

Abstract
The dopamine D3 receptor (D3R) is crucial in the regulation of blood pressure and sodium balance, in that Drd3 gene ablation in mice results in hypertension and failure to excrete a dietary salt load. The mechanism responsible for the renal sodium retention in these mice is largely unknown. We now offer and describe a novel mechanism by which D3R decreases sodium transport in the long term by inhibiting the deubiquitinylating activity of ubiquitin-specific peptidase 48 (USP48), thereby promoting Na+-H+ exchanger (NHE)-3 degradation. We found that stimulation with the D3R-specific agonist PD128907 (1 μM, 30 min) promoted the interaction and colocalization among D3R, NHE3, and USP48; inhibited USP48 activity (−35±6%, vs. vehicle), resulting in increased ubiquitinylated NHE3 (+140±10%); and decreased NHE3 expression (−50±9%) in human renal proximal tubule cells (hRPTCs). USP48 silencing decreased NHE3's half-life (USP48 siRNA t1/2=6.1 h vs. vehicle t1/2=12.9 h), whereas overexpression of USP48 increased NHE3 half-life (t1/2=21.8 h), indicating that USP48 protects NHE3 from degradation via deubiquitinylation. USP48 accounted for ∼30% of the total deubiquitinylating activity in these cells. Extending our studies in vivo, we found that pharmacologic blockade of D3R via the D3R-specific antagonist GR103691 (1 μg/kg/min, 4 d) in C57Bl/6J mice increased renal NHE3 expression (+310±15%, vs. vehicle), whereas an innovative kidney-restricted Usp48 silencing via siRNA (3 μg/d, 7 d) increased ubiquitinylated NHE3 (+250±30%, vs. controls), decreased total NHE3 (−23±2%), and lowered blood pressure (−24±2 mm Hg), compared with that in control mice that received either the vehicle or nonsilencing siRNA. Our data demonstrate a crucial role for the dynamic interaction between D3R and USP48 in the regulation of NHE3 expression and function.—Armando, I., Villar, V. A. M., Jones J. E., Lee, H., Wang, X., Asico L. D., Yu, P., Yang, J., Escano, C. S. Jr., Pascua-Crusan, A. M., Felder, R. A., Jose, P. A. Dopamine D3 receptor inhibits the ubiquitin-specific peptidase 48 to promote NHE3 degradation.
Keywords: renal proximal tubule cells, kidney, blood pressure, C57Bl/6 mice
The renal dopaminergic system, which inhibits sodium transport in the kidney, including the proximal tubule and medullary thick ascending limb (mTAL) of Henle, is responsible for at least 50% of renal sodium excretion in conditions of moderate sodium excess (1–4). This effect is exerted through 2 families of mammalian dopamine receptors: the D1-like dopamine D1/5 receptors (D1R and D5R) and the D2-like dopamine D2–4 receptors (D2R, D3R, and D4R), all of which are expressed in the kidney (5–8). The genetic disruption of any of the dopamine receptor genes results in hypertension (9), whereas dysfunctional D1R (10, 11), D2R (12), D3R (9, 13–15), or D5R (16) results in an impaired ability to excrete an NaCl load. The decreased ability to excrete a sodium load in hypertension is due to enhanced sodium transport per se and/or a failure to respond appropriately to signals that decrease sodium transport (17, 18).
The D3R inhibits renal ion transport and regulates blood pressure, by itself or through its interaction with other dopamine receptors or other G-protein-coupled receptors (GPCRs; refs. 14, 15, 19). The genetic disruption of the dopamine D3 receptor (Drd3) gene (Drd3−/−) in mice results in hypertension and a decreased ability to excrete an acute intravenous sodium load (13). Chronic pharmacologic inhibition of D3R activity causes salt-dependent hypertension (14), whereas the administration of highly selective D3R agonists, such as PD128907, pramipexole, and 7-OH-DPAT (3, 20–22), increases absolute and fractional sodium excretion in rats.
The D3R acutely inhibits Na+-H+ exchanger 3 (NHE3; SLC9A3) activity via PLC/PKC in rat renal proximal tubule cells (23, 24) or via PKA in opossum kidney cells, which have renal proximal tubule cell characteristics (25). However, the ability of D3R to regulate NHE3 function in the long term and the mechanisms involved are not known. We have identified ubiquitin (Ub)-specific peptidase 48 (USP48) as a novel binding partner for the 3rd intracellular loop of the human D3R in a yeast 2-hybrid screen. USP48 is a deubiquitinylating enzyme that removes Ub moieties from proteins. Ubiquitinylation is a reversible post-translational modification that conjugates Ub to lysine in target proteins. This modification has a major role in protein degradation, because the Ub tag directs the proteins for proteasomal degradation. Protein ubiquitinylation is important for NHE3 internalization in yeast (26). Dopamine treatment increases cell surface NHE3 ubiquitinylation in opossum kidney cells (27).
Ubiquitinylation is a dynamic process that is catalyzed by Ub ligases and Ub proteases/deubiquitinylating enzymes. There are at least 7 protease families that can participate in the deconjugation of Ub and Ub-like adducts. One of these groups comprises the Ub-specific proteases (USPs), which is a group of structurally diverse proteases expressed in different eukaryotic organisms and to which different functions have been ascribed (28). USP48 is widely distributed in a variety of tissues in humans, including the testis, pancreas, kidney (proximal and distal tubules, mTAL, and collecting duct), skeletal muscle, placenta, brain, and heart, suggesting that it participates in general deubiquitinylating reactions that may occur at basal levels in many tissues (29). Despite possessing all the residues proposed to be important in the catalytic activity of the enzyme, USP48 showed no activity in an assay in which Ub-β-galactosidase was the substrate (30), indicating that USP48 may have high substrate specificity.
In this study, we tested the hypothesis that agonist-activated D3R interacts with and prevents USP48 from removing the Ub tags on NHE3 and hence regulates NHE3 abundance and function in the long term by promoting its degradation. In this work, we unveil a novel and important biological function of USP48 in human renal proximal tubule cells (hRPTCs), and describe its pivotal role in the homeostatic regulation of Na+ excretion and blood pressure.
MATERIALS AND METHODS
Cell culture
hRPTCs
Immortalized hRPTCs obtained from normotensive Caucasian males were grown in DMEM/F12 supplemented with 10% fetal bovine serum (FBS); epidermal growth factor (EGF; 10 ng/ml); an insulin, transferrin, and selenium cocktail (5 μg/ml each); and dexamethasone (4 ng/ml) in a 37°C incubator with humidified 5% CO2 and 95% air. The cells used were limited to <25 passages to avoid the confounding effects of cellular senescence. The cells tested negative for Mycoplasma infection.
Treatments
The cells were subjected to the following treatments for various experiments. The D3R agonist quinpirole (1 μM) was used for long-term stimulation (24 h), with or without the D3R antagonist GR103691 (1 μM). The D3R agonist PD128907 (1 μM) was used for short-term stimulation (15 and 30 min). Cycloheximide (10 μM) was used to inhibit de novo protein synthesis. Serum-free medium with 0.4 M sucrose was used to inhibit endocytosis of plasma membrane–localized NHE3.
Isolation of the plasma membrane–enriched fraction
hRPTCs were grown in 150-mm dishes to 100% confluence. The cells were serum starved for 1 h, treated with the D3R agonist PD128907, washed with phosphate-buffered saline (PBS), scraped, and pelleted at 1200 g for 5 min. They were lysed in cold hypotonic TE buffer supplemented with protease inhibitors by passing them 20 times through a 23-gauge needle. The cells were then spun at 2000 g to remove the nuclear fraction and intact cells. The supernatant was transferred to another tube and spun anew at 42,000 g for 30 min, to separate the plasma membrane from the cytoplasmic fraction containing intracellular vesicles. The plasma membrane–enriched fraction was resuspended in RIPA buffer and sonicated (three 20-s bursts) on ice before immunoblot analysis.
Immunoblot analysis
Total cell lysates, whole kidney homogenates, and the plasma membrane and cytoplasmic fractions were resolved via 10% SDS-PAGE, electrotransferred onto nitrocellulose support, and immunoblotted with a chicken polyclonal antibody against rodent NHE3, a mouse monoclonal NHE3 antibody, and a rabbit polyclonal antibody against human NHE3, which were provided by Dr. Mark Knepper [U.S. National Heart, Lung, and Blood Institute (NHLBI), National Institutes of Health (NIH), Bethesda, MD, USA]; a rabbit polyclonal USP48 antibody (Bethyl Laboratories, Montgomery, TX, USA); a proprietary rabbit D3R antibody (31); and a mouse monoclonal Ub antibody (Santa Cruz Biotechnology, Inc., Dallas, TX, USA; ref. 32). The immunoreactive bands were visualized with chemiluminescence and autoradiography or via the Odyssey Imaging System (LI-COR Biosciences, Lincoln, NE, USA). Band densitometry was performed with Scion Image software (Scion Corp., Frederick, MD, USA).
Coimmunoprecipitation
Total cell lysates from hRPTCs were prepared in Nonidet P-40 lysis buffer supplemented with protease inhibitors (Halt Protease Inhibitor Cocktail; Thermo Scientific, Rockford, IL, USA). Equal amounts of cell lysates were mixed with equal amounts of the immunoprecipitating antibody and Dynabeads (Invitrogen, Grand Island, NY, USA). The bound proteins were eluted by the addition of Laemmli buffer, resolved via 10% SDS-PAGE, and visualized, as above.
Confocal microscopy
hRPTCs were grown to 50% confluence on poly-d-lysine-coated coverslips (BD Biosciences, San Jose, CA, USA) in 24-well plates. The cells were serum starved for 1 h before treatment with the D3R agonist PD128907 at indicated time points. The cells were then double immunostained with the appropriate primary antibodies, and the corresponding secondary antibodies were tagged with Alexa Fluor 488 or Alexa Fluor 568 (Life Technologies-Molecular Probes, Eugene, OR, USA). The cells were mounted in Fluorogel with Tris buffer (Electron Microscopy Sciences, Hatfield, PA, USA). Wheat germ agglutinin (WGA) labeled with Alexa Fluor 647 and DAPI were used to visualize the plasma membrane and nucleus, respectively. Images were obtained sequentially in separate channels at room temperature, with an IX-70 Laser Confocal Scanning Microscope with Plan Apo, ×60/1.4 NA oil-immersion objective (Olympus, Lake Success, NY, USA). Images were merged by using Olympus Fluoview FV300, version 3C acquisition software, to determine colocalization. Some images were obtained with an LSM 510 microscope with ×63/NA1.4 oil-immersion objective (Zeiss, Thornwood, NY, USA). The images were processed with Zen 2011 software (Zeiss).
Cholera toxin subunit B (CTxB) uptake
hRPTCs grown to 50% confluence on poly-d-lysine-coated coverslips were serum-starved for 1 h before they were labeled with CTxB tagged with Alexa Fluor 647 (1 mM; ref. 33) and treated with the D3R agonist PD128907 (15 min) to induce NHE3 trafficking. The cells were then immunostained for NHE3 and visualized via confocal microscopy.
Sucrose gradient cell fractionation
hRPTCs were grown to 90% confluence in 150-mm dishes. Membrane microdomains were obtained by sucrose gradient centrifugation according to a detergent-free protocol (34, 35). Briefly, the cells were prestarved for 2 h, washed, and pelleted into 1.5 ml of 500 mM sodium carbonate (pH 11), and homogenized with a Dounce homogenizer (10 strokes), a Polytron tissue grinder (three 10-s bursts) (Polytron Technologies, Taoyuan City, Taiwan), and a sonicator equipped with a conical tip (three 30-s bursts). The homogenate was diluted 1:2 with 80% sucrose, overlaid with 6 ml 35% sucrose and 3 ml 5% sucrose, and centrifuged at 160,000 g at 4°C for 18 h. A uniform amount of protein was loaded into each tube. After centrifugation, twelve 1-ml fractions were collected from top to bottom, and aliquots of each fraction were mixed with Laemmli buffer, boiled, and subjected to immunoblot analysis. All experiments were performed at 4°C in the presence of protease inhibitors. To disrupt the lipid rafts, some cells were incubated with 2% methyl-β-cyclodextrin (β-MCD), a cholesterol-depleting reagent, at 37°C for 1 h before fractionation.
RNA extraction, cDNA preparation, and RT-PCR
hRPTCs were homogenized, and total RNA was extracted with the RNeasy RNA Extraction kit (Qiagen, Germantown, MD, USA). Semiquantitative RT-PCR was performed with the Superscript III One-Step RT-PCR kit (Invitrogen), according to the manufacturer's protocol. The primers used for determination of NHE3 mRNA expression were TATGGAGAACCTGGCACACA (mNHE3-F, nt 2085–2105) and TCCTCATCAGGGGAGAACAC (mNHE3-R, nt 2250–2270). β-Actin was used as the housekeeping gene. The products were resolved in 2% agarose gel and visualized with a digital gel documentation system (Alpha Innotech, San Leandro, CA, USA). Band densities were quantified with the Scion software.
USP48 activity assay
USP48 deubiquitinylase activity was determined with the fluorogenic substrate Ub-7-amino-4-methylcoumarin (Ub-AMC; (Boston Biochem, Cambridge, MA, USA), which releases the fluorescent compound AMC during Ub hydrolysis. hRPTCs were transfected for 48 h with the human USP48 gene subcloned in the pcDNA6.2-V5 vector or with USP48-specific siRNA. Transfection of empty vector or vehicle served as the negative control. In another set of experiments, cells previously transfected with pcDNA6.2-V5::hUSP48 or empty vector were treated with PD128907 (15 min). The assay was performed according to the procedure described by Fernández-Montalván et al. (36). Briefly, the treated cells were washed with PBS, pelleted, and resuspended in 50 mM Tris-HCl buffer (pH 7.5), with 1 mM EDTA, 5 mM dithiothreitol, 100 mM NaCl, and 0.1% (w:v) CHAPS and then sonicated and centrifuged before aliquots of the supernatant were incubated in 96-well plates with the substrate Ub-AMC, to a final concentration of 1 μM. The plates were read in a Victor2 Multilabel Counter (Perkin Elmer, Waltham, MA, USA) set at an excitation wavelength of 360 nm and an emission wavelength of 480 nm. Results were expressed as arbitrary units/milligram protein.
Yeast 2-hybrid screen and His pulldown assay
The 3rd intracellular loop of the human D3R (D3R-IC3) was PCR-amplified from a full-length human D3R cDNA. The amplified product was cloned into the pGBKT7 vector such that the expressed protein fragment was fused in-frame with amino acids 1–147 of the GAL4 DNA-binding domain. This fusion protein served as “bait” in a yeast 2-hybrid screen of 2 × 107 clones of a human expressed kidney cDNA library. After rigorous testing to eliminate false positives, USP48 was identified as a D3R-IC3 interacting protein. In another set of experiments, hRPTCs were transiently transfected with pcDNA3.1/TO/hD3R-His or with the empty vector as the negative control. His-tagged D3R was pulled down by using the MagneHis protein purification kit (Promega, San Luis Obispo, CA, USA) and immunoblotted for USP48.
[35S]-Methionine/cysteine (Met/Cys) labeling and pulse–chase assay
hRPTCs transfected for 48 h with the pcDNA6.2-V5::USP48, USP48-specific siRNA, or vehicle as the negative control, were labeled by [35S]-Met/Cys, as described previously (37). Briefly, the cells were starved in Met/Cys-free DMEM (Invitrogen) containing 10% dialyzed FBS for 2 h and pulsed for 3 h with medium containing 300 μCi/ml [35S]-labeled Met/Cys and cycloheximide. The cells were then chased for various durations in full culture medium containing 3 mM unlabeled Met/Cys. At the end of each chase period, the cells were washed 3 times with PBS to remove the unbound label. Cell pellets were lysed with lysis buffer, and NHE3 was immunoprecipitated with anti-NHE3 antibody. Immunoprecipitated protein complexes were eluted with protein sample buffer at 85°C for 15 min and resolved by SDS-PAGE. Dried gels were exposed to X-ray films for autoradiography. The quantified NHE3 bands were normalized to time 0 value. The mean and se of 3 experiments were plotted, and the exponential decays were calculated (OriginPro, Northampton, MA, USA). The decay rate constants and the half-lives of NHE3, which represent the rates of NHE3 degradation, were calculated by individual decay equations.
Mice
Generation of D3R+/− and D3R−/− mice
The studies were conducted in accordance with NIH guidelines for the ethical treatment and handling of animals in research and approved by the University of Maryland Institutional Animal Care and Use Committee. Heterozygous mice (Drd3+/−) on a C57Bl/6J background (>7 generations) were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Drd3−/−, Drd3+/−, and Drd3+/+ mice were identified by DNA genotyping (13). Drd3−/− mice and their wild-type littermates (Drd3+/+) were studied while on a normal (0.8% NaCl) or high (4% NaCl)-salt diet. All animals used in this study were bred and maintained at the institutional animal facility. Blood pressure was measured via the femoral vein in animals under anesthesia (13).
Treatment with GR103691
C57Bl/6J mice were treated with the D3R antagonist GR103691 (1 μg/kg/min) via an osmotic minipump for 4 d while they were ration fed a moderately high-salt diet (1.6%NaCl).
Acute renal-specific USP48 knockdown
Mouse renal cortical Usp48 was silenced by a 7-d renal subcapsular infusion of Usp48-specific siRNA delivered through an osmotic minipump (38–41). Adult male C57Bl/6J mice were uninephrectomized 1 wk before siRNA infusion. The minipumps were implanted in mice anesthetized with pentobarbital (50 mg/kg body weight, i.p.). The osmotic minipumps (100 μl capacity, 0.5 μl/h flow rate for 7 d; Alzet; Durect Corp., Cupertino, CA, USA) were filled with previously validated Usp48-specific siRNA (3 μg/d) or nonsilencing siRNA (3 μg/d) as the control. The siRNA was dissolved in an in vivo transfection reagent (TransIT In Vivo Gene Delivery System; Mirus, Madison, WI, USA) under sterile conditions. The minipumps were fitted with a polyethylene delivery tubing (Alzet 0007701), and the tip of the tubing was inserted into the subcapsular space of the remaining kidney. Surgical glue was applied at the puncture site to hold the tube in place and prevent extrarenal leakage. The osmotic pump was sutured to the abdominal wall to prevent excessive movement for the duration of the implantation. Blood pressure was measured as described above, before and after the 7-d siRNA infusion.
Statistical analysis
Data are expressed as means ± se. Significant differences between 2 groups were determined by Student's t test, and that among groups was determined by 1-way ANOVA followed by the Holm-Sidak post hoc test. Values of P < 0.05 were considered significant. Statistical analysis was performed with Sigma Stat 3.5 (Systat Software, Inc., San Jose, CA, USA).
RESULTS
D3R interacts with NHE3 and regulates its function
We initially evaluated the dynamic interaction between D3R and NHE3 in immortalized hRPTCs. In these cells, both D3R and NHE3 were distributed at the membrane and cytosol in the basal state (Fig. 1A). Stimulation with the D3R agonist PD128907 in hRPTCs resulted in the internalization and accumulation of the receptor and NHE3, mostly at the perinuclear area, and increased colocalization with D3R at 15 and 30 min (Fig. 1A). In agreement with these observations, we found that NHE3 coimmunoprecipitated with D3R basally, an effect that was enhanced after 15 and 30 min of D3R stimulation (Fig. 1B). To demonstrate the specificity of the interaction, we treated the cells with the D3R antagonist GR103691 and observed a 20% reduction in the immunoprecipitated NHE3 (Fig. 1C), a consequence of the D3R being constitutively active. D3R agonist treatment with PD128907 increased the coimmunoprecipitation of NHE3 and D3R, in agreement with that shown in Fig. 1B. The positive and negative effects of D3R agonist and D3R antagonist treatment, respectively, on the coimmunoprecipitation of NHE3 and D3R were no longer observed with cotreatment with both the D3R agonist and the antagonist. We next determined the change in NHE3 at the plasma membrane on D3R agonist stimulation and found that agonist treatment resulted in 50% reduction in the abundance of NHE3 in the plasma membrane (Fig. 1D), indicating either NHE3 endocytosis or degradation. Thus, we also evaluated the extent of NHE3 degradation occurring at the plasma membrane of cells that were pretreated with 0.4 M sucrose to inhibit receptor endocytosis and found that there was no change in NHE3 abundance (Fig. 1E), suggesting that the degradation at the surface is negligible and that the short-term (in minutes) decrease in plasma membrane NHE3 is due to internalization. We next determined whether D3R stimulation also regulates NHE3 expression in the long term. Although acute dopamine treatment was not associated with changes in total cellular NHE3 abundance, long-term treatment with dopamine in opossum kidney cells reduced both total cellular and cell surface NHE3 expression (27). A 24-h treatment with quinpirole (a mixed D2R/D3R agonist) decreased NHE3 protein expression in hRPTCs, an effect that was blocked by cotreatment with the specific D3R antagonist GR103691, indicating a D3R-specific effect (Fig. 1F).
Figure 1.

D3R interaction with NHE3. A) Cellular distribution and colocalization of D3R and NHE3 in hRPTCs treated with the D3R agonist PD128907 (1 μM) at the indicated time points. Images were obtained via laser scanning confocal microscope. D3R was pseudocolored green, and NHE3 was pseudocolored red. Colocalization was observed as discrete yellow areas in merged images. WGA tagged with Alexa Fluor 647 was used to target the lectins on the plasma membrane and DAPI to visualize the nucleus. All experiments were performed 3 times (×600; scale bar=10 μm). B) Top: uniform amounts of the lysate and immunoprecipitating antibody were used for the coimmunoprecipitation of D3R and NHE3 in hRPTCs treated with the D3R agonist PD128907 (1 μM) at the indicated time points. NC, negative control (normal mouse IgG); PC, positive control (regular immunoblot of hRPTC lysate). *P < 0.05 vs. basal; 1-way ANOVA and Holm-Sidak post hoc test (dn=3–4). Bottom: coimmunoprecipitation of NHE3 and D3R in hRPTCs treated with PD128907 for 30 min; immunoprecipitation and immunoblot data are in reverse order, relative to the data in the top panel. Normal mouse IgG was used as the negative control. C) Effect of 30-min treatment with a D3R agonist (PD128907, 1 μM), D3R antagonist (GR103691, 1 μM), or both on the coimmunoprecipitation of D3R and NHE3 in hRPTCs. Immunoblots of the D3R and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) from total cell lysates are shown to indicate uniformity of protein loading. *P < 0.05, vs. control (vehicle); 1-way ANOVA and Holm-Sidak post hoc test (n=3). D, E) Abundance of NHE3 in the plasma membrane of hRPTCs treated with the D3R agonist PD128907 (1 μM/30 min), indicating the extent of NHE3 internalization (D) or degradation in the plasma membrane (E), the latter in cells that were incubated in 0.4 M sucrose to inhibit endocytosis. Actin was used for normalization. *P < 0.05 vs. control; Student's t test (n=3). F) Effect of 24-h treatment with quinpirole (Q, a mixed D2R/D3R agonist, 1 μM) in the presence or absence of the specific D3R antagonist GR103691 (GR, 1 μM) or vehicle (V) on the protein abundance of NHE3 in hRPTCs. Molecular sizes: NHE3, 90 kDa; D3R, ∼60 kDa; GAPDH, 35 kDa. *P < 0.05 vs. basal; 1-way ANOVA and Holm-Sidak post hoc test (n=3–4).
D3R activation promotes NHE3 ubiquitinylation
We next determined whether D3R modulates NHE3 ubiquitinylation in hRPTCs. We found that the basally active D3R promoted NHE3 ubiquitinylation, whereas agonist activation further increased this effect, as demonstrated by the coimmunoprecipitation of NHE3 and Ub on treatment with a D3R agonist (Fig. 2A). Ubiquitinylated NHE3 (NHE3-Ub) decreased in the plasma membrane, indicating its internalization in the cytoplasm (Fig. 2B). Although the NHE3-Ub was equally as abundant in the cytoplasm as in the plasma membrane in the basal state, the amount of NHE3-Ub in the cytoplasm increased ∼3-fold after D3R stimulation for 30 min.
Figure 2.
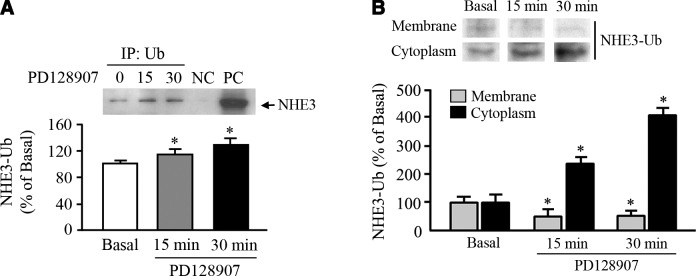
NHE3 ubiquitinylation and NHE3-Ub distribution. Extent of NHE3 ubiquitinylation (A) and cellular distribution of ubiquitinylated NHE3 (B) in the plasma membrane–enriched fractions and cytoplasm containing intravesicular fractions of hRPTCs in response to the D3R agonist PD128907 (1 μM) at the indicated time points. Abundance of ubiquitinylated NHE3 was determined via coimmunoprecipitation with an anti-Ub antibody as the immunoprecipitant. Immunoblots are shown above the corresponding bar graphs. Molecular sizes: NHE3, 90 kDa; NHE3-Ub, 100 kDa. *P < 0.05 vs. basal; 1-way ANOVA and Holm-Sidak post hoc test (n=3–4).
D3R interacts with USP48 and regulates its activity
To determine the mechanism by which the D3R regulates NHE3 ubiquitinylation, we evaluated the dynamic interaction between the D3R and USP48. We identified USP48 as a novel interacting partner of the 3rd intracytoplasmic loop of D3R in a yeast 2-hybrid screen. The ability of USP48 to bind to the receptor was initially confirmed when a 120-kDa band corresponding to human USP48 was pulled-down by D3R-myc,his (Fig. 3A). To corroborate these results, we performed coimmunoprecipitation experiments with an anti-D3R antibody used as the immunoprecipitant. We found that endogenous USP48 and D3R coimmunoprecipitated basally, whereas agonist stimulation enhanced their coimmunoprecipitation in hRPTCs (Fig. 3B). We next evaluated the spatial and temporal aspects of the interaction between endogenous D3R and USP48 and found that D3R and USP48 were distributed at the plasma membrane and cytoplasm under basal conditions (Fig. 3C). D3R activation resulted in the internalization of plasma membrane–bound D3R and NHE3 and enhanced their intracellular colocalization. Since USP48 had no enzymatic activity in an assay with ubiquitinylated β-galactosidase used as the substrate (30), we verified its deubiquitinylase activity with Ub-AMC as the substrate in hRPTCs either overexpressing USP48 (190% overexpression) or depleted of their endogenous USP48 (∼60% knockdown) (Fig. 3D, E). The deubiquitinylase activity increased 2-fold in hRPTCs overexpressing USP48 and decreased by 20% in the USP48-depleted cells compared with that in the control cells, indicating the portion of the total deubiquitinylating activity in these cells that could be ascribed to USP48. Stimulation with the D3R agonist PD128907 resulted in a 35% reduction in the deubiquitinylating activity in cells overexpressing USP48 and a 24% reduction in the activity in nontransfected cells compared to that in vehicle-treated control cells, indicating that agonist-activated D3R inhibits the activity of USP48 (Fig. 3F).
Figure 3.

D3R interaction with USP48. A) His pulldown of endogenous USP48 in hRPTCs with heterologously expressed D3R-myc,His. Cells transfected with the empty vector were used as the negative control (n=3/group). Ab, antibody. B) Coimmunoprecipitation of endogenous D3R and USP48 in hRPTCs treated with the D3R agonist PD128907 (1 μM) at the indicated time points. NC, negative control (rabbit mouse IgG), PC, positive control (regular immunoblot of hRPTC lysate). *P < 0.05 vs. basal; 1-way ANOVA and Holm-Sidak post hoc test (n=3). C) Cellular distribution and colocalization of D3R and USP48 in hRPTCs treated with the D3R agonist PD128907 (1 μM) at the indicated time points. Images were obtained via laser scanning confocal microscope. USP48 was pseudocolored red, and D3R was pseudocolored green. Colocalization was observed as discrete yellow areas in merged images. WGA tagged with Alexa Fluor 647 was used to target the lectins on the plasma membrane and DAPI to visualize the nucleus (×600; scale bar=10 μm, n=3). D) Immunoblots of USP48 in hRPTCs depleted of its endogenous USP48 pool and in cells overexpressing USP48. Overexpression was achieved by transfection of pcDNA6.2-V5::hUSP48, whereas depletion of endogenous USP48 was achieved by transfection of USP48-specific siRNA. Actin was used as the housekeeping protein. E, F) USP48 deubiquitinylating activity in hRPTCs in response to stimulation with the D3R agonist PD128907 (1 μM, 30 min; E) or as a function of USP48 expression (overexpression or siRNA-mediated depletion; F). USP48 assays were performed in duplicate. Molecular sizes: USP48, 120 kDa; actin, 40 kDa. *P < 0 .05 vs. basal; 1-way ANOVA and Holm-Sidak post hoc test (n=3). #P < 0 .05 vs. (−) PD128907 treatment; Student's t test (n=3).
USP48 interacts with NHE3 and increases its degradation
Lipid rafts are highly ordered plasma membrane microdomains that are stabilized by cholesterol to allow the assembly of receptors and signaling molecules. We have reported that the D3R is distributed in both lipid and nonlipid rafts (31). We now report that both USP48 and NHE3 cosegregated basally in lipid and nonlipid rafts in hRPTCs (Fig. 4A). NHE3 has been shown to exist in lipid rafts in rabbit ileal brush borders (42, 43). Pretreatment with the cholesterol-depleting agent β-MCD, which disrupts the lipid rafts, redistributed both proteins to the less buoyant non-lipid-raft fractions. We used flotillin-1 and caveolin-1 as markers of the lipid raft microdomains. We next determined whether NHE3 is internalized in lipid raft microdomains of hRPTCs via the CTxB uptake assay. CTxB targets the lipid raft marker ganglioside GM1 (44, 45). NHE3 partially colocalized with CTxB at the plasma membrane basally and at the cytoplasm on stimulation with the D3R agonist PD128907 (Fig. 4B). USP48 and NHE3 colocalized at the plasma membrane and cytosol in the basal state. We observed a marked increase in their colocalization at the perinuclear area after 15 min of agonist treatment; the extent of colocalization diminished at 30 min after treatment (Fig. 4C). Moreover, NHE3 coimmunoprecipitated with USP48 under basal condition, and the amount of immunoprecipitated NHE3 increased 2.5-fold after 15 min of agonist stimulation (Fig. 4D).
Figure 4.

USP48 interaction with NHE3. A) Distribution of NHE3 and USP48 in lipid raft (fractions 1–6) and non-lipid-raft (fractions 7–12) membrane microdomains of hRPTCs under basal conditions and after treatment with β-MCD (2%), an agent that disrupts lipid rafts by cholesterol depletion. Flotillin-1 and caveolin-1 are lipid raft markers. B) Colocalization of NHE3 and the CTxB, which targets the lipid raft marker ganglioside GM1, in the basal state and after treatment with the D3R agonist PD128907 (1 μM, 15 min). Images were obtained via laser scanning confocal microscope. NHE3 was pseudocolored green, and CTxB was pseudocolored red. Colocalization was observed as discrete yellow areas in merged images (×600, scale bar=10 μm, n=2). C) Cellular distribution and colocalization of NHE3 and USP48 in hRPTCs treated with the D3R agonist PD128907 (1 μM) at the indicated time points. Colocalization was observed as discrete yellow areas in merged images. WGA tagged with Alexa Fluor 647 was used to target the lectins on the plasma membrane and DAPI to visualize the nucleus (×600, scale bar=10 μm, n=3. D) Coimmunoprecipitation of endogenous NHE3 and USP48 in hRPTCs treated with the D3R agonist PD128907 (1 μM) at the indicated time points. An immunoblot is shown above the corresponding bar graph. NC, negative control (rabbit mouse IgG); PC, positive control (regular immunoblot of hRPTC lysate). *P < 0.05 vs. basal; 1-way ANOVA and Holm-Sidak post hoc test (n=3 performed in duplicate). E) Effect of USP48 expression on NHE3 degradation measured using [35S]-Met/Cys labeling. Overexpression was achieved by transfection of pcDNA6.2-V5::hUSP48, whereas depletion of endogenous USP48 was achieved by transfection of USP48-specific siRNA. Radiographs are shown above the line graph. F, G) Total NHE3 and ubiquitinylated NHE3 (NHE3-Ub) in hRPTCs that were either overexpressing (USP48; F) or depleted of USP48 (USP48 siRNA; G). Cells that were transfected with empty vector (EV; F) or nonsilencing siRNA (Mock; G) served as controls. Actin was used for normalization of total NHE3. Molecular sizes: NHE3, 90 kDa; USP48, 120 kDa; Flotillin-1, 50 kDa; Caveolin-1, 25 kDa; Actin, 40 kDa; NHE3-Ub, ∼100 kDa. *P < 0.05 vs. control; Student's t test (n=3).
Because D3R stimulation inhibits USP48 activity and increases NHE3 ubiquitinylation, we next determined the effect of USP48 on NHE3 turnover via pulse–chase experiments with [35S]-Met/Cys labeling. Overexpression of USP48 resulted in decreased spontaneous degradation of NHE3 relative to that observed with vehicle treatment (Fig. 4E). Conversely, siRNA-mediated silencing of USP48 increased the spontaneous degradation of NHE3. The decrease in NHE3 abundance in the presence of vehicle or USP48 overexpression followed a first-order exponential curve. The half-life of NHE3 was 12.9 h under basal condition (vehicle treatment), but increased to 21.8 h when USP48 was overexpressed, and decreased to 6.1 h when USP48 was silenced. We next determined the effect of USP48 overexpression or silencing on the extent of NHE3 ubiquitinylation. Overexpressing USP48 for 72 h decreased the amount of NHE3-Ub by 50% (Fig. 4F). In contrast, USP48 depletion resulted in a 25% reduction of total NHE3, but increased the amount of NHE3-Ub by 112% (Fig. 4G), indicating a role for USP48 on basal NHE3 turnover.
NHE3 colocalizes with the proteasome and lysosome
There are 2 major sites of protein degradation, the lysosome and proteasome, where most ubiquitinylated proteins are degraded. Thus, we evaluated whether NHE3 colocalizes with these sites of degradation. NHE3 colocalized with the proteasome (identified via the p44S10 marker) in the basal state and, to a greater extent, at the perinuclear area on D3R agonist treatment, suggesting that the proteasome is a major site of NHE3 degradation (Fig. 5A). NHE3 did not colocalize with the lysosome (identified via the LAMP-1 marker) basally, but colocalized with it within 15 min of agonist stimulation, suggesting a minor and ephemeral role for the lysosome in NHE3 degradation (Fig. 5B).
Figure 5.

NHE3 localization at the proteasome or lysosome. Cellular distribution and colocalization of NHE3 with the proteasome (A) or lysosome (B) in hRPTCs treated with the D3R agonist PD128907 (1 μM) at the indicated time points. Images were obtained via laser scanning confocal microscope. NHE3 was pseudocolored red; proteasome (via p44S10 marker) or lysosome (via LAMP-1 marker) was pseudocolored red. Colocalization was observed as discrete yellow areas in merged images (×600, scale bar=10 μm, n=3). WGA was used to target the lectins on the plasma membrane and DAPI to visualize the nucleus.
Decreased D3R function increases renal expression of NHE3 and blood pressure
As previously reported, Drd3−/− mice on a normal salt diet have increased systolic and diastolic blood pressure, whether animals are under anesthesia (Fig. 6A) or conscious (13). The high blood pressure in Drd3−/− mice was associated with increased renal protein expression of NHE3 (Fig. 6B), as well as NHE3 abundance in the renal proximal tubules (Fig. 6C). This was, however, not accompanied by a corresponding increase in mRNA expression (Fig. 6D), indicating that the increase in NHE3 protein abundance was not related to increased gene transcription. To rule out compensatory mechanisms that may have developed in the Drd3−/− knockout mice and further assess the role of D3R in the regulation of NHE3 expression, mice were placed on a high-salt diet and were given a subcutaneous infusion of the specific D3R antagonist GR103691 for 4 d. The treatment increased the NHE3 protein expression in the kidney (Fig. 6E) without significant changes in blood pressure, creatinine clearance, or the expression of a sodium transporter, sodium chloride cotransporter (NCC), indicating that D3R function is needed to maintain normal levels of NHE3.
Figure 6.

Effect of Drd3 deletion on blood pressure and renal NHE3 expression. A) Systolic and diastolic blood pressures of Drd3−/− knockout mice and wild-type littermates on a normal salt diet, measured in animals under pentobarbital anesthesia. B) Renal NHE3 expression in Drd3−/− mice and wild-type littermates on a normal salt diet. *P < 0.05 vs. basal; Student's t test (n=3 performed in duplicate). C) NHE3 immunostaining in renal proximal tubules of Drd3−/− mice and wild-type littermates. Left: ×200. Right: ×1000. PT, proximal tubule; G, glomerulus. D) mRNA profiles of NHE3 in the renal cortices of Drd3−/− mice and wild-type littermates. E) NHE3 and NCC expression profiles in C57Bl/6J mice on a high-salt (1.6% NaCl) diet and treated with the D3R antagonist GR103691 (1 μg/kg/min for 4 d via miniosmotic pump) or vehicle. Molecular sizes: NHE3, 90 kDa; NCC, 115 kDa. *P < 0.05 vs. D3R+/+; 1-way ANOVA and Holm-Sidak post hoc test (n=4/group).
Renal subcapsular infusion of Usp48-specific siRNA increases ubiquitinylation of NHE3 and decreases NHE3 expression and blood pressure in mice
To determine the effects of the loss of USP48 in vivo, we infused Usp48-specific siRNA into the left kidney of uninephrectomized mice via minipump for 7 d to selectively down-regulate renal Usp48 expression. This procedure caused a 55% reduction of renal Usp48 expression (Fig. 7A). In agreement with the in vitro data, renal depletion of USP48 resulted in a 30% decrease in total NHE3 abundance (Fig. 7B) and a 50% increase in ubiquitinylated NHE3, when compared to that in the control groups (Fig. 7C), without a change in D3R expression. To determine whether there is a physiological correlate with the decrease in NHE3 expression, we measured the blood pressure in mice before and after the renal subcapsular infusion of vehicle, Usp48-specific siRNA, or nonsilencing “mock” siRNA. We found that both the systolic and diastolic blood pressures were lower by ∼20 mm Hg in USP48-depleted C57Bl/6J mice (Fig. 7D, E), indicating that USP48 plays a role in blood pressure regulation through its effect on NHE3 degradation. Baseline systolic and diastolic blood pressures were not different before siRNA, mock, or vehicle infusion.
Figure 7.

Effect of kidney-restricted USP48 depletion on NHE3 and NHE3-Ub abundance and blood pressure in mice. A–C) Expression profiles of endogenous USP48, NHE3, and ubiquitinylated NHE3 (NHE3-Ub) in renal cortices of C57Bl/6J mice that received renal infusion of Usp48-specific siRNA, nonsilencing “mock” siRNA, or vehicle through an osmotic minipump. GAPDH was used for normalization. C) NHE3-Ub was visualized by immunoprecipitating with anti-NHE3 antibody and immunoblotting for Ub. Immunoblots are shown above the corresponding bar graphs. *P < 0.05 vs. vehicle; 1-way ANOVA and Holm-Sidak post hoc test (n=3–4/group). #P < 0.05 vs. mock siRNA transfection or vehicle; Student's t test (n=3–4/group). D, E) Systolic (D) and diastolic (E) blood pressures of uninephrectomized C67Bl/6J mice that had Usp48-specific siRNA, nonsilencing “mock” siRNA, or vehicle infused into the renal subcapsular space through an osmotic pump. The blood pressure was measured before and after the siRNA infusion. Molecular sizes: USP48, 120 kDa; GSPDH, 35 kDa; NHE3, 90 kDa; NHE3-Ub, 100 kDa.*P < 0.05 vs. other groups; 1-way ANOVA and Holm-Sidak post hoc test (n=3–4/group).
DISCUSSION
D3R regulates NHE3 degradation
The mechanisms regulating NHE3 function in the short-term, which for the most part are related to phosphorylation and trafficking of the exchanger, have been extensively studied (46–48). However, the factors that affect NHE3 expression and consequently function in the long term are much less established and are generally regarded to be dependent on transcriptional regulation. The parathyroid hormone decreases renal NHE3 protein expression by decreasing gene transcription (49), whereas angiotensin II, insulin, and glucocorticoids increase NHE3 protein expression by increasing gene transcription (47, 50, 51). In an opossum kidney cell line, putative mechanisms in which dopamine may regulate NHE3 include a reduction in NHE3 translation, but not transcription, and in NHE3 half-life (52). In the current study, the long-term regulation of NHE3 in hRPTCs and mouse kidney involved the regulation of its degradation.
Both phosphorylation (53) and ubiquitinylation (54, 55) are necessary for slow protein degradation in proteasomes. In yeast, NHE2 and NHE3 at the plasma membrane are ubiquitinylated before internalization, and decreasing the activity of the Ub ligase Rsp5 increases NHE2/3 plasma membrane expression (26). Dopamine has also been reported to increase the ubiquitinylation of cell surface NHE3 in opossum kidney cells (27). Furthermore, ischemia–reperfusion injury is associated with decreased NHE3 expression that is dependent on NHE3 ubiquitinylation and proteasomal degradation (52). We now show that NHE3 is ubiquitinylated in the basal state, suggesting that this post-translational modification is involved in the tonic recycling of the exchanger, whereas D3R stimulation promotes NHE3 ubiquitinylation and its translocation to the cytosol.
The catalytic activity of deubiquitinylating enzymes is regulated by multiple mechanisms, including protein–protein interaction and post-translational modification (56). Although we did not attempt to establish the underlying events in the inhibition of USP48 deubiquitinylating activity, there are at least 2 possible mechanisms involved. First, it has been reported that the protease activity of UCH37 is inhibited by its binding to the chromatin-remodeling complex (57); thus, the D3R–USP48 interaction may induce conformational alterations in the enzyme's active site to decrease its activity. Second, phosphorylation has also been reported to decrease protease activity in other USPs (56). It is conceivable that D3R stimulation results in USP48 phosphorylation, in that USP48 has several predicted phosphorylation sites. We have already reported that the D3R phosphorylates p44/42 MAP kinases in hRPTCs (31).
USP48 is important in the regulation of NHE3 abundance and the control of blood pressure
Dopamine synthesized in renal proximal tubular cells acts as an intrarenal natriuretic hormone by inhibiting NHE3 and Na+,K+-ATPase, among others, resulting in a decrease in sodium reabsorption, both in the proximal tubule and more distal segments of the nephron (1, 6–9, 11, 58). D3R is important in regulating renal sodium transport in vivo. Lack of D3R function in mice, whether from genetic deletion or pharmacologic antagonism of the receptor, was associated with increased renal expression of NHE3. Conversely, D3R stimulation decreased NHE3 expression. An increase in renal NHE3 protein expression was also found in the C56Bl/6 mice on a moderately high-salt diet treated with a D3R antagonist for 4 d. Although Drd3−/− mice are hypertensive (13), short-term D3R antagonist treatment of mice did not affect blood pressure. Therefore, the D3R may constitutively inhibit NHE3 expression independent of blood pressure.
To study the effects of USP48 in vivo, we used a model of selective renal cortical silencing of the gene (38–41). Through this approach, the down-regulation of gene expression was restricted to the kidney, thus precluding the confounding effects of systemic silencing. The intrarenal cortical administration of Usp48-specific siRNA mirrored its effects in vitro. Renal cortical expression of NHE3 decreased, whereas ubiquitinylated NHE3 increased in USP48-depleted mice. These changes resulted in lower systolic and diastolic blood pressures, indicating that USP48 activity through regulation of NHE3 expression has a role in the control of blood pressure. The decrease in blood pressure in our model is in agreement with the decrease in blood pressure shown in mice with deletion of the NHE3 gene (59).
In summary, we have shown that D3R, NHE3, and USP48 cosegregate in lipid raft membrane microdomains, physically interact with one another in hRPTCs, and perhaps form a cohesive signaling network for an efficient propagation of signal transduction (Fig. 8). Activation of D3R results in receptor phosphorylation and internalization (31); ubiquitinylation, internalization, and inhibition of membrane-bound NHE3, directly through protein–protein transregulation or indirectly via PLC and PKC (23); and internalization and inactivation of USP48. The D3R-mediated inactivation of USP48 prevents it from deubiquitinylating the NHE3-Ub, earmarking it presumably for proteasomal degradation. This constitutes a novel mechanism by which D3R regulates NHE3 expression in the long term. Thus, the D3R's effect on USP48 activity reveals a previously undescribed mechanism of regulation of sodium transport and blood pressure and underscores the biological relevance of USP48 in the long-term control of arterial blood pressure.
Figure 8.
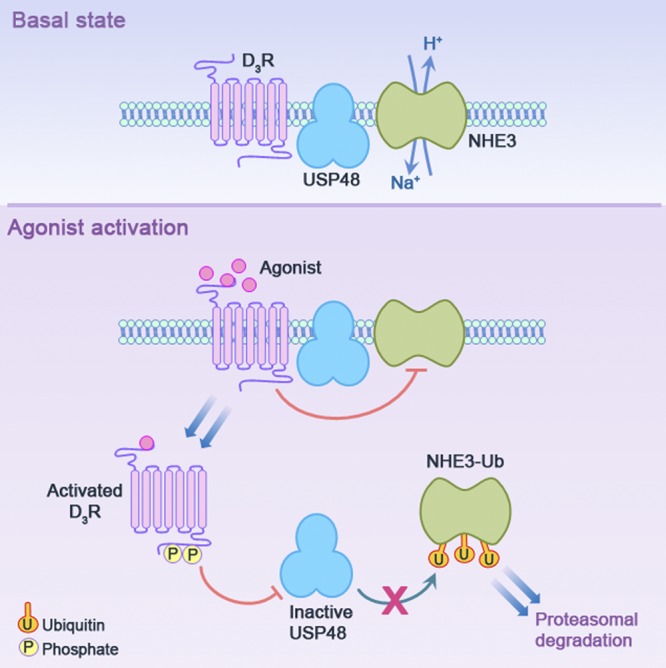
Proposed schema for the dynamic interaction among D3R, USP48, and NHE3 in hRPTCs. D3R, USP48, and NHE3 cofractionate in lipid raft membrane microdomains in the basal state, suggesting the proximity of these key proteins for an effective signal transduction. Ligand occupation of the receptor causes a change in receptor conformation followed by receptor phosphorylation by GRK4 and internalization (31), which effectively attenuates the receptor's responsiveness to subsequent stimulus (receptor desensitization), leading to the internalization of NHE3 and its inactivation by D3R directly, through protein–protein transregulation, or indirectly via PLC and PKC signaling (23) or NHE3 ubiquitinylation (current study). The internalized D3R colocalizes with and inactivates the USP48, thereby preventing the removal of the Ub tags from NHE3-Ub and promoting NHE3-Ub degradation, presumably via the proteasome.
Acknowledgments
This work was supported in part by U.S. National Institutes of Health grants P01HL074940, P01HL068686, R01HL092196, R37HL023081, R01DK039308, and R01DK090918.
The authors declare no conflicts of interest.
Footnotes
- β-MCD
- methyl-β-cyclodextrin
- AMC
- 7-amino-4-methylcoumarin
- CTxB
- cholera toxin subunit B
- D1–5R
- dopamine D1–5 receptor
- Drd3
- dopamine D3 receptor (gene)
- D3R-IC3
- 3rd intracellular loop of the human D3R
- FBS
- fetal bovine serum
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- GPCR
- G-protein-coupled receptor
- hRPTC
- human renal proximal tubule cell
- Met/Cys
- methionine/cysteine
- mTAL
- medullary thick ascending limb
- NCC
- sodium chloride cotransporter
- NHE3
- Na+-H+ exchanger 3
- PBS
- phosphate-buffered saline
- Ub
- ubiquitin
- USP48
- ubiquitin-specific protease 48
- WGA
- wheat germ agglutinin
REFERENCES
- 1. Felder R. A., Seikaly M. G., Cody P., Eisner G. M., Jose P. A. (1990) Attenuated renal response to dopaminergic drugs in spontaneously hypertensive rats. Hypertension 15, 560–569 [DOI] [PubMed] [Google Scholar]
- 2. Chen C. J., Lokhandwala M. F. (1992) An impairment of renal tubular DA-1 receptor function as the causative factor for diminished natriuresis to volume expansion in spontaneously hypertensive rats. Clin. Exp. Hypertens. A. 14, 615–628 [DOI] [PubMed] [Google Scholar]
- 3. Luippold G., Küster E., Joos T. O., Mühlbauer B. (1998) Dopamine D3 receptor activation modulates renal function in anesthetized rats. Naunyn Schmiedebergs Arch. Pharmacol. 358, 690–693 [DOI] [PubMed] [Google Scholar]
- 4. Ladines C. A., Zeng C., Asico L. D., Sun X., Pocchiari F., Semeraro C., Pisegna J., Wank S., Yamaguchi I., Eisner G. M., Jose P. A. (2001) Impaired renal D1-like and D2-like dopamine receptor interaction in the spontaneously hypertensive rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281, R1071–R1078 [DOI] [PubMed] [Google Scholar]
- 5. Sibley D. R. (1999) New insights into dopaminergic receptor function using antisense and genetically altered animals. Annu. Rev. Pharmacol. Toxicol. 39, 313–314 [DOI] [PubMed] [Google Scholar]
- 6. Brismar H., Holtbäck U., Aperia A. (2000) Mechanisms by which intrarenal dopamine and ANP interact to regulate sodium metabolism. Clin. Exp. Hypertens. 22, 303–307 [DOI] [PubMed] [Google Scholar]
- 7. Carey R. M. (1992) Theodore Cooper lecture: renal dopamine system: paracrine regulator of sodium homeostasis and blood pressure. Hypertension 38, 297–302 [DOI] [PubMed] [Google Scholar]
- 8. Hussain T., Lokhandwala M. F. (2003) Renal dopamine receptors and hypertension. Exp. Biol. Med. (Maywood) 228, 134–142 [DOI] [PubMed] [Google Scholar]
- 9. Zeng C., Armando I., Luo Y., Eisner G. M., Felder R. A., Jose P. A. (2008) Dysregulation of dopamine-dependent mechanisms as a determinant of hypertension: studies in dopamine receptor knockout mice. Am. J. Physiol. Heart Circ. Physiol. 294, H551–H569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Felder R. A., Sanada H., Xu J., Yu P. Y., Wang Z., Watanabe H., Asico L. D., Wang W., Zheng S., Yamaguchi I., Williams S. M., Gainer J., Brown N. J., Hazen-Martin D., Wong L. J., Robillard J. E., Carey R. M., Eisner G. M., Jose P. A. (2002) G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc. Natl. Acad. Sci. U. S. A. 99, 3872–38777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harris R. C. (2012) Abnormalities in renal dopamine signaling and hypertension: the role of GRK4. Curr. Opin. Nephrol. Hypertens. 21, 61–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ueda A., Ozono R., Oshima T., Yano A., Kambe M., Teranishi Y., Katsuki M., Chayama K. (2003) Disruption of the type 2 dopamine receptor gene causes a sodium-dependent increase in blood pressure in mice. Am. J. Hypertens. 16, 853–858 [DOI] [PubMed] [Google Scholar]
- 13. Asico L. D., Ladines C., Fuchs S., Accili D., Carey R. M., Semeraro C., Pocchiari F., Felder R. A., Eisner G. M., Jose P. A. (1998) Disruption of the dopamine D3 receptor gene produces renin-dependent hypertension. J. Clin. Invest. 102, 493–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luippold G., Zimmermann C., Mai M., Kloor D., Starck D., Gross G., Mühlbauer B. (2001) Dopamine D3 receptors and salt-dependent hypertension. J. Am. Soc. Nephrol. 12, 2272–2279 [DOI] [PubMed] [Google Scholar]
- 15. Zeng C., Eisner G. M., Felder R. A., Jose P. A. (2006) D3 dopamine receptor and essential hypertension. Curr. Hypertens. Rev. 2, 247–253 [Google Scholar]
- 16. Yang Z., Asico L. D., Yu P., Wang Z., Jones J. E., Escano C. S., Wang X., Quinn M. T., Sibley D. R., Romero G. G., Felder R. A., Jose P. A. (2006) D5 dopamine receptor regulation of reactive oxygen species production, NADPH oxidase, and blood pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R96–R104 [DOI] [PubMed] [Google Scholar]
- 17. Felder R. A., Jose P. A. (2006) Mechanisms of disease: the role of GRK4 in the etiology of essential hypertension and salt sensitivity. Nat. Clin. Pract. Nephrol. 2, 637–650 [DOI] [PubMed] [Google Scholar]
- 18. Ortiz P. A., Garvin J. L. (2001) Intrarenal transport and vasoactive substances in hypertension. Hypertension 38, 621–624 [DOI] [PubMed] [Google Scholar]
- 19. Zeng C., Liu Y., Wang Z., He D., Huang L., Yu P., Zheng S., Jones J. E., Asico L. D., Hopfer U., Eisner G. M., Felder R. A., Jose P. A. (2006) Activation of D3 dopamine receptor decreases angiotensin II type 1 receptor expression in rat renal proximal tubule cells. Circ. Res. 99, 494–500 [DOI] [PubMed] [Google Scholar]
- 20. Kaneko S., Albrecht F., Asico L. D., Eisner G. M., Robillard J. E., Jose P. A. (1992) Ontogeny of DA1 receptor-mediated natriuresis in the rat: in vivo and in vitro correlations. Am. J. Physiol. 263, R631–R638 [DOI] [PubMed] [Google Scholar]
- 21. Kaneko S., Eisner G. M., Jose P. A. (1990) Effect of pramipexole, a dopamine-1/dopamine-2 receptor agonist, on sodium excretion and blood pressure in spontaneously hypertensive rats. J. Auton. Pharmacol. 10, s53–s60 [DOI] [PubMed] [Google Scholar]
- 22. Zeng C., Asico L. D., Yu C., Villar V. A., Shi W., Luo Y., Wang Z., He D., Liu Y., Huang L., Yang C., Wang X., Hopfer U., Eisner G. M., Jose P. A. (2008) Renal D3 dopamine receptor stimulation induces natriuresis via an ETB endothelin receptor mechanism. Kidney Int. 74, 750–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pedrosa R., Gomes P., Hopfer U., Jose P. A., Soares-da-Silva P. (2004) Giα3 protein-coupled dopamine D3 receptor-mediated inhibition of renal NHE3 activity in SHR proximal tubular cells is a PLC-PKC-mediated event. Am. J. Physiol. Renal Physiol. 287, F1059–F1066 [DOI] [PubMed] [Google Scholar]
- 24. Pedrosa R., Gomes P., Zeng C., Hopfer U., Jose P. A., Soares-da-Silva P. (2004) Dopamine D3 receptor-mediated inhibition of Na+/H+ exchanger activity in normotensive and spontaneously hypertensive rat proximal tubular epithelial cells. Br. J. Pharmacol. 142, 1343–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gomes P., Soares-da-Silva P. (2002) D2-like receptor-mediated inhibition of Na+-K+-ATPase activity is dependent on the opening of K+ channels. Am. J. Physiol. Renal Physiol. 283, F114–F123 [DOI] [PubMed] [Google Scholar]
- 26. Flegelova H., Haguenauer-Tsapis R., Sychrova H. (2006) Heterologous expression of mammalian Na/H antiporters in Saccharomyces cerevisiae. Biochim. Biophys. Acta 1760, 504–516 [DOI] [PubMed] [Google Scholar]
- 27. Hu M. C., Di Sole F., Zhang J., McLeroy P., Moe O. W. (2013) Chronic regulation of the renal Na+/H+ exchanger NHE3 by dopamine: Translational and post-translational mechanisms. Am. J. Physiol. Renal Physiol. 304, F1169–F1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilkinson K. D. (1999) Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J. 11, 1245–1256 [DOI] [PubMed] [Google Scholar]
- 29. Lockhart P. J., Hulihan M., Lincoln S., Hussey J., Skipper L., Bisceglio G., Wilkes K., Farrer M. J. (2004) Identification of the human ubiquitin specific protease 31 (USP31) gene: structure, sequence and expression analysis. DNA Seq. 15, 9–14 [DOI] [PubMed] [Google Scholar]
- 30. Quesada V., Diaz-Perales A., Gutierrez-Fernandez A., Garabaya C., Cal S., Lopez-Otin C. L. (2004) Cloning and enzymatic analysis of 22 novel human ubiquitin-specific proteases. Biochem. Biophys. Res. Commun. 314, 54–62 [DOI] [PubMed] [Google Scholar]
- 31. Villar V. A., Jones J. E., Armando I., Palmes-Saloma C., Yu P., Pascua A. M., Keever L., Arnaldo F. B., Wang Z., Luo Y., Felder R. A., Jose P. A. (2009) G protein-coupled receptor kinase 4 (GRK4) regulates the phosphorylation and function of the dopamine D3 receptor. J. Biol. Chem. 284, 21425–21434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li H., Armando I., Yu P., Escano C., Mueller S. C., Asico L., Pascua A., Lu Q., Wang X., Villar V. A., Jones J. E., Wang Z., Periasamy A., Lau Y. S., Soares-da-Silva P., Creswell K., Guillemette G., Sibley D. R., Eisner G., Gildea J. J., Felder R. A., Jose P. A. (2008) Dopamine 5 receptor mediates Ang II type 1 receptor degradation via a ubiquitin-proteasome pathway in mice and human cells. J. Clin. Invest. 118, 2180–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Day C. A., Kenworthy A. K. (2012) Mechanisms underlying the confined diffusion of cholera toxin B-subunit in intact cell membranes. PLoS ONE 7, e34923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Song K. S., Li S., Okamoto T., Quilliam L. A., Sargiacomo M., Lisanti M. P. (1996) Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains: detergent-free purification of caveolae microdomains. J. Biol. Chem. 271, 9690–9697 [DOI] [PubMed] [Google Scholar]
- 35. Yu P., Villar V. A., Jose P. A. (2013) Methods for the study of dopamine receptors within lipid rafts of kidney cells. Methods Mol. Biol. 964, 15–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fernández-Montalván A., Bouwmeester T., Joberty G., Mader R., Mahnke M., Pierrat B., Schlaeppi J. M., Worpenberg S., Gerhartz B. (2007) Biochemical characterization of USP7 reveals post-translational modification sites and structural requirements for substrate processing and subcellular localization. FEBS J. 274, 4256–42570 [DOI] [PubMed] [Google Scholar]
- 37. Ladasky J. J., Boyle S., Seth M., Li H., Pentcheva T., Abe F., Steinberg S. J., Edidin M. (2006) Bap31 enhances the endoplasmic reticulum export and quality control of human class I MHC molecules. J. Immunol. 177, 6172–6181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cuevas S., Zhang Y., Yang Y., Escano C., Asico L., Jones J. E., Armando I., Jose P. A. (2012) Role of renal DJ-1 in the pathogenesis of hypertension associated with increased reactive oxygen species production. Hypertension 59, 446–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Y., Cuevas S., Asico L. D., Escano C., Yang Y., Pascua A. M., Wang X., Jones J. E., Grandy D., Eisner G., Jose P. A., Armando I. (2012) Deficient dopamine D2 receptor function causes renal inflammation independently of high blood pressure. PLoS ONE 7, e38745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Villar V. A., Jones J. E., Armando I., Asico L. D., Escano C. S., Jr., Lee H., Wang X., Yang Y., Pascua-Crusan A. M., Palmes-Saloma C. P., Felder R. A., Jose P. A. (2013) Sorting nexin 1 loss results in D5 dopamine receptor dysfunction in human renal proximal tubule cells and hypertension in mice. J. Biol. Chem. 288, 152–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Villar V. A., Armando I., Sanada H., Frazer L. C., Russo C. M., Notario P. M., Lee H., Comisky L., Russell H. A., Yang Y., Jurgens J. A., Jose P. A., Jones J. E. (2013) Novel role of sorting nexin 5 in renal D1 dopamine receptor trafficking and function: implications for hypertension. FASEB J. 27, 1808–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li X., Galli T., Leu S., Wade J.B., Weinman E.J., Leung G., Cheong A., Louvard D., Donowitz M. (2001) Na+-H+ exchanger 3 (NHE3) is present in lipid rafts in the rabbit ileal brush border: a role for rafts in trafficking and rapid stimulation of NHE3. J. Physiol. 537, 537–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Murtazina R., Kovbasnjuk O., Donowitz M., Li X. (2006) Na+/H+ exchanger NHE3 activity and trafficking are lipid raft-dependent. J. Biol. Chem. 281,17845–17855 [DOI] [PubMed] [Google Scholar]
- 44. Kenworthy A. K., Petranova N., Edidin M. (2000) High-resolution FRET microscopy of cholera toxin B-subunit and GPI-anchored proteins in cell plasma membranes. Mol. Biol. Cell 11, 1645–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mikhalyov I., Samsonov A. (2011) Lipid raft detecting in membranes of live erythrocytes. Biochim. Biophys. Acta. 1808, 1930–1929 [DOI] [PubMed] [Google Scholar]
- 46. Moe O. W. (1997) Sodium-hydrogen exchange in renal epithelia: mechanisms of acute regulation. Curr. Opin. Nephrol. Hypertens. 6, 440–446 [DOI] [PubMed] [Google Scholar]
- 47. Donowitz M., Li X. (2007) Regulatory binding partners and complexes of NHE3. Physiol. Rev. 87, 825–872 [DOI] [PubMed] [Google Scholar]
- 48. McDonough A. A. (2010) Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R851–R861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bezerra C. N., Girardi A. C., Carraro-Lacroix R., Rebouças N. A. (2008) Mechanisms underlying the long-term regulation of NHE3 by parathyroid hormone. Am. J. Physiol. Renal Physiol. 294, F1232–F1237 [DOI] [PubMed] [Google Scholar]
- 50. Klisic J., Hu M. C., Nief V., Reyes L., Fuster D., Moe O. W., Ambühl P. M. (2002) Insulin activates Na+/H+ exchanger 3: biphasic response and glucocorticoid dependence. Am. J. Physiol. Renal Physiol. 283, F532–F539 [DOI] [PubMed] [Google Scholar]
- 51. Queiroz-Leite G. D., Peruzzetto M. C., Neri E. A., Rebouças N. A. (2011) Transcriptional regulation of the Na+/H+ exchanger NHE3 by chronic exposure to angiotensin II in renal epithelial cells. Biochem. Biophys. Res. Commun. 409, 470–4761 [DOI] [PubMed] [Google Scholar]
- 52. Di Sole F., Hu M. C., Zhang J., Babich V., Bobulescu I. A., Shi M., McLeroy P., Rogers T. E., Moe O. W. (2011) The reduction of Na/H exchanger-3 protein and transcript expression in acute ischemia-reperfusion injury is mediated by extractable tissue factor(s). Kidney Int. 80, 822–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Morales P., Díaz E. S., Kong M. (2007) Proteasome activity and its relationship with protein phosphorylation during capacitation and acrosome reaction in human spermatozoa. Soc. Reprod. Fertil. Suppl. 65, 269–273 [PubMed] [Google Scholar]
- 54. Wojcikiewicz R. J. (2004) Regulated ubiquitination of proteins in GPCR-initiated signaling pathways. Trends Pharmacol. Sci. 25, 35–41 [DOI] [PubMed] [Google Scholar]
- 55. Nalepa G., Rolfe M., Harper J. W. (2006) Drug discovery in the ubiquitin-proteasome system. Nat. Rev. Drug Discov. 5, 596–613 [DOI] [PubMed] [Google Scholar]
- 56. Kessler B. M., Edelmann M. J. (2011) PTMs in conversation: activity and function of deubiquitinating enzymes regulated via post-translational modifications. Cell Biochem. Biophys. 60, 21–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yao T., Song L., Jin J., Cai Y., Takahashi H., Swanson S. K., Washburn M. P., Florens L., Conaway R. C., Cohen R. E., Conaway J. W. (2008) Distinct modes of regulation of the Uch37 deubiquitinating enzyme in the proteasome and in the INo980 chromatin-remodeling complex. Mol. Cell. 31, 909–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jose P. A., Eisner G. M., Felder R. A. (1998) Renal dopamine receptors in health and hypertension. Pharmacol. Ther. 80, 149–182 [DOI] [PubMed] [Google Scholar]
- 59. Schultheis P. J., Clarke L. L., Meneton P., Miller M. L., Soleimani M., Gawenis L. R., Riddle T. M., Duffy J. J., Doetschman T., Wang T., Giebisch G., Aronson P. S., Lorenz J. N., Shull G. E. (1998) Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat. Genet. 19, 282–285 [DOI] [PubMed] [Google Scholar]