

Abstract
Neutrophil extravasation occurs across postcapillary venules, structures composed of endothelial cells (ECs), pericytes (PCs), and basement membrane (BM). We constructed composite models of the human postcapillary venule, combining ECs with PCs or PC-deposited BM, to better study this process. Quiescent and tumor necrosis factor α (TNF-α)-activated composites demonstrated in situ-like expression of cadherins, E-selectin, intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), platelet-endothelial cell adhesion molecule 1 (PECAM-1), CD99, and interleukin 8 (IL-8). After TNF-α activation, the ECs supported greater neutrophil adhesion (66.1 vs. 23.7% of input cells) and transmigration (35.1 vs. 7.20% of input cells) than did the PCs, but the composites behaved comparably (no significant difference) to ECs in both assays. TNF-α-activated EC-conditioned medium (CM) increased transmigration across the PCs, whereas TNF-α-activated PC-CM decreased transmigration across the ECs, and culturing on PC-derived BM decreased both adhesion to and transmigration across the ECs. Anti-very late antigen 4 (VLA-4; on neutrophils) inhibited adhesion to TNF-α-activated composites, but not to ECs alone. Anti-CD99 (expressed on all 3 cell types) inhibited transmigration across the composites (14.5% of control) more than across the ECs (39.0% of control), and venular shear stress reduced transmigration across the ECs (17.3% of static) more than across the composites (36.7% of static). These results provide proof of concept that our composite human EC/PC/BM venular construct can reveal new interactions in the inflammatory cascade.—Lauridsen, H. M., Pober, J. S., Gonzalez, A. L. A composite model of the human postcapillary venule for investigation of microvascular leukocyte recruitment.
Keywords: neutrophil migration, ICAM-1, N-cadherin, pericyte, endothelial cell, inflammation, transmigration
Inflammation is a complex process that can be a protective component of the host defense, a pathologic process leading to tissue injury, or both. Controlling inflammation to limit tissue injury has become a major therapeutic target of modern medicine. The initiation of inflammation is marked by the transmigration of circulating leukocytes, predominantly neutrophils in the acute phase, through the postcapillary venule wall into peripheral tissue. Molecular therapeutic targets that are central to this process have generally been revealed in in vitro systems and then validated mostly in mouse models. Unfortunately, over the past decade, in vitro model systems of human postcapillary venules have not kept up with in vivo investigations. Specifically, standard models of the venular structure for investigation of inflammation have largely been limited to leukocyte–endothelial cell (EC) interactions, ignoring the complexity contributed by other components of the microvessel wall—namely, pericytes (PCs) and the condensed extracellular matrix shared by ECs and PCs, referred to as basement membrane (BM). These 3 principal constituents of the postcapillary venular wall communicate with one another to form an interdependent system that mediates vessel integrity and function (1–4). Therefore, a composite replicate of the human microvascular structure could remedy this deficit and further advance in vitro studies. Intermural communications can occur through intercellular contact (EC–EC, PC–PC, and EC–PC)–, soluble-, and BM component–mediated signals. Cadherins, a family of junctional proteins, are principally responsible for the formation of intercellular adherens junctions. EC–EC junctions are organized by vascular endothelial (VE)-cadherin, whereas neural (N)-cadherin is essential for EC–PC interactions (2, 5, 6). BM proteins, such as type IV collagen and laminin, influence leukocyte recruitment directly (BM–leukocyte interactions) and indirectly (BM–EC and BM–PC interactions)—for example, by causing EC and PC cytoskeleton reorganization and alterations in cell migration (7, 8).
Dynamic changes in the postcapillary venule in response to proinflammatory signals, referred to as activation, are necessary for efficient recruitment of leukocytes. The prototypic inflammatory cytokine tumor necrosis factor α (TNF-α) induces EC synthesis and display of adhesion molecules, such as E-selectin (CD62E), intercellular adhesion molecule 1 (ICAM-1; CD54), and vascular cell adhesion molecule 1 (VCAM-1; CD104), that interact with circulating leukocytes. Sequential interactions of these EC-expressed molecules with circulating neutrophils can initiate extravasation during a series of processes described in a paradigm known as the leukocyte adhesion cascade (9). In humans, cytokine-activated ECs expressing elevated levels of E-selectin capture and tether circulating neutrophils via binding of localized L-selectin (CD62L) on the tips of neutrophil microvilli (10, 11). Blood flow–induced shear stress causes cyclic disruption and reformation of these selectin interactions, resulting in neutrophil rolling along the EC luminal surface (10). Transiently adherent and rolling neutrophils encounter EC-displayed chemokines, principally interleukin 8 (IL-8; CXCL-8) in humans, that increase the affinity of leukocyte integrins for their ligands. Chemokine-activated neutrophils firmly adhere to ECs via integrin binding. Neutrophil-expressed lymphocyte function-associated antigen 1 (LFA-1; also designated αLβ2 integrin or CD11a/CD18) and macrophage 1 (Mac-1) antigen (also designated αMβ2 integrin or CD11b/CD18) both bind to EC-expressed ICAM-1, and neutrophil-expressed very late antigen 4 (VLA-4; also designated α4β1 integrin or CD49d/CD29) binds to EC-expressed VCAM-1 (10, 12, 13). Integrin-mediated adhesion to the EC surface is maintained as the activated neutrophil spreads and migrates to a preferential site for transmigration. In mice, transmigration occurs preferentially in low laminin, collagen IV, and nidogen-2 expression regions (14). In humans, the site of transmigration is also most likely influenced by the underlying BM, although this association has not been conclusively described. At these sites, neutrophils breach the EC monolayer by moving through the lateral border recycling compartment composed of a population of small vesicles rich in platelet-endothelial cell adhesion molecule 1 (PECAM-1; CD31) and CD99 adhesion molecules, in a series of homotypic adhesions (15). ECs additionally guide neutrophils by extending microvilli-like projections, rich in ICAM-1 and VCAM-1, along the direction of diapedesis (12). During this process, adhesion molecules on the leukocytes, such as Mac-1 or VLA-4, may also engage BM components. Within the BM, neutrophils encounter PCs, which like ECs, may synthesize and display chemokines, but do not express comparable levels of adhesion molecules. Specifically, PCs display markedly lower levels of ICAM-1 (16, 17) and VCAM-1 and do not express selectins. PCs preferentially guide neutrophils to sites characterized by larger inter-PC gaps, the formation of which is thought to be influenced by the previous steps of transmigration (16).
Until recently, cultured human PCs have not been readily available, and consequently, neutrophil–EC interactions have been much more extensively studied than have neutrophil–PC or neutrophil–BM interactions. Our recent advance in isolating and culturing PCs from placental microvessels has created the opportunity to extend these studies to other venular wall components (18). Herein, we describe a more complete model of the human venular wall using human placental microvascular PCs and BM components synthesized by these cells in conjunction with human EC cultures (Fig. 1). We characterize this model and present the results from a series of assays in which we study and manipulate cell contact-, soluble-, and BM-mediated signals in human microvasculature to identify key vascular mechanisms that regulate inflammation. We demonstrate that EC- and PC-soluble signals, including the release of IL-8, are sufficient to overcome the barriers to leukocyte transmigration associated with PCs and the inhibitory nature of PC-deposited protein in the shared microvascular BM. Further, initial studies of functional blocking of neutrophil adhesion and transmigration with monoclonal antibodies suggest that the use of an EC/PC/BM composite model, which more comprehensively replicates human in vivo postcapillary venules, will yield new and perhaps more relevant results for parsing out inflammatory mechanisms and identifying potential targets for therapeutic intervention.
Figure 1.
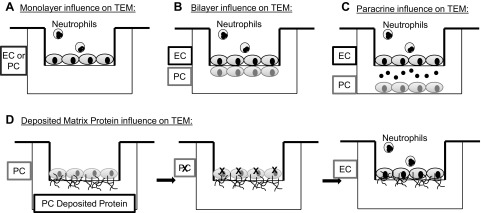
Schematic of a decomposed microvessel model. A) The influence of a singular cell type, either EC or PC, on neutrophil transmigration can be isolated by seeding it in the apical chamber of the porated polycarbonate transwell membrane to form a confluent monolayer. Neutrophil transmigration is a result of the exclusive interactions of that cell type. B) To determine cell–cell contact-mediated alterations in neutrophil transmigration, the PCs were seeded on the basal face of the chamber, and the ECs were seeded in the apical chamber. Processes extended from both cell types through the pores in the membrane, forming adhesions. C) Soluble paracrine signaling effects were isolated by removing cellular contact. PCs were cultured below the transwell insert within the well, such that diffusion of soluble molecules altered the interactions. The model can also be set up with the PCs in the apical chamber and ECs in the well. D) PCs were seeded on the apical side of the transwell membrane, where they deposited protein. The PC were then removed, leaving the protein intact, and the ECs are seeded on top of the protein on the apical face of the transwell cell culture insert.
MATERIALS AND METHODS
Cell culture
PCs were isolated by explant outgrowth from microvessels obtained from anonymized human placenta, according to the methods of Maier et al. (18). ECs and PCs were cultured in tissue culture–treated T75 flasks in M199 medium (Gibco, Grand Island, NY, USA) supplemented with 20% FBS (Hyclone Laboratories, Inc., Logan, UT, USA) and 1% penicillin-streptomycin (Gibco). ECs were harvested from human umbilical veins, according to the procedures described by Gimbrone et al. (19), and cultured in T75 flasks coated with 0.1% gelatin (Sigma-Aldrich, St. Louis, MO, USA). In brief, the resulting PC populations were positive for chondroitin sulfate proteoglycan 4 (NG2); CD90; CD146, α-smooth muscle actin; platelet derived growth factor β (PDGFR-β); and calponin. These cells stained negatively for markers of smooth muscle cells, endothelial cells, and leukocytes, including transgelin (SM22-α), smooth muscle myosin heavy chain, CD31, CD34, CD45, and CD14. EC monolayers were cultured in PC media supplemented with 10 μg/ml endothelial cell growth supplement (ECGS; Collaborative Biomedical Products, Bedford, MA, USA). Confluent T75 flasks were trypsinized with 0.25% trypsin EDTA (Gibco) and resuspended in their respective media for seeding on either gelatin-coated 25 mm diameter glass coverslips (VWR International, Radnor, PA, USA), for neutrophil adhesion studies, or on polycarbonate transwell inserts (Corning, Corning, NY, USA) for neutrophil transmigration studies, as described below.
Neutrophil isolation
Neutrophils were isolated from blood obtained from healthy human volunteers, as approved by Yale Human Investigations Committee (Protocol 0902004786). Blood was drawn into a syringe containing citrate phosphate dextrose (CPD) and dextran and immediately mixed thoroughly. After a 45–60 min separation period, plasma was removed and the red blood cells were discarded. After 6 min of centrifugation, the supernatant was removed, and the cell pellet was resuspended in a Histopaque/PBS solution (Sigma-Aldrich). The cell suspension was centrifuged, the supernatant was discarded, and the pellet was resuspended in Mg+ and Ca+ PBS and glucose at a concentration of 1 × 106 cells/ml, as determined by hemocytometer counts.
Neutrophil membrane labeling
Neutrophils were labeled with a PKH26 red fluorescence cell linker kit (Sigma-Aldrich) to stain the neutrophil membranes according to the manufacturer's recommendation. Stained neutrophils were used exclusively for imaging studies and not quantitative transmigration rates.
Adhesion assays
Comparison of EC and PC monolayers with an EC/PC bilayer
ECs and PCs were seeded on 24-well plate polycarbonate transwell inserts (Corning), as EC or PC monolayers or EC/PC bilayers, and remained in culture for 3–5 d. The cells were stimulated with 10 ng/ml of rhTNF-α (R&D Systems, Minneapolis, MN, USA) for 4 h before being assessed for neutrophil adhesion. Adhesion assays were performed with freshly isolated neutrophils, as described previously. For blocking studies, neutrophils were incubated in anti-CD49d (R15.7; eBiosciences, San Diego CA, USA), anti-CD18 (Millipore, Danvers, MA, USA), anti-CD99 (YG32 mAb, DiNonA, Seoul, South Korea, USA), and anti-αvβ3 (Millipore) blocking antibody on ice for 15 min with constant agitation. The neutrophils were then diluted to a concentration of 100,000 cells/ml in Mg+, and Ca+ PBS and glucose and a total of 14,000 neutrophils were added to the apical chamber of the transwell insert. The transwells with neutrophils were incubated for 10 min at 37°C, after which they were inverted for ∼5 min, to allow any nonadherent neutrophils to fall off the surface. The cells were fixed with 4% paraformaldehyde, and the neutrophils were stained with a mouse anti-human CD45 (BD Biosciences, San Jose, CA, USA) primary antibody and a FITC secondary antibody (Sigma-Aldrich). Adherent and positively stained cells were counted to yield adhesion counts.
Soluble paracrine and protein adhesion assays
Either ECs or PCs were seeded on gelatin-coated 25-mm glass coverslips. The medium was changed daily until the cells reached confluence. Monolayers were stimulated with 10 ng/ml of rhTNF-α (R&D Systems) for 4 h before being assessed for neutrophil adhesion. Adhesion assays were performed on the coverslips by injecting a neutrophil solution (100 μl of a solution containing 1×106 cells/ml and 900 μl PBS) into a Sykes-Moore adhesion chamber containing the EC or PC coverslips. Neutrophils were injected into the chamber and allowed to adhere for 500 s. The number of neutrophils in contact with the EC or PC monolayer was counted and the chamber inverted for an additional 500 s. The remaining neutrophils in contact with the monolayer after inversion were counted and expressed as the percentage of adherent cells.
Transmigration studies
Transwell setup
Polycarbonate permeable supports (24 mm diameter) with 3.0-μm pores served as the foundation for the culture system (Corning). Cells were seeded onto the inserts in 3 different conformations corresponding to different model systems (Fig. 1). In all 3 submodels, cultured cells were trypsinized with 0.25% trypsin EDTA (Gibco) and resuspended in medium to be seeded. The cell suspension was added to the transwell surface to form a meniscus to ensure localized cell seeding. After 4 h, sufficient medium was added to cover the transwell in a tissue culture well plate. Simultaneously, the same volume of the cell suspension was added to a gelatin-coated glass coverslip (25 mm diameter; VWR International, Radnor, PA) placed in a 6-well plate, in order to monitor growth; similarly, medium was added after 4 h to sustain cell growth.
Model 1: isolated EC or PC monolayer influence on neutrophil transmigration.
EC or PC cell suspensions were added to the apical chamber of transwell membranes in welled plates and on coverslips as described above. The cells remained in culture for between 3 and 6 d until a confluent monolayer was formed, as demonstrated by the glass coverslip control. Medium was changed every other day.
Model 2: EC–PC bilayer contact influence on neutrophil transmigration.
Initially, PC cell suspensions were prepared as described above. Transwell inserts were sterilely inverted in an open Petri dish, so that the basal face was accessible. PC cell suspension was added to the basal face of the transwell, and the inverted inserts were carefully moved to a tissue culture incubator (37°C; 5% CO2) for ∼4 h, to ensure PC adhesion to the polycarbonate membrane. An observational PC glass coverslip was also seeded. After this incubation period, an EC cell suspension was prepared. The inverted transwells seeded with PCs were placed in a biosafety cabinet and returned to their proper orientation in a tissue culture plate. EC suspension was added to the apical chamber, as well as to a glass coverslip for observation (medium added 4 h later). EC medium was added to fill the apical and basal portions of the transwell. Cells remained in culture for between 3 and 6 d, as determined by cell growth on control coverslips, with the medium changed every other day.
Model 3: soluble factor influence on neutrophil transmigration.
ECs or PCs were seeded on gelatin-coated 25-mm glass slides, as described for adhesion studies, at the same time that EC and PC monolayers in the apical chamber of transwell inserts were seeded. EC coverslips were placed in wells with PC transwells and vice versa. All wells were filled with EC medium, which was changed every other day for between 3 and 6 d until the cells on both the coverslip and the transwell were confluent (observational coverslips were set up to monitor transwell cell proliferation).
Model 4: influence of PC-deposited extracellular matrix on neutrophil transmigration.
Transwells were seeded with PCs in the apical chamber, as described previously for monolayers. The transwell was filled with PC complete medium supplemented with sterile ascorbic acid, to a final concentration of 0.1 mM ascorbic acid. Glass coverslips were again seeded with the same cell solution for monitoring purposes. PCs remained in culture for up to 5 wk before a decellularization solution composed of 10 mM ammonium hydroxide (J. T. Baker, Phillipsburg, NJ, USA) and 0.5% Triton-X 100 (Sigma-Aldrich) was applied for 5 to 8 min with physical agitation, to remove the PC monolayers while preserving the protein deposits. Cell removal from observational glass coverslips was used as a gauge of the progress of decellularization. The transwells and coverslips were washed 3 times with sterile PBS (Gibco). Decellularized substrates were then either fixed for analysis, or EC cell suspension from confluent T75 flasks were seeded on top of the protein deposited by the PCs, as described for EC monolayer studies. ECs remained in culture in EC medium, which was changed every other day until confluent monolayers were formed.
Transmigration assays
Transmigration assays were completed with all 4 models once cells reached confluence. Cells were stimulated for 4 h with 10 ng/ml of rhTNF-α. After human neutrophils were isolated, tissue culture inserts were transferred to 6-well plates coated with a thin layer of 2% agarose. Approximately 3 × 106 neutrophils were added to each well and were then allowed to transmigrate through the activated monolayer for 1 h. Neutrophils that successfully migrated were collected from the bottom of the well and counted with a hemocytometer. Transmigration assays with functional blocking of CD99 were completed using YG32 mAb (DiNonA), similar to previously published methods (20). In brief, after 4 h of TNF-α stimulation, cell monolayers or bilayers were incubated for 15 min in a 20 μg/ml YG32 solution. The cells were then washed vigorously 2 times with PBS, before being transferred to agarose wells. The transmigration then proceeded, as described previously.
Transmigration under flow
ECs and ECs/PCs were cultured on transwell membranes and stimulated with TNF-α for 4 h. After stimulation, the polycarbonate membranes were removed from the plastic housing of the transwell insert and the membrane placed between a circular flow chamber for 35-mm coverslips (Glycotech, Gaithersburg, MD, USA) and a custom-made polycarbonate catchment chamber. The catchment chamber was a hollow cylindrical polycarbonate base that enabled a vacuum to be formed with the Glycotech chamber, but also allowed for neutrophils to transmigrate and to be caught in the center of the apparatus. The apparatus was connected to a syringe pump and placed under a low-heat gun to warm the sample. A homogenous solution of human neutrophils in medium at a concentration of ∼67,500/ml was pumped across the membrane over the course of 1 h, ultimately exposing the membrane to 3 × 106 neutrophils. Transmigrated neutrophils were collected and counted with a hemocytometer.
Scanning electron microscopy (SEM)
Transwells and coverslips for each model were fixed with 3% paraformaldehyde and dehydrated with ethanol and HDMS before being mounted for SEM. Samples were coated with either carbon or chrome and imaged on an SU-70 scanning electron microscope using a 5-kV beam (Hitachi, Tokyo, Japan).
Fluorescent staining and imaging
Cells on transwell inserts or coverslips were rinsed in PBS and fixed in 3% paraformaldehyde. The cells were blocked in a 2% bovine serum albumin (BSA; Sigma-Aldrich) solution before the addition of the primary antibody. Cells were stained for ICAM-1, with a mouse anti-human ICAM-1 (CD54) antibody (R&D Systems) conjugated with an FITC secondary antibody (Sigma-Aldrich), or for N-cadherin, with a rabbit anti-human N-cadherin primary antibody (Abcam, Cambridge, MA, USA) and an anti-rabbit TRITC secondary antibody (Abcam). Mouse anti-human VCAM-1 (R&D Systems and Santa Cruz Biotechnology, Santa Cruz, CA, USA) and goat anti-human PECAM-1 (Santa Cruz Biotechnology) were conjugated with an FITC secondary antibody (Sigma-Aldrich) and goat-anti-rabbit AlexaFluor 467 (Life Technologies, Grand Island, NY, USA), respectively. To stain for CD99, an antigen retrieval protocol was completed. In brief, the seeded substrates were fixed and blocked and then boiled for 10 min in a citrate buffer. After boiling, CD99 was labeled with a rabbit anti-human CD99 primary antibody (Spring Bioscience, Pleasanton, CA, USA) and subsequently conjugated with an FITC secondary antibody (Sigma-Aldrich). E-Selectin was fluorescently labeled with a mouse anti-human E-selectin antibody conjugated to fluorescein (R&D Systems). Protein samples were stained for laminin with a rabbit anti-human laminin primary antibody (Abcam) and an FITC secondary (Abcam) antibody or a mouse anti-human collagen 1 primary antibody (Abcam). Samples were mounted on glass slides with Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA, USA) and imaged on an LSM 510 laser scanning confocal microscope (Zeiss, Thornwood, NY, USA). Image analysis and postprocessing were completed using ZEN software (Zeiss) and ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA). For LI-COR analysis of laminin and collagen, the aforementioned laminin body and a mouse anti-human collagen I primary antibody (Abcam) conjugated with a goat anti-rabbit 800CW IRDye (LI-COR Biosciences, Lincoln, NE, USA) and a goat anti-mouse 680LT IRDye (LI-COR Biosciences). The samples were analyzed on a LI-COR Odyssey scanner, and the integrated intensities were determined with associated software (LI-COR Biosciences).
Sodium fluorescein (Na-F) permeability assay
EC and PC monolayers and EC/PC bilayers were cultured on transwells and were treated or not with TNF-α for 4 h, as described above. Na-F was then added to the apical well of each chamber at a final concentration of 10 μg/ml. Bare transwells were set up in a similar fashion as the control. All the transwells were incubated for 1 h at 37°C/5%CO2, after which medium was collected from the apical and basal chambers. Na-F concentration in the collected media samples was determined with a SpectraMax M5 fluorescence plate reader (Molecular Devices, Sunnyvale, CA, USA). Clearance was calculated as described previously by Nakagawa et al. (21).
ELISA
ELISAs for IL-8 expression in cell supernatant collected from control and TNF-α-stimulated (4 h) monolayers of ECs or PCs cultured on glass coverslips was determined via an ELISA. Supernatant from transwells was collected in a similar manner, to ensure the same volume of supernatant per cell density. IL-8 secretion was determined with a human IL-8 ELISA Ready-SET-Go kit (eBiosciences), according to the manufacturer's protocol; 4-parameter nonlinear regression of the data was completed.
Statistical analysis
All statistical analyses were completed with Prism 5 (GraphPad, San Diego, CA, USA). Significance was determined with either a 1-way ANOVA with the Tukey post hoc test or a 2-tailed unpaired t test, as appropriate. Significance was set at P < 0.05. All conditions had a sample size of ≥3 samples, using neutrophils from ≥3 distinct human donors. Outliers beyond 2 sd from the mean were excluded from analysis.
RESULTS
Characterization of an in vitro EC and PC bilayer composite model of the postcapillary venular wall
We have developed an in vitro composite model of a PC-dense postcapillary venular wall (shown schematically in Fig. 1), similar to that formed in sites like the central nervous system. We used a combination of SEM and confocal fluorescence microscopy, as shown in Fig. 2, to characterize the morphologic features of our composite structures. Elliptical ECs spread to fully cover the transwell membrane. The cytoplasmic portions of the EC body retained their physiological thin structure around the nucleus (Fig. 2A). Measuring the maximum height of the ECs, as determined by confocal imaging, revealed an average maximum thickness of 6.98 ± 1.15 μm (mean±sem); the thinnest portions around the cell border could not be measured reliably. Physiological EC heights ranged from 2.90 to 7.11 μm, confirming that the cultured ECs were comparable to in vivo standards (22). Likewise, SEM imaging demonstrates that the cultured PCs (Fig. 2B) retained an elongated structure commonly seen in vivo and expressed a high degree of linear organization that could support the vessel circumferentially in vivo (16). Measurements of PCs imaged with SEM and confocal microscopy also revealed dimensions similar to those observed in vivo. The average PC length was determined to be 175 ± 60 μm (mean±sem), within the previously reported range of 150–200 μm, and the average PC width was determined to be 18.3 ± 5.4 μm, again within the published range of 10–25 μm (10). SEM imaging of the transwell cross section demonstrated processes that extended through the porated membrane and made contact with the opposing cells (Fig. 2C). This is corroborated by images of subconfluent transwells on which individual extensions can be more easily visualized (Fig. 2D).
Figure 2.

Formation of an interconnected EC/PC bilayer in vitro. A) SEM imaging of an EC monolayer on polycarbonate transwells (TW). The ECs spread to form a thin monolayer while extending processes to create intercellular contacts. B) SEM imaging of PCs on polycarbonate TW demonstrated a highly ordered arrangement of cells, as is seen in the perivascular tissue in vivo. C) Cross section of a transwell shows EC and PC processes contacting one another in the membrane's pores. D) Nonconfluent transwells revealed individual processes, in this case from a PC, extending through pores to the opposite side. Scale bar = 20 μm. E, F) PECAM (red) and DAPI (blue) nuclear stain on EC (E) and PC (F) monolayers. G, H) VE-cadherin (red) and DAPI nuclear (blue) staining of EC (G) and PC (H) monolayers. VE-cadherin localized to the intercellular borders, as expected from in vivo and previous in vitro studies. PCs lacked VE-cadherin, as shown by the absence of staining. I, J) N-cadherin (red) was diffusely expressed across both the EC (I) and PC (J) monolayers. DAPI (blue) nuclear stain was also present. K) Z-stack image of the EC/PC bilayer stained for N-cadherin (red) and DAPI (blue) demonstrated that N-cadherin was present on the processes in pores. L, M) CD99 (green) and DAPI (blue) staining on EC (L) and PC (M) monolayers. N) Z-stack image of the EC/PC bilayer stained for ICAM-1 (green) demonstrated that ICAM-1 was present on processes in pores. The intensity of ICAM-1 expression was artificially increased during image capture, to equalize EC and PC expression levels. White arrows: EC and PC extensions positively stained for N-cadherin (K) and ICAM-1 (N). Scale bars = 10 μm.
Immunofluorescence microscopy was used to identify and localize PECAM-1, members of the cadherin family of junctional proteins (VE-cadherin and N-cadherin), CD99, and ICAM-1 under quiescent conditions. Cellular presentation of these molecules demonstrates that monolayers and the composite model not only conform to in vivo morphologies, but also are capable of intercellular communications that could functionally interact with neutrophils. PECAM-1 and VE-cadherin localization to EC–EC borders confirms the formation of cohesive monolayers (Fig. 2E, G), replicating the structure of the luminal layer of the human postcapillary venule responsible for limiting vascular leakage (6, 23). Localization of VE-cadherin to the inter-EC junctions is consistent with the formation of adherens junctions characteristic of the EC lining postcapillary venules. Neither PECAM-1 nor VE-cadherin was expressed by the PCs (Fig. 2F, H), however, although both ECs and PCs diffusely expressed N-cadherin (Fig. 2E, F). In addition, Z-stack confocal images demonstrated that some N-cadherin molecules localized at the EC–PC junctures in the intermediate regions between the 2 cellular layers (Fig. 2K; white arrows denote EC and PC extensions positively stained for N-cadherin), consistent with N-cadherin-organized adherens junctions linking ECs and PCs. Under quiescent conditions, however, we found that CD99 and ICAM-1 were expressed on both the ECs and PCs, suggesting that CD99 is involved in neutrophil-PC interactions, mimicking the previously described CD99-dependent interactions observed between neutrophils and ECs (Fig. 2L, M). Moreover, Z-stack imaging further confirmed that ICAM-1 was expressed on extensional processes connecting the EC and PC monolayer (Fig. 2N; white arrows denote EC and PC extensions positively stained for ICAM-1).
We also assessed the deposition of extracellular BM proteins in our system. The ability of PCs to secrete proteins in our model system was verified through SEM and fluorescence imaging (Fig. 3). PCs cultured on both sterile glass coverslips and polycarbonate transwells deposited tracks of proteins, ultimately forming long fibrils. These fibrils contained several different proteins (referred to as total protein; Fig. 3A) and were easily visualized in scanning electron micrographs. To ensure that 2 major constituents, collagen and laminin, were contained in the total protein deposit, we quantitatively determined the relative amounts of collagen and laminin through LI-COR imaging and subsequent determination of integrated intensities (Fig. 3B). The integrated intensity of collagen, the primary matrix protein constituent of the vascular wall, was significantly (P<0.001) higher than that of laminin, as determined by an unpaired, 2-tailed t test. Both collagen and laminin were heterogeneously distributed over the decellularized substrate (Fig. 3C, D), resembling BM composition in vivo. Z-stack confocal images enabled the thickness of the protein layer to be determined, showing an average thickness of 3.7 μm on top of the polycarbonate transwells. Although an accepted value of human BM thickness in the postcapillary venule has not been described in the literature, our measurement is consistent with values determined for the generalized capillary BM (24).
Figure 3.
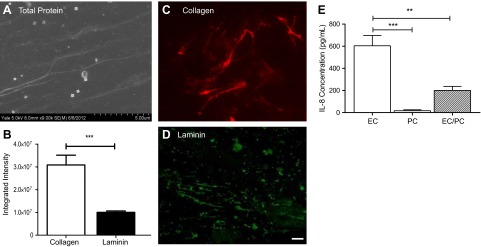
Quiescent cultures replicate vascular synthesis roles. A) SEM imaging of PC-deposited protein on glass demonstrates fibrous tracks, after substrate decellularization. B) LI-COR analysis of collagen and laminin content in PC-deposited protein on glass coverslips after decellularization. ***P < 0.001; unpaired 2-tailed t test. C, D) Collagen-specific (red; C) and laminin-specific (green; D) fluorescent stains, displaying the physiological heterogeneous protein deposition. Scale bars = 10 μm. E) IL-8 concentrations in the EC monolayer, PC monolayer, and EC/PC bilayer standardized to an equal volume of supernatant per cell. IL-8 was significantly higher in the EC monolayer than in the PC monolayer and the EC/PC bilayer. **P < 0.01, ***P < 0.001; 1-way ANOVA with the Tukey post hoc test.
IL-8, although lacking in mice, is the principal human chemokine known to induce neutrophil activation and chemotaxis (25). Under quiescent culture conditions, low levels of IL-8 are produced by EC and PC monolayers, as well as the EC/PC bilayer (Fig. 3E). IL-8 concentrations generated by the quiescent EC monolayer are higher than the concentrations generated by the PC monolayer and by the EC/PC bilayer, on an averaged basis. The reduced output by the bilayer represents the average IL-8 concentration standardized to the number of cells in culture; thus, the high IL-8 concentrations that were produced were reduced by the low IL-8 concentrations from the PCs, as the bilayer was composed of approximately twice as many cells. The IL-8 concentration generated by the EC/PC bilayer, however, was lower than the mean of the EC and PC monolayers, suggesting that IL-8 production by the composite model of the venular wall was diminished. Alternatively, binding of soluble IL-8 to the increased cell surface area of elaborated BM could decrease the level of IL-8 assayed in the medium (23).
EC/PC composite model response to TNF-α activation
We next investigated the response of the EC/PC bilayer to inflammatory stimulation. After TNF-α activation, VE-cadherin was no longer confined to the EC border, consistent with increased permeability of the bilayer (refs. 26, 27 and Fig. 4A, B), due to opening of the adherens junctions. VE-cadherin remained absent from the PCs after TNF-α-activation (Fig. 4C, D). In contrast to VE-cadherin, TNF-α-activation of the EC/PC bilayer did not appear to alter N-cadherin expression on the ECs or the PCs (Fig. 4E–H). ICAM-1 intensity increased after TNF-α-activation on both ECs and PCs; ICAM-1 staining on stimulated ECs was diffuse, whereas on PCs, it presented in a more punctate manner (Fig. 4I–L). Similarly, VCAM-1 expression was diffuse and dim on both the ECs and the PCs under quiescent conditions (Fig. 4M, O). TNF-α-activation subsequently increased VCAM-1 expression on the ECs, but unlike the changes observed in ICAM-1, this increase was more pronounced on some ECs than on others (Fig. 4N). We do not know the basis of this heterogeneous response. TNF-α activation of PCs, however, uniformly increased punctate staining of VCAM-1 (Fig. 4P). CD99 expression was diffuse on the quiescent ECs and PCs (Fig. 4Q, S). CD99 appeared unaltered after TNF-α activation of the ECs, but may decrease slightly on the PCs (Fig. 4R, T). E-selectin was weakly expressed on the quiescent ECs, but not on the quiescent PCs (Fig. 4U, W). After TNF-α activation, the ECs strongly expressed E-selectin and, unexpectedly, a few PCs expressed high levels of E-selectin as well (Fig. 4X). It is unclear at this time whether this represents synthesis by the PCs or transfer from the ECs.
Figure 4.

EC/PC bilayer response to TNF-α-activation. A–X) Confocal imaging of quiescent (−TNF-α) and TNF-α-activated (+TNF-α) bilayers demonstrated the change in junctional (A–H) and surface molecule adhesion expression (I–X) on ECs (left panels) and PCs (right panels). Scale bars = 10 μm. Y) All culture conditions showed an increase in IL-8 levels after TNF-α-activation. IL-8 concentration in EC/PC bilayer was significantly higher than that in the EC or PC monolayer in stimulated conditions. **P < 0.01, ***P < 0.001 as indicated; #P < 0.05 vs. quiescent cells; 1-way ANOVA with Tukey post hoc test. Z) Permeability of EC and PC monolayers and EC/PC bilayers under quiescent and TNF-α-activated conditions as determined by clearance of Na-F. All 3 systems showed similar leakiness under basal conditions, but only EC monolayers demonstrated a significant augmentation in cellular permeability after TNF-α-activation. The TNF-α-activated EC/PC bilayer was significantly less permeable than either the EC or PC monolayers. *P < 0.05, ***P < 0.001, 1-way ANOVA and Tukey post hoc test; **P < 0.01, unpaired t test.
The production of IL-8 increased significantly after 4 h of TNF-α-activation. As shown in Fig. 4Y, TNF-α activation resulted in increased IL-8 production in both isolated EC and PC monolayers and in the EC/PC bilayers. This increase was greatest in the case of the EC/PC bilayer, which, as noted above, had lower basal expression, indicating a potential coculture synergy. The permeability of the EC and PC monolayers and the EC/PC bilayers, measured by Na-F flux, appeared comparable at baseline. However, changes in the permeability of the different cellular constructs varied with TNF-α activation (Fig. 4Z). As expected, TNF-α activation significantly (P<0.01) increased the permeability of the EC monolayer when compared with the quiescent culture conditions. In contrast, the permeability of the EC/PC bilayer was not increased by TNF-α activation. Flux across the PC monolayers may have increased but did not reach statistical significance. Of particular note, the TNF-α-activated EC/PC construct was significantly less permeable than the EC (P<0.05) and PC (P<0.001) monolayers.
TNF-α stimulated neutrophil adhesion and diapedesis
We next evaluated the capacity of neutrophils to adhere to and migrate across our bilayer model and compared this capacity to adhesion and transmigration across both the EC and the PC monolayers. Previous methods for investigating leukocyte adhesion by using Sykes-Moore chambers (17) were incompatible with our EC/PC bilayer model because of the optical properties of the transwell. We therefore developed a similar protocol, which requires fluorescent staining after neutrophil adhesion to visualize the cells without interfering with their ability to adhere. After TNF-α activation for 4 h, neutrophil adhesion to the EC/PC composite bilayers was comparable to adhesion to the ECs, and these levels were significantly higher than adhesion to the PC monolayers (Fig. 5A). Studies using standard Sykes-Moore chambers confirmed the lower level neutrophil adhesion to TNF-α-activated PCs compared to TNF-α-activated ECs. On EC coverslips, 66.1% of neutrophils adhered, whereas only 23.7% of neutrophils adhered to PC monolayers (P<0.001, unpaired 2-tailed t test).
Figure 5.
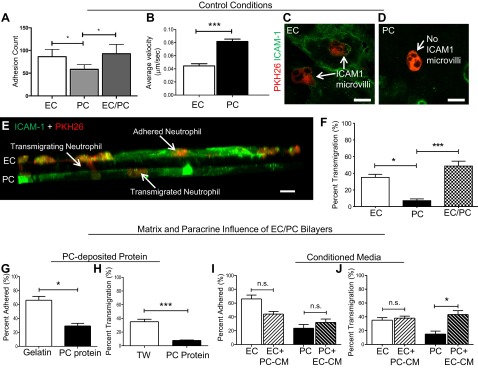
Altered transmigration through the composite model, as compared to that through the EC and PC monolayers. A) Neutrophil adhesion to EC and PC monolayers and the EC/PC bilayer after TNF-α activation for 4 h (all cells cultured on polycarbonate transwells). Neutrophil adhesion to EC monolayers and EC/PC bilayers was significantly higher than adhesion to PC monolayers. *P < 0.05; unpaired 2-tailed t test. B) PCs (n=20) had a significantly higher velocity than ECs (n=43). ***P < 0.001; unpaired t test. C) Confocal microscopic image of EC-expressed ICAM-1 microvilli (green) surrounding a human neutrophil (red). Scale bar = 10 μm. D) Confocal microscopic image of PC-expressed ICAM-1 lacking microvilli (green) around a human neutrophil (red). Scale bar = 10 μm. E) PKH26 staining of human neutrophils (red) and their interactions with EC and PC bilayers (stained with ICAM-1, green, with intensity adjusted to equalize EC and PC monolayers). Neutrophils interacted with either the EC layer, actively transmigrating through the pores, or with the PC layer. Scale bar = 10 μm. F) Neutrophil transmigration percentages through EC and PC monolayers compared to an EC/PC bilayer. Transmigration through PC monolayers was significantly reduced compared to that through EC monolayers and the EC/PC bilayer. There was no significant difference between the EC monolayer and the bilayer. *P < 0.01, ***P < 0.001; 2-tailed unpaired t test. G) Comparison of neutrophil adhesion on ECs cultured on coverslips coated with gelatin vs. PC-deposited protein. *P < 0.05; unpaired t test (n=6/condition). H) Neutrophil transmigration through EC monolayers cultured on PC protein showed significantly attenuated migration compared to migration through EC monolayers grown in coverslips (n=6/condition). ***P < 0.001; unpaired t test. I) Neutrophil adhesion on EC monolayers was significantly decreased. Adhesion increased nonsignificantly on PC monolayers after the inclusion of CM from the alternate cell type. **P < 0.007; unpaired t test. J) Inclusion of EC-CM significantly increased neutrophil transmigration through the TNF-α-stimulated PC monolayer. *P < 0.05, 1-way ANOVA and Tukey post hoc test, as well as an unpaired t test.
Given this initial difference in neutrophil adhesion to EC and PC monolayers, we further investigated the behavior of neutrophils while they were attached to the surface of the 2 different vascular cell types. On TNF-α-activated EC monolayers, neutrophil lateral motility was significantly (P<0.001) reduced when compared to migration on PC monolayers (Fig. 5B). Although the exact cause of this difference in speed is not known, both average velocities measured in vitro were within the range of neutrophil velocities seen in vivo in mouse cremaster models (16). Average neutrophil crawling velocity on PCs in mice is known to increase with time. Early neutrophils identify advantageous gaps for transmigration, which are subsequently used by later neutrophils (16). Thus, the mean velocity of the PCs could increase as a result of faster neutrophil migration following earlier and slower neutrophils.
Confocal imaging demonstrates that ECs and PCs displayed ICAM-1 differently in the presence of neutrophils. PKH26-stained neutrophils (Fig. 5C) became encompassed by ICAM-1-rich microvilli on the ECs, a previously described aspect of EC-neutrophil interactions (12). In contrast, TNF-α-activated PCs did not interact with neutrophils in a similar way (Fig. 5D). This is not simply due to lesser levels of ICAM-1 expression, as prior studies with ICAM-1-transduced PCs also failed to encompass bound neutrophils in this manner (17). We next tracked the diapedesis of the PKH26-labeled neutrophils through an ICAM-1-labeled TNF-α-activated bilayer (Fig. 5E) and compared the results to transmigration through EC or PC monolayers. As seen with adhesion, the percentage of neutrophils undergoing diapedesis was dependent on the vascular cell type that formed the monolayer: migration through a TNF-α-activated EC monolayer occurred more readily than migration through a TNF-α-activated PC monolayer (Fig. 5F; transmigration percentages were determined in the unlabeled cells). On average, 35.1% of neutrophils successfully moved through an EC monolayer, whereas only ∼7.2% of neutrophils transmigrated through a PC monolayer over the course of 1 hour. Overall, these data suggest that ECs are more permissive to leukocyte extravasation, whereas PCs tend to act as a barrier. Surprisingly, the ability of neutrophils to transmigrate the composite venule model was not inhibited by the presence of the PCs. The extent of neutrophil migration through the bilayer appeared greater than migration through an EC monolayer, averaging ∼48.8% (Fig. 5F), although this difference did not reach statistical significance. In contrast, the increase over the extent of transmigration through a PC monolayer was significant (P<0.001). Since, as shown in Fig. 4Z, TNF-α-activated bilayers were less permeable than similarly activated PC monolayers, the enhanced neutrophil transmigration through TNF-α-activated bilayers did not correlate with and thus cannot be simply attributed to differences in the permeability of the various cellular constructs.
Analysis of functional interactions among components of our EC/PC composite bilayers
To better understand the differences observed in neutrophil interactions with EC/PC composite constructs when compared with isolated monolayers, we examined how signals may be transmitted among the different components of the composite, including the PC-deposited BM and both EC and PC-generated soluble signals.
Changes in cellular substrate
Comparing neutrophil adhesion to ECs grown on gelatin-coated glass, the standard substrate for in vitro EC-leukocyte interaction studies (28), to adhesion to ECs grown on PC-deposited protein-coated glass indicated that PC-deposited protein provided a mechanism to which the PCs contribute as a barrier to neutrophil extravasation. The mean neutrophil adhesion percentage on the TNF-α-activated EC monolayer decreased from 66.1 to 29.2% as a result of EC culture on PC protein (Fig. 5G). Similarly, the percentage of neutrophil transmigration was significantly (P<0.001) attenuated when ECs were cultured on PC-deposited protein. Transmigration levels were reduced 5-fold from the 35.1% transmigration seen when ECs were cultured on polycarbonate transwell inserts (Fig. 5H). The reduction in neutrophil transmigration cannot be explained solely by a reduction in neutrophil adhesion to the cellular monolayer. In the absence of PC protein, approximately two-thirds of adherent neutrophils successfully transmigrated across the TNF-α-activated EC monolayer. The addition of the protein layer reduced to approximately one-quarter the fraction of adherent neutrophils that successfully transmigrated. This reduction in transmigration could be the result of neutrophils being trapped within the protein layer before exiting the system within the 1 h time span. Neutrophil tracking in fixed EC/PC bilayers, for example, demonstrated that ∼2% of neutrophils were trapped between the ECs and PCs, a percentage that would probably increase in pathological conditions involving excess BM, as in fibrosis. These data are impossible to confirm in humans in vivo although, in light of previous animal studies (29) and the influence of the extracellular microenvironment seen throughout the body in all biological processes, our model provides an excellent opportunity to investigate matrix protein–based signal regulation of leukocyte recruitment.
Soluble influences
On average, 66.1% of the settled neutrophils adhered to TNF-α-activated EC monolayers, which was significantly higher than the average percentage adhering to the PC monolayer (23.7%). When neutrophil adhesion studies were repeated on EC monolayers incubated with TNF-α-activated PC-conditioned medium (CM) there was a significant decrease in neutrophil adhesion to approximately two-thirds of the previous value (EC+PC-CM mean=44.1%; Fig. 5I). The opposite was true of levels of neutrophil adhesion to PC monolayers incubated with TNF-α-activated EC supernatant (PC+EC soluble mean=32.1%; Fig. 5J). Soluble signaling effectively altered neutrophil adhesion to TNF-α-activated ECs and PCs to equalize the previously disproportionate responses. The difference in neutrophil adhesion to ECs and PCs decreased from 42.5% in the absence of soluble signaling to 12.0% with soluble signaling.
Neutrophil transmigration through the EC and PC monolayers in the presence of soluble paracrine signals similarly reduced the stark differences between EC and PC support of neutrophil extravasation (Fig. 5J). As previously noted, transmigration through the PC monolayer was relatively low compared with that through the EC monolayer after 4 h of TNF-α activation. The difference between these 2 conditions was diminished when soluble signals from the other cell type were included. The incubation of ECs with TNF-α-activated PC-CM did not result in a significant change to EC support of transmigration, probably because of the continued release of high concentrations of IL-8 from the EC monolayer and the innate ability of activated ECs to guide the neutrophils across the cellular monolayer in an ICAM-1-dependent manner. The addition of the TNF-α-activated EC-CM to the PCs resulted in increased neutrophil transmigration. Since the composite behaved more like ECs than PCs, these results suggest that EC signals dominate in regulating leukocyte adhesion and diapedesis in the composite model.
Evaluation of specific molecular interactions using the composite EC/PC bilayer model
Having shown that components of the composite model influence behavior of other components, we speculated that our composite model may be useful for the discovery of novel interactions not observed with EC monolayers. We conducted several proof-of-concept studies using conditions known to affect neutrophil transmigration across EC monolayers, to demonstrate that the composite model can reveal functional differences.
Functional blocking antibodies
Our composite system is capable of supporting functional antibody-blocking experiments directed against adhesion molecules on the neutrophil or on the vascular cells. Functional blocking of neutrophil adhesion was tested with antibodies that react with CD99, CD18 (β2-integrin subunit), and CD49d (α4-integrin subunit), on neutrophils before adding them to TNF-α-activated EC and PC monolayers or TNF-α-activated EC/PC bilayers. Adhesion counts were standardized to the control. Functional blocking of the integrin αvβ3, an integrin responsible for adhesion to vitronectin, was used as a negative control for neutrophil adhesion to cellular substrates, as ECs do not provide an αvβ3 ligand for neutrophils to bind. Neutrophil adhesion to the ECs (Fig. 6A) appeared to be reduced after treatment with anti-CD99, anti-CD18, and anti-CD49d, but this change in adhesion reached statistical significance only for anti-CD18 (P<0.05). As expected, neutrophil adhesion to the ECs was not altered with anti-αvβ3, which served as a negative control. Functional blocking of neutrophil binding to PC monolayers was not achieved with any of the antibodies tested, although there was a slight decrease in neutrophil adhesion when CD49d binding was inhibited (Fig. 6B). Given the previous imaging results that demonstrate that, in general, PCs express surface adhesion molecules at a lower level than do ECs, the reduced effect of surface adhesion molecule inhibition is not surprising. Adhesion to the EC/PC bilayer with functional antibodies results in a unique set of results that did not follow the patterns of ECs alone, highlighting the importance of investigating inflammatory mechanisms with a bilayer model rather than a monolayer model (Fig. 6C). Whereas CD99 inhibition reduced adhesion to the EC monolayer, adhesion to the EC/PC bilayer composite after functional blocking of neutrophil-CD99 resulted in no change in neutrophil adhesion. Anti-CD18 antibody once again reduced neutrophil adhesion to the ECs to a significantly lower level (P<0.05) compared with the control, but unlike the EC monolayer alone, neutrophil adhesion after CD49d inhibition was also significantly reduced (P<0.05). As a negative control, inhibiting neutrophil-expressed αvβ3 resulted in no change in neutrophil adhesion to the EC/PC bilayer.
Figure 6.

Transmigration under antibody inhibition and flow conditions. A–C) Neutrophil adhesion to EC monolayers (A), PC monolayers (B), and EC/PC bilayers (C) was significantly blocked with functional antibodies (significant differences determined by a series of unpaired, 2-tailed t tests relative to control). All adhesions were completed with cells cultured on transwell (TW) membranes. Neutrophils were either not functionally inhibited (control) or were incubated with anti-CD99, anti-CD18, anti-CD49d, and anti-αvβ3. Neutrophil adhesion to PCs did not significantly decrease with functional blocking, although there were small changes with the anti-CD49d antibody. Neutrophil adhesion to the EC/PC bilayer was significantly reduced when the neutrophils were functionally inhibited with anti-CD18 and anti-CD49d. *P < 0.05, **P < 0.01; 1-way ANOVA and Tukey post hoc test, as well as an unpaired t test. D) CD99 inhibition studies demonstrated that transmigration was significantly reduced through the EC monolayer and the EC/PC bilayer. *P < 0.05, ***P < 0.001. E) TW membranes seeded with EC and EC/PC bilayers (shown here) were removed from the plastic casing and placed on top of the catchment chamber with a Glycotech circular flow device, to enable flow across the membrane. F) Comparison of neutrophil transmigration rates under static and flow conditions after TNF-α stimulation for 4 h. P values indicate significance when conditions were compared to the static condition for the same cell type based on 1-way ANOVA and Tukey post hoc test. G) PKH26-labeled neutrophils in static and flow transmigration conditions were localized via fluorescence microscopy and were determined to be attached to the EC monolayer, between the EC and PC layers, or to the PC monolayer. Counts were standardized to the number of cells present on the EC layer. Statistics based on 1-way ANOVA and Tukey post hoc test, as well as the paired t test.
Functional inhibition of neutrophil transmigration through the monolayers and bilayers was also assessed by using the anti-CD99 blocking antibody, blocking the CD99 presented on the vascular ECs and PCs (Fig. 6D). As anti-CD99 antibody can inhibit neutrophils as well as vascular cells, vascular cellular constructs were washed vigorously with PBS before the addition of neutrophils, to remove soluble unbound antibody. Blocking CD99 binding on the EC monolayers and EC/PC bilayers resulted in statistically significant reductions in neutrophil transmigration. Although there was a decrease in transmigration through the PC monolayer alone, the change was not significant. Transmigration through the EC monolayer was significantly reduced (P<0.05) relative to the control, but an even larger reduction was seen through the EC/PC bilayer (P<0.001). Whereas the EC and PC monolayers were reduced to roughly one-third of their original value, transmigration through the blocked bilayer was only 14.5% of the original value. The EC/PC bilayer model could therefore identify potential anti-inflammatory molecules that may otherwise be considered ineffective when tested on the EC monolayer alone.
Flow-induced changes in neutrophil transmigration
Blood flow-induced shear stresses, which can be sensed by adherent neutrophils or an EC monolayer, are yet another aspect of vascular neutrophil recruitment. In a final series of experiments, we adapted our composite model to impose laminar shear stress. As illustrated in Fig. 6E, we formed composite bilayers in a static transwell system and then placed it into a customized Glycotech parallel flow system with fluid flow rates mimicking those of microvascular flow (shear stress, 2 dyn/cm2). Therefore, neutrophil transmigration across TNF-α-stimulated cellular structures (EC monolayers and EC/PC bilayers) under flow was tested. As shown in Fig. 5B, the location of transmigrating neutrophils (PKH-26 labeled) within the composite model after 1 h under static and flow conditions was comparable. In both cases, most of the cells remained associated with the EC monolayer, with ∼18% of the cells associated with the PC layer, and an even smaller fraction were located between the 2 layers (BM; Fig. 6F). However, as shown in Fig. 6C, the application of shear more profoundly reduced net transmigration on the EC monolayers (to 17.3% of static) than on the EC/PC composites (to 31.7% of static), producing a statistically significant difference between the 2 systems. Thus, even though PCs are not exposed to shear stresses, their effects on ECs may alter the manner in which the latter are affected by shear stress.
DISCUSSION
It has been demonstrated in vivo (16), and confirmed in this study in vitro, that ECs and PCs are not equally permissive of neutrophil extravasation. Moreover, the postcapillary venule microenvironment is defined by a complex and dynamic milieu of intercellular contacts, surface molecule expression, cytokine release, and protein secretion. It is the cumulative intramural signaling that ultimately determines the success of the neutrophils in exiting the vasculature during the inflammatory response. We present a novel, human cell–based model developed to gain additional insight into comprehensive microvascular regulation of human inflammation via a series of modular assays that would not be possible in previous EC-monolayer models or in animal models. This method will ultimately identify complex and interdependent biological signaling mechanisms and molecular interactions while also identifying new targets for therapeutic intervention in inappropriate inflammation and inflammatory pathologies.
Our model is an advance over previous systems based on EC monolayers, in that it combines human umbilical vein ECs with human placental PCs. We chose to replicate the high PC-to-EC (1:1) ratio found in the eye, brain, and skin. We confirm the formation of an interconnected EC/PC bilayer model composed of a confluent EC monolayer connected to a dense PC network. This composite layer retains its physiological synthesis functions to secrete several of the proteins found in the human BM and release IL-8, a potent mediator of inflammation. Unlike many synthetic biomimetic surfaces, the inclusion of cell-generated proteins and chemical signals creates an in vitro environment more capable of capturing the in vivo complexities, such as integrin-binding sites, that are lost when simple peptide chains are used in place of whole proteins (30).
Of significance, the elements in our composite model system act cooperatively to respond to inflammatory signals. Dynamic changes in the presentation of leukocyte adhesion molecules (LAMs) such as ICAM-1, VCAM-1, CD99, and E-selectin on vascular mural cells in vitro have been confirmed after activation with TNF-α. Confocal imaging revealed that the ECs and PCs responded to activation differently with regard to surface adhesion molecule expression. This disparity was reflected in our functional studies of neutrophil adhesion and transmigration. The ECs supported neutrophil adhesion to and transmigration through a cellular monolayer to a much greater extent than did the PCs. The ability of the ECs to support leukocyte interactions was significantly attenuated after functional inhibition of neutrophil CD18, a change that was not present with the PC monolayers. Acting as a cooperative unit, however, adhesion to and transmigration through the EC/PC bilayer was not diminished despite the PCs, and under TNF-α-activated conditions, neutrophil adhesion to and transmigration through the EC/PC composite more closely mirrored that seen with an EC monolayer than with a PC monolayer. In light of the lesser permeability of the TNF-α-treated EC/PC bilayers compared to the EC or PC monolayers, the high levels of neutrophil transmigration through the EC/PC bilayer is consistent with an active and cooperative vascular response in the composite model that is lacking in monolayer models. Functional blocking of neutrophil adhesion to EC/PC bilayer also confirms that neutrophils do in fact interact uniquely with the bilayer system compared with the EC monolayer alone. In addition to reduced adhesion after neutrophil CD18 blocking, functional blocking of neutrophil CD49d resulted in a statistically significant decrease in adhesion to the EC/PC bilayer that was not present with the EC monolayer. Transmigration studies revealed similar disparities and potentially identified a role for CD99 in post-EC transmigration. Functional inhibition of CD99 on ECs and PCs resulted in diminished transmigration across all conditions, but was most dramatic across the EC/PC bilayer.
The modular nature of our model permitted therapeutic target-, protein-, paracrine-, and shear-specific influences to be parsed from a more complex system in a manner that would not be possible in an animal model. This capability is particularly beneficial in the realm of assay development for biomedical research, as the postcapillary–venular microenvironment changes significantly as inflammatory disease progresses. For example, changes in the BM associated with fibrosis or changes in PC density with diabetic retinopathy can be captured in this model, thereby providing more insight into disease treatment with an in vitro model. As demonstrated, each of the aforementioned intramural controls can alter the vascular response to inflammation. Although the ECs were more capable of supporting neutrophil adhesion to and transmigration through their cellular monolayer when compared to the PCs and their monolayers, both cells were susceptible to functional inhibition with blocking antibodies. Blocking transmigration with a CD99 antibody results in significant decreases in neutrophil transmigration through the EC monolayer and the EC/PC bilayer. The decrease and the degree of significance are larger in the EC/PC bilayer compared with the EC monolayer, which implies that CD99 may also be involved in post-EC extravasation and that the bilayer is most likely capable of identifying previously overlooked therapeutic agents that are not potent enough in an exclusively EC experimental system. Similarly, inflammatory response changed with the protein and soluble paracrine contributions of the vessel as a complete system. PC-deposited protein reduced neutrophil adhesion and transmigration, possibly identifying a direct protein–neutrophil interaction as well as an indirect protein–EC mechanism. IL-8 production was also regulated uniquely by the EC/PC monolayer and did not simply reflect an additive effect of the EC and PC monolayers. The inclusion of high EC production of IL-8 increased the PCs' ability to support neutrophil transmigration, most likely a critical factor in increasing the vascular permeability during inflammation. Last, it is possible to translate this static system to a dynamic one through alterations to previously used flow devices. Modifying the Glycotech flow chamber to contain a catchment chamber enabled the role of shear in transmigration to be experimentally controlled as well, thereby yielding control over 4 unique parameters in one experimental model. Unlike many previous flow chambers that can only examine neutrophil adhesion under flow, the porated membrane in our flow model easily allowed for neutrophil transmigration, such that neutrophils that transmigrated under flow could be collected, counted, and/or used for additional studies. The addition of flow to more fully replicate the human venule environment facilitates further investigation into the ability of the vascular wall to respond to physiological stress and modulate leukocyte recruitment; for example, the larger reduction in neutrophil transmigration across the EC monolayer (to 17.3% of static) was diminished in the EC/PC composite (to 36.7% of static). Ultimately, neutrophil transmigration was less dramatically influenced by fluid shear stress when neutrophils attempted to cross an EC/PC bilayer, than when crossing the EC monolayer alone. This suggests that the composite structure provides cell–cell or soluble signals that enhance the binding and migratory abilities of neutrophils to the luminal ECs of the postcapillary venule.
In summary, we have developed a new, more comprehensive model of the human postcapillary venule, and our initial studies produced several new findings. 1) PCs respond to TNF-α to support increased neutrophil adhesion and transmigration, but the levels of both activities are far lower than those observed with TNF-α-activated ECs. 2) The EC/PC bilayer composite supports neutrophil adhesion and transmigration at levels comparable to those observed with EC monolayers, suggesting that PCs do not constitute a barrier to neutrophil capture and egress when they interact with ECs. 3) EC-CM significantly increases neutrophil adhesion to TNF-α-activated PCs. 4) ECs are less supportive of TNF-α-induced increases of neutrophil binding and transmigration when grown on PC-derived matrix. 5) Blocking neutrophil CD18 reduces neutrophil binding to both EC monolayers and EC/PC bilayers, but blocking CD49d integrin produces a significant effect only on neutrophil adhesion to the composite model. 6) Blocking CD99 inhibits trans-PC and trans-EC/PC migration of neutrophils in addition to its previously known effect in trans-EC migration. 7) Application of venular shear stress reduces the level of neutrophil transmigration across EC monolayers more profoundly than across EC/PC bilayers.
These experiments, using only 1 inflammatory cytokine at 1 early time point, constitute proof of concept that our composite model can be used to reveal phenomena not observed with conventional EC monolayers. These studies represent only a small fraction of the research capacity of this system for understanding the interplay between vascular biology and immunology.
Acknowledgments
The authors thank Dr. Kathryn Miller-Jensen for allowing them to use the LI-COR Scanner, Dr. Thomas Manes for help with immunofluorescence, and Dr. Cheryl Maier for discussions and review of the manuscript.
Facilities used were supported by the Yale Institute for Nanoscience and Quantum Engineering and National Science Foundation (NSF), Materials Research Science and Engineering Center (MRSEC), Division of Materials Research (DMR) 1119826. A.L.G. and J.S.P. were supported by U.S. National Institutes of Health grant HL051014, with additional support for A.L.G. coming from the Dubinsky New Initiative grant and The Hartwell Foundation.
Footnotes
- BM
- basement membrane
- CM
- conditioned medium
- EC
- endothelial cell
- EC-CM
- endothelial cell–conditioned medium
- ICAM-1
- intercellular adhesion molecule 1
- IL-8
- interleukin 8
- Mac-1
- macrophage 1
- Na-F
- sodium fluorescein
- N-cadherin
- neural cadherin
- PC
- pericyte
- PC-CM
- pericyte-conditioned medium
- PECAM-1
- platelet endothelial cell adhesion molecule 1
- SEM
- scanning electron midcroscopy
- TNF-α
- tumor necrosis factor-α
- VCAM-1
- vascular cell adhesion molecule 1
- VE-cadherin
- vascular endothelial cadherin
- VLA-4
- very late antigen 4
REFERENCES
- 1. Armulik A., Genové G., Betsholtz C. (2011) Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2, 193–215 [DOI] [PubMed] [Google Scholar]
- 2. Armulik A., Abramsson A., Betsholtz C. (2005) Endothelial/pericyte interactions. Circ. Res. 97, 512–523 [DOI] [PubMed] [Google Scholar]
- 3. Hirschi K., D'Amore P. (1996) Pericytes in the microvasculature. Cardiovasc. Res. 32, 687–698 [PubMed] [Google Scholar]
- 4. Brey E., McIntire L., Johnston C., Reece G., Patrick J. (2004) Three-dimensional, quantitative analysis of desmin and smooth muscle alpha actin expression during angiogenesis. Ann. Biomed. Eng. 32, 1100–1107 [DOI] [PubMed] [Google Scholar]
- 5. Gentil-dit-Maurin A., Oun S., Almagro S., Bouillot S., Courcon M., Linnepe R., Vestweber D., Huber P., Tillet E. (2010) Unraveling the distinct distributions of VE- and N-cadherins in endothelial cells: a key role for p120-catenin. Exp. Cell Res. 316, 2587–2599 [DOI] [PubMed] [Google Scholar]
- 6. Oas R., Nanes B., Esimai C., Vincent P., Garcia A., Kowalczyk A. (2013) p120 catenin and b-catenin differentially regulate cadherin adhesive function. Mol. Biol. Cell 24, 704–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu W., Chen C. (2005) Engineering biomaterials to control cell function. Mater. Today 8, 28–35 [Google Scholar]
- 8. Francis M., Uriel S., Brey E. (2008) Endothelial cell-matrix interactions in neovascularization. Tissue Eng. B Rev. 14, 19–32 [DOI] [PubMed] [Google Scholar]
- 9. Muller W. (2002) Leukocyte-endothelial cell interactions in the inflammatory response. Lab. Invest. 82, 521–533 [DOI] [PubMed] [Google Scholar]
- 10. Ley K., Laudanna C., Cybulsky M., Nourshargh S. (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689 [DOI] [PubMed] [Google Scholar]
- 11. Burns A., Walker D., Brown E., Thurmon L., Bowden R., Keese C., Simon S., Entman M., Smith W. (1997) Neutrophil transendothelial migration is independent of tight junctions and occurs preferentially at tricellular corners. J. Immunol. 159, 2893–2903 [PubMed] [Google Scholar]
- 12. Carman C., Springer T. (2004) A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J. Cell Biol. 167, 377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ding Z., Babensee J., Simon S., Lu H., Perrard J., Bullard D., Dai X., Bromley S., Dustin M., Entman M., Smith C., Ballantyne C. (1999) Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J. Immunol. 163, 5029–5038 [PubMed] [Google Scholar]
- 14. Voisin M.-B., Pröbstl D., Nourshargh S.: Venular basement membranes ubiquitously express matrix protein low-expression regions: characterization in multiple tissues and remodeling during inflammation. Am. J. Pathol. 176, 482–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muller W. (2009) Mechanisms of transendothelial migration of leukocytes. Circ. Res. 105, 223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Proebstl D., Voisin M.-B., Woodfin A., Whiteford J., D'Acquisto F., Jones G., Rowe D., Nourshargh S. (2012) Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo1. J. Exp. Med. 209, 1219–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ayres-Sander C., Lauridsen H., Maier C., Sava P., Pober J., Gonzalez A. (2013) Transendothelial migration enables subsequent transmigration of neutrophils through underlying pericytes. PLoS ONE 8, e60025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maier C., Shepherd B., Yi T., Pober J. (2010) Explant, outgrowth, propagation and characterization of human pericytes. Microcirculation 17, 367–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gimbrone M., Jr., Cotran R., Folkman J. (1974) Human vascular endothelial cells in culture. Growth and DNA synthesis. J. Cell. Biol. 60, 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lou O., Alcaide P., Luscinskas F. W., Muller W. A. (2007) CD99 is a key mediator of the transendothelial migration of neutrophils. J. Immunol. 178, 1136–1143 [DOI] [PubMed] [Google Scholar]
- 21. Nakagawa S., Deli M., Kawaguchi H., Shimizudani T., Shimino T., Kittel A., Tanaka K., Niwa M. (2009) A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem. Int. 54, 253–263 [DOI] [PubMed] [Google Scholar]
- 22. King J., Hamil T., Creighton J., Wu S., Bhat P., McDonald F., Stevens T. (2004) Structural and functional characteristics of lung macro- and microvascular endothelial cell phenotypes. Microvasc. Res. 67, 139–151 [DOI] [PubMed] [Google Scholar]
- 23. Nelson C., Pirone D., Tan J., Chen C. (2004) Vascular endothelial-cadherin regulates cytoskeletal tension, cell spreading, and focal adhesions by stimulating RhoA. Mol. Biol. Cell 15, 2943–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Merimee T., Sipertsein M., Hall J., Fineberg S. (1970) Capillary basement membrane structure: a comparative study of diabetics and sexual ateliotic dwarfs. J. Clin. Invest. 49, 2161–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baggiolini M., Clark-Lewis I. (1992) Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 307, 97–101 [DOI] [PubMed] [Google Scholar]
- 26. Nwariaku F., Chang J., Zhu X., Liu Z., Duffy S., Halaihel N., Terada L., Turnage R. (2002) The role of p38 map kinase in tumor necrosis factor-induced redistribution of vascular endothelial cadherin and increased endothelial permeability. Shock 18, 82–85 [DOI] [PubMed] [Google Scholar]
- 27. Hermant B., Bibert S., Concord E., Dublet B., Weidenhaupt M., Vernet T., Gulino-Debrac D. (2003) Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J. Biol. Chem. 278, 14002–14012 [DOI] [PubMed] [Google Scholar]
- 28. Marin V., Kaplanski G., Gres S., Farnarier C., Bongrand P. (2001) Endothelial cell culture: protocol to obtain and cultivate human umbilical endothelial cells. J. Immunol. Methods 254, 183–190 [DOI] [PubMed] [Google Scholar]
- 29. Bixel M., Petri B., Khandoga A., Khandoga A., Zarbock A., Wolburg-Buchholz K., Wolburg H., Sorokin L., Zeuschner D., Maerz S., S B., Kromback F., Vestweber D. (2010) CD99 and CD99L2 act at the same site as, but independently of, PECAM-1 during leukocyte diapedesis. Blood 116, 1172–1184 [DOI] [PubMed] [Google Scholar]
- 30. Petrie T., Capadona J., Reyes C., AJ G. (2006) Integrin specificity and enhanced cellular activities associated with surfaces presenting a recombinant fibronectin fragment compared to RGD supports. Biomaterials 27, 5459–5470 [DOI] [PubMed] [Google Scholar]