

Abstract
Interstitial lung diseases (ILDs) are characterized by injury, inflammation, and scarring of alveoli, leading to impaired function. The etiology of idiopathic forms of ILD is not understood, making them particularly difficult to study due to the lack of appropriate animal models. Consequently, few effective therapies have emerged. We developed an inbred mouse model of ILD using vanadium pentoxide (V2O5), the most common form of a transition metal found in cigarette smoke, fuel ash, mineral ores, and steel alloys. Pulmonary responses to V2O5, including dose-dependent increases in lung permeability, inflammation, collagen content, and dysfunction, were significantly greater in DBA/2J mice compared to C57BL/6J mice. Inflammatory and fibrotic responses persisted for 4 mo in DBA/2J mice, while limited responses in C57BL/6J mice resolved. We investigated the genetic basis for differential responses through genetic mapping of V2O5-induced lung collagen content in BXD recombinant inbred (RI) strains and identified significant linkage on chromosome 4 with candidate genes that associate with V2O5-induced collagen content across the RI strains. Results suggest that V2O5 may induce pulmonary fibrosis through mechanisms distinct from those in other models of pulmonary fibrosis. These findings should further advance our understanding of mechanisms involved in ILD and thereby aid in identification of new therapeutic targets.—Walters, D. M., White, K. M., Patel, U., Davis, M. J., Veluci-Marlow, R. M., Bhupanapadu Sunkesula, S. R., Bonner, J. C., Martin, J. R., Gladwell, W., Kleeberger, S. R. Genetic susceptibility to interstitial pulmonary fibrosis in mice induced by vanadium pentoxide (V2O5).
Keywords: lung injury, inflammation, collagen, haplotype, mapping
Interstitial lung diseases (ILDs) are characterized by parenchymal injury, inflammation, and scarring of the alveoli, leading to impaired lung function. Many ILDs result in interstitial fibrosis for which there are no effective therapies, but prognosis varies depending on cause and pathological pattern. ILDs include more than 200 lung disorders, caused by a variety of stimuli including occupational and environmental exposures (1). However, a number of ILDs, including the prototypical idiopathic pulmonary fibrosis (IPF), have no known cause, making them difficult to model.
Whether of known or unknown etiology, pulmonary fibrosis is a complex trait thought to involve interactions between genetic and environmental factors. Gene-environment interactions in pulmonary fibrosis are supported by observed susceptibility differences in individuals with similar exposures, familial cases, and differential susceptibility among inbred strains of mice exposed to fibrogenic agents such as bleomycin (2), silica (3, 4), and asbestos (5). While extensive research using animal exposures to these known fibrogenic agents has increased understanding of pathology associated with specific exposures, effective therapies to halt the fibrotic process in idiopathic interstitial pneumonias (IIPs) have not been found and may be partially due to unknown environmental factors. Bleomycin has been employed to test numerous antifibrotic agents, yet none of these compounds have been shown to improve patient survival (6, 7). Therefore, alternative models that reflect human exposures associated with IIPs may provide a more relevant context in which to investigate fibrogenic mechanisms and potential therapies.
A number of ILDs are associated with metal exposures (8, 9), and epidemiological studies have consistently found associations between IPF and exposure to metal dusts [odds ratio (OR) 2.44] and cigarette smoking (OR 1.58) (10). In fact, exposure to metal dusts in a Japanese population was very strongly associated with IPF (OR 9.55; ref. 11). Although specific components of metal dusts or cigarette smoke that lead to ILD have not been identified, transition metals are hypothesized to be likely candidates due to their potential to induce cellular damage through production of reactive oxygen species(ROS). In addition, chronic exposure to high levels of vanadium compounds is associated with pneumonitis (12).
Vanadium poses an inhalation risk at high concentrations in occupational settings (13) but is a ubiquitous transition metal found in air pollution particles and cigarettes (14). Vanadium pentoxide (V2O5) is the most common chemical form of vanadium and is used extensively as an alloy in steel for strength at high temperatures and as an industrial catalyst (15). In animals, V2O5 exposure induces a variety of pulmonary responses, including inflammation, fibroblast proliferation, interstitial and/or peribronchiolar fibrosis, and epithelial cell hyperplasia, depending on the species and strain of animal (16–18). In humans, acute occupational exposure to vanadium has been predominantly associated with decrements in lung function indicative of obstructive disease and bronchitis-like symptoms (19–23). Although a direct link between vanadium exposure and ILD has not been established in human populations, animal studies and suggestive evidence from human occupational exposures imply that vanadium exposure may contribute to some cases of ILD in susceptible individuals.
The current study describes a novel model of fibrosing alveolitis using V2O5 to investigate the gene-environment interactions in pulmonary fibrogenesis. Here we describe V2O5-induced lung injury and inflammation with interstitial fibrogenesis in a strain-dependent manner. Furthermore, the strain-dependent differences in susceptibility suggest a genetic contribution to V2O5-induced pulmonary fibrosis, which was analyzed using a set of recombinant inbred strains. The pathophysiological characteristics and environmental relevance of this model should be valuable to identify mechanisms involved in fibrogenesis that have not been previously identified in other models.
MATERIALS AND METHODS
Animals
Male C57BL/6J (B6), DBA/2J (D2), and 49 BXD recombinant inbred (RI) strains of mice (5–7 wk) were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and housed in an environmentally controlled animal facility with a 12:12 light:dark cycle and provided food and water ad libitum throughout the experiments. Mice were acclimated for at least 5 d prior to study. All animal procedures were approved by the Institutional Animal Care and Use Committees of East Carolina University and the U.S. National Institute of Environmental Health Sciences.
V2O5 exposure
Mice were lightly anesthetized with isoflurane gas, and doses of V2O5 particles (Sigma-Aldrich, Milwaukee, WI, USA) suspended in PBS or PBS alone were administered by oropharyngeal aspiration (24) of ∼50 μl on study d 0 and 7. For the dose-response study, 0, 0.1, 0.5, 1, 2, or 4 mg/kg V2O5 was aspirated, and mice were euthanized on study d 14 (n=6–8 mice/dose). For kinetic studies, 4 mg/kg V2O5 was aspirated and mice were euthanized on study d 1, 2, 4, 6, 8, 10, 12, 14, 21, 28, 56, 84, and 112 (n=4–8/group). BXD RI strains were aspirated with 4 mg/kg V2O5 or PBS on study d 0 and 7 and euthanized on study d 14 (n=4–8/group).
Lung function measurements
Baseline lung function measurements were made after V2O5 or vehicle using a forced oscillation technique on the flexiVent system (Scireq, Montreal, QC, Canada). Animals were anesthetized (∼400 mg/kg tribromoethanol, i.p.), tracheotomized, and ventilated with room air at 10 ml/kg and 150 breaths/min with positive end expiratory pressure of 3 cmH2O. The animals were then paralyzed with pancuronium bromide (0.8 mg/kg, i.p.), and respiratory mechanics were measured using a script to ensure consistent timing of perturbations relative to total lung capacity maneuvers to standardize volume history. Measures of airway newtonian resistance (RN) and tissue elastance (H) were obtained using the constant phase model, and static compliance (Cst) was measured using ramped pressure inflation and deflation.
Bronchoalveolar lavage
Immediately after lung function measurements, mice were exsanguinated, the left bronchus was clamped, and the right lung was lavaged with 4 successive aliquots of 26.25 ml/kg cold HBSS. Recovered bronchoalveolar lavage fluid (BALF) was centrifuged at 500 g for 10 min at 4°C. Supernatant from the first BALF aliquot was used for measurement of total protein. All cells from the same individual were pooled, and total cell counts were made with a hemacytometer. Slides were prepared by cytocentrifugation of 20,000 cells at 600 rpm for 5 min (Shandon Cytospin III; Thermo Fisher Scientific, Waltham, MA, USA) and stained (Richard-Allan, Kalamazoo, MI, USA). BALF cell differential counts were performed on 300 cells/slide using standard morphological criteria.
Quantitation of protein in BALF
Total protein concentration in BALF (an index of lung permeability and lung injury) was measured using a Bradford protein assay (Bio-Rad, Hercules, CA, USA) according to the manufacturer's recommendations and read at 595 nm on a spectrophotometer (Beckman-Coulter, Brea, CA, USA). Concentration was calculated from a standard curve.
Measurement of lung collagen
Lavaged right lung lobes were homogenized in RIPA lysis buffer plus protease inhibitor cocktail and PMSF (Sigma-Aldrich). Lung homogenates were centrifuged at 10,000 g for 10 min at 4°C. Soluble collagen was measured by adding 500 μl Sircol Dye Reagent (Biocolor, Carrickfergus, UK) to 50 μl of lung homogenate supernatant. Samples and standards were agitated for 30 min, then centrifuged at 10,000 g for 10 min to pellet collagen-bound dye. Supernatants were discarded, and pellets were dissolved in 1 ml 0.5 M NaOH. Absorbance was read at 540 nm, and sample concentrations calculated from a standard curve.
Histology
Left lungs were inflated with 10% neutral buffered formalin (Azer Scientific, Morgantown, PA, USA) for 24–72 h before cutting into 3 sections and undergoing standard processing. Cross sections (5 μm) were placed in xylene to remove parrafin, hydrated through an ethanol gradient, and stained with Masson's Trichrome to allow visualization of collagen.
PCR fibrosis array
Total lung RNA was isolated from the lower right lung lobe of 3 mice/group euthanized 10 and 21 d after V2O5 (4 mg/kg) or PBS using a RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's recommendations. Total RNA (1.5 μg) was reverse-transcribed to cDNA using SABioscience's RT2 First Strand Kit (Qiagen). Mouse Fibrosis PCR Arrays (Qiagen) were run on an Applied Biosystems StepOnePlus Real-Time PCR System (ABI, Foster City, CA, USA) following kit instructions. Data were analyzed using SABiosciences' web-based PCR array analysis tool (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php).
Quantitative real-time (R-T) PCR
The same total lung RNA isolated for the PCR-array was reverse transcribed using a QuantiTect reverse transcription kit (Qiagen). Several genes found to be differentially expressed, including Ccl11 [chemokine (C-C motif) ligand 11], Il1b (interleukin 1β), Lox (lysyl oxidase), and Mmp3 (matrix metallopeptidase 3) as well as Gapdh (internal control) were verified in B6 and D2 mice by quantitative R-T PCR using QuantiTect primer assays and SYBR green master mix (Qiagen). In addition, mRNA expression for genes found in the significant chromosome 4 quantitative trait locus [QTL; Cdk5rap2 (CDK5 regulatory subunit associated protein 2), Tle1 (transducin-like enhancer of split 1), Frmd3 (FERM domain containing 3), Kdm4c (lysine [K]-specific demethylase 4C), and Ptprd (protein tyrosine phosphatase, receptor type D)] was measured in lungs from B6 and D2 mice after vehicle and V2O5 treatment. R-T PCR was used to obtain cycle threshold (Ct) values for target and internal control cDNA levels. Data are expressed as the average ± se ΔCt of 3 animals/group, where ΔCt is defined as Ct target − Ct internal control.
Transforming growth factor β (TGF-β) ELISA
TGF-β protein levels were measured in the same lung homogenates as collagen content using the Human/Mouse TGF-β ELISA Ready-SET-Go! kit (eBioscience; Affymetrix, San Diego, CA, USA) according to the manufacturer's instructions. The assay is specific for activated TGF-β; however, further acid activation to measure total TGF-β levels produced readings below the level of detection suggesting that the detergent in the RIPA lysis buffer had already caused dissociation of the latency-associated peptide. Therefore, data represent total TGF-β protein levels.
Genetic linkage analyses
The mean PBS response for each BXD RI strain was subtracted from individual V2O5-induced responses, and the difference was used for V2O5-induced cell and collagen phenotypes for genetic mapping. Correlations between cell and collagen phenotypes were evaluated using the correlation matrix function in GeneNetwork (http://www.genenetwork.org/webqtl/main.py). GeneNetwork was also used to identify QTLs for V2O5-induced increases in lung collagen content, total BALF cells, and cell differentials. Genome-wide scans for QTLs were done with the entire RI data set and parental phenotypes. More than 7600 informative markers and ∼3800 markers with unique strain distribution patterns exist for the BXD RI set. Details of analysis procedures were published previously (25).
Statistical analyses
All data are presented as means ± se for each group. Two-way ANOVA was used to evaluate dose and strain effects in dose-response studies, while treatment (V2O5 or PBS) and strain effects were evaluated at each time point for the kinetics and BXD RI studies. Bonferroni post hoc testing was used to determine statistical differences between groups, and P < 0.05 was accepted as statistically significant. SigmaStat 3.0 software (SPSS Science, Chicago, IL, USA) was used for all statistical analyses.
RESULTS
Dose response
Dose-dependent increases in mean BALF protein were found in B6 and D2 mice 7 d after the second of 2 V2O5 doses (Fig. 1A). In both strains, protein concentrations were significantly elevated compared to PBS controls after 0.5–4.0 mg/kg V2O5. However, the response in D2 mice was significantly greater than in B6 mice after 2 and 4 mg/kg V2O5.
Figure 1.
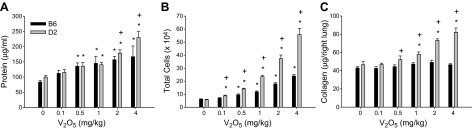
Responses to increasing doses of V2O5 in B6 and D2 mice for protein content in BALF (A), total number of cells recovered in BALF (B), and lung collagen content (C). Bars represent group means ± se; n = 7–8 mice/group. *P < 0.05 vs. 0 mg/kg V2O5 within strain; +P < 0.05 vs. B6 within dose; n = 6–8 mice/group.
Similarly, mean numbers of BALF cells increased dose-dependently in both strains; however, significantly more cells were found in D2 mice compared to B6 mice at all V2O5 doses (Fig. 1B). Macrophages accounted for ∼90% of BALF cells (range 88.7% – 94.3%) across all groups (Table 1); however, numbers of macrophages were significantly higher in D2 mice at all V2O5 doses. Numbers of lymphocytes were also significantly elevated in D2 compared to B6 mice for the highest 3 V2O5 doses. No change from control collagen content at any V2O5 dose was found in B6 mice, whereas V2O5 induced a significant increase in lung collagen at 1, 2, and 4 mg/kg V2O5 in D2 mice (Fig. 1C).
Table 1.
Differential cell counts from BALF 14 d after increasing doses of V2O5
| Strain | V2O5 (mg/kg) | Macrophages | Epithelial cells | Neutrophils | Eosinophils | Lymphocytes | Monocytes |
|---|---|---|---|---|---|---|---|
| B6 | 0.0 | 56,905 ± 2900 | 3842 ± 321 | 56 ± 36 | 0 ± 0 | 535 ± 81 | 92 ± 62 |
| D2 | 0.0 | 53,433 ± 3112 | 2978 ± 500 | 24 ± 24 | 120 ± 120 | 589 ± 245 | 44 ± 29 |
| B6 | 0.1 | 65,419 ± 3760 | 4887 ± 696 | 212 ± 88 | 213 ± 93* | 950 ± 277 | 105 ± 70 |
| D2 | 0.1 | 80,680 ± 5109*,# | 4201 ± 352 | 206 ± 146 | 387 ± 152 | 1330 ± 431 | 383 ± 103# |
| B6 | 0.5 | 88,599 ± 7747* | 5354 ± 915 | 723 ± 148 | 398 ± 43* | 1268 ± 305 | 88 ± 57 |
| D2 | 0.5 | 127,278 ± 5685*,# | 7130 ± 1009* | 475 ± 246 | 840 ± 254* | 3568 ± 823* | 1022 ± 299# |
| B6 | 1.0 | 107,313 ± 7735* | 7024 ± 605* | 1048 ± 232 | 489 ± 136* | 1433 ± 252 | 193 ± 142 |
| D2 | 1.0 | 213,687 ± 12929*,# | 9657 ± 1769* | 568 ± 219 | 2405 ± 1030* | 6238 ± 1842*,# | 1730 ± 452*,# |
| B6 | 2.0 | 160,721 ± 8994* | 9636 ± 1170* | 1568 ± 696* | 1477 ± 511* | 3314 ± 361 | 855 ± 321 |
| D2 | 2.0 | 342,627 ± 28302*,# | 11,626 ± 1120* | 1877 ± 1357 | 7667 ± 2282* | 15,601 ± 3655*,# | 2744 ± 672* |
| B6 | 4.0 | 214,854 ± 13167* | 9339 ± 290* | 1219 ± 682 | 798 ± 283* | 11,110 ± 1553* | 1967 ± 375* |
| D2 | 4.0 | 494,420 ± 45762*,# | 16,158 ± 1673*,# | 4007 ± 858*,# | 9495 ± 3362*,# | 29,884 ± 3665*,# | 3535 ± 742* |
P < 0.05 vs. 0 mg/kg V2O5 within strain
P < 0.05 vs. B6 within dose.
Dose-dependent increases in airway RN and tissue H were also found in D2 mice but did not change significantly in B6 mice (Fig. 2A, B). Quasi-static Cst was significantly lower compared to PBS control at the 2 highest doses of V2O5 in D2 mice. Significantly lower Cst at baseline and at all doses of V2O5 were found in D2 mice compared to B6 mice (Fig. 2C).
Figure 2.
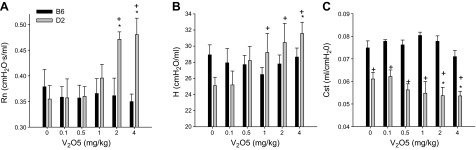
Changes in lung function with increasing doses of V2O5 in B6 and D2 mice. A) Newtonian resistance, RN, B) Tissue elastance, H. C) Static compliance, Cst. Bars represent group means ± se; n = 6–8 mice/group. *P < 0.05 vs. control (0 mg/kg V2O5) within strain; +P < 0.05 vs. B6 within dose; n = 6–8 mice/group.
Overall, V2O5-treated B6 lung sections were similar to vehicle controls, although at the highest dose of V2O5, mild peribronchiolar/perivascular inflammation was found in 2 of 7 lungs (Fig. 3). In D2 mice, patchy areas of alveolar inflammation with increased collagen staining appeared after 0.5 mg/kg, and the area and severity of inflammation and alveolar collagen deposition increased with dose. Subpleural regions appeared to be most affected, and at the highest doses, alveolar epithelial type II cell hyperplasia and infrequent fibrotic nodules were found.
Figure 3.

V2O5-induced changes in lung histology in B6 and D2 mice. V2O5 had little effect on B6 mice at all doses except 4 mg/kg, where minimal peribronchiolar/perivascular inflammation was found (arrow). In contrast, exposure to 0.5 mg/kg V2O5 caused focal inflammation, alveolar wall thickening, and collagen deposition in D2 mice and became more extensive with increasing dose, especially in subpleural regions (arrows). Infrequent fibrotic nodules surrounded by hypertrophic epithelium were seen in D2 mice after 4 mg/kg V2O5 (arrowhead). Masson's Trichrome staining of 5 μm paraffin sections. Original magnification: ×100; insets, ×400.
Time course
A small but significant increase in BALF protein was found in B6 mice until 6 d after V2O5 (Fig. 4A), and a second V2O5 aspiration did not increase BALF protein above controls. Compared to B6 mice, significantly greater BALF protein was found in D2 mice, peaking 2–3 d after the first and second V2O5 doses. Protein concentrations remained significantly elevated over controls to 21 d in D2 mice.
Figure 4.
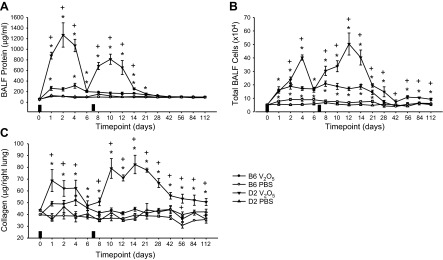
Time course of responses to 4 mg/kg V2O5 in B6 and D2 mice. V2O5-induced increases in BALF protein levels (A), total number of cells recovered in BALF (B), and lung collagen content (C) to a much greater extent in D2 mice than in B6 mice. Solid rectangles on x axis indicate the days when V2O5 was aspirated. *P < 0.05 vs. PBS control within strain; +P < 0.05 vs. B6 mice within same treatment group; n = 4–8 mice/group.
Total numbers of BALF cells increased similarly in B6 and D2 mice 1 d after V2O5 (Fig. 4B), and they remained near this level until d 21 in B6 mice; cell numbers continued to rise in D2 and peaked 4–5 d after V2O5. Unlike BALF protein, cell numbers were enhanced after a second exposure and did not return to controls at 112 d after V2O5 in D2 mice. The majority of BALF cells were macrophages at all times except d 1 (Table 2). The greatest numbers of neutrophils were found 1–2 d after V2O5 and remained significantly elevated above controls through d 14 in both strains, although peaks were significantly higher in D2 mice. Numbers of eosinophils were greatest 2 d after V2O5 in both strains of mice, and the increase was significant in B6 mice only on d 6. The greatest numbers of lymphocytes were found 5–6 d after V2O5 in D2 mice and remained significantly elevated above controls until d 21; numbers in B6 mice were significantly increased only on d 10. Numbers of epithelial cells and monocytes were variable in both strains, and no consistent V2O5 or strain effects were found.
Table 2.
Time course of differential cell counts from bronchoalveolar lavage fluid after 4 mg/kg V2O5
| Strain | Treatment | Day | Macrophages | Epithelial cells | Neutrophils | Eosinophils | Lymphocytes | Monocytes |
|---|---|---|---|---|---|---|---|---|
| B6 | PBS | 1 | 74,568 ± 6335 | 2851 ± 923 | 269 ± 127 | 175 ± 65 | 323 ± 180 | 29 ± 29 |
| V2O5 | 1 | 136,850 ± 17,242* | 2591 ± 606 | 17,795 ± 7363* | 3490 ± 1519 | 796 ± 392 | 354 ± 122 | |
| D2 | PBS | 1 | 53,608 ± 8521 | 1227 ± 438 | 639 ± 318 | 232 ± 178 | 328 ± 263 | 216 ± 129 |
| V2O5 | 1 | 48,573 ± 9115# | 3375 ± 789 | 87,191 ± 18,880*,# | 7806 ± 2917* | 1120 ± 409* | 4435 ± 2543 | |
| B6 | PBS | 2 | 94,982 ± 6775 | 704 ± 235 | 458 ± 299 | 290 ± 186 | 632 ± 442 | 114 ± 73 |
| V2O5 | 2 | 144,811 ± 20,196 | 1252 ± 407 | 26,553 ± 6536* | 4434 ± 2681 | 632 ± 195 | 391 ± 204 | |
| D2 | PBS | 2 | 47,055 ± 4249 | 2175 ± 413 | 2088 ± 1994 | 79 ± 52 | 675 ± 388 | 116 ± 81 |
| V2O5 | 2 | 120,126 ± 36,488* | 4131 ± 2064# | 98,993 ± 19,045*,# | 7451 ± 1647* | 2381 ± 817*,# | 3793 ± 1118*,# | |
| B6 | PBS | 4 | 87,693 ± 10,628 | 600 ± 155 | 43 ± 28 | 108 ± 60 | 626 ± 200 | 94 ± 94 |
| V2O5 | 4 | 157,155 ± 14,987 | 622 ± 358 | 9938 ± 2522* | 2056 ± 1049 | 2211 ± 1306 | 517 ± 292 | |
| D2 | PBS | 4 | 42,701 ± 1274 | 1879 ± 483 | 147 ± 67 | 0 ± 0 | 225 ± 63 | 47 ± 47 |
| V2O5 | 4 | 323,328 ± 19,661*,# | 3422 ± 1489 | 54,751 ± 22,372*,# | 5163 ± 1395*,# | 3334 ± 964* | 2085 ± 883 | |
| B6 | PBS | 6 | 87,914 ± 10,786 | 704 ± 286 | 190 ± 97 | 110 ± 79 | 342 ± 165 | 205 ± 100 |
| V2O5 | 6 | 161,864 ± 11,882* | 447 ± 220 | 3575 ± 1459* | 1185 ± 375* | 1552 ± 347 | 1377 ± 1068* | |
| D2 | PBS | 6 | 55,915 ± 9388 | 3205 ± 1117# | 21 ± 21 | 32 ± 32 | 425 ± 222 | 90± 74 |
| V2O5 | 6 | 148,895 ± 18,494* | 2567 ± 653 | 3826 ± 1102* | 1108 ± 174*,# | 4317 ± 1110*,# | 1430 ± 276* | |
| B6 | PBS | 8 | 76,569 ± 8710 | 1100 ± 402 | 0 ± 0 | 106 ± 106 | 297 ± 184 | 261 ± 31 |
| V2O5 | 8 | 174,992 ± 14,137* | 1458 ± 301 | 25,297 ± 12,328* | 3167 ± 890 | 1000 ± 355 | 2419 ± 1001* | |
| D2 | PBS | 8 | 64,423 ± 7863 | 2654 ± 653 | 685 ± 427 | 125 ± 77 | 238 ± 238 | 0 ± 0 |
| V2O5 | 8 | 208,504 ± 31,004* | 2257 ± 1073 | 82,521 ± 30,037*,# | 5211 ± 3282 | 3429 ± 1244*,# | 2007 ± 865 | |
| B6 | PBS | 10 | 73,567 ± 6493 | 838 ± 289 | 60 ± 60 | 0 ± 0 | 89 ± 62 | 89 ± 89 |
| V2O5 | 10 | 154,389 ± 17,799* | 1500 ± 655 | 26,187 ± 9583* | 1574 ± 771 | 2283 ± 538* | 2505 ± 838* | |
| D2 | PBS | 10 | 48,852 ± 6635 | 6939 ± 2551# | 285 ± 172 | 63 ± 42 | 111 ± 61 | 0 ± 0 |
| V2O5 | 10 | 278,422 ± 41,807*,# | 12,121 ± 10,148*,# | 38,392 ± 12,957* | 5413 ± 1771* | *3016 ± 1108 | 763 ± 420# | |
| B6 | PBS | 12 | 64,322 ± 7381 | 519 ± 261 | 578 ± 578 | 0 ± 0 | 1242 ± 576 | 839 ± 477 |
| V2O5 | 12 | 160,214 ± 11,386 | 329 ± 221 | 7497 ± 6491* | 467 ± 467 | 1211 ± 315 | 699 ± 319 | |
| D2 | PBS | 12 | 47,679 ± 7410 | 2319 ± 794 | 492 ± 389 | 46 ± 46 | 46 ± 46 | 44 ± 44 |
| V2O5 | 12 | 463,124 ± 81,903*,# | 2460 ± 972# | 23,143 ± 11,122*,# | 4302 ± 1360*,# | 7199 ± 2540*,# | 2987 ± 1111 | |
| B6 | PBS | 14 | 71,730 ± 9969 | 539 ± 178 | 21 ± 21 | 0 ± 0 | 350 ± 103 | 217 ± 115 |
| V2O5 | 14 | 177,479 ± 17,122* | 1072 ± 522 | 2270 ± 1326 | 1440 ± 883 | 1740 ± 429 | 1625 ± 785 | |
| D2 | PBS | 14 | 47,969 ± 7509 | 4347 ± 1282# | 502 ± 180 | 128 ± 91 | 698 ± 599 | 419 ± 405 |
| V2O5 | 14 | 357,179 ± 58,672*,# | 19,975 ± 11,651 | 16,721 ± 14,213*,# | 4451 ± 1638*,# | 2819 ± 1474*,# | 1668 ± 1028 | |
| B6 | PBS | 21 | 75,254 ± 8887 | 381 ± 151 | 54 ± 54 | 0 ± 0 | 713 ± 330 | 385 ± 149 |
| V2O5 | 21 | 139,441 ± 16,420* | 1269 ± 506 | 189 ± 71 | 328 ± 199 | 2217 ± 459 | 932 ± 189 | |
| D2 | PBS | 21 | 45,451 ± 2588# | 1679 ± 614 | 81 ± 43 | 35 ± 35 | 284 ± 85 | 281 ± 96 |
| V2O5 | 21 | 187,834 ± 12,863*,# | 2599 ± 798 | 75 ± 75 | 421 ± 234 | 3672 ± 1463*,# | 1024 ± 309 | |
| B6 | PBS | 28 | 41,733 ± 9489 | 1250 ± 270 | 21 ± 21 | 0 ± 0 | 121 ± 95 | 0 ± 0 |
| V2O5 | 28 | 69,504 ± 13,042 | 1679 ± 641 | 0 ± 0 | 0 ± 0 | 256 ± 162 | 435 ± 225 | |
| D2 | PBS | 28 | 55,419 ± 8060 | 1173 ± 235 | 0 ± 0 | 0 ± 0 | 223 ± 153 | 60 ± 60 |
| V2O5 | 28 | 139,519 ± 32,980*,# | 5847 ± 1564*,# | 0 ± 0 | 0 ± 0 | 2472 ± 2472 | 494 ± 494 | |
| B6 | PBS | 42 | 38,427 ± 2234 | 488 ± 488 | 94 ± 63 | 133 ± 94 | 233 ± 233 | 0 ± 0 |
| V2O5 | 42 | 38,696 ± 3080 | 2688 ± 1930 | 94 ± 55 | 413 ± 413 | 381 ± 249 | 229 ± 144 | |
| D2 | PBS | 42 | 48,808 ± 4006 | 2977 ± 1163 | 44 ± 44 | 46 ± 46 | 0 ± 0 | 0 ± 0 |
| V2O5 | 42 | 69,867 ± 14,870# | 2867 ± 816 | 146 ± 94 | 50 ± 50 | 48 ± 48 | 148 ± 94 | |
| B6 | PBS | 56 | 46,079 ± 5268 | 6777 ± 4116 | 83 ± 49 | 29 ± 29 | 110 ± 66 | 46 ± 46 |
| V2O5 | 56 | 46,969 ± 8680 | 7825 ± 4610 | 0 ± 0 | 460 ± 460 | 335 ± 211 | 35 ± 35 | |
| D2 | PBS | 56 | 36,596 ± 3878 | 3496 ± 925 | 31 ± 31 | 0 ± 0 | 308 ± 120 | 194 ± 117 |
| V2O5 | 56 | 101,039 ± 16,545*,# | 6528 ± 1271 | 0 ± 0 | 0 ± 0 | 200 ± 200 | 567 ± 186*,# | |
| B6 | PBS | 84 | 57,538 ± 3895 | 6233 ± 168 | 338 ± 84 | 96 ± 55 | 296 ± 51 | 0 ± 0 |
| V2O5 | 84 | 56,108 ± 9472 | 5238 ± 1155 | 256 ± 60 | 140 ± 55 | 133 ± 133 | 0 ± 0 | |
| D2 | PBS | 84 | 57,629 ± 3505 | 5929 ± 214 | 96 ± 55 | 0 ± 0 | 96 ± 55 | 0 ± 0 |
| V2O5 | 84 | 87,571 ± 6865*,# | 15,254 ± 3958*,# | 1150 ± 197*,# | 617 ± 98*,# | 408 ± 307 | 0 ± 0 | |
| B6 | PBS | 112 | 48,846 ± 1123 | 5596 ± 200 | 96 ± 55 | 375 ± 115 | 88 ± 51 | 0 ± 0 |
| V2O5 | 112 | 44,906 ± 7175 | 7104 ± 747 | 173 ± 78 | 431 ± 192 | 510 ± 210 | 0 ± 0 | |
| D2 | PBS | 112 | 52,733 ± 1578 | 8125 ± 188 | 308 ± 53 | 517 ± 48 | 817 ± 221 | 0 ± 0 |
| V2O5 | 112 | 73,142 ± 7285*,# | 17,126 ± 2099*,# | 943 ± 184*,# | 858 ± 347 | 431 ± 250 | 0 ± 0 |
P < 0.05 vs. 0 mg/kg V2O5 within strain;
P < 0.05 vs. B6 within dose.
V2O5 induced significant increases in lung collagen content, and responses were enhanced after the second exposure in D2 mice and remained elevated above controls until the end of the study (112 d), whereas collagen content in B6 mice was not significantly increased after V2O5 except at 6 d (Fig. 4C). Collagen content in D2 mice was greater than V2O5-treated B6 mice throughout the study.
Lungs from PBS-exposed mice of both strains were histologically normal at all times (Fig. 5). Mild peribronchiolar/perivascular inflammation was most prominent 1–2 d after V2O5 in B6 mice, and small focal areas of alveolar inflammation and atelectasis were found in <10% of the sections until 28 d, after which all V2O5-induced abnormalities resolved. Much greater parenchymal effects were found in lungs from V2O5-treated D2 mice, including intrapulmonary hemorrhage, vascular congestion, edema, and mixed infiltration of inflammatory cells 1 d after V2O5. By d 4, predominantly mononuclear organizing pneumonitis was found, which started to resolve by d 6. However, the second V2O5 aspiration caused heterogeneous lesions with thickened alveolar septae, early interstitial fibrosis, and type II alveolar epithelial hypertrophy/hyperplasia, as well as mixed granulocytic/mononuclear inflammation, hemorrhage, and alveolar consolidation. Much of the affected area was subpleural, with some central foci interspersed with normal tissue. Most granulocytic inflammation had resolved after 21 d, but large numbers of mononuclear cells remained in the alveoli with increased collagen staining in these areas. Despite resolution of granulocytic inflammation, gross morphological changes were visible with constriction of the lung surface from 28 to 112 d. Increased cellularity, alveolar consolidation, and fibrosis were present to 84 d. Although inflammation and fibrotic tissue had resolved by 112 d, collapsed alveoli remained.
Figure 5.
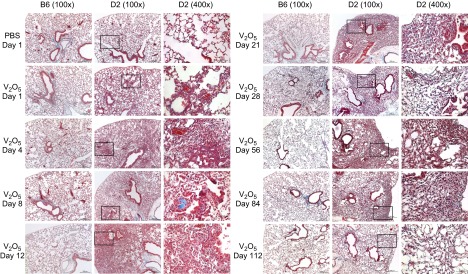
Time course of V2O5-induced lung histology in B6 and D2 mice. V2O5 -induced mild inflammatory foci and alveolar septal thickening, which completely resolved by 56 d in B6 mice. In D2 mice, V2O5 induced early lung injury with alveolar hemorrhage and edema followed by inflammation, septal thickening, and interstitial fibrosis, as well as epithelial type II metaplasia. Structurally remodeled alveoli persisted to 4 mo after V2O5. Masson's Trichrome staining of 5 μm paraffin sections. Original magnification: ×100 and ×400.
mRNA expression of many conventional fibrosis-associated genes was reduced in D2 mice compared to B6 mice 10 d after V2O5, including Ccr2 [chemokine (C-C motif) receptor 2], Eng (endoglin), Il1b, Itga1 (integrin α 1), Jun, Lox, Mmp3, Mmp8, Mmp9, Smad6 (SMAD family 6), and Smad7 (Table 3). Relative to B6 mice, greater expression of profibrotic genes, such as Ccl11 and Ctgf (connective tissue growth factor), as well as Fasl (Fas ligand), Il1a, and Serpina1a [serine (or cysteine) peptidase inhibitor, clade A, member 1A], but less expression of Mmp3, Plau (plasminogen activator, urokinase), and Tgif1, was found in D2 mice 21 d after V2O5. V2O5-induced changes in gene expression detected in the PCR array were verified by RT-PCR for Ccl11, Il1b, Lox, and Mmp3 (Supplemental Fig. S1). Because Tgfb1 mRNA levels were also lower in D2 mice compared to B6 mice, we measured protein levels of this important profibrotic mediator and found that protein levels were also significantly lower in D2 mice compared to B6 mice. While a treatment effect was not found in either strain, a temporal effect was found in B6 mice with increased levels at 21 d (Fig. 6).
Table 3.
Fold up- or down-regulation of V2O5-induced fibrosis-related genes in DBA/2J (D2) compared to C57BL/6J (B6) mice
| Gene | Description | d 10 | d 21 |
|---|---|---|---|
| Acta2 | Actin, α 2, smooth muscle | −2.84 | 1.52 |
| Bcl2 | B cell leukemia/lymphoma 2 | −3.47 | 1.49* |
| Bmp7 | Bone morphogenetic protein 7 | −2.25 | 1.19 |
| Cav1 | Caveolin 1, caveolae protein | −2.77 | 1.89* |
| Ccl11 | Chemokine (C-C motif) ligand 11 (eotaxin) | −1.41 | 3.75* |
| Ccl12 | Chemokine (C-C motif) ligand 12 | −2.29 | 1.49 |
| Ccl3 | Chemokine (C-C motif) ligand 3 (Mip1-α) | −2.90 | −1.33 |
| Ccr2 | Chemokine (C-C motif) receptor 2 (Mcp1 receptor) | −4.35* | −1.32 |
| Col1a2 | Collagen, type I, α 2 | 1.78 | 1.90* |
| Ctgf | Connective tissue growth factor | 1.12 | 2.38* |
| Cxcr4 | Chemokine (C-X-C motif) receptor 4 | −2.77 | 1.50* |
| Dcn | Decorin | −2.26 | −1.35 |
| Edn1 | Endothelin 1 | −3.47 | 1.88* |
| Egf | Epidermal growth factor | −2.27 | 1.19 |
| Eng | Endoglin | −3.49* | 1.49* |
| Fasl | Fas ligand (TNF superfamily, member 6) | −3.74 | 2.94* |
| Grem1 | Gremlin 1 | −2.88 | 1.15 |
| Hgf | Hepatocyte growth factor | −2.19 | −1.33 |
| Il13ra2 | Interleukin 13 receptor α 2 | −2.25 | −1.11 |
| Il1a | Interleukin 1 α | −4.62* | 2.36* |
| Il1b | Interleukin 1 β | −11.06* | 1.19 |
| Il4 | Interleukin 4 | −2.21 | 1.15 |
| Il5 | Interleukin 5 | −2.19 | −1.09 |
| Inhbe | Inhibin β E | −3.55 | −1.69 |
| Itga1 | Integrin α 1 | −2.86* | −1.35 |
| Itga2 | Integrin α 2 | −2.91 | −1.34 |
| Itgb3 | Integrin β 3 | −2.22 | 1.20 |
| Itgb5 | Integrin β 5 | −3.47 | −1.68* |
| Jun | Jun oncogene | −3.48* | −1.33 |
| Lox | Lysyl oxidase | −3.49* | −1.34 |
| Mmp13 | Matrix metallopeptidase 13 (interstitial collagenase) | −2.60 | 1.20 |
| Mmp14 | Matrix metallopeptidase 14 (membrane-inserted) | −2.26 | −1.34 |
| Mmp3 | Matrix metallopeptidase 3 (stromelysin 1) | −7.12* | −3.41* |
| Mmp8 | Matrix metallopeptidase 8 (neutrophil collagenase) | −14.61* | 1.49* |
| Mmp9 | Matrix metallopeptidase 9 (type IV collagenase) | −5.91* | −1.07 |
| Myc | Myelocytomatosis oncogene | −2.31 | 1.50* |
| Plat | Plasminogen activator, tissue | −3.65 | 1.88* |
| Plau | Plasminogen activator, urokinase | −2.21 | −2.11* |
| Serpina1a | Serine (or cysteine) peptidase inhibitor, clade A, member 1A | −2.19 | 2.55* |
| Serpine1 | Serine (or cysteine) peptidase inhibitor, clade E, member 1 | −2.91 | −1.08 |
| Smad2 | SMAD family member 2 | −2.19 | 1.50 |
| Smad6 | SMAD family member 6 | −4.40* | 1.51* |
| Smad7 | SMAD family member 7 | −5.52* | 1.17 |
| Snai1 | Snail homolog 1 (Drosophila) | −4.42* | 1.50 |
| Tgfb1 | Transforming growth factor, β 1 | −2.20 | −1.34 |
| Tgfb3 | Transforming growth factor, β 3 | −2.76 | −1.06 |
| Tgif1 | TGFB-induced factor homeobox 1 | −2.26 | −2.69* |
| Thbs2 | Thrombospondin 2 | −2.18 | 1.50* |
| Timp4 | Tissue inhibitor of metalloproteinase 4 | −1.73 | 2.38 |
P < 0.05 for V2O5-treated D2 mice vs. V2O5-treated B6 mice.
Figure 6.

Total TGF-β protein levels in lung tissue homogenates from B6 and D2 mice 10 and 21 d after aspiration of PBS or 4mg/kg V2O5. Bars represent means + se of 4 mice/group. +P < 0.001 vs. B6 mice within time point and treatment group; ‡P < 0.001 vs. d 10 within strain and treatment.
V2O5 effects in BXD RI mice
We next investigated the genetic basis for differential inflammatory and fibrotic responses in B6 and D2 mice using 49 BXD RI strains. We found treatment and strain effects (Fig. 7) for BALF cell numbers and lung collagen content between RI strains 14 d after PBS and 4 mg/kg V2O5. PBS control BALF cell numbers ranged from 1.9 × 104 in BXD11 to 24.1 × 104 in BXD55, while V2O5 responses ranged from 8.6 × 104 in BXD11 to 73.2 × 104 in BXD9. Collagen content in the right lung also varied with strain in controls from 19.5 μg in BXD34 to 60.0 μg in BXD44, and in response to V2O5 from 28.5 μg in BXD24 to 117.2 μg in BXD44. Because of the strain-dependent differences in controls, the mean PBS response for each strain was subtracted from individual V2O5-induced response, and this difference was used for genetic mapping of V2O5-induced phenotypes. Total cells, macrophages, and lymphocytes significantly correlated with each other, but we found no correlation between V2O5-induced increases in collagen content and any cell type (Fig. 8A).
Figure 7.

BXD RI strain distribution for PBS and V2O5-induced BALF total cell numbers (A) and lung collagen content (B). Each BXD RI strain was exposed to 4 mg/kg V2O5 or PBS, and phenotypes were measured at study d 14. Bars represent means ± se of 4–8 mice/strain in each treatment group. *P < 0.05 vs. PBS treatment within strain.
Figure 8.

A) Matrix shows significant correlations between V2O5-induced total cells, macrophages, and lymphocytes, but a lack of correlation between collagen and any cell type for 49 BXD RI strains. B) Genome-wide interval map of V2O5-induced changes in lung collagen content across 49 BXD RI strains using the WebQTL function of GeneNetwork to identify quantitative trait loci. The x axis represents the length of each chromosome, the left y axis indicates the log of the odds ratio (LOD, blue line), and the right y axis indicates the degree to which either C57BL/6J (red line) or DBA/2J (green line) alleles increase phenotypic values. Numbers along the top of the figure are chromosome numbers. The upper horizontal line indicates the significant LOD threshold (P<0.05), and the lower horizontal line indicates the suggestive LOD threshold. C) Detailed linkage map of chromosome 4 shows the significant quantitative trait locus for V2O5-induced changes in lung collagen content in the RI strains.
Genome-wide association mapping identified no significant QTLs for V2O5-induced changes in BALF cells, although there was a suggestive QTL on chromosome 12 for macrophages (data not shown). Association mapping for V2O5-induced changes in collagen content (Fig. 8B) identified a statistically significant QTL on chromosome 4 (Fig. 8C). Suggestive QTLs were found on chromosomes 2 and 9 (Fig. 8B). We then queried the haplotype structure of the QTL and searched for candidate genes (http://phenome.jax.org/db/q?rtn=snp/ret1). All BXD RIs have one of 2 haplotypes that were found in the chromosome 4 QTL (Fig. 9A): 29 RIs are resistant B6 haplotype and 19 are susceptible D2 haplotype. Relative to the B6 haplotype RI strains, mean V2O5-induced lung collagen content was significantly greater (P=0.002) in D2 haplotype RI strains (Fig. 9B). Genes within the QTL include Dbc1 (deleted in breast cancer 1), Cdk5rap2, Megf9 (multiple EGF-like-domains 9), Tle1, Aldoart1 (aldolase 1 A, retrogene 1), Rasef (RAS and EF hand domain containing), Frmd3, Kdm4c, and Ptprd. Two haplotypes and 3 haplotypes were found in the chromosomes 2 and 9 QTLs, respectively (Supplemental Fig. S2). Significantly greater V2O5-induced collagen content was found in D2-like strains compared to B6-like strains.
Figure 9.

Haplotype structure for the chromosome 4 quantitative trait locus for susceptibility to V2O5-induced changes in lung collagen content in BXD RI strains of mice. A) Haplotype with single-nucleotide polymorphism (SNP) location, gene names, and gene function in RI strains with similar haplotype. Parental B6 (left) and D2 (right) mice flank the RI strains. Cs, synonymous coding SNP; I, intronic SNP. B) Mean lung collagen content in RI strains with the B6 haplotype (n=29 strains) and strains with the D2 haplotype (n=19 strains).
Gene expression levels of 5 candidate genes within the collagen QTL on chromosome 4 were measured using mRNA from lungs of B6 and D2 mice at d 10 and 21 (Supplemental Fig. S3). All genes were expressed at relatively low levels (indicated by high Ct values) and Frmd3 was not detected in any of the samples. In D2 mice, V2O5 exposure increased Kdm4c expression at d 10, while Tle1 was increased at d 21 compared to controls. In contrast, Ptprd was decreased in B6 mice at d 21 and was undetectable in V2O5-treated D2 mice.
DISCUSSION
Interstitial lung diseases include a large number of respiratory conditions with various etiologies, pathological courses, and prognoses. Idiopathic forms of ILD are particularly difficult to study because, in the absence of a known cause and natural history, it has not been possible to develop an appropriate animal model. Existing models have taken advantage of known fibrogenic agents to study mechanisms of disease pathogenesis. While understanding of a number of fibrogenic pathways has been advanced with these models, few have successfully identified effective therapeutic targets for human disease (6). In the current study, we used vanadium to develop a novel, alternative model of pulmonary fibrosis to explore the genetic basis for fibrosis susceptibility. Differential lung injury, inflammation, and fibrogenic responses between responsive D2 and resistant B6 mice after identical V2O5 exposure demonstrated genetic background is an important determinant of susceptibility.
Functional measures indicative of restrictive disease were found in the fibrosis susceptible D2 strain, but significant increases in airway resistance were also evident. Increased airway resistance is consistent with V2O5-induced bronchitis-like phenotypes in rodents and primates including airway hyperresponsiveness, inflammation, mucus cell hyperplasia, and airway smooth muscle thickening (16–18, 26). Studies of human exposure to vanadium-rich particles in occupational settings have also reported functional and symptom changes indicative of obstructive disease (19–23). However, restrictive and obstructive lung diseases are not mutually exclusive, as seen in cases of combined pulmonary fibrosis and emphysema (27). Increased airway resistance in D2 mice may reflect airway smooth muscle and/or basement membrane thickening, or airway epithelial hyperplasia/hypertrophy. Although these phenotypes were not quantitatively measured, hypertrophy and hyperplasia of airway epithelial cells were found in D2 mice exposed to V2O5. Interestingly, these hyperplastic cells did not stain positive for mucus in the 2 strains we examined (data not shown). This contrasts with studies in other strains of mice and rats, in which V2O5 exposure induced goblet cell metaplasia, mucous production, thickening of airway smooth muscle, and peribronchiolar fibrosis (17, 28).
Elevated numbers of BALF macrophages were predominant in both strains of mice after V2O5, however they persisted throughout the study only in D2 mice. Alveolar macrophage phenotype (M1, M2) is thought to be essential in regulating inflammatory and repair processes after lung injury (29). Alternatively activated alveolar macrophages (M2) were shown be critical for fibrogenesis in the bleomycin model and are the prevalent phenotype in BALF from patients with IPF (30). However, additional subphenotypes and other mechanisms that determine resolution or continued progression of fibrosis are needed to explain progressive fibrosis in IPF compared to fibrosis resolution in mouse models.
Vanadium compounds affect host defense activities including phagocytic ability, production of ROS, enzyme activity, resistance to infection, and production and response to immunomodulatory cytokines (31–35). Previous reports of reduced phagocytic activity of macrophages and increased ROS and cytokine production after vanadium exposure may have contributed to the V2O5-induced injury and inflammation we observed, particularly in D2 mice. Changes in cytokine production after exposure to vanadium varies depending on the vanadium compound and cytokines investigated (36–38). A Th2 profile has been hypothesized to play a role in IPF and in bleomycin-induced lung fibrosis (39–43), although whether a Th2 response is required for fibrogenesis is unclear (44). In our study, Th2 cytokines measured in the fibrosis array were either undetectable (Il10 and Il13) or were down-regulated (Il4, Il5, Il13ra2) in D2 mice after 10 d and were not different from B6 at 21 d. Furthermore, in lung fibroblasts, V2O5 stimulates a Type I immune response (45). The lack of a significant Th2-like response suggests the immune response to V2O5 may differ from other fibrogenic models. It is also possible that the differences in immune responses may be time dependent, and further investigation is necessary to resolve these differences.
We also found greater numbers of eosinophils and elevated expression of eosinophil-specific chemotactic factor eotaxin (Ccl11; ref. 46) in D2 mice relative to B6 mice. Increased eosinophil numbers have been reported after exposure to metal-containing particles and may play a role in particle-induced exacerbation and/or induction of asthma-like parameters such as airway hyperresponsiveness (47–49). Increased eosinophil and neutrophil numbers in BALF from IPF patients have been reported to correlate with decrements in diffusing capacity and reduced survival time (50–54), suggesting that these cells may have a role in pulmonary fibrosis. In addition, inflammation is a key feature of a number of other ILDs that result in fibrosis.
Interestingly, mRNA expression of most fibrosis-associated genes was decreased in V2O5-treated D2 mice at 10 d, a time at which models such as bleomycin switch from inflammatory to fibrotic phenotypes (55). Although a significant increase in collagen levels at 10 d was found in D2 mice, there was also cellular inflammation, likely due to the second V2O5 exposure at d 7. The extensive decreased gene expression observed in the V2O5 mouse model at 10 d is consistent with in vitro studies of human lung fibroblasts in which V2O5 suppressed 3–4 times more genes than were induced (56). The list of genes that were down-regulated 10 d after V2O5 in D2 mice compared to B6 mice included a number of genes that are typically up-regulated after bleomycin challenge and thought to be key to pulmonary fibrosis such as Mmps and Tgfb1. TGF-β protein levels were also lower in D2 compared to B6 mice, although V2O5 treatment did not affect levels in either strain. Notably, Smad6 and Smad7, known to inhibit Tgfb signaling (57–59), were significantly up-regulated in B6 mice, but down-regulated in D2 mice suggesting that a lack of Tgfb inhibition may contribute to development of fibrosis in the V2O5 model.
At 21 d, when cell numbers decreased but collagen levels were still near their peak, only 5 genes were significantly up-regulated more than 2-fold, and 3 were down-regulated. Up-regulated genes included Ccl11, Ctgf, Fasl, Il1a, and Serpina1a. Ccl11 and Il1a are inflammatory genes and would be expected to contribute to the mixed eosinophilic/neutrophilic inflammation seen after V2O5 exposure; yet, granulocytic inflammation had largely resolved by 21 d. However, increased levels of Ccl11 have been associated with models of fibrotic conditions of the lung (60–63). Ctgf has previously been reported to be increased in response to V2O5 exposure in human lung fibroblasts (56) and is thought to be important in early wound healing (64, 65). Serpina1a, a mouse ortholog of α1 antitrypsin, is a protease inhibitor and its induction, along with continued down-regulation of Mmp3, may contribute to accumulation of extracellular matrix. Finally, the Fas-Fas ligand system has been shown to be up-regulated in IPF lung tissue leading to apoptosis of epithelial cells (66). Epithelial cell apoptosis was observed at early time points, but later apoptotic cells likely represented recruited inflammatory cells, while notable fibroblast apoptosis was not observed at any time (data not shown).
The wide range of values for BALF cells and lung collagen content across 49 BXD RI strains suggested that cellular inflammation and fibrosis are complex traits involving multiple genetic factors. Because the role of inflammation in pulmonary fibrosis is controversial in some ILDs such as IPF, we asked whether inflammatory cell phenotypes were related to lung collagen content. The lack of a correlation between collagen and cell numbers could be a temporal effect as significantly greater cellular inflammation was found in D2 mice at early time points, but it is also possible that inflammation and fibrosis may be controlled by different sets of genes as suggested by the lack of concordance of QTLs between the phenotypes.
The lack of a fibrotic response and associated functional changes in B6 mice was surprising, as they are highly susceptible to pulmonary fibrosis induced by bleomycin, silica, and asbestos (2–5), suggesting that V2O5 may work through different mechanisms than other known fibrogenic agents. In our genome-wide linkage analyses to identify the genetic basis for the differential responses between strains, we found a significant QTL for the collagen phenotype on chromosome 4 that was largely driven by D2 alleles. A number of gene candidates were found within the QTL, including Kdm4c (or Jmjd2c, jumonji domain containing protein 2C) which was expressed at higher levels in D2 mice at 21 d. This histone methylase has been associated with cancer proliferation (67). Interestingly, the methylase has also been shown to selectively interact with HIF-1α (68), which has a role in inflammation and fibrosis (69). Another gene candidate in the chromosome 4 QTL is protein-tyrosine phosphatase receptor-type δ (Ptprd). Ptprd has been implicated as a tumor suppressor gene in lung carcinogenesis (70, 71), and its decreased expression by V2O5 to undetectable levels in D2 mice at 21 d may contribute to the ongoing epithelial hypertrophy/hyperplasia observed in D2 mice. Polymorphisms in PTPRD have also been associated with pediatric asthma (72) and may have a role in other complex lung diseases. The B6 and D2 haplotypes that contain these genes and others (see above) associated with differential V2O5-induced lung fibrosis in the RI strains. However, because these genes are in linkage disequilibrium with each other, additional studies will be necessary to characterize their roles in this fibrosis model. Significant QTLs for the cell phenotypes were not identified, which may suggest that numerous genes may have a relatively small impact on these phenotypes.
In summary, the pulmonary effects of V2O5, including lung injury, inflammation, and fibrosis, are dependent on genetic background. This V2O5 model is useful for investigating fibrotic mechanisms and therapeutic targets, as it appears to be more persistent than the more commonly used bleomycin model and likely works through different mechanisms. Because vanadium is a component of cigarette smoke and metal dusts, which have been associated with ILDs such as IPF, it also has the advantage of being an environmentally/occupationally relevant agent. Whether continued exposures would produce even more stable or progressive fibrosis in susceptible strains presents an interesting area for further research, while genetic studies to address variant alleles across numerous strains of mice are underway.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the U.S. National Institute of Environmental Health Sciences (NIEHS) Histology Core facility and Joani Zary Oswald (Brody School of Medicine Histology Core, East Carolina University) for histological services, and Dr. Paul Strausbauch for pathology advice. The authors also thank Dr. Jared M. Brown for use of laboratory equipment and technical advice, and M. Anthony Phipps for technical assistance.
This research was supported (in part) by the Intramural Research Program of the U.S. National Institutes of Health, NIEHS, and East Carolina University.
The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- B6
- C57BL/6J
- BALF
- bronchoalveolar lavage fluid
- Ccl11
- chemokine (C-C motif) ligand 11
- Cdk5rap2
- CDK5 regulatory subunit associated protein 2
- Cst
- static compliance
- D2
- Ctgf, connective tissue growth factor; DBA/2J
- Fasl
- Fas ligand
- Frmd3
- FERM domain containing 3
- H
- elastance
- IIP
- idiopathic interstitial pneumonia
- Il1b
- interleukin 1β
- ILD
- interstitial lung disease
- IPF
- idiopathic pulmonary fibrosis
- Kdm4c
- lysine [K]-specific demethylase 4C
- Lox
- lysyl oxidase
- Mmp
- matrix metallopeptidase
- OR
- odds ratio
- Ptprd
- protein tyrosine phosphatase, receptor type D
- RN
- newtonian resistance
- QTL
- quantitative trait locus
- RI
- recombinant inbred
- ROS
- reactive oxygen species
- R-T PCR
- real-time PCR
- Serpina1a
- serine (or cysteine) peptidase inhibitor, clade A, member 1A
- TGF-β
- transforming growth factor β
- Tle1
- transducin-like enhancer of split 1
REFERENCES
- 1. Olsen A. L., Schwartz M. I., Roman J. (2010) Interstitial lung disease. In Breathing in America (Schraufnagel D. E., ed) pp. 99–108, American Thoracic Society, New York [Google Scholar]
- 2. Paun A., Lemay A. M., Tomko T. G., Haston C. K. (2013) Association analysis reveals genetic variation altering bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 48, 330–336 [DOI] [PubMed] [Google Scholar]
- 3. Davis G. S., Leslie K. O., Hemenway D. R. (1998) Silicosis in mice: effects of dose, time, and genetic strain. J. Environ. Pathol. Toxicol. Oncol. 17, 81–97 [PubMed] [Google Scholar]
- 4. Ohtsuka Y., Wang X. T., Saito J., Ishida T., Munakata M. (2006) Genetic linkage analysis of pulmonary fibrotic response to silica in mice. Eur. Respir. J. 28, 1013–1019 [DOI] [PubMed] [Google Scholar]
- 5. Brody A. R., Warshamana G. S., Liu J. Y., Tsai S. Y., Pociask D. A., Brass D. M., Schwartz D. (2002) Identifying fibrosis susceptibility genes in two strains of inbred mice. Chest 121, 31S. [PubMed] [Google Scholar]
- 6. Moeller A., Ask K., Warburton D., Gauldie J., Kolb M. (2008) The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol. 40, 362–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Loomis-King H., Flaherty K. R., Moore B. B. Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis. Curr. Opin. Pharmacol. 13, 377–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nemery B. (1990) Metal toxicity and the respiratory tract. Eur. Respir. J. 3, 202–219 [PubMed] [Google Scholar]
- 9. Taskar V., Coultas D. (2008) Exposures and idiopathic lung disease. Semin. Respir. Crit. Care Med. 29, 670–679 [DOI] [PubMed] [Google Scholar]
- 10. Taskar V. S., Coultas D. B. (2006) Is idiopathic pulmonary fibrosis an environmental disease? Proc. Am. Thorac. Soc. 3, 293–298 [DOI] [PubMed] [Google Scholar]
- 11. Miyake Y., Sasaki S., Yokoyama T., Chida K., Azuma A., Suda T., Kudoh S., Sakamoto N., Okamoto K., Kobashi G., Washio M., Inaba Y., Tanaka H. (2005) Occupational and environmental factors and idiopathic pulmonary fibrosis in Japan. Ann. Occup. Hyg. 49, 259–265 [DOI] [PubMed] [Google Scholar]
- 12. World Health Organization (2002) Air Quality Guidelines, 2nd Ed., pp 162–172, WHO Regional Publications, Copenhagen, Denmark [Google Scholar]
- 13. World Health Organization/International Agency for Research on Cancer (2006) Vanadium pentoxide. In Cobalt in Hard Metals and Cobalt Sulfate, Gallium Arsenide, Indium Phosphide and Vanadium Pentoxide. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 86, pp 227–292, International Agency for Research on Cancer, Lyon, France; [PMC free article] [PubMed] [Google Scholar]
- 14. Adachi A., Asai K., Koyama Y., Matsumoto Y., Kobayashi T. (1998) Vanadium content of cigarettes. Bull. Environ. Contam. Toxicol. 61, 276–280 [DOI] [PubMed] [Google Scholar]
- 15. Schuler D., Chevalier H. J., Merker M., Morgenthal K., Ravanat J. L., Sagelsdorff P., Walter M., Weber K., McGregor D. (2011) First steps towards an understanding of a mode of carcinogenic action for vanadium pentoxide. J. Toxicol. Pathol. 24, 149–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. National Toxicology Program (2002) NTP toxicology and carcinogensis studies of vanadium pentoxide (CAS No. 1314-62-1) in F344/N rats and B6C3F1 mice (inhalation). Natl. Toxicol. Program Tech. Rep. Ser. 507, 1–343 [PubMed] [Google Scholar]
- 17. Bonner J. C., Rice A. B., Moomaw C. R., Morgan D. L. (2000) Airway fibrosis in rats induced by vanadium pentoxide. Am. J. Physiol. Lung Cell. Mol. Physiol. 278, L209–L216 [DOI] [PubMed] [Google Scholar]
- 18. Bonner J. C., Lindroos P. M., Rice A. B., Moomaw C. R., Morgan D. L. (1998) Induction of PDGF receptor-alpha in rat myofibroblasts during pulmonary fibrogenesis in vivo. Am. J. Physiol. 274, L72–L80 [DOI] [PubMed] [Google Scholar]
- 19. Irsigler G. B., Visser P. J., Spangenberg P. A. (1999) Asthma and chemical bronchitis in vanadium plant workers. Am. J. Ind. Med. 35, 366–374 [DOI] [PubMed] [Google Scholar]
- 20. Hauser R., Elreedy S., Hoppin J. A., Christiani D. C. (1995) Airway obstruction in boilermakers exposed to fuel oil ash. A prospective investigation. Am. J. Respir. Crit. Care Med. 152, 1478–1484 [DOI] [PubMed] [Google Scholar]
- 21. Lees R. E. (1980) Changes in lung function after exposure to vanadium compounds in fuel oil ash. Br. J. Ind. Med. 37, 253–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sjoberg S. G. (1955) Vanadium bronchitis from cleaning oil-fired boilers. AMA Arch. Ind. Health 11, 505–512 [PubMed] [Google Scholar]
- 23. Woodin M. A., Liu Y., Neuberg D., Hauser R., Smith T. J., Christiani D. C. (2000) Acute respiratory symptoms in workers exposed to vanadium-rich fuel-oil ash. Am. J. Ind. Med. 37, 353–363 [DOI] [PubMed] [Google Scholar]
- 24. Walters D. M., Kleeberger S. R. (2008) Mouse models of bleomycin-induced pulmonary fibrosis. Curr. Protoc. Pharmacol. Chapter 5, Unit 5.46 [DOI] [PubMed] [Google Scholar]
- 25. Howden R., Liu E., Miller-DeGraff L., Keener H. L., Walker C., Clark J. A., Myers P. H., Rouse D. C., Wiltshire T., Kleeberger S. R. (2008) The genetic contribution to heart rate and heart rate variability in quiescent mice. Am. J. Physiol. Heart Circ. Physiol. 295, H59–H68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Knecht E. A., Moorman W. J., Clark J. C., Hull R. D., Biagini R. E., Lynch D. W., Boyle T. J., Simon S. D. (1992) Pulmonary reactivity to vanadium pentoxide following subchronic inhalation exposure in a non-human primate animal model. J. Appl. Toxicol. 12, 427–434 [DOI] [PubMed] [Google Scholar]
- 27. Cottin V., Cordier J. F. (2009) The syndrome of combined pulmonary fibrosis and emphysema. Chest 136, 1–2 [DOI] [PubMed] [Google Scholar]
- 28. Yu D., Walters D. M., Zhu L., Lee P. K., Chen Y. (2011) Vanadium pentoxide (V(2)O(5)) induced mucin production by airway epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 301, L31–L39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alber A., Howie S. E., Wallace W. A., Hirani N. (2012) The role of macrophages in healing the wounded lung. Int. J. Exp. Pathol. 93, 243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gibbons M. A., MacKinnon A. C., Ramachandran P., Dhaliwal K., Duffin R., Phythian-Adams A. T., van Rooijen N., Haslett C., Howie S. E., Simpson A. J., Hirani N., Gauldie J., Iredale J. P., Sethi T., Forbes S. J. (2011) Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am. J. Respir. Crit. Care Med. 184, 569–581 [DOI] [PubMed] [Google Scholar]
- 31. Grabowski G. M., Paulauskis J. D., Godleski J. J. (1999) Mediating phosphorylation events in the vanadium-induced respiratory burst of alveolar macrophages. Toxicol. Appl. Pharmacol. 156, 170–178 [DOI] [PubMed] [Google Scholar]
- 32. Waters M. D., Gardner D. E., Coffin D. L. (1974) Cytotoxic effects of vanadium on rabbit alveolar macrophages in vitro. Toxicol. Appl. Pharmacol. 28, 253–263 [DOI] [PubMed] [Google Scholar]
- 33. Cohen M. D., Yang Z., Zelikoff J. T., Schlesinger R. B. (1996) Pulmonary immunotoxicity of inhaled ammonium metavanadate in Fisher 344 rats. Fundam. Appl. Toxicol. 33, 254–263 [DOI] [PubMed] [Google Scholar]
- 34. Cohen M. D., Becker S., Devlin R., Schlesinger R. B., Zelikoff J. T. (1997) Effects of vanadium upon polyl: C-induced responses in rat lung and alveolar macrophages. J. Toxicol. Environ. Health 51, 591–608 [DOI] [PubMed] [Google Scholar]
- 35. Cohen M. D. (2004) Pulmonary immunotoxicology of select metals: aluminum, arsenic, cadmium, chromium, copper, manganese, nickel, vanadium, and zinc. J. Immunotoxicol. 1, 39–69 [DOI] [PubMed] [Google Scholar]
- 36. Chong I. W., Shi M. M., Love J. A., Christiani D. C., Paulauskis J. D. (2000) Regulation of chemokine mRNA expression in a rat model of vanadium-induced pulmonary inflammation. Inflammation 24, 505–517 [DOI] [PubMed] [Google Scholar]
- 37. Cohen M. D., McManus T. P., Yang Z., Qu Q., Schlesinger R. B., Zelikoff J. T. (1996) Vanadium affects macrophage interferon-gamma-binding and -inducible responses. Toxicol. Appl. Pharmacol. 138, 110–120 [DOI] [PubMed] [Google Scholar]
- 38. Pierce L. M., Alessandrini F., Godleski J. J., Paulauskis J. D. (1996) Vanadium-induced chemokine mRNA expression and pulmonary inflammation. Toxicol. Appl. Pharmacol. 138, 1–11 [DOI] [PubMed] [Google Scholar]
- 39. Shimizu Y., Kuwabara H., Ono A., Higuchi S., Hisada T., Dobashi K., Utsugi M., Mita Y., Mori M. (2006) Intracellular Th1/Th2 balance of pulmonary CD4(+) T cells in patients with active interstitial pneumonia evaluated by serum KL-6. Immunopharmacol. Immunotoxicol. 28, 295–304 [DOI] [PubMed] [Google Scholar]
- 40. Jakubzick C., Kunkel S. L., Puri R. K., Hogaboam C. M. (2004) Therapeutic targeting of IL-4- and IL-13-responsive cells in pulmonary fibrosis. Immunol. Res. 30, 339–349 [DOI] [PubMed] [Google Scholar]
- 41. Costabel U., Guzman J. (2001) Bronchoalveolar lavage in interstitial lung disease. Curr. Opin. Pulm. Med. 7, 255–261 [DOI] [PubMed] [Google Scholar]
- 42. Belperio J. A., Dy M., Burdick M. D., Xue Y. Y., Li K., Elias J. A., Keane M. P. (2002) Interaction of IL-13 and C10 in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 27, 419–427 [DOI] [PubMed] [Google Scholar]
- 43. Chung M. P., Monick M. M., Hamzeh N. Y., Butler N. S., Powers L. S., Hunninghake G. W. (2003) Role of repeated lung injury and genetic background in bleomycin-induced fibrosis. Am. J. Respir. Cell Mol. Biol. 29, 375–380 [DOI] [PubMed] [Google Scholar]
- 44. Hirata H., Arima M., Fukushima Y., Ishii Y., Tokuhisa T., Fukuda T. (2008) Effects of Th2 pulmonary inflammation in mice with bleomycin-induced pulmonary fibrosis. Respirology 13, 788–798 [DOI] [PubMed] [Google Scholar]
- 45. Antao-Menezes A., Turpin E. A., Bost P. C., Ryman-Rasmussen J. P., Bonner J. C. (2008) STAT-1 signaling in human lung fibroblasts is induced by vanadium pentoxide through an IFN-beta autocrine loop. J. Immunol. 180, 4200–4207 [DOI] [PubMed] [Google Scholar]
- 46. Rothenberg M. E., Ownbey R., Mehlhop P. D., Loiselle P. M., van de Rijn M., Bonventre J. V., Oettgen H. C., Leder P., Luster A. D. (1996) Eotaxin triggers eosinophil-selective chemotaxis and calcium flux via a distinct receptor and induces pulmonary eosinophilia in the presence of interleukin 5 in mice. Mol. Med. 2, 334–348 [PMC free article] [PubMed] [Google Scholar]
- 47. Dreher K., Jaskot R., Kodavanti U., Lehmann J., Winsett D., Costa D. (1996) Soluble transition metals mediate the acute pulmonary injury and airway hyperreactivity induced by residual oil fly ash particles. Chest 109, 33S–34S [DOI] [PubMed] [Google Scholar]
- 48. Kodavanti U. P., Hauser R., Christiani D. C., Meng Z. H., McGee J., Ledbetter A., Richards J., Costa D. L. (1998) Pulmonary responses to oil fly ash particles in the rat differ by virtue of their specific soluble metals. Toxicol. Sci. 43, 204–212 [DOI] [PubMed] [Google Scholar]
- 49. Walters D. M., Breysse P. N., Wills-Karp M. (2001) Ambient urban Baltimore particulate-induced airway hyperresponsiveness and inflammation in mice. Am. J. Respir. Crit. Care Med. 164, 1438–1443 [DOI] [PubMed] [Google Scholar]
- 50. Boomars K. A., Wagenaar S. S., Mulder P. G., van Velzen-Blad H., van den Bosch J. M. (1995) Relationship between cells obtained by bronchoalveolar lavage and survival in idiopathic pulmonary fibrosis. Thorax 50, 1087–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fujimoto K., Kubo K., Yamaguchi S., Honda T., Matsuzawa Y. (1995) Eosinophil activation in patients with pulmonary fibrosis. Chest 108, 48–54 [DOI] [PubMed] [Google Scholar]
- 52. Hallgren R., Bjermer L., Lundgren R., Venge P. (1989) The eosinophil component of the alveolitis in idiopathic pulmonary fibrosis. Signs of eosinophil activation in the lung are related to impaired lung function. Am. Rev. Respir. Dis. 139, 373–377 [DOI] [PubMed] [Google Scholar]
- 53. Kinder B. W., Brown K. K., Schwarz M. I., Ix J. H., Kervitsky A., King T. E., Jr. (2008) Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest 133, 226–232 [DOI] [PubMed] [Google Scholar]
- 54. Schwartz D. A., Helmers R. A., Dayton C. S., Merchant R. K., Hunninghake G. W. (1991) Determinants of bronchoalveolar lavage cellularity in idiopathic pulmonary fibrosis. J. Appl. Physiol. 71, 1688–1693 [DOI] [PubMed] [Google Scholar]
- 55. Chaudhary N. I., Schnapp A., Park J. E. (2006) Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am. J. Respir. Crit. Care Med. 173, 769–776 [DOI] [PubMed] [Google Scholar]
- 56. Ingram J. L., Antao-Menezes A., Turpin E. A., Wallace D. G., Mangum J. B., Pluta L. J., Thomas R. S., Bonner J. C. (2007) Genomic analysis of human lung fibroblasts exposed to vanadium pentoxide to identify candidate genes for occupational bronchitis. Respir. Res. 8, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hayashi H., Abdollah S., Qiu Y., Cai J., Xu Y. Y., Grinnell B. W., Richardson M. A., Topper J. N., Gimbrone M. A., Jr., Wrana J. L., Falb D. (1997) The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 89, 1165–1173 [DOI] [PubMed] [Google Scholar]
- 58. Imamura T., Takase M., Nishihara A., Oeda E., Hanai J., Kawabata M., Miyazono K. (1997) Smad6 inhibits signalling by the TGF-beta superfamily. Nature 389, 622–626 [DOI] [PubMed] [Google Scholar]
- 59. Nakao A., Afrakhte M., Moren A., Nakayama T., Christian J. L., Heuchel R., Itoh S., Kawabata M., Heldin N. E., Heldin C. H., ten Dijke P. (1997) Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 389, 631–635 [DOI] [PubMed] [Google Scholar]
- 60. Fulkerson P. C., Fischetti C. A., Rothenberg M. E. (2006) Eosinophils and CCR3 regulate interleukin-13 transgene-induced pulmonary remodeling. Am. J. Pathol. 169, 2117–2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huaux F., Gharaee-Kermani M., Liu T., Morel V., McGarry B., Ullenbruch M., Kunkel S. L., Wang J., Xing Z., Phan S. H. (2005) Role of Eotaxin-1 (CCL11) and CC chemokine receptor 3 (CCR3) in bleomycin-induced lung injury and fibrosis. Am. J. Pathol. 167, 1485–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Puxeddu I., Bader R., Piliponsky A. M., Reich R., Levi-Schaffer F., Berkman N. (2006) The CC chemokine eotaxin/CCL11 has a selective profibrogenic effect on human lung fibroblasts. J. Allergy Clin. Immunol. 117, 103–110 [DOI] [PubMed] [Google Scholar]
- 63. Emad A., Emad Y. (2007) Relationship between eosinophilia and levels of chemokines (CCL5 and CCL11) and IL-5 in bronchoalveolar lavage fluid of patients with mustard gas-induced pulmonary fibrosis. J. Clin. Immunol. 27, 605–612 [DOI] [PubMed] [Google Scholar]
- 64. Alfaro M. P., Deskins D. L., Wallus M., DasGupta J., Davidson J. M., Nanney L. B., M A. G., Gannon M., Young P. P. (2013) A physiological role for connective tissue growth factor in early wound healing. Lab. Invest. 93, 81–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pi L., Shenoy A. K., Liu J., Kim S., Nelson N., Xia H., Hauswirth W. W., Petersen B. E., Schultz G. S., Scott E. W. (2013) CCN2/CTGF regulates neovessel formation via targeting structurally conserved cystine knot motifs in multiple angiogenic regulators. FASEB J. 26, 3365–3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kuwano K., Miyazaki H., Hagimoto N., Kawasaki M., Fujita M., Kunitake R., Kaneko Y., Hara N. (1999) The involvement of Fas-Fas ligand pathway in fibrosing lung diseases. Am. J. Respir. Cell Mol. Biol. 20, 53–60 [DOI] [PubMed] [Google Scholar]
- 67. Berry W. L., Janknecht R. (2013) KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 73, 2936–2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Luo W., Chang R., Zhong J., Pandey A., Semenza G. L. (2012) Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc. Natl. Acad. Sci. U. S. A. 109, E3367–E3376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kottmann R. M., Kulkarni A. A., Smolnycki K. A., Lyda E., Dahanayake T., Salibi R., Honnons S., Jones C., Isern N. G., Hu J. Z., Nathan S. D., Grant G., Phipps R. P., Sime P. J. (2012) Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-beta. Am. J. Respir. Crit. Care Med. 186, 740–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ding L., Getz G., Wheeler D. A., Mardis E. R., McLellan M. D., Cibulskis K., Sougnez C., Greulich H., Muzny D. M., Morgan M. B., Fulton L., Fulton R. S., Zhang Q., Wendl M. C., Lawrence M. S., Larson D. E., Chen K., Dooling D. J., Sabo A., Hawes A. C., Shen H., Jhangiani S. N., Lewis L. R., Hall O., Zhu Y., Mathew T., Ren Y., Yao J., Scherer S. E., Clerc K., Metcalf G. A., Ng B., Milosavljevic A., Gonzalez-Garay M. L., Osborne J. R., Meyer R., Shi X., Tang Y., Koboldt D. C., Lin L., Abbott R., Miner T. L., Pohl C., Fewell G., Haipek C., Schmidt H., Dunford-Shore B. H., Kraja A., Crosby S. D., Sawyer C. S., Vickery T., Sander S., Robinson J., Winckler W., Baldwin J., Chirieac L. R., Dutt A., Fennell T., Hanna M., Johnson B. E., Onofrio R. C., Thomas R. K., Tonon G., Weir B. A., Zhao X., Ziaugra L., Zody M. C., Giordano T., Orringer M. B., Roth J. A., Spitz M. R., Wistuba I. I., Ozenberger B., Good P. J., Chang A. C., Beer D. G., Watson M. A., Ladanyi M., Broderick S., Yoshizawa A., Travis W. D., Pao W., Province M. A., Weinstock G. M., Varmus H. E., Gabriel S. B., Lander E. S., Gibbs R. A., Meyerson M., Wilson R. K. (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kohno T., Otsuka A., Girard L., Sato M., Iwakawa R., Ogiwara H., Sanchez-Cespedes M., Minna J. D., Yokota J. (2010) A catalog of genes homozygously deleted in human lung cancer and the candidacy of PTPRD as a tumor suppressor gene. Genes Chromosomes Cancer 49, 342–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shyur S. D., Wang J. Y., Lin C. G., Hsiao Y. H., Liou Y. H., Wu Y. J., Wu L. S. (2008) The polymorphisms of protein-tyrosine phosphatase receptor-type delta gene and its association with pediatric asthma in the Taiwanese population. Eur. J. Hum. Genet. 16, 1283–1288 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.