthesis [73–79].
(a) Effect of DHA
| Affected pathway | Mechanism of action |
|---|---|
| Nonamyloidogenic processing | sAPPα ↑ |
| ADAM 17 protein stability ↑ | |
|
| |
| Amyloidogenic processing | Aβ↓ |
| β-Secretase activity ↓ | |
| Endosomal BACE1 ↓ | |
| γ-Secretase activity ↓ | |
| PS1 shift: raft → non-raft | |
|
| |
| Aβ Oligomerization and toxicity | Aβ Fibrillation ↓ |
| Soluble toxic oligomers ↓ | |
| Aβ Phagocytosis ↑ | |
|
| |
| Cholesterol homeostasis | HMG-CoA reductase activity ↓ |
| Cholesterol de novo synthesis ↓ | |
| Cholesterol shift: raft → non-raft | |
|
| |
| Other non-APP-mediated pathways/mechanisms | SorLA/R11 ↑, a sorting protein reduced in AD |
| Neuronal differentiation ↑ | |
| Protection against synaptic loss, Synaptogenesis ↑ | |
| Neurogenesis ↑ | |
| Inflammation ↓ | |
| Reactive oxidative species ↓ | |
|
| |
(b) Effect of DHA derivates
| Affected pathway | Mechanism of action | |
|---|---|---|
| NPD1 | Nonamyloidogenic processing | sAPPα ↑ |
| ADAM 10 maturation ↑ | ||
| Amyloidogenic processing | Aβ↓ | |
| sAPPβ↓ | ||
| BACE 1 protein level ↓ | ||
| Aβ Toxicity | Neuroprotective and antiapoptotic | |
| Soluble toxic oligomers ↓ | ||
These findings indicate a possible therapeutic use of DHA in preventing, modulating, or improving AD progression. Clinical trials, however, delivered ambiguous results concerning DHA supplementation and cognitive functions suggesting a limited benefit depending on the disease stage and ApoE allele genotype [116–118]. Further investigations with great cohorts have to clarify whether DHA has more preventing than therapeutic effects by including only patients in the earliest stages of memory decline. More recently, an advanced DHA formulation containing several diet-derived molecules to enhance DHA activity resulted in a number of clinical trials that resulted in various degrees of cognitive benefit. The benefit was most pronounced in patients with very mild to mild AD but less in mild to moderate AD patients [119, 120]. Most recently, at the ADPD conference 2012, Scheltens reported on an open-label extension study in very mild to mild AD patients with continuous increase in memory performance over the study period of 48 weeks [121].
4. Sphingo- and Glycosphingolipids
Sphingolipids are a heterogeneous group of lipids, structurally based on the 18-carbon amino alcohol sphingosine which is synthesized from palmitoyl-CoA and serine by serine palmitoyl-CoA transferase (SPT), representing the rate-limiting step in sphingolipid synthesis and shown to be regulated by AICD [122]. Ceramide (Cer) is an important branching point for the synthesis of different sphingolipid subspecies. For example, sphingomyelin (SM) is generated out of Cer by SM-synthase, whereas the neutral sphingomyelinase (nSMase) catalyzes the turnover of SM to Cer. Glycosylation of Cer results in the production of glycosphingolipids which can be further processed to gangliosides. The cerebroside synthase adds a galactose moiety to Cer which is the first step towards the formation of sulfatides. Finally, the decomposition of Cer is provided by the ceramidase receiving sphingosine which itself can be phosphorylated to sphingosine-1-phosphate (S1P). First evidence of sphingolipid metabolism being involved in neurodegenerative diseases was derived from investigations of lysosomal storage diseases (LSDs). This group of inherited metabolic disorders is characterized by accumulation of different sphingolipids due to dysfunction or deficiency of the corresponding lysosomal enzymes. Importantly, affected patients clinically develop progressive cognitive decline resulting in early dementia. Additionally, AD-related pathologies like Aβ accumulation and hyperphosphorylation of tau, leading to neurofibrillary tangles, can be observed [123, 124]. Beside these similarities between AD and LSD, several studies of AD postmortem brains indicate that the sphingolipid metabolism is altered during AD progression, further substantiated by biochemical studies, linking sphingolipids to APP processing [125–131] (Figure 2).
Figure 2.
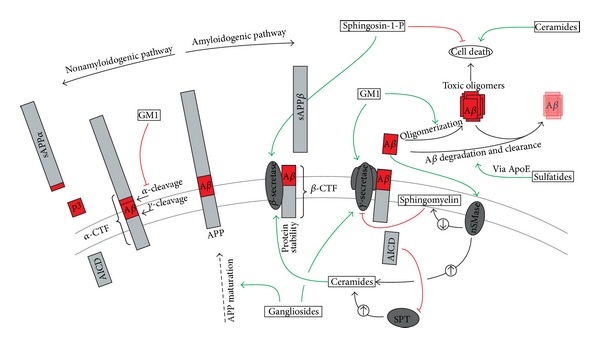
Schematic illustration of the effects of sphingolipids and glycosphingolipids on APP processing. Interestingly, APP processing in return affects the metabolic pathways of sphingolipids. For example, it has been shown that AICD regulates the sphingolipid de novo synthesis by decreasing the expression of the Serinepalmitoyl-CoA-Transferase (SPT) or that Aβ itself directly increases the activity of the sphingomyelin degrading enzyme Sphingomyelinase (SMase), resulting in complex regulatory cycles which are dysregulated in the case of Alzheimer's disease.
4.1. Ceramide
The majority of postmortem brain tissue analysis found elevated Cer levels in the grey and white matters of AD patients. These alterations were observed even in early stages of AD hypothesizing that these might promote the development of the disease [125, 128, 131, 132]. In line with these findings, gene expression abnormalities of the key enzymes that control sphingolipid metabolism were found in AD patients: enzymes involved in glycosphingolipid synthesis (e.g., UDP-glucose ceramide glucosyltransferase) were altered accompanied by changes in enzymes resulting in the accumulation of Cer (e.g., serine palmitoyltransferase, neutral sphingomyelinase, and acid sphingomyelinase) [131, 133]. Recently, Mielke et al. reported in a 9-year-follow-up study that even elevated baseline serum Cer levels are associated with a higher risk (up to 10-fold) of developing AD [134], indicating that serum Cer is associated with incident AD.
In contrast to the results obtained from AD postmortem brains, the analysis of AD mouse models revealed inconsistent data. Cer levels are elevated in the cerebral cortex of APPSL/PS1Ki transgenic mice, whereas the corresponding single-transgenic mice did not show this alteration [135]. Furthermore, in APPSL/PS1M146L transgenic mice, in which the time course of pathology is closer to that seen in most currently available models, Cer did not accumulate in disease-associated brain regions (cortex and hippocampus) [136]. These different findings might be attributed to the various mouse models used in these studies [135, 136]. The authors suggest that APPSL/PS1Ki mice, compared to the other mouse models, produce exceeding amounts of N-truncated Aβx-42, also found in AD brains [135].
It is well established that ceramides induce apoptosis and exhibit neurotoxic properties [137–139]. Interestingly, Aβ toxicity is linked to Cer-dependent apoptotic pathways. Lee et al. observed an Aβ-induced elevation of nSMase and consequently increased Cer levels which resulted in remarkable cell death. Inhibiting this pathway abolished the Aβ-triggered cascade [140]. In this context, activation of nSMase by Aβ42 but not Aβ40 has been reported [66, 141] (Figure 2). Recently, a potential novel mechanism of ceramide-enriched exosomes released by Aβ-treated astrocytes was proposed to be responsible for Aβ-induced apoptosis. Thereby, nSMase2 was essential for charging these exosomes with Cer [142, 143]. Beside the involvement of Cer in Aβ toxicity, Cer has been shown to alter APP processing and Aβ production. Increasing Cer levels by either direct Cer administration or stimulation of endogenous biosynthesis by nSMase resulted in enhanced Aβ production. Elevated Aβ levels are attributed to Cer-induced protein stabilization of β-secretase BACE1, whereas γ-secretase is not affected [144]. Further studies elucidated the underlying mechanism of increased BACE1 stability: elevated Cer level caused upregulation of the acetyltransferases, ATase1, and ATase2, acetylating BACE1 protein and thus protecting the nascent protein from degradation [145]. Taken together, it seems likely that Cer is the driving force in a circulus vitiosus: increasing Cer levels lead to an intensified Aβ production whereupon Aβ is responsible for Cer accumulation.
Noteworthy in this context, S1P is discussed as a possible counterpart. S1P is considered to be neuroprotective and important for neuronal differentiation [146, 147]. Remarkably, one study reported reduced S1P levels in frontotemporal grey matter of AD patients [131], which implicates a possible role in AD. However, recent findings reported an increased proteolytic activity of BACE1 by direct interaction with S1P [148]. Therefore, additional studies addressing S1P are important to clarify the significance of S1P in APP processing and AD.
4.2. Sphingomyelin
Sphingomyelin is an important component of mammalian cell membranes, particularly enriched in myelin sheets and especially represented in the brain [149]. Considering the observations outlined in the preceding chapter it is suggested that SM concentrations in AD brains might be decreased. Analysis of postmortem brain, however, show inconsistent results. Although two studies reported elevated SM levels in AD brains compared to age-related controls [150, 151], two studies described a significant decrease [131, 132]. Further studies only revealed a modest reduction in severe AD postmortem brains and no significant differences in earlier stages of the disease [125]. Moreover, recent investigations found increased SM levels in the cerebrospinal fluid (CSF) of individuals suffering from prodromal AD, whereas there was only a slight but not significant decrease in mild and moderate AD groups [152]. Interestingly, a current epidemiological study correlated higher plasma SM levels with slower cognitive decline among AD patients, illustrating SM as potential sensitive blood-based biomarker for disease progression [153]. Importantly, nSMase was reported to be upregulated in AD brains [133], resulting in increased SM breakdown. Additionally, in cell culture studies, nSMase activity is known to be elevated in presenilin familial Alzheimer's disease mutations (PS-FAD) causing early onset AD [66], pointing towards a possible role of SMases in sporadic late onset as well as in familial early onset AD pathology. Interestingly, SM itself is discussed to alter APP processing. Increasing SM by either direct treatment of cells with SM or inhibition of nSMase resulted in diminished Aβ levels [66]. In this context, it is again worth mentioning that Aβ42 itself regulates SM homeostasis as described previously.
4.3. Gangliosides
Gangliosides are a family of sialic acid containing glycosphingolipids, highly expressed in neuronal and glial membranes, where they play important roles for development, proliferation, differentiation, and maintenance of neuronal tissues and cells [154, 155]. The first step towards the formation of gangliosides is the glycosylation of Cer by glucosylceramide-synthase (GCS). According to their number of sialic acid residues, gangliosides are separated in four different ganglio-series: o-series, a-series, b-series, and c-series. Importantly, the most common brain gangliosides belong either to the a-series (GM1 and GD1a) or b-series (GD1b and GT1b). The ganglioside GM3 serves as a common precursor for a- and b-series gangliosides. The GD3-synthase (GD3S) catalyzes the synthesis of GD3 by adding sialic acid to GM3, segregating the a- and b-series of gangliosides [156] and therefore controlling the levels of the major brain gangliosides. Gangliosides are also able to interact with further membrane lipids like SM and cholesterol, thereby, being involved in the formation of lipid rafts [157]. Interestingly, postmortem studies of AD brains suggest a strong connection between ganglioside homeostasis and AD pathology. In a previous work, Kracun et al. found all major brain gangliosides to be reduced in the temporal and frontal cortex and in nucleus basalis of Meynert, whereas simple gangliosides GM2 and GM3 were elevated in parietal and frontal cortex [158]. GM3 elevation was further supported by a recent study. Here, the authors also described an increase in glucosylceramide levels, the precursor for ganglioside synthesis [159]. Additionally, Gottfries et al. reported a significant reduction of gangliosides in the grey matter of early onset AD subjects compared to late onset AD and control individuals. Nevertheless, a decrease in total gangliosides was also observed in brains of late onset AD patients, however, to a smaller extent [160]. A more recent study found elevated levels of GM1 and GM2 in lipid raft fractions of the temporal and frontal cortex of AD brains [161]. Moreover, the analysis of AD transgenic mouse models suggests an altered ganglioside metabolism in AD. Barrier et al. compared different transgenic mice with age-matched wildtype controls. While all mice expressing APP (SL) showed an increase in GM2 and GM3 in the cerebral cortex and a moderate decrease in complex b-series gangliosides, only APP/PS1Ki transgenic mice exhibited a loss of complex a-series gangliosides, GT1a, GD1a, and GM1 [162]. In summary, ganglioside metabolism seems to be highly affected during disease progression. While more complex gangliosides appear to be depleted, simple gangliosides, like GM1 and GM3 are increased. As mentioned previously GM3 is an important precursor for a- and b-series gangliosides suggesting a disease-dependent alteration in the biosynthesis of these ganglioside series. Indeed, we found a close link between APP processing products and ganglioside metabolism. The activity of GD3S, the key enzyme converting a- to b-series gangliosides, is significantly reduced by two separate and additive mechanisms. On one hand, GD3S activity is inhibited by the binding of Aβ peptides to GM3, consequently reducing substrate availability and preventing the conversion of GM3 to GD3. On the other hand, gene transcription of GD3S is downregulated and mediated by the APP intracellular domain (AICD), thus, resulting in GD3 depletion and GM3 accumulation [163].
Furthermore, especially GM1 is related to several AD-specific pathomechanisms like altered APP processing, aggregation, and cytotoxicity. GM1 was found to decrease sAPPα levels and to increase Aβ generation, whereas sAPPβ levels were unchanged [164]. This suggests increased γ-secretase and decreased α-secretase activities without affecting β-secretase. Indeed, as recently described, the direct administration of gangliosides to purified γ-secretase resulted in an increased enzyme activity. Moreover, a shift towards the formation of Aβ42 peptides was observed [165]. Interestingly, Aβ decreases membrane fluidity by binding GM1. As a consequence, amyloidogenic APP processing was stimulated, proposing an Aβ-triggered, GM1-mediated, vicious cycle [166]. Further mechanisms linking gangliosides to Aβ production were described by Tamboli et al. Inhibition of GCS, the committed step towards ganglioside formation, significantly decreased Aβ formation. An altered APP maturation and cell surface transport, leading to less access of APP to amyloidogenic processing in the endosomal compartments, were proposed as the underlying mechanisms [167].
Furthermore, GM1 seems to be particularly important as a “seed” for amyloid plaque formation. Thereby, GM1 interacts with Aβ, resulting in GAβ complexes [168]. This complex tends to aggregate more easily due to changing the secondary structure of Aβ towards β-sheet formation [169, 170]. Interestingly, Mahfoud et al. described a sphingolipid binding domain of Aβ, which is also contained in HIV-1 and prion proteins [171]. Further investigations displayed accumulation and aggregation of Aβ on cell membranes especially in GM1-enriched lipid rafts, resulting in enhanced cytotoxicity [172, 173]. Additionally, enhanced cytotoxicity of Aβ fibrils was observed after release of GM1 from damaged neurons, indicating a possible aggravation mechanism [174]. Importantly, GAβ complexes also occur in AD brains and aged mice [175]. Synaptosomes, prepared from aged mouse brains, exhibited GM1 clusters, showing high ability to initiate Aβ aggregation [176]. Remarkably, Aβ deposition was observed to begin mainly at the presynaptic neuronal membranes in AD brains, suggesting a possible role of GAβ in the early pathogenesis of AD [177, 178].
4.4. Sulfatides
Another sphingolipid subgroup, possibly involved in AD pathogenesis, are sulfatides. They are generated from Cer by adding a galactose moiety, catalyzed by the ceramide galactosyltransferase (CGT). Finally, synthesis of sulfatides is provided by cerebroside sulfotransferase (CST), which transfers a sulfate group to the galactosyl moiety. The degradation of sulfatides takes place in the lysosomal compartment, where arylsulfatase A (ASA) hydrolyzes the sulfate group. ASA deficiency leads to accumulation of sulfatides and to the clinical picture of metachromatic leukodystrophy. Sulfatides are especially enriched in myelin sheaths making up about 5% of the myelin lipids. Hence, they are particularly produced by oligodendrocytes and Schwann cells. Nevertheless, sulfatides have also been detected in neurons and astrocytes, however, in a lower amount [179]. Interestingly, AD pathology is known to induce focal demyelination and degeneration of oligodendrocytes [180].
Importantly, Han et al. described an extraordinary depletion of sulfatide levels in all analyzed brain regions of AD subjects. Sulfatides were depleted up to 93% in grey matter and up to 58% in white matter. These alterations were already observed in the earliest stages of the disease [125]. Additionally, a previous study found decreased sulfatide levels in the white matter of the frontal lobe only in patients with late onset AD compared to age-matched controls and early onset AD patients [160]. Furthermore, Bandaru et al. also described decreased sulfatide content in the white matter; however, they did not confirm the alterations in the grey matter [150]. In contrast, further studies found no significant changes between control and AD brains [132, 159]. Worth mentioning, a 40% reduction in CSF sulfatide levels was detected among AD patients. In this context, sulfatide/phosphatidylinositol ratio was proposed as potential clinical biomarker for early AD diagnosis [181]. However, data obtained by analyzing transgenic mouse models in respect to alterations in sulfatide levels are more inconsistent. Although Barrier et al. found no significant changes between wildtype mice and double transgenic APPSL/PS1Ki mice or single-transgenic PS1Ki mice [135], a more recent study found significantly decreased sulfatide levels in the forebrain of these mouse models [159].
Noteworthy, there is a possible link between sulfatide homeostasis and ApoE trafficking. First, the intercellular sulfatide transport in the brain is mediated by ApoE-containing lipoproteins. Interestingly, human ApoE4 carrying transgenic mice presented the highest sulfatide depletion in the brain in comparison to wildtype ApoE or human ApoE3 transgenic mice [181]. Furthermore, a recent study showed a significant, age-related decrease of sulfatides in APP transgenic mice, which was completely abolished by ApoE knockout [182]. These findings offer a possible explanation for decreased sulfatide levels in AD brains. On the other hand, sulfatides seem to be involved in the ApoE-dependent Aβ clearance. Direct supplementation of sulfatides to cultured cells dramatically reduced Aβ levels. This observation was most likely ascribed to a modification of Aβ clearance through an endocytotic pathway. Thereby, sulfatides facilitated the ApoE-mediated Aβ clearance, especially of ApoE4 containing vesicles [183]. Nevertheless, the importance of sulfatide-dependent molecular mechanisms, being involved in AD pathogenesis, is still ambiguous. Therefore, further investigations are necessary to clarify the role of sulfatides in APP processing and AD.
5. Lipids and Tau Pathology
Beside amyloid plaques, neurofibrillary tangles (NFTs), consisting of hyperphosphorylated tau proteins, are considered to be a pivotal pathological hallmark of AD [184–186]. Although this review focuses on the impact of lipids on APP processing, it is worth mentioning that tau pathology is also affected by an altered lipid homeostasis. Tau proteins belong to the family of microtubule-associated proteins, important for the assembly of tubulin monomers into microtubules, to stabilize the neuronal microtubule network which is essential for maintaining cell shape and axonal transport [187]. In AD, this function is disrupted, due to the hyperphosphorylation of serine/threonine residues of tau. This abnormal phosphorylation promotes the release of tau proteins from microtubules, its disassembly, and self-assembly into paired helical filaments (PHFs), a major component of NFTs, and, as a consequence, provokes microtubule disruption [188–191]. Supporting the connection between lipids and tau, Kawarabayashi et al. reported an accumulation of phosphorylated tau in brain-extracted lipid rafts of an AD mouse model [192]. In addition, tau phosphorylation by cyclin-dependent kinase 5 (CDK5) was observed in lipid rafts after short-term incubation of SH-SY5Y cells with Aβ peptides [193]. Interestingly, Niemann-Pick type C (NPC), an inherited lysosomal storage disease with an abnormal intracellular accumulation of cholesterol, also exhibits tau pathology [194, 195]. Noteworthy, these alterations are more pronounced in neurons containing higher cholesterol levels [194, 196]. In line with these findings elevated Aβ and total tau levels were observed in the cerebrospinal fluid of NPC patients [197]. In an NPC mouse model, Sawamura et al. illustrated a possible explanation for these hyperphosphorylated tau forms. They found an intensified activation of the mitogen-activated protein kinase (MAPK)-pathway, one of several kinases phosphorylating tau physiologically in the brain [198]. Not only in NPC model mice but also in further mouse models, the cholesterol and ApoE status were associated with increased tau phosphorylation [199–201]. In contrast, statin treatment reduced NFT burden in normocholesterolemic and hypercholesterolemic mice. This effect, however, was rather attributed to the anti-inflammatory properties than to the cholesterol lowering aspect of statins [202]. Additionally, simvastatin treatment of hypercholesterolemic subjects without dementia revealed a significant phospho-tau-181 decrease in the CSF, whereas no differences in total tau or Aβ levels were observed [203]. Importantly, membrane cholesterol levels are closely linked to the Aβ-induced calpain activation and tau toxicity [204]. Calpain is a calcium-dependent cysteine protease responsible for the generation of the neurotoxic 17 kDa tau [205, 206]. Decreasing membrane cholesterol in mature neurons reduced their susceptibility to the Aβ-induced calpain activation, 17 kDa tau production, and cell death, whereas elevated membrane cholesterol levels enhanced this Aβ-triggered cascade in young neurons [204]. Like calpain, AMP-activated protein kinase (AMPK), a serine/threonine kinase, is also activated by elevated calcium levels. Interestingly, in addition to its important function in regulating cholesterol homeostasis, emerging studies revealed AMPK as a potential tau phosphorylating enzyme [207, 208]. AMPK-induced abnormal tau phosphorylation inhibited microtubule binding of tau [207]. In line with these findings Vingtdeux et al. reported an accumulation of activated AMPK in cerebral neurons of AD brains [209]. Other authors, however, even ascribed AMPK an inhibiting function in tau phosphorylation by downregulating glycogen synthase kinase-3β (GSK3β) activity, one of the main tau phosphorylating kinases [210, 211]. Beside cholesterol, ω-3 fatty acids are also discussed to influence tau pathology. In a 3xTg-AD mouse model, Green et al. demonstrated lowered levels of intraneuronal tau and reduced tau phosphorylation after 3 to 9 months of DHA supplementation [112]. Similar results were also observed by Ma et al. after fish oil administration to 3xTg-AD mice [212]. In both studies, the reduced phosphorylation was attributed to an inhibition of the c-Jun N-terminal kinase (JNK). In contrast, low ω3 intake with a decreased ω3 : ω6 ratio leads to an aggravation of tau pathology in these transgenic mice [213]. Furthermore, some studies revealed a colocalization of sphingolipids and gangliosides with PHFs proposing a possible relation between sphingolipid metabolism and tau [214, 215]. Indeed, inhibition of the serine palmitoyl transferase (SPT), the rate-limiting enzyme in sphingolipid synthesis, and the first step towards ceramide synthesis resulted in a reduced tau hyperphosphorylation in an AD mouse model [216]. After treatment of differentiated PC12 cells with ceramide derivates (N-acetylsphingosine, and N-hexanoylsphingosine), Xie and Johnson reported a significant reduction in tau levels without affecting tau phosphorylation. This was attributed to an increased expression of calpain I and thus stimulated tau protein degradation [217]. Taken together, not only APP processing but also tau pathology is influenced by lipids. Although the general view for APP processing seems more consistent, further investigations linking tau to altered lipid homeostasis should follow.
6. Conclusion
Summing it up, lipids are tightly linked to AD. It has been shown that cholesterol increases amyloidogenic pathways and decreases non amyloidogenic pathways followed by an enhanced Aβ production and aggregation. Opposite effects were observed for DHA, suggesting a potential beneficial role for DHA or PUFAs in AD. For sphingolipids and glycosphingolipids, a more complex situation in respect to AD is reported. Although lipids like SM, S1P, and sulfatides seem to be protective by enhancing Aβ clearance or decreasing Aβ production, other glycosphingolipids like gangliosides or ceramides increase Aβ toxicity or Aβ oligomerization. Interestingly, it has been shown that, in return, APP processing also affects lipid metabolism, resulting in complex regulatory feed-back cycles, which seem to be dysregulated in AD. In line, several studies suggest an altered lipid metabolism in human AD brains. However, controversial effects are reported in different brain regions and tissues, making more detailed analysis with new lipidomic approaches and higher numbers necessary.
Supplementary Material
Supplementary material provides a tabular overview of the proposed mechanisms of cholesterol on APP processing with selected publications included.
References
- 1.Dyrks T, Weidemann A, Multhaup G, et al. Identification, transmembrane orientation and biogenesis of the amyloid A4 precursor of Alzheimer’s disease. EMBO Journal. 1988;7(4):949–957. doi: 10.1002/j.1460-2075.1988.tb02900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anliker B, Müller U. The functions of mammalian amyloid precursor protein and related amyloid precursor-like proteins. Neurodegenerative Diseases. 2006;3(4-5):239–246. doi: 10.1159/000095262. [DOI] [PubMed] [Google Scholar]
- 3.Herms J, Anliker B, Heber S, et al. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO Journal. 2004;23(20):4106–4115. doi: 10.1038/sj.emboj.7600390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magara F, Müller U, Li ZW, et al. Genetic background changes the pattern of forebrain commissure defects in transgenic mice underexpressing the β-amyloid-precursor protein. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(8):4656–4661. doi: 10.1073/pnas.96.8.4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinbach JP, Müller U, Leist M, Li ZW, Nicotera P, Aguzzi A. Hypersensitivity to seizures in β-amyloid precursor protein deficient mice. Cell Death and Differentiation. 1998;5(10):858–866. doi: 10.1038/sj.cdd.4400391. [DOI] [PubMed] [Google Scholar]
- 6.Tyan, -H S, Shih, et al. Amyloid precursor protein (APP) regulates synaptic structure and function. Molecular and Cellular Neuroscience. 2012;51(1-2):43–52. doi: 10.1016/j.mcn.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee KJ, Moussa CEH, Lee Y, et al. Beta amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. Neuroscience. 2010;169(1):344–356. doi: 10.1016/j.neuroscience.2010.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoe HS, Fu Z, Makarova A, et al. The effects of amyloid precursor protein on postsynaptic composition and activity. Journal of Biological Chemistry. 2009;284(13):8495–8506. doi: 10.1074/jbc.M900141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esch FS, Keim PS, Beattie EC, et al. Cleavage of amyloid β peptide during constitutive processing of its precursor. Science. 1990;248(4959):1122–1124. doi: 10.1126/science.2111583. [DOI] [PubMed] [Google Scholar]
- 10.Mattson MP, Guo ZH, Geiger JD. Secreted form of amyloid precursor protein enhances basal glucose and glutamate transport and protects against oxidative impairment of glucose and glutamate transport in synaptosomes by a cyclic GMP-mediated mechanism. Journal of Neurochemistry. 1999;73(2):532–537. doi: 10.1046/j.1471-4159.1999.0730532.x. [DOI] [PubMed] [Google Scholar]
- 11.Meziane H, Dodart JC, Mathis C, et al. Memory-enhancing effects of secreted forms of the β-amyloid precursor protein in normal and amnestic mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(21):12683–12688. doi: 10.1073/pnas.95.21.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ring S, Weyer SW, Kilian SB, et al. The secreted β-amyloid precursor protein ectodomain APPsα is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. Journal of Neuroscience. 2007;27(29):7817–7826. doi: 10.1523/JNEUROSCI.1026-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asai M, Hattori C, Szabó B, et al. Putative function of ADAM9, ADAM10, and ADAM17 as APP α-secretase. Biochemical and Biophysical Research Communications. 2003;301(1):231–235. doi: 10.1016/s0006-291x(02)02999-6. [DOI] [PubMed] [Google Scholar]
- 14.Buxbaum JD, Liu KN, Luo Y, et al. Evidence that tumor necrosis factor α converting enzyme is involved in regulated α-secretase cleavage of the Alzheimer amyloid protein precursor. Journal of Biological Chemistry. 1998;273(43):27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 15.Lammich S, Kojro E, Postina R, et al. Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(7):3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan R, Blenkowski MJ, Shuck ME, et al. Membrane-anchored aspartyl protease with Alzheimer’s disease β-secretase activity. Nature. 1999;402(6761):533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- 17.Vassar R, Bennett BD, Babu-Khan S, et al. β-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 18.Hook VYH, Toneff T, Aaron W, Yasothornsrikul S, Bundey R, Reisine T. β-amyloid peptide in regulated secretory vesicles of chromaffin cells: evidence for multiple cysteine proteolytic activities in distinct pathways for β-secretase activity in chromaffin vesicles. Journal of Neurochemistry. 2002;81(2):237–256. doi: 10.1046/j.1471-4159.2002.00794.x. [DOI] [PubMed] [Google Scholar]
- 19.Hu X, Hicks CW, He W, et al. Bace1 modulates myelination in the central and peripheral nervous system. Nature Neuroscience. 2006;9(12):1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- 20.Willem M, Garratt AN, Novak B, et al. Control of peripheral nerve myelination by the β-secretase BACE1. Science. 2006;314(5799):664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- 21.Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. γ-secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(11):6382–6387. doi: 10.1073/pnas.1037392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Strooper B, Saftig P, Craessaerts K, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391(6665):387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 23.Grimm MO, Tomic I, Hartmann T. Potential external source of Aβ in biological samples. Nature Cell Biology. 2002;4:E164–E165. doi: 10.1038/ncb0702-e164b. [DOI] [PubMed] [Google Scholar]
- 24.Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 25.Grziwa B, Grimm MOW, Masters CL, Beyreuther K, Hartmann T, Lichtenthaler SF. The transmembrane domain of the amyloid precursor protein in microsomal membranes is on both sides shorter than predicted. Journal of Biological Chemistry. 2003;278(9):6803–6808. doi: 10.1074/jbc.M210047200. [DOI] [PubMed] [Google Scholar]
- 26.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochemical and Biophysical Research Communications. 1984;120(3):885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 27.Jarrett JT, Berger EP, Lansbury PT., Jr. The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32(18):4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 28.Cao X, Sudhof TC. A Transcriptively active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293(5527):115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 29.Wang S, Wang R, Chen L, Bennett DA, Dickson DW, Wang DS. Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. Journal of Neurochemistry. 2010;115(1):47–57. doi: 10.1111/j.1471-4159.2010.06899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang JY, Hafez DM, James BD, Bennett DA, Marr RA. Altered NEP2 expression and activity in mild cognitive impairment and Alzheimer’s disease. Journal of Alzheimer’s Disease. 2012;28(2):433–441. doi: 10.3233/JAD-2011-111307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker LC, Diamond MI, Duff KE, Hyman BT. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurology. 2013;70(3):304–310. doi: 10.1001/jamaneurol.2013.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nature Genetics. 2009;41(10):1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferrari R, Moreno JH, Minhajuddin AT, et al. Implication of common and disease specific variants in CLU, CR1, and PICALM. Neurobiology of Aging. 2012;33(8):1846.e7–1846.e18. doi: 10.1016/j.neurobiolaging.2012.01.110. [DOI] [PubMed] [Google Scholar]
- 34.Reitz C, Jun G, Naj A, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ϵ4, and the risk of late-onset Alzheimer disease in African Americans. Journal of the American Medical Association. 2013;309(14):1483–1492. doi: 10.1001/jama.2013.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nuutinen T, Suuronen T, Kauppinen A, Salminen A. Clusterin: a forgotten player in Alzheimer’s disease. Brain Research Reviews. 2009;61(2):89–104. doi: 10.1016/j.brainresrev.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 36.Schrijvers EMC, Koudstaal PJ, Hofman A, Breteler MMB. Plasma clusterin and the risk of Alzheimer disease. Journal of the American Medical Association. 2011;305(13):1322–1326. doi: 10.1001/jama.2011.381. [DOI] [PubMed] [Google Scholar]
- 37.May PC, Lampert-Etchells M, Johnson SA, Poirier J, Masters JN, Finch CE. Dynamics of gene expression for a hippocampal glycoprotein elevated in Alzheimer’s disease and in response to experimental lesions in rat. Neuron. 1990;5(6):831–839. doi: 10.1016/0896-6273(90)90342-d. [DOI] [PubMed] [Google Scholar]
- 38.Falgarone G, Chiocchia G. Chapter 8: clusterin: a multifacet protein at the crossroad of inflammation and autoimmunity. Advances in Cancer Research. 2009;104:139–170. doi: 10.1016/S0065-230X(09)04008-1. [DOI] [PubMed] [Google Scholar]
- 39.Zhang H, Kim JK, Edwards CA, Xu Z, Taichman R, Wang CY. Clusterin inhibits apoptosis by interacting with activated Bax. Nature Cell Biology. 2005;7(9):909–915. doi: 10.1038/ncb1291. [DOI] [PubMed] [Google Scholar]
- 40.Yerbury JJ, Poon S, Meehan S, et al. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB Journal. 2007;21(10):2312–2322. doi: 10.1096/fj.06-7986com. [DOI] [PubMed] [Google Scholar]
- 41.Kim WS, Fitzgerald ML, Kang K, et al. Abca7 null mice retain normal macrophage phosphatidylcholine and cholesterol efflux activity despite alterations in adipose mass and serum cholesterol levels. Journal of Biological Chemistry. 2005;280(5):3989–3995. doi: 10.1074/jbc.M412602200. [DOI] [PubMed] [Google Scholar]
- 42.Ikeda Y, Abe-Dohmae S, Munehira Y, et al. Posttranscriptional regulation of human ABCA7 and its function for the apoA-I-dependent lipid release. Biochemical and Biophysical Research Communications. 2003;311(2):313–318. doi: 10.1016/j.bbrc.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 43.Chan SL, Kim WS, Kwok JB, et al. ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. Journal of Neurochemistry. 2008;106(2):793–804. doi: 10.1111/j.1471-4159.2008.05433.x. [DOI] [PubMed] [Google Scholar]
- 44.Kivipelto M, Helkala EL, Laakso MP, et al. Apolipoprotein E ε4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Annals of Internal Medicine. 2002;137(3):149–155. doi: 10.7326/0003-4819-137-3-200208060-00006. [DOI] [PubMed] [Google Scholar]
- 45.Pappolla MA, Bryant-Thomas TK, Herbert D, et al. Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology. 2003;61(2):199–205. doi: 10.1212/01.wnl.0000070182.02537.84. [DOI] [PubMed] [Google Scholar]
- 46.Solomon A, Kivipelto M, Wolozin B, Zhou J, Whitmer RA. Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later. Dementia and Geriatric Cognitive Disorders. 2009;28(1):75–80. doi: 10.1159/000231980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lütjohann D, Papassotiropoulos A, Björkhem I, et al. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. Journal of Lipid Research. 2000;41(2):195–198. [PubMed] [Google Scholar]
- 48.Hur JY, Welander H, Behbahani H, et al. Active γ-secretase is localized to detergent-resistant membranes in human brain. FEBS Journal. 2008;275(6):1174–1187. doi: 10.1111/j.1742-4658.2008.06278.x. [DOI] [PubMed] [Google Scholar]
- 49.Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ. Exclusively targeting β-secretase to lipid rafts by GPI-anchor addition up-regulates β-site processing of the amyloid precursor protein. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(20):11735–11740. doi: 10.1073/pnas.1635130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kojro E, Gimpl G, Lammich S, März W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(10):5815–5820. doi: 10.1073/pnas.081612998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beel AJ, Sakakura M, Barrett PJ, Sanders CR. Direct binding of cholesterol to the amyloid precursor protein: an important interaction in lipid-Alzheimer’s disease relationships? Biochimica et Biophysica Acta. 2010;1801(8):975–982. doi: 10.1016/j.bbalip.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barrett PJ, Song Y, van Horn WD, et al. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science. 2012;336(6085):1168–1171. doi: 10.1126/science.1219988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grimm MOW, Grimm HS, Tomic I, Beyreuther K, Hartmann T, Bergmann C. Independent inhibition of Alzheimer disease β- and γ-secretase cleavage by lowered cholesterol levels. Journal of Biological Chemistry. 2008;283(17):11302–11311. doi: 10.1074/jbc.M801520200. [DOI] [PubMed] [Google Scholar]
- 54.Osenkowski P, Ye W, Wang R, Wolfe MS, Selkoe DJ. Direct and potent regulation of γ-secretase by its lipid microenvironment. Journal of Biological Chemistry. 2008;283(33):22529–22540. doi: 10.1074/jbc.M801925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalvodova L, Kahya N, Schwille P, et al. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. Journal of Biological Chemistry. 2005;280(44):36815–36823. doi: 10.1074/jbc.M504484200. [DOI] [PubMed] [Google Scholar]
- 56.Hosaka A, Araki W, Oda A, Tomidokoro Y, Tamaoka A. Statins reduce amyloid β-peptide production by modulating amyloid precursor protein maturation and phosphorylation through a cholesterol-independent mechanism in cultured neurons. Neurochemical Research. 2013;38(3):589–600. doi: 10.1007/s11064-012-0956-1. [DOI] [PubMed] [Google Scholar]
- 57.Winkler E, Kamp F, Scheuring J, Ebke A, Fukumori A, Steiner H. Generation of Alzheimer disease-associated amyloid beta42/43 peptide by gamma-secretase can be inhibited directly by modulation of membrane thickness. Journal of Biological Chemistry. 2012;287(25):21326–21334. doi: 10.1074/jbc.M112.356659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haass C, Schlossmacher MG, Hung AY, et al. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359(6393):322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 59.Dahlgren KN, Manelli AM, Blaine Stine W, Jr., Baker LK, Krafft GA, Ladu MJ. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. Journal of Biological Chemistry. 2002;277(35):32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 60.Schneider A, Schulz-Schaeffer W, Hartmann T, Schulz JB, Simons M. Cholesterol depletion reduces aggregation of amyloid-beta peptide in hippocampal neurons. Neurobiology of Disease. 2006;23(3):573–577. doi: 10.1016/j.nbd.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 61.Zhou X, Xu J. Free cholesterol induces higher beta-sheet content in Abeta peptide oligomers by aromatic interaction with Phe19. PLoS ONE. 2012;7(9) doi: 10.1371/journal.pone.0046245.e46245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abramov AY, Ionov M, Pavlov E, Duchen MR. Membrane cholesterol content plays a key role in the neurotoxicity of β-amyloid: implications for Alzheimer’s disease. Aging Cell. 2011;10(4):595–603. doi: 10.1111/j.1474-9726.2011.00685.x. [DOI] [PubMed] [Google Scholar]
- 63.Ferrera P, Mercado-Gómez O, Silva-Aguilar M, Valverde M, Arias C. Cholesterol potentiates β-amyloid-induced toxicity in human neuroblastoma cells: involvement of oxidative stress. Neurochemical Research. 2008;33(8):1509–1517. doi: 10.1007/s11064-008-9623-y. [DOI] [PubMed] [Google Scholar]
- 64.Sponne I, Fifre A, Koziel V, Oster T, Olivier JL, Pillot T. Membrane cholesterol interferes with neuronal apoptosis induced by soluble oligomers but not fibrils of amyloid-beta peptide. The FASEB Journal. 2004;18(7):836–838. doi: 10.1096/fj.03-0372fje. [DOI] [PubMed] [Google Scholar]
- 65.Arispe N, Doh M. Plasma membrane cholesterol controls the cytotoxicity of Alzheimer’s disease AβP (1-40) and (1-42) peptides. FASEB Journal. 2002;16(12):1526–1536. doi: 10.1096/fj.02-0829com. [DOI] [PubMed] [Google Scholar]
- 66.Grimm MOW, Grimm HS, Pätzold AJ, et al. Regulation of cholesterol and sphingomyelin metabolism by amyloid-β and presenilin. Nature Cell Biology. 2005;7(11):1118–1123. doi: 10.1038/ncb1313. [DOI] [PubMed] [Google Scholar]
- 67.Canepa E, Borghi R, Via J, et al. Cholesterol and amyloid-β: evidence for a cross-talk between astrocytes and neuronal cells. Journal of Alzheimer’s Disease. 2011;25(4):645–653. doi: 10.3233/JAD-2011-110053. [DOI] [PubMed] [Google Scholar]
- 68.Grimm MOW, Tschäpe JA, Grimm HS, Zinser EG, Hartmann T. Altered membrane fluidity and lipid raft composition in presenilin-deficient cells. Acta Neurologica Scandinavica. 2006;114(185):27–32. doi: 10.1111/j.1600-0404.2006.00682.x. [DOI] [PubMed] [Google Scholar]
- 69.Umeda T, Mori H, Zheng H, Tomiyama T. Regulation of cholesterol efflux by amyloid β secretion. Journal of Neuroscience Research. 2010;88(9):1985–1994. doi: 10.1002/jnr.22360. [DOI] [PubMed] [Google Scholar]
- 70.van de Parre TJL, Guns PJDF, Fransen P, et al. Attenuated atherogenesis in apolipoprotein E-deficient mice lacking amyloid precursor protein. Atherosclerosis. 2011;216(1):54–58. doi: 10.1016/j.atherosclerosis.2011.01.032. [DOI] [PubMed] [Google Scholar]
- 71.Nunes A, Pressey SNR, Cooper JD, Soriano S. Loss of amyloid precursor protein in a mouse model of Niemann-Pick type C disease exacerbates its phenotype and disrupts tau homeostasis. Neurobiology of Disease. 2011;42(3):349–359. doi: 10.1016/j.nbd.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 72.Liu Q, Zerbinatti CV, Zhang J, et al. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007;56(1):66–78. doi: 10.1016/j.neuron.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hashimoto M, Shahdat HM, Yamashita S, et al. Docosahexaenoic acid disrupts in vitro amyloid β1-40 fibrillation and concomitantly inhibits amyloid levels in cerebral cortex of Alzheimer’s disease model rats. Journal of Neurochemistry. 2008;107(6):1634–1646. doi: 10.1111/j.1471-4159.2008.05731.x. [DOI] [PubMed] [Google Scholar]
- 74.Grimm MOW, Kuchenbecker J, Grosgen S, et al. Docosahexaenoic acid reduces amyloid β production via multiple pleiotropic mechanisms. Journal of Biological Chemistry. 2011;286(16):14028–14039. doi: 10.1074/jbc.M110.182329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao Y, Calon F, Julien C, et al. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARγ-mediated mechanisms in Alzheimer’s disease models. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0015816.e15816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lukiw WJ, Cui JG, Marcheselli VL, et al. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. Journal of Clinical Investigation. 2005;115(10):2774–2783. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hjorth E, Zhu M, Toro VC, et al. Omega-3 Fatty Acids Enhance Phagocytosis of Alzheimer's Disease-Related Amyloid-beta42 by Human Microglia and Decrease Inflammatory Markers. Journal of Alzheimer's Disease. 2013;35(4):697–713. doi: 10.3233/JAD-130131. [DOI] [PubMed] [Google Scholar]
- 78.Labrousse VF, Nadjar A, Joffre C, et al. Short-term long chain omega3 diet protects from neuroinflammatory processes and memory impairment in aged mice. PLoS One. 2012;7(5) doi: 10.1371/journal.pone.0036861.e36861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma QL, Teter B, Ubeda OJ, et al. Omega-3 fatty acid docosahexaenoic acid increases SorLA/LR11, a sorting protein with reduced expression in sporadic Alzheimer’s disease (AD): relevance to AD prevention. Journal of Neuroscience. 2007;27(52):14299–14307. doi: 10.1523/JNEUROSCI.3593-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sparks DL, Scheff SW, Hunsaker JC, III, Liu H, Landers T, Gross DR. Induction of Alzheimer-like β-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Experimental Neurology. 1994;126(1):88–94. doi: 10.1006/exnr.1994.1044. [DOI] [PubMed] [Google Scholar]
- 81.Shie FS, Jin LW, Cook DG, Leverenz JB, LeBoeuf RC. Diet-induced hypercholesterolemia enhances brain Aβ accumulation in transgenic mice. NeuroReport. 2002;13(4):455–459. doi: 10.1097/00001756-200203250-00019. [DOI] [PubMed] [Google Scholar]
- 82.Refolo LM, Malester B, LaFrancois J, et al. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiology of Disease. 2000;7(4):321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- 83.Refolo LM, Pappolla MA, LaFrancois J, et al. A cholesterol-lowering drug reduces β-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiology of Disease. 2001;8(5):890–899. doi: 10.1006/nbdi.2001.0422. [DOI] [PubMed] [Google Scholar]
- 84.Kurata T, Miyazaki K, Kozuki M, et al. Atorvastatin and pitavastatin reduce senile plaques and inflammatory responses in a mouse model of Alzheimer's disease. Journal of Neurology Research. 2012;34(6):601–610. doi: 10.1179/1743132812Y.0000000054. [DOI] [PubMed] [Google Scholar]
- 85.Sato N, Shinohara M, Rakugi H, Morishita R. Dual effects of statins on Aβ metabolism: upregulation of the degradation of APP-CTF and Aβ clearance. Neurodegenerative Diseases. 2012;10(1–4):305–308. doi: 10.1159/000334534. [DOI] [PubMed] [Google Scholar]
- 86.Fassbender K, Simons M, Bergmann C, et al. Simvastatin strongly reduces levels of Alzheimer’s disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(10):5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cibickova L, Radomir H, Stanislav M, et al. The influence of simvastatin, atorvastatin and high-cholesterol diet on acetylcholinesterase activity, amyloid beta and cholesterol synthesis in rat brain. Steroids. 2009;74(1):13–19. doi: 10.1016/j.steroids.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 88.Park IH, Hwang EM, Hong HS, et al. Lovastatin enhances Aβ production and senile plaque deposition in female Tg2576 mice. Neurobiology of Aging. 2003;24(5):637–643. doi: 10.1016/s0197-4580(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 89.Haag MDM, Hofman A, Koudstaal PJ, Stricker BHC, Breteler MMB. Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam Study. Journal of Neurology, Neurosurgery and Psychiatry. 2009;80(1):13–17. doi: 10.1136/jnnp.2008.150433. [DOI] [PubMed] [Google Scholar]
- 90.Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Archives of Neurology. 2000;57(10):1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 91.Wolozin B, Wang SW, Li NC, Lee A, Lee TA, Kazis LE. Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Medicine. 2007;5(article 20) doi: 10.1186/1741-7015-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rea TD, Breitner JC, Psaty BM, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Archives of Neurology. 2005;62(7):1047–1051. doi: 10.1001/archneur.62.7.1047. [DOI] [PubMed] [Google Scholar]
- 93.Arvanitakis Z, Schneider JA, Wilson RS, et al. Statins, incident Alzheimer disease, change in cognitive function, and neuropathology. Neurology. 2008;70(19):1795–1802. doi: 10.1212/01.wnl.0000288181.00826.63. [DOI] [PubMed] [Google Scholar]
- 94.Feldman HH, Doody RS, Kivipelto M, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010;74(12):956–964. doi: 10.1212/WNL.0b013e3181d6476a. [DOI] [PubMed] [Google Scholar]
- 95.Sano M, Bell KL, Galasko D, et al. A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology. 2011;77(6):556–563. doi: 10.1212/WNL.0b013e318228bf11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pawlosky RJ, Hibbeln JR, Novotny JA, Salem N., Jr. Physiological compartmental analysis of α-linolenic acid metabolism in adult humans. Journal of Lipid Research. 2001;42(8):1257–1265. [PubMed] [Google Scholar]
- 97.Wassall SR, Stillwell W. Polyunsaturated fatty acid-cholesterol interactions: domain formation in membranes. Biochimica et Biophysica Acta. 2009;1788(1):24–32. doi: 10.1016/j.bbamem.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 98.Gamoh S, Hashimoto M, Sugioka K, et al. Chronic administration of docosahexaenoic acid improves reference memory-related learning ability in young rats. Neuroscience. 1999;93(1):237–241. doi: 10.1016/s0306-4522(99)00107-4. [DOI] [PubMed] [Google Scholar]
- 99.Katakura M, Hashimoto M, Shahdat HM, et al. Docosahexaenoic acid promotes neuronal differentiation by regulating basic helix-loop-helix transcription factors and cell cycle in neural stem cells. Neuroscience. 2009;160(3):651–660. doi: 10.1016/j.neuroscience.2009.02.057. [DOI] [PubMed] [Google Scholar]
- 100.Dagai L, Peri-Naor R, Birk RZ. Docosahexaenoic acid significantly stimulates immediate early response genes and neurite outgrowth. Neurochemical Research. 2009;34(5):867–875. doi: 10.1007/s11064-008-9845-z. [DOI] [PubMed] [Google Scholar]
- 101.Calon F, Lim GP, Yang F, et al. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron. 2004;43(5):633–645. doi: 10.1016/j.neuron.2004.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Soderberg M, Edlund C, Kristensson K, Dallner G. Fatty acid composition of brain phospholipids in aging and in Alzheimer’s disease. Lipids. 1991;26(6):421–425. doi: 10.1007/BF02536067. [DOI] [PubMed] [Google Scholar]
- 103.Montine TJ, Morrow JD. Fatty acid oxidation in the pathogenesis of Alzheimer’s disease. American Journal of Pathology. 2005;166(5):1283–1289. doi: 10.1016/S0002-9440(10)62347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Markesbery WR, Kryscio RJ, Lovell MA, Morrow JD. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Annals of Neurology. 2005;58(5):730–735. doi: 10.1002/ana.20629. [DOI] [PubMed] [Google Scholar]
- 105.Kalmijn S, Launer LJ, Ott A, Witteman JCM, Hofman A, Breteler MMB. Dietary fat intake and the risk of incident dementia in the Rotterdam study. Annals of Neurology. 1997;42(5):776–782. doi: 10.1002/ana.410420514. [DOI] [PubMed] [Google Scholar]
- 106.Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Diet and risk of dementia: does fat matter? The Rotterdam study. Neurology. 2002;59(12):1915–1921. doi: 10.1212/01.wnl.0000038345.77753.46. [DOI] [PubMed] [Google Scholar]
- 107.Barberger-Gateau P, Letenneur L, Deschamps V, Pérès K, Dartigues JF, Renaud S. Fish, meat, and risk of dementia: cohort study. British Medical Journal. 2002;325(7370):932–933. doi: 10.1136/bmj.325.7370.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Archives of Neurology. 2003;60(7):940–946. doi: 10.1001/archneur.60.7.940. [DOI] [PubMed] [Google Scholar]
- 109.Schaefer EJ, Bongard V, Beiser AS, et al. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and alzheimer disease: the framingham heart study. Archives of Neurology. 2006;63(11):1545–1550. doi: 10.1001/archneur.63.11.1545. [DOI] [PubMed] [Google Scholar]
- 110.Whalley LJ, Deary IJ, Starr JM, et al. n-3 Fatty acid erythrocyte membrane content, APOE ε4, and cognitive variation: an observational follow-up study in late adulthood. American Journal of Clinical Nutrition. 2008;87(2):449–454. doi: 10.1093/ajcn/87.2.449. [DOI] [PubMed] [Google Scholar]
- 111.Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39(3):409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 112.Green KN, Martinez-Coria H, Khashwji H, et al. Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-β and tau pathology via a mechanism involving presenilin 1 levels. Journal of Neuroscience. 2007;27(16):4385–4395. doi: 10.1523/JNEUROSCI.0055-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lim GP, Calon F, Morihara T, et al. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. Journal of Neuroscience. 2005;25(12):3032–3040. doi: 10.1523/JNEUROSCI.4225-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hashimoto M, Hossain S, Shimada T, Shido O. Docosahexaenoic acid-induced protective effect against impaired learning in amyloid β-infused rats is associated with increased synaptosomal membrane fluidity. Clinical and Experimental Pharmacology and Physiology. 2006;33(10):934–939. doi: 10.1111/j.1440-1681.2006.04467.x. [DOI] [PubMed] [Google Scholar]
- 115.Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(22):8491–8496. doi: 10.1073/pnas.0402531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Freund-Levi Y, Eriksdotter-Jönhagen M, Cederholm T, et al. ω-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: omegAD study—a randomized double-blind trial. Archives of Neurology. 2006;63(10):1402–1408. doi: 10.1001/archneur.63.10.1402. [DOI] [PubMed] [Google Scholar]
- 117.Kotani S, Sakaguchi E, Warashina S, et al. Dietary supplementation of arachidonic and docosahexaenoic acids improves cognitive dysfunction. Neuroscience Research. 2006;56(2):159–164. doi: 10.1016/j.neures.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 118.Chiu CC, Su KP, Cheng TC, et al. The effects of omega-3 fatty acids monotherapy in Alzheimer’s disease and mild cognitive impairment: a preliminary randomized double-blind placebo-controlled study. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2008;32(6):1538–1544. doi: 10.1016/j.pnpbp.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 119.Scheltens P, Kamphuis PJGH, Verhey FRJ, et al. Efficacy of a medical food in mild Alzheimer’s disease: a randomized, controlled trial. Alzheimer’s and Dementia. 2010;6(1):1–10. doi: 10.1016/j.jalz.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 120.Scheltens P, Twisk JW, Blesa R, et al. Efficacy of Souvenaid in mild Alzheimer's disease: results from a randomized, controlled trial. Journal of Alzheimer's Disease. 2012;31(1):225–236. doi: 10.3233/JAD-2012-121189. [DOI] [PubMed] [Google Scholar]
- 121.Scheltens P. Alzheimer’s and Parkinson’s diseases: mechanisms, clinical strategies, and promising treatments of neurodegenerative diseases. Proceedings of the 11th International Conference AD/PD; 2013; Florence, Italy. Karger; [Google Scholar]
- 122.Grimm MOW, Grsgen S, Rothhaar TL, et al. Intracellular APP domain regulates serine-palmitoyl-CoA transferase expression and is affected in alzheimer’s disease. International Journal of Alzheimer’s Disease. 2011;2011:8 pages. doi: 10.4061/2011/695413.695413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tarasiuk J, Kapica-Topczewska K, Kulakowska A, et al. Increased concentration of the CSF Tau protein and its phosphorylated form in the late juvenile metachromatic leukodystrophy form: a case report. Journal of Neural Transmission. 2012;119(7):759–762. doi: 10.1007/s00702-012-0826-7. [DOI] [PubMed] [Google Scholar]
- 124.Keilani S, Lun Y, Stevens AC, et al. Lysosomal dysfunction in a mouse model of sandhoff disease leads to accumulation of ganglioside-bound amyloid-β peptide. Journal of Neuroscience. 2012;32(15):5223–5236. doi: 10.1523/JNEUROSCI.4860-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Han X, Holtzman DM, McKeel DW, Jr., Kelley J, Morris JC. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. Journal of Neurochemistry. 2002;82(4):809–818. doi: 10.1046/j.1471-4159.2002.00997.x. [DOI] [PubMed] [Google Scholar]
- 126.Taki T. An approach to glycobiology from glycolipidomics: ganglioside molecular scanning in the brains of patients with Alzheimer's disease by TLC-blot/matrix assisted laser desorption/ionization-time of flight MS. Biological & Pharmaceutical Bulletin. 2012;35(10):1642–1647. doi: 10.1248/bpb.b12-00400. [DOI] [PubMed] [Google Scholar]
- 127.Pernber Z, Blennow K, Bogdanovic N, Mansson JE, Blomqvist M. Altered distribution of the gangliosides GM1 and GM2 in Alzheimer's disease. Dementia and Geriatric Cognitive Disorders. 2012;33(2-3):174–188. doi: 10.1159/000338181. [DOI] [PubMed] [Google Scholar]
- 128.Filippov V, Song MA, Zhang K, et al. Increased ceramide in brains with alzheimer’s and other neurodegenerative diseases. Journal of Alzheimer’s Disease. 2012;29(3):537–547. doi: 10.3233/JAD-2011-111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hejazi L, Wong JWH, Cheng D, et al. Mass and relative elution time profiling: two-dimensional analysis of sphingolipids in Alzheimer’s disease brains. Biochemical Journal. 2011;438(1):165–175. doi: 10.1042/BJ20110566. [DOI] [PubMed] [Google Scholar]
- 130.Martín V, Fabelo N, Santpere G, et al. Lipid alterations in lipid rafts from Alzheimer’s disease human brain cortex. Journal of Alzheimer’s Disease. 2010;19(2):489–502. doi: 10.3233/JAD-2010-1242. [DOI] [PubMed] [Google Scholar]
- 131.He X, Huang Y, Li B, Gong CX, Schuchman EH. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiology of Aging. 2010;31(3):398–408. doi: 10.1016/j.neurobiolaging.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cutler RG, Kelly J, Storie K, et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(7):2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Katsel P, Li C, Haroutunian V. Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: a shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochemical Research. 2007;32(4-5):845–856. doi: 10.1007/s11064-007-9297-x. [DOI] [PubMed] [Google Scholar]
- 134.Mielke MM, Bandaru VVR, Haughey NJ, et al. Serum ceramides increase the risk of Alzheimer disease: the Women's Health and Aging Study II. Neurology. 2012;79(7):633–641. doi: 10.1212/WNL.0b013e318264e380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Barrier L, Ingrand S, Fauconneau B, Page G. Gender-dependent accumulation of ceramides in the cerebral cortex of the APPSL/PS1Ki mouse model of Alzheimer’s disease. Neurobiology of Aging. 2010;31(11):1843–1853. doi: 10.1016/j.neurobiolaging.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 136.Barrier L, Fauconneau B, Nol A, Ingrand S. Ceramide and related-sphingolipid levels are not altered in disease-associated brain regions of APPSL and APPSL/PS1APPM146L mouse models of Alzheimer’s disease: relationship with the lack of neurodegeneration? International Journal of Alzheimer’s Disease. 2011;2011:10 pages. doi: 10.4061/2011/920958.920958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dawson G, Goswami R, Kilkus J, Wiesner D, Dawson S. The formation of ceramide from sphingomyelin is associated with cellular apoptosis. Acta Biochimica Polonica. 1998;45(2):287–297. [PubMed] [Google Scholar]
- 138.Toman RE, Movsesyan V, Murthy SK, Milstien S, Spiegel S, Faden AI. Ceramide-induced cell death in primary neuronal cultures: upregulation of ceramide levels during neuronal apoptosis. Journal of Neuroscience Research. 2002;68(3):323–330. doi: 10.1002/jnr.10190. [DOI] [PubMed] [Google Scholar]
- 139.Zhang X, Wu J, Dou Y, et al. Asiatic acid protects primary neurons against C2-ceramide-induced apoptosis. European Journal of Pharmacology. 2012;679(1–3):51–59. doi: 10.1016/j.ejphar.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 140.Lee JT, Xu J, Lee JM, et al. Amyloid-β peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. Journal of Cell Biology. 2004;164(1):123–131. doi: 10.1083/jcb.200307017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zinser EG, Hartmann T, Grimm MOW. Amyloid beta-protein and lipid metabolism. Biochimica et Biophysica Acta. 2007;1768(8):1991–2001. doi: 10.1016/j.bbamem.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 142.Wang G, Dinkins M, He Q, et al. Astrocytes secrete exosomes enriched with proapoptotic ceramide and prostate apoptosis response 4 (PAR-4): potential mechanism of apoptosis induction in Alzheimer disease (AD) Journal of Biological Chemistry. 2012;287(25):21384–21395. doi: 10.1074/jbc.M112.340513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Trajkovic K, Hsu C, Chiantia S, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319(5867):1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- 144.Puglielli L, Ellis BC, Saunders AJ, Kovacs DM. Ceramide stabilizes β-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid β-peptide biogenesis. Journal of Biological Chemistry. 2003;278(22):19777–19783. doi: 10.1074/jbc.M300466200. [DOI] [PubMed] [Google Scholar]
- 145.Mi HK, Puglielli L. Two Endoplasmic Reticulum (ER)/ER golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. Journal of Biological Chemistry. 2009;284(4):2482–2492. doi: 10.1074/jbc.M804901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Edsall LC, Pirianov GG, Spiegel S. Involvement of sphingosine 1-phosphate in nerve growth factor-mediated neuronal survival and differentiation. Journal of Neuroscience. 1997;17(18):6952–6960. doi: 10.1523/JNEUROSCI.17-18-06952.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rius RA, Edsall LC, Spiegel S. Activation of sphingosine kinase in pheochromocytoma PC12 neuronal cells in response to trophic factors. FEBS Letters. 1997;417(2):173–176. doi: 10.1016/s0014-5793(97)01277-5. [DOI] [PubMed] [Google Scholar]
- 148.Takasugi N, Sasaki T, Suzuki K, et al. BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. Journal of Neuroscience. 2011;31(18):6850–6857. doi: 10.1523/JNEUROSCI.6467-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ben-David O, Futerman AH. The role of the ceramide acyl chain length in neurodegeneration: involvement of ceramide synthases. NeuroMolecular Medicine. 2010;12(4):341–350. doi: 10.1007/s12017-010-8114-x. [DOI] [PubMed] [Google Scholar]
- 150.Bandaru VVR, Troncoso J, Wheeler D, et al. ApoE4 disrupts sterol and sphingolipid metabolism in Alzheimer’s but not normal brain. Neurobiology of Aging. 2009;30(4):591–599. doi: 10.1016/j.neurobiolaging.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pettegrew JW, Panchalingam K, Hamilton RL, Mcclure RJ. Brain membrane phospholipid alterations in Alzheimer’s disease. Neurochemical Research. 2001;26(7):771–782. doi: 10.1023/a:1011603916962. [DOI] [PubMed] [Google Scholar]
- 152.Kosicek M, Zetterberg H, Andreasen N, Peter-Katalinic J, Hecimovic S. Elevated cerebrospinal fluid sphingomyelin levels in prodromal Alzheimer’s disease. Neuroscience Letters. 2012;516(2):302–305. doi: 10.1016/j.neulet.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 153.Mielke MM, Haughey NJ, Bandaru VVR, et al. Plasma sphingomyelins are associated with cognitive progression in alzheimer’s disease. Journal of Alzheimer’s Disease. 2011;27(2):259–269. doi: 10.3233/JAD-2011-110405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sheikh KA, Sun J, Liu Y, et al. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(13):7532–7537. doi: 10.1073/pnas.96.13.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yamashita T, Wada R, Sasaki T, et al. A vital role for glycosphingolipid synthesis during development and differentiation. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(16):9142–9147. doi: 10.1073/pnas.96.16.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lahiri S, Futerman AH. The metabolism and function of sphingolipids and glycosphingolipids. Cellular and Molecular Life Sciences. 2007;64(17):2270–2284. doi: 10.1007/s00018-007-7076-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387(6633):569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 158.Kracun I, Rosner H, Drnovsek V, Heffer-Lauc M, Cosovic C, Lauc G. Human brain gangliosides in development, aging and disease. International Journal of Developmental Biology. 1991;35(3):289–295. [PubMed] [Google Scholar]
- 159.Chan RB, Oliveira TG, Cortes EP, et al. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. Journal of Biological Chemistry. 2012;287(4):2678–2688. doi: 10.1074/jbc.M111.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gottfries CG, Karlsson I, Svennerholm L. Membrane components separate early-onset Alzheimer’s disease from senile dementia of the Alzheimer type. International Psychogeriatrics. 1996;8(3):365–372. doi: 10.1017/s1041610296002736. [DOI] [PubMed] [Google Scholar]
- 161.Molander-Melin M, Blennow K, Bogdanovic N, Dellheden B, Månsson JE, Fredman P. Structural membrane alterations in Alzheimer brains found to be associated with regional disease development; increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent-resistant membrane domains. Journal of Neurochemistry. 2005;92(1):171–182. doi: 10.1111/j.1471-4159.2004.02849.x. [DOI] [PubMed] [Google Scholar]
- 162.Barrier L, Ingrand S, Damjanac M, Rioux Bilan A, Hugon J, Page G. Genotype-related changes of ganglioside composition in brain regions of transgenic mouse models of Alzheimer’s disease. Neurobiology of Aging. 2007;28(12):1863–1872. doi: 10.1016/j.neurobiolaging.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 163.Grimm MOW, Zinser EG, Grösgen S, et al. Amyloid precursor protein (app) mediated regulation of ganglioside homeostasis linking alzheimer’s disease pathology with ganglioside metabolism. PLoS ONE. 2012;7(3) doi: 10.1371/journal.pone.0034095.e34095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zha Q, Ruan Y, Hartmann T, Beyreuther K, Zhang D. GM1 ganglioside regulates the proteolysis of amyloid precursor protein. Molecular Psychiatry. 2004;9(10):946–952. doi: 10.1038/sj.mp.4001509. [DOI] [PubMed] [Google Scholar]
- 165.Holmes O, Paturi S, Ye W, Wolfe MS, Selkoe DJ. Effects of membrane lipids on the activity and processivity of purified γ-secretase. Biochemistry. 2012;51(17):3565–3575. doi: 10.1021/bi300303g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Peters I, Igbavboa U, Schütt T, et al. The interaction of beta-amyloid protein with cellular membranes stimulates its own production. Biochimica et Biophysica Acta. 2009;1788(5):964–972. doi: 10.1016/j.bbamem.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tamboli IY, Prager K, Barth E, Heneka M, Sandhoff K, Walter J. Inhibition of glycosphingolipid biosynthesis reduces secretion of the β-amyloid precursor protein and amyloid β-peptide. Journal of Biological Chemistry. 2005;280(30):28110–28117. doi: 10.1074/jbc.M414525200. [DOI] [PubMed] [Google Scholar]
- 168.Yanagisawa K, Odaka A, Suzuki N, Ihara Y. GM1 ganglioside-bound amyloid β-protein (Aβ): a possible form of preamyloid in Alzheimer’s disease. Nature Medicine. 1995;1(10):1062–1066. doi: 10.1038/nm1095-1062. [DOI] [PubMed] [Google Scholar]
- 169.Kakio A, Nishimoto SI, Yanagisawa K, Kozutsumi Y, Matsuzaki K. Interactions of amyloid β-protein with various gangliosides in raft-like membranes: importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry. 2002;41(23):7385–7390. doi: 10.1021/bi0255874. [DOI] [PubMed] [Google Scholar]
- 170.Utsumi M, Yamaguchi Y, Sasakawa H, Yamamoto N, Yanagisawa K, Kato K. Up-and-down topological mode of amyloid β-peptide lying on hydrophilic/hydrophobic interface of ganglioside clusters. Glycoconjugate Journal. 2009;26(8):999–1006. doi: 10.1007/s10719-008-9216-7. [DOI] [PubMed] [Google Scholar]
- 171.Mahfoud R, Garmy N, Maresca M, Yahi N, Puigserver A, Fantini J. Identification of a common sphingolipid-binding domain in Alzheimer, prion, and HIV-1 proteins. Journal of Biological Chemistry. 2002;277(13):11292–11296. doi: 10.1074/jbc.M111679200. [DOI] [PubMed] [Google Scholar]
- 172.Wakabayashi M, Okada T, Kozutsumi Y, Matsuzaki K. GM1 ganglioside-mediated accumulation of amyloid β-protein on cell membranes. Biochemical and Biophysical Research Communications. 2005;328(4):1019–1023. doi: 10.1016/j.bbrc.2005.01.060. [DOI] [PubMed] [Google Scholar]
- 173.Lemkul JA, Bevan DR. Lipid composition influences the release of Alzheimer’s amyloid β-peptide from membranes. Protein Science. 2011;20(9):1530–1545. doi: 10.1002/pro.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Okada T, Wakabayashi M, Ikeda K, Matsuzaki K. Formation of toxic fibrils of Alzheimer’s amyloid β-protein-(1-40) by monosialoganglioside GM1, a neuronal membrane component. Journal of Molecular Biology. 2007;371(2):481–489. doi: 10.1016/j.jmb.2007.05.069. [DOI] [PubMed] [Google Scholar]
- 175.Hayashi H, Kimura N, Yamaguchi H, et al. A seed for Alzheimer amyloid in the brain. Journal of Neuroscience. 2004;24(20):4894–4902. doi: 10.1523/JNEUROSCI.0861-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yamamoto N, Matsubara T, Sato T, Yanagisawa K. Age-dependent high-density clustering of GM1 ganglioside at presynaptic neuritic terminals promotes amyloid β-protein fibrillogenesis. Biochimica et Biophysica Acta. 2008;1778(12):2717–2726. doi: 10.1016/j.bbamem.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 177.Probst A, Langui D, Ipsen S, Robakis N, Ulrich J. Deposition of β/A4 protein along neuronal plasma membranes in diffuse senile plaques. Acta Neuropathologica. 1991;83(1):21–29. doi: 10.1007/BF00294426. [DOI] [PubMed] [Google Scholar]
- 178.Bugiani O, Giaccone G, Verga L, et al. Alzheimer patients and Down patients: abnormal presynaptic terminals are related to cerebral preamyloid deposits. Neuroscience Letters. 1990;119(1):56–59. doi: 10.1016/0304-3940(90)90754-w. [DOI] [PubMed] [Google Scholar]
- 179.Eckhardt M. The role and metabolism of sulfatide in the nervous system. Molecular Neurobiology. 2008;37(2-3):93–103. doi: 10.1007/s12035-008-8022-3. [DOI] [PubMed] [Google Scholar]
- 180.Mitew S, Kirkcaldie MTK, Halliday GM, Shepherd CE, Vickers JC, Dickson TC. Focal demyelination in Alzheimer’s disease and transgenic mouse models. Acta Neuropathologica. 2010;119(5):567–577. doi: 10.1007/s00401-010-0657-2. [DOI] [PubMed] [Google Scholar]
- 181.Han X, Cheng H, Fryer JD, Fagan AM, Holtzman DM. Novel role for apolipoprotein E in the central nervous system: modulation of sulfatide content. Journal of Biological Chemistry. 2003;278(10):8043–8051. doi: 10.1074/jbc.M212340200. [DOI] [PubMed] [Google Scholar]
- 182.Cheng H, Zhou Y, Holtzman DM, Han X. Apolipoprotein E mediates sulfatide depletion in animal models of Alzheimer’s disease. Neurobiology of Aging. 2010;31(7):1188–1196. doi: 10.1016/j.neurobiolaging.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zeng Y, Han X. Sulfatides facilitate apolipoprotein E-mediated amyloid-β peptide clearance through an endocytotic pathway. Journal of Neurochemistry. 2008;106(3):1275–1286. doi: 10.1111/j.1471-4159.2008.05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Grundke-Iqbal I, Iqbal K, Tung YC. Abnormal phosphorylation of the microtubule-associated protein τ (tau) in Alzheimer cytoskeletal pathology. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(13):44913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Grundke-Iqbal I, Iqbal K, Quinlan M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. Journal of Biological Chemistry. 1986;261(13):6084–6089. [PubMed] [Google Scholar]
- 186.Lee VMY, Balin BJ, Otvos L, Jr., Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science. 1991;251(4994):675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 187.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proceedings of the National Academy of Sciences of the United States of America. 1975;72(5):1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochemical Journal. 1997;323(part 3):577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Alonso ADC, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(12):6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Del C. Alonso A, Grundke-Iqbal I, Iqbal K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nature Medicine. 1996;2(7):783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- 191.Alonso ADC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(12):5562–5566. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kawarabayashi T, Shoji M, Younkin LH, et al. Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. Journal of Neuroscience. 2004;24(15):3801–3809. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hernandez P, Lee G, Sjoberg M, MacCioni RB. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Aβ25-35: involvement of lipid rafts. Journal of Alzheimer’s Disease. 2009;16(1):149–156. doi: 10.3233/JAD-2009-0933. [DOI] [PubMed] [Google Scholar]
- 194.Distl R, Treiber-Held S, Albert F, Meske V, Harzer K, Ohm TG. Cholesterol storage and tau pathology in Niemann-Pick type C disease in the brain. Journal of Pathology. 2003;200(1):104–111. doi: 10.1002/path.1320. [DOI] [PubMed] [Google Scholar]
- 195.Love S, Bridges LR, Case CP. Neurofibrillary tangles in Niemann-Pick disease type C. Brain. 1995;118(part 1):119–129. doi: 10.1093/brain/118.1.119. [DOI] [PubMed] [Google Scholar]
- 196.Distl R, Meske V, Ohm TG. Tangle-bearing neurons contain more free cholesterol than adjacent tangle-free neurons. Acta Neuropathologica. 2001;101(6):547–554. doi: 10.1007/s004010000314. [DOI] [PubMed] [Google Scholar]
- 197.Mattsson N, Zetterberg H, Bianconi S, et al. γ-Secretase-dependent amyloid-β is increased in Niemann-Pick type C: a cross-sectional study. Neurology. 2011;76(4):366–372. doi: 10.1212/WNL.0b013e318208f4ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sawamura N, Gong JS, Garver WS, et al. Site-specific Phosphorylation of Tau Accompanied by Activation of Mitogen-activated Protein Kinase (MAPK) in Brains of Niemann-Pick Type C Mice. Journal of Biological Chemistry. 2001;276(13):10314–10319. doi: 10.1074/jbc.M009733200. [DOI] [PubMed] [Google Scholar]
- 199.Glöckner F, Meske V, Lütjohann D, Ohm TG. Dietary cholesterol and its effect on tau protein: a study in apolipoprotein e-deficient and P301L human tau mice. Journal of Neuropathology and Experimental Neurology. 2011;70(4):292–301. doi: 10.1097/NEN.0b013e318212f185. [DOI] [PubMed] [Google Scholar]
- 200.Rahman A, Akterin S, Flores-Morales A, et al. High cholesterol diet induces tau hyperphosphorylation in apolipoprotein e deficient mice. FEBS Letters. 2005;579(28):6411–6416. doi: 10.1016/j.febslet.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 201.Bhat NR, Thirumangalakudi L. Increased tau phosphorylation and impaired brain insulin/IGF signaling in mice fed a high fat/high cholesterol diet. Journal of Alzheimer's Disease. 2013 doi: 10.3233/JAD-2012-121030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Boimel M, Grigoriadis N, Lourbopoulos A, et al. Statins reduce the neurofibrillary tangle burden in a mouse model of tauopathy. Journal of Neuropathology and Experimental Neurology. 2009;68(3):314–325. doi: 10.1097/NEN.0b013e31819ac3cb. [DOI] [PubMed] [Google Scholar]
- 203.Riekse RG, Li G, Petrie EC, et al. Effect of statins on Alzheimer’s disease biomarkers in cerebrospinal fluid. Journal of Alzheimer’s Disease. 2006;10(4):399–406. doi: 10.3233/jad-2006-10408. [DOI] [PubMed] [Google Scholar]
- 204.Nicholson AM, Ferreira A. Increased membrane cholesterol might render mature hippocampal neurons more susceptible to β-Amyloid-induced calpain activation and tau toxicity. Journal of Neuroscience. 2009;29(14):4640–4651. doi: 10.1523/JNEUROSCI.0862-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates β-amyloid-induced neurodegeneration. Journal of Neuroscience. 2005;25(22):5365–5375. doi: 10.1523/JNEUROSCI.1125-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ferreira A, Bigio EH. Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Molecular Medicine. 2011;17(7-8):676–685. doi: 10.2119/molmed.2010.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Thornton C, Bright NJ, Sastre M, Muckett PJ, Carling D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid β-peptide exposure. Biochemical Journal. 2011;434(3):503–512. doi: 10.1042/BJ20101485. [DOI] [PubMed] [Google Scholar]
- 208.Cai Z, Yan LJ, Li K, Quazi SH, Zhao B. Roles of AMP-activated protein kinase in Alzheimer’s disease. NeuroMolecular Medicine. 2012;14(1):1–14. doi: 10.1007/s12017-012-8173-2. [DOI] [PubMed] [Google Scholar]
- 209.Vingtdeux V, Davies P, Dickson DW, Marambaud P. AMPK is abnormally activated in tangle-and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathologica. 2011;121(3):337–349. doi: 10.1007/s00401-010-0759-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Reynolds CH, Betts JC, Blackstock WP, Nebreda AR, Anderton BH. Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3β . Journal of Neurochemistry. 2000;74(4):1587–1595. doi: 10.1046/j.1471-4159.2000.0741587.x. [DOI] [PubMed] [Google Scholar]
- 211.Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochemical and Biophysical Research Communications. 2009;380(1):98–104. doi: 10.1016/j.bbrc.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ma QL, Yang F, Rosario ER, et al. β-Amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. Journal of Neuroscience. 2009;29(28):9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Julien C, Tremblay C, Phivilay A, et al. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiology of Aging. 2010;31(9):1516–1531. doi: 10.1016/j.neurobiolaging.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 214.Gellermann GP, Appel TR, Davies P, Diekmann S. Paired helical filaments contain small amounts of cholesterol, phosphatidylcholine and sphingolipids. Biological Chemistry. 2006;387(9):1267–1274. doi: 10.1515/BC.2006.157. [DOI] [PubMed] [Google Scholar]
- 215.Tooyama I, Yamada T, Kim SU, McGeer PL. Immunohistochemical study of A2B5-positive ganglioside in postmortem human brain tissue of Alzheimer disease, amyotrophic lateral sclerosis, progessive supranuclear palsy and control cases. Neuroscience Letters. 1992;136(1):91–94. doi: 10.1016/0304-3940(92)90655-q. [DOI] [PubMed] [Google Scholar]
- 216.Geekiyanage H, Upadhye A, Chan C. Inhibition of serine palmitoyltransferase reduces Abeta and tau hyperphosphorylation in a murine model: a safe therapeutic strategy for Alzheimer’s disease. Neurobiol Aging. 2013;34(8):2037–2051. doi: 10.1016/j.neurobiolaging.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Xie HQ, Johnson GVW. Ceramide selectively decreases tau levels in differentiated PC12 cells through modulation of calpain I. Journal of Neurochemistry. 1997;69(3):1020–1030. doi: 10.1046/j.1471-4159.1997.69031020.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material provides a tabular overview of the proposed mechanisms of cholesterol on APP processing with selected publications included.