

Abstract
Rationale
Foam cell formation due to excessive accumulation of cholesterol by macrophages is a pathological hallmark of atherosclerosis, the major cause of morbidity and mortality in Western societies. Liver X nuclear receptors (LXRs) regulate the expression of the adenosine triphosphate-binding cassette (ABC) transporters, including ABCA1 and ABCG1. ABCA1 and ABCG1 facilitate the efflux of cholesterol from macrophages and regulate high-density lipoprotein (HDL) biogenesis. Increasing evidence supports the role of microRNA (miRNAs) in regulating cholesterol metabolism through ABC transporters.
Objective
We aimed to identify novel miRNAs that regulate cholesterol metabolism in macrophages stimulated with LXR agonists.
Methods and Results
To map the miRNA expression signature of macrophages stimulated with LXR agonists, we performed a miRNA profiling microarray analysis in primary mouse peritoneal macrophages stimulated with LXR ligands. We report that LXR ligands increase miR-144 expression in macrophages and mouse livers. Overexpression of miR-144 reduces ABCA1 expression and attenuates cholesterol efflux to ApoA1 in macrophages. Delivery of miR-144 oligonucleotides to mice attenuates ABCA1 expression in the liver, reducing HDL levels. Conversely, silencing of miR-144 in mice increases the expression of ABCA1 and plasma HDL levels. Thus, miR-144 appears to regulate both macrophage cholesterol efflux and HDL biogenesis in the liver.
Conclusions
1) miR-144 regulates cholesterol metabolism via suppressing ABCA1 expression; and 2) modulation of miRNAs may represent a potential therapeutical intervention for treating dyslipidemia and atherosclerotic vascular disease.
Keywords: ABCA1, cholesterol efflux, HDL, microRNAs, cardiovascular research, lipids and lipoprotein metabolism, cholesterol homeostatis
INTRODUCTION
Foam cell formation due to excessive accumulation of cholesterol by macrophages is a pathological hallmark of atherosclerosis, the major cause of morbidity and mortality in Western societies 1, 2. Macrophages cannot limit the uptake of cholesterol and therefore depend on cholesterol efflux pathways for preventing their transformation into foam cells 1, 2. Several ABC transporters, including ABCA1 and ABCG1, facilitate the efflux of cholesterol from macrophages. ABCA1 and ABCG1 are thought to act in sequence to lipidate nascent and then mature HDL to generate larger α-HDL particles destined for clearance by the liver 3-5. Mutations in the Abca1 gene cause Tangier disease, which is characterized by defects in cholesterol efflux and cholesterol ester accumulation in macrophages, and increase the risk of developing atherosclerosis 6-8. In the liver, ABCA1 also plays a critical role in the biogenesis of HDL and its deficiency leads to a dramatic reduction of plasma HDL levels.
The expression of both ABCA1 and ABCG1 are up-regulated in states of cholesterol excess by LXR 9. LXRs are activated by oxysterol metabolites of cholesterol and play key roles in regulating multiple components of the reverse cholesterol transport (RCT) pathway, cholesterol conversion to bile acid, and intestinal cholesterol absorption 5, 10. Moreover, LXR also regulates cellular cholesterol homeostasis by activating the transcription of the inducible degrader of the LDLr (IDOL), an E3 ubiquitin ligase that triggers ubiquitination of the LDLr on its cytoplasmatic domain, thereby targeting it for degradation 11.
Over the past decade, it has become progressively more clear that a large class of small noncoding RNAs, known as microRNAs (miRNAs), function as important regulators of a wide range of cellular processes by modulating gene expression 12. Generally, miRNAs regulate gene expression post-transcriptionally by base-pairing to target mRNAs 13. In animals, most investigated miRNAs form imperfect hybrids with sequences in the 3’-untranslated region (3’UTR), with the miRNA 5’-proximal “seed” region (positions 2-8) providing most of the pairing specificity 13, 14. Imperfections in the central portion of miRNA-mRNA duplexes preclude RNAi-like cleavage. Instead, the miRNA association results in translational repression, frequently accompanied by a considerable degradation of the mRNA by a non-RNAi mechanism 13, 14. To date, several miRNAs have been described to regulate lipid metabolism including miR-122, miR-370, miR-378/378*, miR-758, miR-106 and miR-33 15-23. Recently, our group and others identified miR-33a/b as intronic miRNAs located within the sterol response element binding protein (SREBP) genes, Srebp1 and Srebp223. These loci encode for the membrane-bound transcription factors SREBP1 and SREBP2, which activate the synthesis of fatty acids and the synthesis and uptake of cholesterol. Coincident with the transcription of Srebp1 and Srebp2, the embedded miR-33a and miR-33b are transcribed, and these negative regulators act to repress a number of genes involved in regulating cellular cholesterol export and fatty acid oxidation including Abca1, Abcg1, Niemann-Pick C1 (Npc1), carnitine palmitoyltransferase 1A (Cpt1a), carnitine O-octanyl transferase (Crot), hydroxyacyl-CoA dehydrogenase-3-ketoacyl-CoA thiolase-enoyl-CoA hydratase (trifunctional protein) β-subunit (Hadhb) and 5’ AMP-activated protein kinase (Ampk)23, 24. Antagonists of miR-33 in mice increase liver and macrophage ABCA1 expression and promote RCT and regression of atherosclerosis 25. Of note, inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers very low-density lipoproteins (VLDL) triglycerides (TG) 26. Altogether these data suggest that antagonism of endogenous miR-33 may be useful as a therapeutic strategy for treating metabolic syndrome and atherosclerosis.
In addition to miR-33, miR-758 and miR-106b have recently been shown to regulate the expression of ABCA1 at the post-transcriptional level 19, 22. miR-758 is down-regulated after cholesterol loading in macrophages and in the liver of mice fed a high fat diet 22. Over-expression of miR-758 and miR-106b reduce ABCA1 expression in macrophage, hepatic and neuronal cell lines 19, 22. Thus, the post-transcriptional regulation of ABCA1 expression by miRNAs appears to be complex and mediated by multiple miRNAs.
In the current study, we present evidence that ABCA1 is post-transcriptionally regulated by miR-144 in vitro and in vivo. miR-144 over-expression inhibits ABCA1 expression in different cell lines including hepatocytes and monocyte/macrophages, thereby attenuating cholesterol efflux to apolipoprotein A1 (ApoA1). Most importantly, in vivo delivery of miR-144 to mice represses ABCA1 expression in the liver, reducing circulating HDL levels. Conversely, silencing of miR-144 in mice increases the expression of ABCA1 and plasma HDL levels. Thus, miR-144 appears to regulate both macrophage cholesterol efflux and HDL biogenesis in the liver. We also report that LXR ligands increase miR-144 in macrophages and mouse livers and that ABCA1 is a target of LXR-induced miR-144. These data reveal how an inducible miRNA makes up a negative feedback loop to ensure a tight regulation of cholesterol homeostasis.
METHODS
Due to space limits, detailed description of the materials and methods is presented in the supplemental material
Animals
Male C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and LXRα,β null mice were kindly provided by David Mangelsdorf. Eight-week-old male C57BL/6 mice were randomized into 4 groups (n= 24 mice): non-targeting control mimic (Con-mir, n=6), miR-144-mimic (miR-144, n=6), inhibitor negative control sequence (Con-inh, n=6) and miR-144-inhibitors (Inh-miR-144). mirVana miRNA mimics and inhibitors (Life Technologies) were complexed with Invivofectamine 2.0 reagent (Invitrogen) and injected intravenously twice (every three days) at 7 mg/kg dose. All animals were kept under constant temperature and humidity in a 12 h controlled dark/light cycle. Mice were fed a standard pellet diet. In another set of experiments mice were treated with LXR synthetic agonist T090 (10 mg/kg body weight) by oral gavage two days after injection with Con-inh or inh-miR-144. After six days mice were fasted for 12-14 h before blood samples were collected by retroorbital venous plexus puncture. Plasma cholesterol levels and lipoprotein fractionation were analyzed as described below. All animal experiments were approved by the Institutional Animal Care Use Committee of New York University Medical Center.
miRNA microarray analysis
Mouse peritoneal macrophages were stimulated with 3 μmol/L T090 for 24 h. Total RNA was extracted using TRIzol (Invitrogen) and microRNA was purified from 40 μg of total RNA using the miRNA Isolation Kit (Qiagen). The purity and integrity of both the total RNA sample and the enriched miRNA was verified using the Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). Mouse peritoneal macrophage miRNAs were amplified and hybridized to Illumina expression profiling microarrays according to the manufacturer’s directions. Quadruplicates for each condition were used for microarray analysis. Raw data were normalized and analyzed by GeneSpring GX software version 11.5 (Agilent Technologies). MicroRNAs showing an altered expression in T090 treatment compared to control were identified using a t-test unpaired with p value <0.2 and fold change >= 1.5, asymptotic p-value computation and no multiple testing correction. The 1.5-fold-change threshold was chosen on the basis of its use in previously published articles employing these particular types of microarrays. Hierarchical cluster analysis on average value per condition was performed using average linkage and Pearson uncentered correlation as a measure of similarity.
miR-144 mimic and anti-miR-144 transfection
Mouse peritoneal macrophages, J774, THP-1, HepG2, Huh-7, Hepa, and EAhy926 cells were transfected with 40 nM miRIDIAN miRNA mimic (miR-144) or with 60 nM miRIDIAN miRNA inhibitor (Inh-miR-144) (Dharmacon) utilizing RNAimax (Invitrogen). For co-transfection experiments with miR-33 and miR-144 mimics, we used 2.5 nM each. All experimental control samples were treated with an equal concentration of a non-targeting control mimic sequence (Con-miR) or inhibitor negative control sequence (Con-inh), for use as controls for non-sequence-specific effects in miRNA experiments. Verification of miR-144 over-expression and knockdown was determined using qPCR, as described above.
miR-144 and anti-miR-144 particle delivery in vivo
The mirVana miRNA inhibitors and mimics (Life Technologies) were complexed with Invivofectamine 2.0 reagent (Invitrogen) to form the nanoparticles suitable for in vivo applications. miRNA oligonucleotides (3 mg/ml in water, 750 μL) were mixed with manufacturer’s complexation buffer (750 μL), then Invivofectamine 2.0 reagent was added (1500 μL). After 30 min incubation at 50°C, dialysis was performed in 4 L of PBS to remove excessive salts and solvents. The resulting miRNA mimic/inhibitor concentration was 0.7 mg/ml, and after a 200 μl injection in the tail vein of a 20 g mouse, it resulted in a 7 mg/kg body weight miRNA dose (~ 10 nmols per animal).
Cholesterol efflux assays
J774 macrophages were transfected with either a control mimic (Con-miR), a miR-144 mimic, a control inhibitor (Con-inh) or an anti-miR-144 inhibitor (Dharmacon) at 40 nM and seeded at a density of 1 × 106 cells per well one day prior to loading with 0.5 μCi/ml 3H-cholesterol for 24 h. Then, cells were washed twice with PBS and incubated in RPMI supplemented with 2 mg/ml fatty-acid free BSA (FAFA-media) in the presence of ACAT inhibitor (2 μmol/L) for 2 h prior to addition of 50 μg/ml human ApoA1 in FAFA-media with or without the indicated treatments. Supernatants were collected after 6 h and expressed as a percentage of total cell 3H-cholesterol content (total effluxed 3H-cholesterol+cell-associated 3H-cholesterol). In another set of experiments we transfected Huh-7 cells with control mimic (Con-miR), miR-144 mimic, miR-33 mimic or a combination of miR-144/miR-33 mimics at 5 nM final concentration and performed the cholesterol efflux assays as described above.
Lipid analysis and lipoprotein profile measurement
Mice were fasted for 12-14 h before blood samples were collected by retro-orbital venous plexus puncture. Plasma was separated by centrifugation and stored at -80°C. Total plasma cholesterol and HDL-cholesterol were enzymatically measured with the Amplex red cholesterol assay kit (Molecular Probes, Invitrogen), according to the manufacture’s instructions. The lipid distribution in plasma lipoprotein fractions were assessed by fast-performance liquid chromatography (FPLC) gel filtration with 2 Superose 6 HR 10/30 columns (Pharmacia).
Primary hepatocyte and Kupffer cells isolation
Primary hepatocyte and Kupffer cells were isolated using standard protocols 27.
Statistical analysis
All data are expressed as ±SEM. Statistical differences were measured by either a Student t test or 2-way ANOVA with Bonferroni correction for multiple comparisons when appropriate. A value of P≤0.05 was considered statistically significant. Data analysis was performed using GraphPad Prism 5.0a software (GraphPad, San Diego, CA).
An expanded materials and method section is available as Online Supplement.
RESULTS
miR-144 is up-regulated in LXR-activated macrophages and in the liver of mice fed with high fat diet
To determine whether LXR-induced miRNAs can regulate cholesterol metabolism at post-transcriptional levels, we undertook an unbiased genome-wide screen of miRNAs modulated by LXR ligands. We identified 32 miRNAs differentially regulated in mouse peritoneal macrophages treated with T0901317 (T090), a synthetic LXR ligand (Figure 1A). Interestingly some of the miRNAs induced by T090, including miR-370 and miR-122, were previously identified as important regulators of lipid metabolism 15, 16, 18, 28. To determine the potential candidates involved in regulating cholesterol metabolism, we analyzed the predicted target genes using a combination of bioinformatic tools for miRNA target predictions [TargetScan (http://www.targetscan.org) and miRanda (http://www.microrna.org)], gene ontology [Panther (http://www.pantherdb.org/) and protein-protein interactions [String (http://string-db.org/)]. We found miR-144 as a strong predicted candidate to regulate genes involved in metabolic processes including lipid metabolism (Online Figure IB-ID). Protein-protein interaction analysis showed that miR-144 predicted targets might play an important role in regulating lipid homeostasis (Figure 1E and F). Interestingly, ABCA1 was a strong predicted target with 7 potential binding sites in the 3’UTR (Online Figure IA) with some of them conserved in most vertebrates (Online Figure IB).
Figure 1. miR-144 expression is up-regulated in LXR-treated macrophages and has multiple lipid-related predicted target genes.
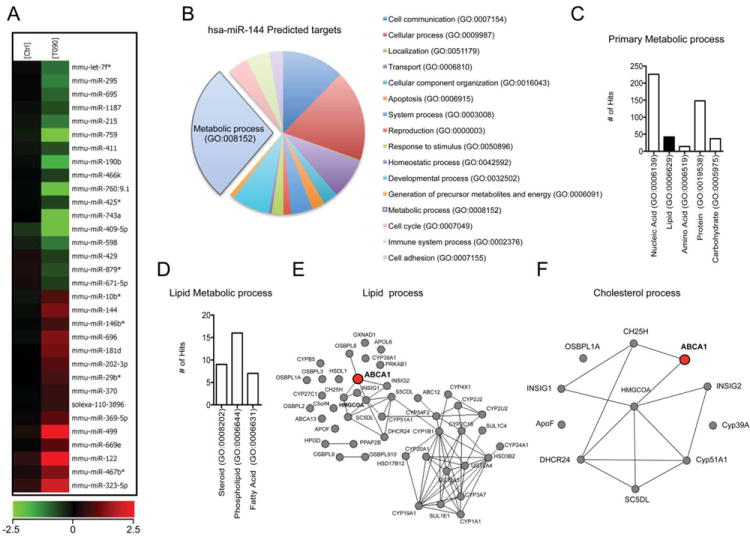
(A) miRNA array analysis of miRNAs up-regulated (red) or down-regulated (green) from peritoneal macrophages treated with T090 (n=4). (B to D) Gene ontology analysis of the miR-144 predicted target genes using Panther software. (E to F) Protein-protein interaction analysis of the miR-144 predicted targets using String software and Navigator 2.2.
We next confirmed the induction of miR-144 by LXR ligands using qRT-PCR analysis. As seen in Figure 2A, mouse peritoneal macrophages, human monocytes (THP-1) and human hepatic (Huh-7) cells treated with T090 showed an increased expression of miR-144. Similar results were obtained using another LXR ligand (GW3965) in THP-1 and Huh-7 cells (Online Figure IIA) and after cholesterol loading with acetylated low-density lipoproteins (Ac-LDL) in mouse peritoneal macrophages (Online Figure IIB). The maximum stimulation of miR-144 expression was observed at 0.1 μM of T090 and 3μM of GW3965 in human THP-1 and Huh-7 cells (Online Figure IIC and IID). In addition to miR-144, miR-451, which is encoded in the same genomic cluster, was also induced upon stimulation with T090 in mouse primary macrophages, THP-1 and Huh-7 (Online Figure IIE). To examine whether LXR ligands regulated the expression of these miRNAs at the transcriptional level, we examined the expression of primary transcripts containing the stem loop of miR-144 and miR-451. As shown in Figure 2B, LXR ligands increased the expression of pri-miR-144/451. Furthermore, the up-regulation of miR-144 and pri-miR-144/451 (primary miRNA transcript) by LXR agonists was impaired in macrophages isolated from LXRα,β null mice (Figure 2C and D), suggesting a transcriptional regulation of miR-144 and miR-451 by LXR. Finally, we also found that the miR-144/451 promoter activity was induced in Huh-7 cells treated with T090 (Figure 2E). Collectively, these data demonstrated that treatment of macrophages and hepatic cells with a LXR agonist resulted in higher expression of miR-144.
Figure 2. LXR induces miR-144 transcriptional expression.
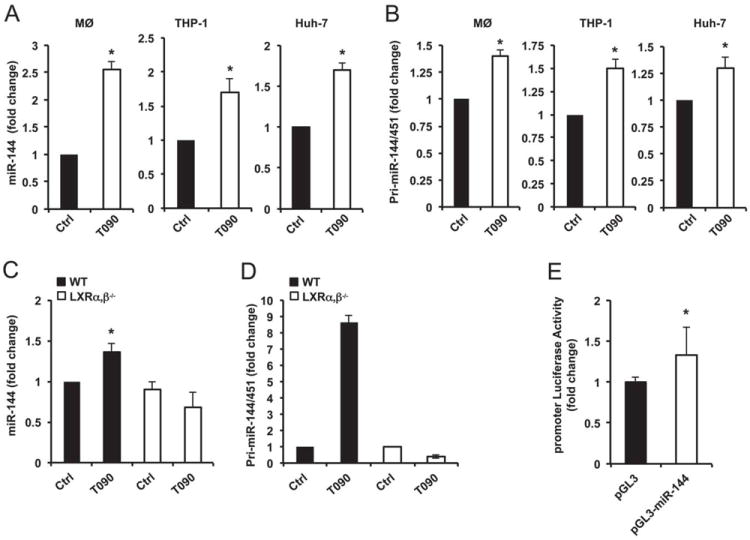
(A) qRT-PCR analysis of miR-144 expression in mouse peritoneal macrophages (MØ, left panel), THP-1 cells (middle panel) and Huh-7 cells (right panel) treated with T090. (B) qRT-PCR analysis of pri-miR-144/451 expression in mouse peritoneal macrophages (left panel), THP-1 cells (middle panel) and Huh-7 cells (right panel) treated with T090. (C and D) qRT-PCR analysis of miR-144 (C) and pri-miR-144/451 (D) expression from wild-type (WT) and LXRα,β-/- macrophages treated with T090. (E) miR-144/451 promoter-luciferase reporter activity in Huh-7 cells treated with T090. Data are expressed as relative expression and correspond to mean±SEM of 3 independent experiments. *Significantly different from cells without treatment (Ctrl), P≤0.05.
Next, we examined the in vivo expression of miR-144 in mice. miR-144 was widely expressed in mouse tissues and is particularly abundant in the liver, spleen and aorta (Online Figure IIIA). To determine the cell type that expressed a higher amount of miR-144 in the liver, we isolated primary hepatocytes and kupffer cells by isopycnic centrifugation. As shown in Online Figure IIIB, hepatocytes express significantly higher levels of miR-144 compared with Kupffer cells. We further analyzed whether miR-144 was regulated by dietary cholesterol. To this end, we measured its expression in mice fed either a chow or high-fat diet for 5 weeks. As expected, treatment of C57BL6 mice with a high fat diet increased body weight and plasma cholesterol and TG levels (Online Figure IIIC). Interestingly, hepatic miR-144 levels are regulated by high fat diet in vivo (Online Figure IIIC, right panel).
miR-144 regulates ABCA1 expression in macrophages and hepatic cells
We next determined the effect of miR-144 over-expression and inhibition on ABCA1 mRNA and protein expression. Transfection of mouse peritoneal macrophages with miR-144 increased its expression 350-fold (not shown) and significantly inhibited ABCA1 mRNA levels (Figure 3A, left panel). Moreover ABCG1 was also down-regulated at the mRNA level in macrophages transfected with miR-144 (Figure 3A, right panel). Even though ABCG1 is not a direct target of miR-144, the inhibition of RXRβ (data not shown), which is a predicted target for miR-144, by this miRNA may influence ABCG1 expression. To further assess the effect of miR-144 on ABCA1 and ABCG1 protein expression, we treated mouse peritoneal macrophages with Ac-LDL (to enrich cholesterol) or T090 (to directly stimulate expression of the two genes). Transfection of mouse peritoneal macrophages with miR-144 mimics but not a control miRNA (Con-miR) strongly decreased the stimulation of ABCA1 (Figure 3B, left panel). In contrast to ABCA1, ABCG1 expression was slightly reduced in miR-144 over-expressing macrophages. Similar effects were observed in human THP-1 cells (Figure 3B, right panel). miR-144 also repressed ABCA1 mRNA and protein expression in hepatic and endothelial cells, indicating that its effects are not cell type specific (Online Figure IVA and IVB). We further determined the role of endogenous miR-144 on ABCA1 expression in mouse peritoneal macrophages and human THP-1 cells. Importantly, inhibition of miR-144 by anti-miR-144 antisense oligonucleotides (Inh-miR-144) increased the expression of ABCA1 in both cell types (Figure 3C).
Figure 3. miR-144 levels regulate ABCA1 expression.
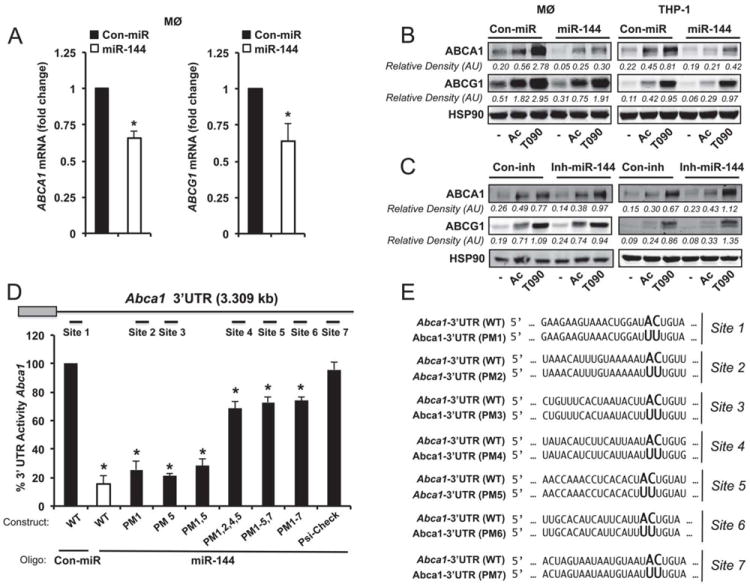
(A) qRT-PCR analysis of ABCA1, and ABCG1 expression in mouse peritoneal macrophages transfected with miR-144 mimics. Data are expressed as relative expression and correspond to the mean±SEM from 3 independent experiments. *Significantly different from cells transfected with control mimic (Con-miR), *P ≤ 0.05. (B and C) Western blot analysis of ABCA1, ABCG1 and HSP90 in mouse peritoneal macrophages and THP-1 cells transfected with (B) Con-miR or miR-144, or (C) control inhibitor (Con-inh) or anti-miR-144 in the presence or absence of Ac-LDL (Ac) or T090. Data correspond to a representative experiment among three that gave similar results. Values of the band densitometry analysis are shown. (D) Luciferase-reporter activity in COS-7 cells transfected with Con-miR or miR-144 (40 nM) mimic and human Abca1 3’UTR containing the indicated point mutations (PM) in the miR-144 target sites. Data are expressed as mean % of 3’UTR activity of Con-miR ±SEM and are representative of ≥3 experiments. *Significantly different from cells cotransfected with Con-miR and wild-type (WT) 3’UTR. P≤ 0.05. (E) Human Abca1 3’UTR containing the indicated point mutations (PM) in the miR-144 target sites.
To assess the effects of miR-144 on the 3’UTR of human Abca1, we used a luciferase reporter construct. MiR-144 (40 nM) markedly repressed Abca1 3’UTR activity (Figure 3D). Mutation of the miR-144 target sites relieved miR-144 repression of the Abca1 3’UTR activity, consistent with a direct interaction of miR-144 with these sites (Figure 3D and E). Site 2 and 4 appear to be the most important sites for miR-144 repression since their mutations are required for the significant derepression of Abca1 3’UTR activity by miR-144. We further confirmed these results using the mouse Abca1 3’ UTR construct, which has two highly conserved predicted miR-144 binding sites. As seen in Online Figure V, overexpression of miR-144 significantly reduced the Abca1 3’UTR activity and specific point mutations in the miR-144 binding sites abolish its inhibitory effect.
miR-33 and miR-144 have an additive effect on ABCA1 protein expression
We have previously reported that miR-33, an intronic miRNA encoded in the Srebp genes, regulates ABCA1 expression. To determine whether or not miR-144 and miR-33 have an additive effect on ABCA1 expression, we co-transfected Huh-7 cells with miR-33 and miR-144 mimics at a very low dose (5 nM). As seen in Figure 4A, co-transfection of both miRNAs slightly reduced the expression of ABCA1 compared with cells transfected with miR-33 or miR-144 alone. We also directly tested the effect of both miRNAs on Abca1 3’UTR activity. Co-transfection of miR-33 and miR-144 resulted in a 60% decrease in luciferase activity whereas miR-144 or miR-33 alone suppressed the 3’UTR activity 30-40% (Figure 4B). To determine the effect of both miRNAs on cellular cholesterol efflux, we transfected Huh-7 cells with miR-33, miR-144 or with a combination of both. miR-33 and miR-144 inhibited cholesterol efflux but the combination of both miRNAs do not further reduced the cholesterol export in this human hepatic cell line (Figure 4C). This could be explained because we used a very low dose of miRNA mimics (5 nM) in this experiment and because T090 induces the endogenous expression of miR-144 in hepatic cells and not in COS-7 cells were the 3’UTR assays were performed.
Figure 4. miR-33 and miR-144 cooperate to regulate ABCA1 expression.
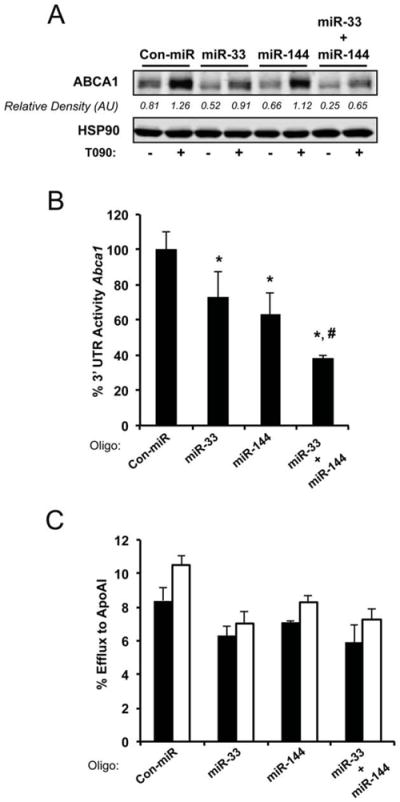
(A) Western blot analysis of Huh-7 cells transfected with 5nM non-targeting control mimic (Con-miR), miR-33, miR-144 and miR-33/miR-144 (upper panel). Data correspond to a representative experiment among three that gave similar results. Quantification of specific bands was conducted by densitometric analysis and is shown as the bottom of each band. (B) Activity of the luciferase reporter construct fused to the 3’UTR of Abca1 in COS-7 cells. Cells were transfected with Con-miR or miR-33, or miR-144, or miR-33 and miR-144 together. All microRNA constructs were transfected at 5νM final concentration. Relative luciferase activity is presented and data are the mean±SEM of 3 independent experiments in triplicate. *Significantly different from cells cotransfected with Con-miR and Abca1 3’UTR. P≤ 0.05. # Significantly different from cells cotransfected with miR-33 or miR-144 and Abca1 3’UTR. P≤ 0.05. (C) Total cholesterol efflux to ApoA1 in Huh-7 cells stimulated with T090 and expressing control miR (Con-miR), miR-33, miR-144 or miR-33 and miR-144. Data are the mean±SEM of 2 independent experiments performed in triplicate.
We further explored the cooperativity of miR-33 and miR-144 in regulating ABCA1 expression by analyzing the miR-33 levels in the setting where miR-144 is inhibited and vice versa. As seen in Online Figure VIA, inhibition of miR-144 does not alter miR-33 levels and miR-33 inhibition does not change miR-144 expression. Moreover, the inhibitory effect of miR-33 and miR-144 on ABCA1 expression is independent of the endogenous expression of both miRNAs (Online Figure VIB). Altogether these results suggest that miR-144 and miR-33 may cooperate to regulate the expression of ABCA1 in vitro.
miR-144 expression regulates cellular cholesterol efflux and HDL levels in vivo
ABCA1 plays a critical role in regulating cellular cholesterol efflux to ApoA1. To determine whether miR-144 modulates the efflux of cellular cholesterol, we transfected J774 murine macrophages with miR-144 and then incubated the cells with [3H]-cholesterol in the presence of Ac-LDL and T090 to induce ABCA1. As expected, miR-144 over-expression inhibited ABCA1 expression (Figure 5A, upper panel) and attenuated cholesterol efflux to ApoA1 (Figure 5A, bottom panel). Importantly, antagonism of endogenous miR-144 increased ABCA1 expression (Figure 5B, upper panel) and cellular cholesterol efflux to ApoA1 (Figure 5B, bottom panel). Thus, manipulation of cellular miR-144 levels alters macrophage cholesterol efflux, a critical step in the RCT pathway for the delivery of excess cholesterol to the liver.
Figure 5. Modulation of miR-144 regulates macrophage cholesterol efflux.
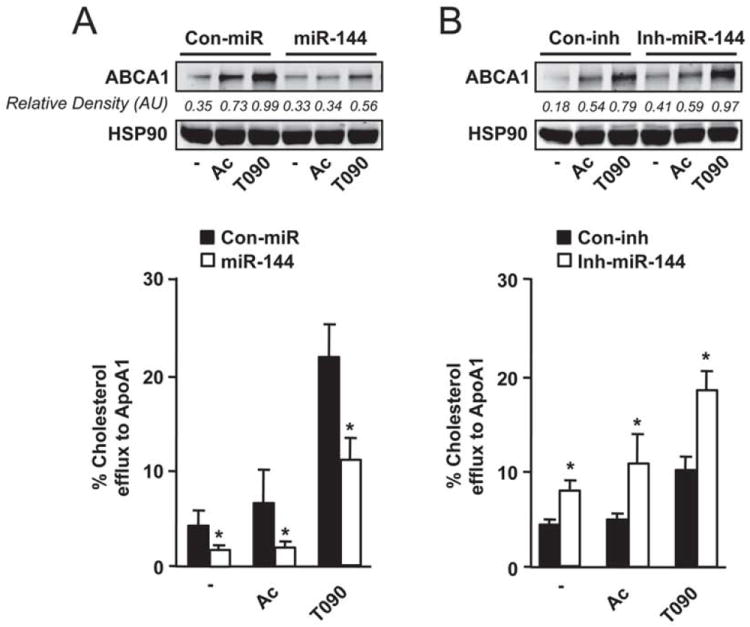
(A) Total cholesterol efflux to ApoA1 in J774 mouse macrophages stimulated with Ac-LDL (Ac) or T090 and expressing control miR (Con-miR) or miR-144. Upper panel shows the ABCA1 protein expression assessed by western blotting in the same conditions. (B) Total cholesterol efflux to ApoA1 in J774 mouse macrophages stimulated with Ac-LDL (Ac) or T090 and expressing control inhibitor (Con-inh) or Inh-miR-144. Upper panel shows ABCA1 protein expression assessed by western blotting in the same conditions. Data are the mean±SEM of 3 independent experiments performed in triplicate. *Significantly different from cells transfected with Con-miR (A) or Con-inh (B). P≤ 0.05.
In addition to regulating cellular cholesterol efflux, ABCA1 plays a key role in regulating HDL biogenesis in the liver. Thus, we studied the effects of miR-144 levels in vivo by injecting mice with miR-144 mimic particles. Efficient over-expression 250 fold-increase (not shown) was confirmed using qRT-PCR in the liver. Consistent with our in vitro results, miR-144 significantly reduced ABCA1 mRNA and protein expression in the liver (Figure 6A and B). We also found a decrease of ABCG1 mRNA and protein expression (Figure 6A and B). Other lipid-related genes, including SR-BI, a cognate receptor for HDL in the liver, CD36 and NPC1 were not affected in mice treated with miR-144 particles (Figure 6B). The inhibition of ABCA1 expression after 6 days of treatment with miR-144 particles leads to a significant reduction in total cholesterol and HDL cholesterol levels without changes in TG or cholesterol distribution in other lipoproteins (Figure 6C and D). To put these latter results in a more physiological context, we inhibited the expression of miR-144 using particles conjugated with miR-144 antisense oligonucleotides. The data show that miR-144 inhibition in basal conditions resulted in a significant reduction of miR-144 levels (Figure 7A) and an increase of liver ABCA1 protein expression without changes at the mRNA level (Figure 7B and C upper panel). In another group of mice, we induced the endogenous expression of miR-144 by oral gavage with T090. Under this condition, miR-144 was induced 2.3-fold and inhibited 2.1-fold after the injection of anti-miR-144 oligonucleotides (Figure 7A). miR-144 inhibition significantly increased ABCA1 protein and mRNA expression (Figure 7B and C bottom panel). We further analyzed the effects of anti-miR-144 treatment on total cholesterol, HDL cholesterol and TG plasma levels. In vivo delivery of anti-miR-144 particles resulted in an increase of plasma HDL cholesterol levels, suggesting that endogenous expression of miR-144 is important in regulating lipoprotein metabolism (Figure 7D). The lipoprotein fractionation analysis also demonstrated that the increased plasma HDL cholesterol was independent of the cholesterol distribution in other lipoproteins (Figure 7E). As expected plasma TG levels were slightly increased after T090 treatment but no differences were not affected by anti-miR-144 treatment (Figure 7D, right panel). Altogether, these results establish that miR-144 overexpression or inhibition reduces and increases circulating HDL, respectively.
Figure 6. Delivery of miR-144 oligonucleotides significantly reduces liver ABCA1 expression and HDL levels in mice.
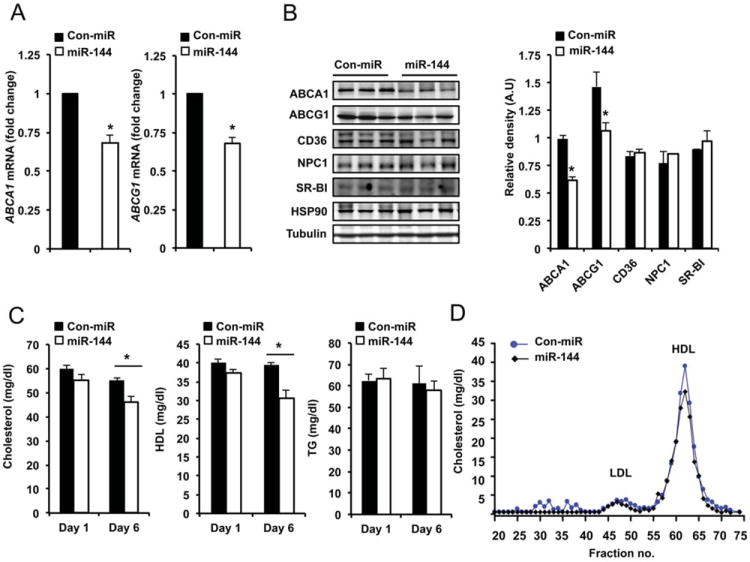
(A) qRT-PCR analysis of ABCA1 and ABCG1 expression from mouse livers treated with miR-144 mimics. Data are the mean±SEM from 6 mice per group. *Significantly different from mice injected with non-targeting control mimic (Con-miR) conjugated particles. P≤ 0.05. (B) Analysis of hepatic gene expression 6 days after injection of Con-miR or miR-144. Western blot analysis of liver tissue from three representative mice per group. Quantification analysis is shown in the right panel. Data are the mean±SEM from 6 mice per group. *Significantly different from mice injected with Con-miR conjugated particles. P≤ 0.05. (C) Total cholesterol (left panel), HDL cholesterol (middle panel) and TG (right panel) levels in mice treated with Con-miR or miR-144 mimics (n=6). *Significantly different from mice injected with non-targeting Con-miR conjugated particles. P≤ 0.05. (D) Lipoprotein profile from mice treated with Con-miR or miR-144 mimics (n=6). Cholesterol distribution across plasma fractions was analyzed by FPLC.
Figure 7. Inhibition of miR-144 increases hepatic ABCA1 expression and raises HDL plasma levels.
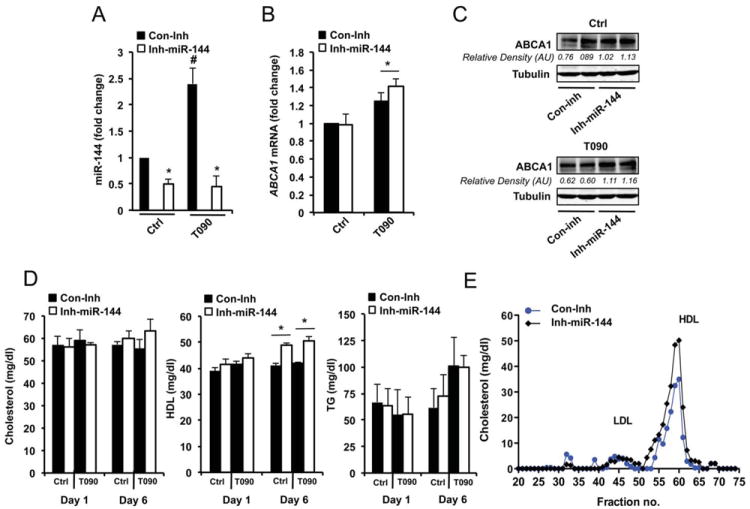
(A) qRT-PCR analysis of miR-144 levels from mouse livers treated with miR-144 inhibitor (Inh-miR-144) conjugated particles. Data are expressed as relative expression and correspond to the mean±SEM from 6 mice per group. *Significantly different from mice injected with control inhibitor (Con-inh), #significantly different from mice untreated and injected with Con-Inh or treated with T090 and injected with Inh-miR-144 compared with mice treated with T090 and injected with Con-Inh, P≤0.05. (B) qRT-PCR analysis of hepatic ABCA1 gene expression mouse liver treated or untreated with T090 and control inhibitor (Con-inh) or Inh-miR-144. Significantly different from mice injected with Con-inh, P≤0.05. (C) Western blot analysis of ABCA1 expression from liver tissue of mice injected with Con-inh or Inh-miR-144 and treated with vehicle (upper panel) or T090 (bottom panel). Quantification of specific bands was conducted by densitometric analysis and is shown at the bottom of each band. (D) Total cholesterol, HDL cholesterol and TG plasma levels in mice injected with Con-miR or miR-144 mimics and treated or not with T090 (n=6). Data are the mean ± SEM from 6 mice per group. Significantly different from mice injected with Con-inh, *P ≤ 0.05. (E) Lipoprotein profile analysis from mice treated with Con-inh or Inh-miR-144 (n=6). Cholesterol distribution across plasma fractions was analyzed by FPLC.
DISCUSSION
Since their discovery in Caenorhabditis elegans, miRNAs have emerged as critical fine tuners of many biological processes 29, 30. Recent advances in the understanding of lipid metabolism have revealed that miRNAs, particularly miR-122 and miR-33, play major roles in regulating cholesterol and fatty acid metabolism 15, 16, 20, 21, 23. We and others provided identification of a highly conserved miRNA family, miR-33a/b, within the intronic sequences of the Srebp genes in organisms ranging from Drosophila to humans 20, 21, 23. The 3’UTR of Abca1 contains 3 highly conserved binding sites for miR-33a/b, and the expression of ABCA1 mRNA and protein is strongly repressed by miR-33a/b over-expression in a variety of cell types. In addition to miR-33, other miRNAs including miR-758, miR-106b and miR-26 have been shown to regulate the expression of ABCA1 at the post-transcriptional level 19, 22, 31. In the present study, we report a novel miRNA, miR-144, that regulates the expression of ABCA1 in macrophages, hepatocytes and endothelial cells. Over-expression of miR-144 inhibits ABCA1 expression and reduces cellular cholesterol efflux in macrophages. Importantly, in vivo manipulation of miR-144 levels in the liver regulates plasma HDL levels. Since HDL levels correlate inversely with coronary artery disease (CAD), anti-miR-144 treatment may be useful to prevent atherosclerosis.
Compared with miR-33a/b, which are located within introns of Srebp genes and regulated by host genes, miR-144 is an intergenic miRNA located in the same locus as miR-451. Here we show that the primary transcript (pri-miR-144/451) expression is regulated by LXR ligands, however whether or not LXR regulates pri-miR-144/451 expression by direct interaction with its promoter remains to be answered. Preliminary data from our laboratory show that the miR-144/451 promoter has two predicted binding sites for SREBP transcription factors. Since SREBP1c expression is activated by LXR, it is plausible that LXR activates SREBP1c and subsequently miR-144/451 expression. Another possibility is that the increase in ABCA1 and ABCG1 expression by LXR agonists causes a depletion of cellular cholesterol, leading to an increase in SREBP2 and SREBP1a activity. Further experiments are warranted to understand the molecular mechanism that regulates the expression of miR-144/451 at the transcriptional level. Similarly, Ou et al have recently demonstrated that miR-613, a LXR-induced miRNA, targets LXRα and played an important role in the autoregulation of the human Lxrα gene 32. Interestingly, miR-613 transcriptional expression is activated by SREBP-1c, a LXR direct target. In a similar manner, it has also been shown that miRNAs regulate the expression of adhesion molecules (SELE and ICAM-1) in response to tumor necrosis factor (TNF) through a negative feedback loop to control inflammatory responses 33. This kind of regulation, in which miRNA-directed target repression acts to oppose the overall outcome of an induced biological response, is likely to be important for fine-tuning cellular processes.
One of the most interesting aspects of miRNA biology is that one miRNA often regulates multiple genes that are involved in a specific signaling cascade or cellular mechanism, making miRNAs potent biological regulators 13. However, defining the gene targets through which a miRNA functions is probably also the most tedious aspect of miRNA research. A given miRNA can be predicted to target several hundred genes, and 60% of mRNAs have predicted binding sites for one or multiple miRNAs in their 3’UTR 13, 14. Under baseline conditions, miRNAs appear to act as moderate regulators that act as a rheostat to fine-tune gene expression, but under conditions of stress or disease, they appear to exert more pronounced functions. To date, it has been reported that at least four miRNAs are able to regulate ABCA1 expression in several cell lines and tissues 19, 22, 23. The contribution of each one in modulating ABCA1 expression will be determined by the abundance of each miRNA in different tissues and the biological stimuli that regulate their expression. For instance, miR-33 and miR-758 are downregulated under cholesterol loading conditions to increase the expression of ABCA1 and promote cholesterol export. By contrast, LXR stimulation increases miR-144 and ABCA1 expression to fine-tune cellular cholesterol efflux in macrophages. The regulation of ABCA1 by miR-33, miR-758, miR-106b and miR-144 could also be influenced by the relative expression of other miR-33, miR-758, miR-106b and miR-144 mRNA targets that can compete for the binding in their 3’UTRs. This could be even more complex with the recent identification of competing endogenous RNAs (ceRNAs) 34, 35. These RNA transcripts share the miRNA response element (MRE) with the target genes and can regulate each other by competing for miRNA binding. Nevertheless, our data using miR-144 antisense oligonucleotides suggests that the endogenous levels of miR-144 in macrophages and hepatic cell lines are important in regulating ABCA1 expression and cellular cholesterol efflux. Finally, because hepatic ABCA1 is critical for the generation of plasma HDL, it seems likely that a combination therapy that includes both miR-144 and miR-33 antisense oligonucleotides might result in increased HDL levels, thus improving the prognosis for patients with cardiovascular disease.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
ABCA1 plays a critical role in regulating cellular cholesterol efflux in macrophages and liver and hepatic ABCA1 expression is required for plasma HDL formation.
Treatment of mice and non-human primates with miRNA antisense oligonucleotides such as miR-33 ASOs, can increase HDL cholesterol levels levels and promote atherosclerosis regression.
What New Information Does This Article Contribute?
We identified a new microRNA (miR-144) that regulates cholesterol metabolism in macrophages and human hepatic cell lines.
Inhibition of miR-144 in macrophages increases ABCA1 expression and cholesterol efflux to ApoA1.
Inhibition of miR-144 in vivo increases hepatic ABCA1 expression and plasma HDL levels.
MicroRNAS have recently emerged as having an important role in regulating macrophage cholesterol efflux and reverse cholesterol transport. Using an unbiased genome-wide screen, we identified miR-144 as a novel regulator of cholesterol metabolism in vitro and in vivo. We show that overexpression of miR-144 reduces ABCA1 expression and attenuates cholesterol efflux to ApoA1 in macrophages. In contrast, endogenous inhibition of miR-144 expression increases ABCA1 expression and cholesterol efflux to ApoA1. Most importantly, delivery of miR-144 oligonucleotides to mice attenuates ABCA1 expression in the liver, reducing HDL levels. Conversely, silencing of miR-144 in mice increases the expression of ABCA1 and plasma HDL levels. Thus, genetic manipulation of this pathway could represent a novel therapeutic intervention to increase reverse cholesterol transport and ameliorate atherosclerotic vascular disease.
Acknowledgments
We gratefully acknowledge Liang Guo for helping us with the primary hepatocyte isolation
SOURCES OF FUNDING
This work was supported by grants from the National Institutes of Health (R01HL107953 and R01HL106063 to C.F.-H., R01HL105945, to Y.S., P30NS069329, to J.K and AG028383 to P.T.N) and the Spanish Ministry of I+D (grant SAF2008-00057 to AC). C.M.R is supported by a postdoctoral fellowship from the American Heart Association (12POST9780016), D.C-S by the Deutsche Forschunqsqemeinschaft and A.G.S and A.W by Capes Foundation, Ministry of Education of Brazil, Brazil. N.R is supported by a postdoctoral fellowship from Spanish Ministry of I+D.
Nonstandard Abbreviations
- ABCA1
Adenosine triphosphate-binding cassette transporter A1
- ABCG1
Adenosine triphosphate-binding cassette transporter G1
- Ac-LDL
Acetylated low-density lipoprotein
- AMPKα
5’ AMP-activated protein kinase
- ApoA1
Apolipoprotein A1
- ASO
Antisense oligonucleotides
- CAD
Coronary artery disease
- ceRNAs
Competing endogenous RNAs
- CPT1a
Carnitine palmitoyltransferase 1A
- CROT
Carnitine O-octanyl transferase
- HADHB
Hydroxyacyl-CoA dehydrogenase-3-ketoacyl-CoA thiolase-enoyl-CoA hydratase (trifunctional protein) β-subunit
- HDL
High-density lipoprotein
- IDOL
Inducible degrader of the LDLr
- miRNAs
MicroRNAs
- NPC1
Niemann-Pick C1
- LXR
Liver X nuclear receptors
- qRT-PCR
Quantitative real-time polymerase chain reaction
- RCT
Reverse cholesterol transport
- RXRβ
Retinoid X receptor beta
- SREBP
Sterol response element binding protein
- T090
T0901317
- TG
Triglycerides
- 3’UTR
3’-untranslated region
- VLDL
Very low-density lipoproteins
Footnotes
DISCLOSURES
None.
References
- 1.Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 2.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gelissen IC, Harris M, Rye KA, Quinn C, Brown AJ, Kockx M, Cartland S, Packianathan M, Kritharides L, Jessup W. Abca1 and abcg1 synergize to mediate cholesterol export to apoa-i. Arterioscler Thromb Vasc Biol. 2006;26:534–540. doi: 10.1161/01.ATV.0000200082.58536.e1. [DOI] [PubMed] [Google Scholar]
- 4.Tall AR. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J Intern Med. 2008;263:256–273. doi: 10.1111/j.1365-2796.2007.01898.x. [DOI] [PubMed] [Google Scholar]
- 5.Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. Hdl, abc transporters, and cholesterol efflux: Implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–375. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Bodzioch M, Orso E, Klucken J, Langmann T, Bottcher A, Diederich W, Drobnik W, Barlage S, Buchler C, Porsch-Ozcurumez M, Kaminski WE, Hahmann HW, Oette K, Rothe G, Aslanidis C, Lackner KJ, Schmitz G. The gene encoding atp-binding cassette transporter 1 is mutated in tangier disease. Nat Genet. 1999;22:347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 7.Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J, Jr, Hayden MR. Mutations in abc1 in tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 8.Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denefle P, Assmann G. Tangier disease is caused by mutations in the gene encoding atp-binding cassette transporter 1. Nat Genet. 1999;22:352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 9.Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor lxr alpha. Proc Natl Acad Sci U S A. 2000;97:12097–12102. doi: 10.1073/pnas.200367697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beaven SW, Tontonoz P. Nuclear receptors in lipid metabolism: Targeting the heart of dyslipidemia. Annu Rev Med. 2006;57:313–329. doi: 10.1146/annurev.med.57.121304.131428. [DOI] [PubMed] [Google Scholar]
- 11.Zelcer N, Hong C, Boyadjian R, Tontonoz P. Lxr regulates cholesterol uptake through idol-dependent ubiquitination of the ldl receptor. Science. 2009;325:100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ambros V. The functions of animal micrornas. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 13.Bartel DP. Micrornas: Target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by micrornas: Are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 15.Elmen J, Lindow M, Silahtaroglu A, Bak M, Christensen M, Lind-Thomsen A, Hedtjarn M, Hansen JB, Hansen HF, Straarup EM, McCullagh K, Kearney P, Kauppinen S. Antagonism of microrna-122 in mice by systemically administered lna-antimir leads to up-regulation of a large set of predicted target mrnas in the liver. Nucleic Acids Res. 2008;36:1153–1162. doi: 10.1093/nar/gkm1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP. Mir-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Gerin I, Bommer GT, McCoin CS, Sousa KM, Krishnan V, MacDougald OA. Roles for mirna-378/378* in adipocyte gene expression and lipogenesis. Am J Physiol Endocrinol Metab. 2010;299:E198–206. doi: 10.1152/ajpendo.00179.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iliopoulos D, Drosatos K, Hiyama Y, Goldberg IJ, Zannis VI. Microrna-370 controls the expression of microrna-122 and cpt1alpha and affects lipid metabolism. J Lipid Res. 2010;51:1513–1523. doi: 10.1194/jlr.M004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim J, Yoon H, Ramirez CM, Lee SM, Hoe HS, Fernandez-Hernando C. Mir-106b impairs cholesterol efflux and increases abeta levels by repressing abca1 expression. Exp Neurol. 2012;235:476–483. doi: 10.1016/j.expneurol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marquart TJ, Allen RM, Ory DS, Baldan A. Mir-33 links srebp-2 induction to repression of sterol transporters. Proc Natl Acad Sci U S A. 2010;107:12228–12232. doi: 10.1073/pnas.1005191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Naar AM. Microrna-33 and the srebp host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramirez CM, Davalos A, Goedeke L, Salerno AG, Warrier N, Cirera-Salinas D, Suarez Y, Fernandez-Hernando C. Microrna-758 regulates cholesterol efflux through posttranscriptional repression of atp-binding cassette transporter a1. Arterioscler Thromb Vasc Biol. 2011;31:2707–2714. doi: 10.1161/ATVBAHA.111.232066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C. Mir-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davalos A, Goedeke L, Smibert P, Ramirez CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U, Pastor-Pareja JC, Esplugues E, Fisher EA, Penalva LO, Moore KJ, Suarez Y, Lai EC, Fernandez-Hernando C. Mir-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci U S A. 2011;108:9232–9237. doi: 10.1073/pnas.1102281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ. Antagonism of mir-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils JM, Ray TD, Sheedy FJ, Goedeke L, Liu X, Khatsenko OG, Kaimal V, Lees CJ, Fernandez-Hernando C, Fisher EA, Temel RE, Moore KJ. Inhibition of mir-33a/b in non-human primates raises plasma hdl and lowers vldl triglycerides. Nature. 2011;478:404–407. doi: 10.1038/nature10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Froh M, Konno A, Thurman RG. Isolation of liver Kupffer cells. Curr Protoc Toxicol. 2003;Chapter 14(Unit14.4) doi: 10.1002/0471140856.tx1404s14. [DOI] [PubMed] [Google Scholar]
- 28.Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjarn M, Hansen HF, Berger U, Gullans S, Kearney P, Sarnow P, Straarup EM, Kauppinen S. Lna-mediated microrna silencing in non-human primates. Nature. 2008;452:896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 29.Lee RC, Feinbaum RL, Ambros V. The c. Elegans heterochronic gene lin-4 encodes small rnas with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 30.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in c. Elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 31.Sun D, Zhang J, Xie J, Wei W, Chen M, Zhao X. Mir-26 controls lxr-dependent cholesterol efflux by targeting abca1 and arl7. FEBS Lett. 586:1472–1479. doi: 10.1016/j.febslet.2012.03.068. [DOI] [PubMed] [Google Scholar]
- 32.Ou Z, Wada T, Gramignoli R, Li S, Strom SC, Huang M, Xie W. Microrna hsa-mir-613 targets the human lxralpha gene and mediates a feedback loop of lxralpha autoregulation. Mol Endocrinol. 2011;25:584–596. doi: 10.1210/me.2010-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suarez Y, Wang C, Manes TD, Pober JS. Cutting edge: Tnf-induced micrornas regulate tnf-induced expression of e-selectin and intercellular adhesion molecule-1 on human endothelial cells: Feedback control of inflammation. J Immunol. 2010;184:21–25. doi: 10.4049/jimmunol.0902369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karreth FA, Tay Y, Perna D, Ala U, Tan SM, Rust AG, DeNicola G, Webster KA, Weiss D, Perez-Mancera PA, Krauthammer M, Halaban R, Provero P, Adams DJ, Tuveson DA, Pandolfi PP. In vivo identification of tumor- suppressive pten cernas in an oncogenic braf-induced mouse model of melanoma. Cell. 2011;147:382–395. doi: 10.1016/j.cell.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tay Y, Kats L, Salmena L, Weiss D, Tan SM, Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, Lieberman J, Rigoutsos I, Pandolfi PP. Coding-independent regulation of the tumor suppressor pten by competing endogenous mrnas. Cell. 2011;147:344–357. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.