

Abstract
The estrogen-related receptor-α (ERRα) regulates mitochondrial biogenesis and glucose and fatty acid oxidation during differentiation in skeletal myocytes. However, whether ERRα controls metabolic remodeling during skeletal muscle regeneration in vivo is unknown. We characterized the time course of skeletal muscle regeneration in wild-type (M-ERRαWT) and muscle-specific ERRα−/− (M-ERRα−/−) mice after injury by intramuscular cardiotoxin injection. M-ERRα−/− mice exhibited impaired regeneration characterized by smaller myofibers with increased centrally localized nuclei and reduced mitochondrial density and cytochrome oxidase and citrate synthase activities relative to M-ERRαWT. Transcript levels of mitochondrial transcription factor A, nuclear respiratory factor-2a, and peroxisome proliferator-activated receptor (PPAR)-γ coactivator (PGC)-1β, were downregulated in the M-ERRα−/− muscles at the onset of myogenesis. Furthermore, coincident with delayed myofiber recovery, we observed reduced muscle ATP content (−45% vs. M-ERRαWT) and enhanced AMP-activated protein kinase (AMPK) activation in M-ERRα−/− muscle. We subsequently demonstrated that pharmacologic postinjury AMPK activation was sufficient to delay muscle regeneration in WT mice. AMPK activation induced ERRα transcript expression in M-ERRαWT muscle and in C2C12 myotubes through induction of the Esrra promoter, indicating that ERRα may control gene regulation downstream of the AMPK pathway. Collectively, these results suggest that ERRα deficiency during muscle regeneration impairs recovery of mitochondrial energetic capacity and perturbs AMPK activity, resulting in delayed myofiber repair.—LaBarge, S., McDonald, M., Smith-Powell, L., Auwerx, J., Huss, J. M. Estrogen-related receptor-α (ERRα) deficiency in skeletal muscle impairs regeneration in response to injury.
Keywords: nuclear receptors, mitochondria, myogenic differentiation, gene expression, AMP-activated protein kinase
Adult skeletal muscle possesses the capacity to repair itself in response to mechanical damage resulting from normal use and direct injury. The regeneration of skeletal muscle is an essential homeostatic response to continuously varying contractile demands and muscle turnover that preserves muscle mass and function. Although skeletal muscle is primarily specialized for locomotion, it also controls whole-body metabolic homeostasis. Loss of muscle mass and contractile function reduces mobility and disrupts energy balance, thereby increasing susceptibility to obesity and insulin resistance. Muscle regenerative capacity declines with age and is severely impaired in many disease contexts, including the muscular dystrophies and type 2 diabetes (1, 2). A key process that enables skeletal muscle regeneration is the differentiation of myoblasts, derived from satellite cells that reside in the muscle basal lamina. Impaired regeneration often involves satellite cell depletion or defects in myoblast differentiation. Characterization of molecular programs involved in controlling skeletal muscle regeneration is critical to understanding how processes are dysregulated in disease.
Muscle regeneration proceeds through defined interrelated phases (3, 4). Damaged muscle fibers undergo necrosis and release myokines to initiate the process. In response to inflammatory signals, macrophages infiltrate the injury site, clear tissue debris, and activate resident satellite cells to proliferate and differentiate into myoblasts. The expanded myoblast population can then differentiate and fuse with existing myofibers or form new myofibers. Last, nascent myofibers restore fiber-type-specific contractile and metabolic programs in response to neuromuscular inputs. Use of a well-characterized model of injury-regeneration enables examination of novel signaling and transcriptional networks integral to its regulation.
Mitochondria biogenesis is essential for normal skeletal muscle regeneration. The gene programs regulating mitochondrial metabolism and specialized contractile function are temporally coordinated during myoblast differentiation. Mitochondrial enzyme expression and activity rapidly rise at the beginning of differentiation, accompanied by a shift from glycolytic to oxidative ATP synthesis (5). Treatments that prevent normal mitochondrial DNA (mtDNA) or protein synthesis block myocyte differentiation (6). Likewise, in skeletal muscle, mitochondrial β-oxidation and tricarboxylic acid (TCA) cycle enzyme activities and maximum respiration capacity decline immediately after injury, but begin to recover in parallel with the onset of myogenesis, and return to baseline before complete regeneration (7, 8). Inhibition of mitochondria protein synthesis with chloramphenicol after skeletal muscle damage prevents regeneration (9). These studies demonstrate the importance of restoring energy-generating capacity before the recovery of contractile function. Control of mitochondrial biogenesis involves coordinating interdependent processes, including transcription of mitochondrial and nuclear-encoded genes and mtDNA replication (10). Transcript profiling indicates that mitochondrial biogenesis–related factors, such as nuclear respiratory factor (NRF)-1 and NRF-2, mitochondrial transcription factor A (TFAM), mitofusins (Mfns), and fission genes are up-regulated with myogenic regulatory factors early in regeneration (9). However, the upstream signaling and muscle-specific role of transcriptional pathways that regulate metabolic gene networks are not well understood.
The nuclear receptor estrogen-related receptor α (ERRα) is an important transcription factor involved in metabolic regulation (11–15). ERRα complexed with a peroxisome proliferator-activated receptor (PPAR)-γ coactivator 1 (PGC-1) coactivator, such as PGC-1α or PGC-1β, activates transcription of target genes through direct binding to its consensus response element (16, 17). The role of ERRα in regulating metabolic gene programs has been examined in highly oxidative tissues, such as brown adipose tissue (BAT), heart, and skeletal muscle, that show elevated ERRα expression levels, using a whole-body ERRα−/− model (18). ERRα−/− mice exhibit decreased adaptive thermogenesis from reduced mitochondrial levels within BAT (19). ERRα−/− mice have smaller hearts with decreased expression of mitochondrial energetic gene programs, resulting in reduced energetic reserve capacity and accelerated failure in response to increased workload (20). ERRα has been found at the promoter regions of genes involved in fuel sensing, substrate uptake, metabolism, contractile work, and other metabolic transcription factors in cardiac skeletal muscle (12, 21). Collectively, these studies highlight the potential involvement of ERRα in transducing metabolic signals to regulate energetic gene programs.
Few studies have been performed to elucidate the biological processes controlled by ERRα in skeletal muscle. In C2C12 myocytes, we and others showed that endogenous ERRα expression increases during differentiation in parallel with the PGC-1 coactivators, suggesting that ERRα activates metabolic, sarcomeric, or other gene programs involved in specialized myocyte function during myogenesis (22–24). Indeed, ERRα overexpression in C2C12 myocytes promotes differentiation concurrent with increased mitochondrial content and up-regulation of mitochondrial enzyme genes and sarcomeric protein expression, whereas ERRα inhibition impairs differentiation and expression of these genes (22). Likewise, differentiated myotubes, derived from ERRα−/− mouse primary myoblasts, are smaller and have reduced mitochondrial enzyme expression and lower glucose and fatty acid oxidative capacity in later stages of differentiation. Although these findings support a potential role for ERRα in regulating skeletal muscle growth and regeneration, their physiological relevance has not been explored in vivo. The global ERRα−/− mice have altered lipid metabolism and antioxidant enzyme activity in tissues that influence skeletal muscle metabolism and growth, including white adipose tissue and macrophages (18, 25, 26). Thus, in the current study we explored the effect of skeletal muscle–specific ERRα deletion on myofiber formation and metabolic recovery during regeneration.
On the basis of previous studies that show that ERRα promotes differentiation and regulates metabolic gene programs and mitochondrial biogenesis in myocytes, we hypothesized that ERRα plays a role in skeletal muscle regeneration (22–24). To investigate how ERRα regulates myogenesis in vivo, we generated mice harboring muscle-specific ERRα deletion (M-ERRα−/−) and characterized skeletal muscle regeneration in response to cardiotoxin injury.
MATERIALS AND METHODS
Animals
Animal experiments were performed in accordance with the City of Hope Institutional Animal Care and Use Committee. Mice were housed in humidity-controlled rooms with a 12-h light-dark cycle and given standard chow and water ad libitum. Mice carrying the Esrra allele with loxP sites flanking exons 3 and 4 were obtained from the Institute Clinique de la Souris (ICS)/Mouse Clinical Institute (MCI; Strasbourg, France). Skeletal muscle–specific disruption of the Esrra locus was achieved by breeding with muscle creatine kinase (MCK)-Cre mice that express Cre recombinase driven by the Mck promoter (ref. 27; The Jackson Laboratory, Bar Harbor, ME, USA).
Muscle regeneration
Muscle damage was induced in the tibialis anterior (TA) by intramuscular injection of cardiotoxin (CTX; Latoxan, Valence, France). The lower hindlimbs of isoflurane-anesthetized animals were shaved, and the TA was injected with 20 μl of 40 μM CTX (left limb) or PBS (right limb) 0.5 cm below the knee. Twenty 14- to 18-wk-old male mice of each genotype [muscle-specific ERRα wild type (M-ERRαWT) or M-ERRα−/−] and at each time point (d 1, 3, 7, and 15) were used for histologic and metabolic analyses (160 mice). In the AMP-activated protein kinase (AMPK) activation trials after CTX injury, wild-type (WT) mice (n=6/time point) were administered (i.p.) 0.9% saline or 500 mg/kg 5-aminoimidazole-4-carboxamide-1-β-d-ribosyl monophosphate (AICAR; Toronto Research Chemicals, Toronto, ON, Canada) once daily on d 3 or 3–6, and histology was assessed on post-CTX d 7 and 15. For tissue collection, animals were anesthetized with ketamine and xylazine and euthanized by cervical dislocation. Dissected TA muscles were either frozen in isopentane for histology or freeze clamped in N2(l) for biochemical and molecular analyses.
Cell culture and transfection assays
C2C12 myocytes were maintained in culture and differentiated into myotubes as described previously (22). The −800.ERRa.Luc and −600.ERRa.Luc reporters were cloned into pGL3.Basic (Promega, Madison, WI, USA) from the 0.8-CAT (−811/+12) and 0.6-CAT (−593/+12) plasmids, provided by Christina Teng (National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA; ref. 28). The −1.27.mtCK2.pGL3 reporter was cloned from human sMtCK-CAT provided by Arnold Strauss (Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA). We obtained pcDNA3.1–dominant-negative AMPK (AMPK-DN) from David Carling (Imperial College London, London, UK). Calcium phosphate was used in transient transfections in subconfluent C2C12 myoblasts 16 h after plating (14). Luciferase activity was assayed, either 24 h later in myoblasts or after changing to differentiation medium for the indicated period in myotubes. For AMPK activation, d 2 myotubes were treated with 0.3 mM AICAR or 40 μM compound C (Calbiochem, EMD Millipore, Billerica, MA, USA) and assayed 24 h later. Luciferase activity was analyzed with the Dual-Glo Luciferase Assay System (Promega). Firefly luciferase was normalized to Renilla luciferase driven by the pRL.CMV vector and is reported relative to the pGL3.Basic vector. At least 4 independent trials plated in triplicate were performed for each experiment.
Histology
TA muscles were sectioned on a CM3050 S cryostat (Leica, Bannockburn, IL, USA) at −20°C. Histochemistry was conducted on fresh 10-μm sections taken from the middle of the damaged area by standardized methods (29). Hematoxylin and eosin (H&E) staining was performed on 10% formalin-fixed muscle sections with Mayer's H&E Y (Sigma-Aldrich, St. Louis, MO, USA). Cytochrome c oxidase (COX) activity was detected by incubation with 0.1% cytochrome c, 0.02% catalase, and 0.1% 3,3′diaminobenzidine (SigmaFast; Sigma-Aldrich) in 0.2 M phosphate buffer for 2 h at 37°C. The sections were washed with deionized water, dehydrated in a graded ethanol series and xylene exchange, and mounted with Permount (ThermoScientific, Rockford, IL, USA). Succinate dehydrogenase (SDH) activity was detected by incubating the sections with 166 mM succinate and 1.2 mM nitrotetrazolium blue (Sigma-Aldrich) in 0.2 M phosphate buffer for 1 h at 37°C. The sections were washed in a graded acetone series, rehydrated, and mounted in glycerol gel.
For immunohistochemistry (IHC), muscle sections were fixed in −20°C acetone or 10% formalin, permeabilized, and blocked with 0.15% Triton X-100 and 2% normal serum, followed by overnight incubation at 4°C with primary antibody. The primary antibody was detected by using biotinylated goat secondary antibody specific to the host species with either ABC detection with ImmPACT NovaRED (Vector Laboratories, Burlingame, CA, USA) or streptavidin-conjugated AlexFluor488 (Invitrogen- Life Technologies, Carlsbad, CA, USA). ERRα (ab16363) and PGC-1α (ab54481) were purchased from Abcam (Cambridge, MA, USA). The CD11b (M1/70.15.11.5.2) and Pax7 (PAX7) antibodies were obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA, USA). Digital images were taken with an AX70 microscope (Olympus, Lake Success, NY, USA). Morphometric analysis was performed with ImagePro Plus (Media Cybernetics, Bethesda, MD, USA) with fiber cross-sectional area (CSA) and central nuclei (CN) measured in 500–1000 fibers/animal from multiple animals.
Electron microscopy
Mice were anesthetized and muscles fixed in situ by cardiac perfusion with PBS containing heparin followed by 2.5% glutaraldehyde/2.5% paraformaldehyde in PBS. Dissected muscles were cut into 1–2 mm3 pieces containing the injected area and were postfixed in osmium tetroxide, embedded in Epon 812, sectioned, and stained by using standard methods (30). Tissue sections were visualized with a Twin Tecnai 120 KV transmission electron microscope (TEM; FEI, Hillsboro, OR, USA).
Muscle biochemical analyses
Citrate synthase (CS) activity was measured by coupling conversion of oxaloacetate to acetyl-CoA with reduction of 5,5′-dithiosbis(2-nitrobenzoic acid) (DTNB; Sigma-Aldrich; ref 31). Frozen muscle (∼5 mg) was disrupted in 250 μl cold 50 mM Tris (pH 7.0) and 150 mM KCl by Dounce homogenization, and soluble protein concentration was determined by BCA Protein Assay (ThermoScientific). Protein (20 μg) was added to 200 μl reaction mix (0.1 mM DTNB, 0.25% Triton X-100, 0.5 mM oxaloacetate, and 0.25 mg/ml acetyl-CoA in 0.1 M Tris, 7.4), and product formation was measured at 412 nm over 300 s on an Infinite M200 PRO plate reader (Tecan, Männedorf, Switzerland). Results were calculated from a constant rate and an extinction coefficient (εB) 13.6 mM−1 · cm−1. For ATP measurements, tissue (1–2 mg) was homogenized in 200 μl cold TCA (0.5%) and neutralized by 30-fold dilution in 20 mM Tris (pH 8.0). Extracts (10 μl) were assayed in triplicate with an Enliten ATP Assay kit with a Turner Biosystems Veritas microplate luminometer (Promega). ATP concentrations were calculated from a standard curve (ATP range, 10−14–10−7 M) and normalized to total protein. Tissue ATP and AMP content was determined in KOH-neutralized TCA (3% in PBS) extracts from CTX-treated TA (post-treatment d 7 and 15) by UV-HPLC, according to published conditions (32) and normalized to protein concentration in extracts.
For AMPK activity assays, homogenates were prepared from muscles (40 mg) powdered under N2(l) in AMPK buffer (210 mM sucrose, 1 mM EDTA, 5 mM sodium pyrophosphate, 50 mM NaF, 1 mM DTT, 2 mM PMSF, and 50 mM HEPES, pH 7.4; ref. 33). Isoform-specific AMPK activity was determined in immunoprecipitates from 200 μg of supernatant protein in a total volume of 0.5 ml of AMPK buffer after overnight incubation at 4°C with 1.0 μg of goat IgG against either AMPKα1 or AMPKα2 in 20 μl of protein A/G-agarose beads (Santa Cruz Biotechnology, Dallas, TX, USA). AMPK activity was determined by measuring the incorporation of [32P]-γ-ATP into the SAMS peptide substrate (33).
Quantitative real-time PCR
RNA was isolated from muscle by Trizol (Invitrogen-Life Technologies). Reverse transcription and real-time PCR reactions (performed in triplicate) and analyses were performed (20). Standard curves were run for each primer set, and results were normalized to GAPDH or 36B4. All results represent the ratio of values in CTX samples to PBS samples (CTX/PBS). The primer sets are as follows: ERRα, forward ccaatgagtgtgagatcacc, reverse ccgtttgtacttctgccgtc; ERRγ, forward agaccagtctaaattagcaggc, reverse cctcatgtaacacatcctgaa; PGC-1α, forward ccctgccattgttaagacc, reverse tgctgctgttcctgttttc; PGC-1β, forward tctgacgtggacgagctttc, reverse cttgctgttggggaggatgt; PRC, forward tggacgcctcccttatatccc, reverse tgtgagcagcgacatttcattc (PrimerBank ID 124487054c1); PDK4, forward agggaggtcgagctgttctc, reverse ggagtgttcactaagcggtca; TFAM, forward aggcttggaaaaatctgtctc, reverse tgctcttcccaagacttcatt; Gabpa, forward ttgggtggtttgggtaatgaag, reverse aggctggtcaatggtaactatct (PrimerBank ID 34328119a2); PAX7, forward ctcagtgagttcgattagccg, reverse agacggttccctttgtcgc (PrimerBank ID 130502943b3); STK11, forward ttgggccttttctccgag, reverse caggtcccccatcaggtact; Erk1, forward caggtgttcgacgtagggc, reverse tctggtgctcaaaaggactga; AMPKα1, forward agagggccgcaataaaagat, reverse tgttgtacaggcagctgagg; AMPKα2, forward tgatcagcactccgacagac, reverse tctctggcttcaggtcccta; AMPKβ1, forward aggcccaagatcctcatgga, reverse gggggctttatcattcgcttc; AMPKβ2, forward accatctctatgcactgtcca, reverse cagcgtggtgacatacttctt; AMPKγ3, forward accagctcagaaagaacctgt, reverse gtggccttcgggaatgtgg; Mfn1, forward tgcaatcttcggccagttact, reverse ctcggatgctattcgatcaagtt (PrimerBank ID 244793487b2); Mfn2, forward ccaactccaagtgtccgctc, reverse gtccagctccgtggtaacatc (PrimerBank ID 120407047b3); Gapdh, forward tgtgtccgtcgtggatctga, reverse ttgctgttgaagtcgcaggag (RTPrimerDB ID 7880); m36B4, forward atccctgacgcaccgccgtga, reverse tgcatctgcttggagcccacgtt.
Immunoblot analysis
Total muscle protein was prepared by sonication in ice-cold TNS buffer (20 mM Tris, 7.5, 50 mM NaCl, 250 mM sucrose, 20 mM NaF, 1 mM DTT, 1 mM sodium pyrophosphate, 0.2 mM sodium orthovanadate, 1 μM PMSF, and 1× SigmaFast), followed by solubilization with Triton X-100 (1%) for 30 min at 4°C. Protein concentration was determined by BCA assay. Protein electrophoresis, membrane transfer, and antibody detection were performed as described elsewhere (34). Commercially available primary antibodies included TFAM (Calbiochem, San Diego, CA, USA); GA-binding protein transcription factor, α subunit (Gabpa; Abcam); Mfn2 (Santa Cruz Biotechnology); and phospho-AMPKα (Thr172), AMPKα, phospho-ACC (Ser79), acyl-CoA carboxylase (ACC), phospho-Erk1/2 (Thr202)/Tyr204), Erk1/2, phospho-LKB1 (Ser428), and LKB1 (Cell Signaling, Danvers, MA, USA). Densitometry analysis was performed with ImageJ v1.44 software (U.S. National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All data are presented as means ± sem. Differences between the means were calculated with 2-way ANOVA with Bonferroni post hoc analysis or by Student's paired or unpaired t test. Statistical significance was set at P ≤ 0.05.
RESULTS
Generation of conditional muscle ERRα-deficient mice
To investigate the role of ERRα in skeletal muscle regeneration, we generated mice with muscle-specific disruption of the Esrra gene (Supplemental Fig. S1). The M-ERRα−/− mice were derived from mice homozygous for the floxed Esrra allele (M-ERRαWT) crossed with MCK-Cre transgenic mice (ref. 27 and Supplemental Fig. S1A). The data demonstrating recombination efficiency and tissue specificity are shown in Supplemental Fig. S1B and those demonstrating selective ablation of ERRα transcript and protein expression in heart and skeletal muscles in Supplemental Fig. S1C, D. Consistent with the phenotype of global ERRα−/− mice (18–20, 25), the M-ERRα−/− mice exhibited no differences in survival, body mass, skeletal muscle growth, or structure compared to the M-ERRαWT mice (Supplemental Fig. S1D).
Impaired regeneration in M-ERRα−/− skeletal muscles
To examine the regeneration phenotype of ERRα-deficient muscles, we used an injury protocol involving intramuscular injection of CTX or PBS into the opposing TA muscles of the M-ERRαWT and M-ERRα−/− mice. Muscle histology was examined at d 1, 3, 7, and 15 after injection (Fig. 1A). Although no difference in muscle damage was evident at d 1 and 3, at postinjury d 7 and 15 the regenerating M-ERRα−/− muscles exhibited reduced myofiber CSA, compared to the M-ERRαWT muscles (Fig. 1B). Furthermore, the CN index (CN n/myofibers), a marker of regeneration, was significantly higher in the injured M-ERRα−/− muscles on d 15 of regeneration (Fig. 1C). Specifically, the M-ERRα−/− TA had a greater proportion of myofibers with multiple CN (34.5±5.3%, M-ERRα−/− vs. 22.1±4.0%, M-ERRαWT), but few fibers with no CN (1.5±0.8%, M-ERRα−/− vs. 9.1±2.4%, M-ERRαWT). This profile indicates that many myofibers in the M-ERRα−/− TA were at an earlier stage, still undergoing myoblast fusion, compared to the M-ERRαWT TA, in which fusion and myofiber formation was complete. Thus, skeletal muscle regeneration is delayed in M-ERRα−/− mice.
Figure 1.
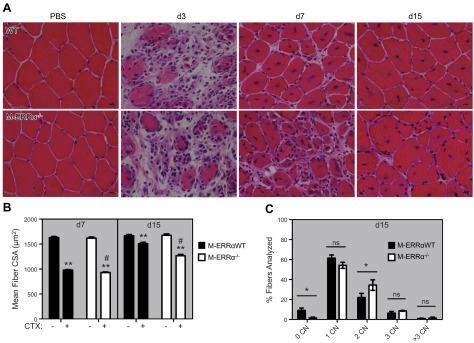
Impaired regeneration in M-ERRα−/− mice. A) H&E staining of regenerating TA from M-ERRαWT (top panels) and M-ERRα−/− (bottom panels) mice. Number of days after CTX injury is indicated at top (×400). Scale bars = 10 μm. B) Mean myofiber CSA of PBS (−)- or CTX (+)-injected TA muscles on postinjury d 7 and 15 in M-ERRαWT (solid bars) and M-ERRα−/− (open bars) mice. Data are expressed as the mean ± sem (n=3–4 mice/time point/genotype) **P ≤ 0.01 vs. PBS-treated muscles. #P ≤ 0.05 vs. M-ERRαWT CTX-treated muscles. C) CN index in regenerating M-ERRαWT (solid bars) and M-ERRα−/− (open bars) TA at d 15 after CTX injection. Data are expressed as the means ± sem (n=3–4 mice/time point/genotype). *P ≤ 0.05.
On the basis of the findings that ERRα is necessary for rapid myofiber regeneration, we wanted to determine the precise onset of ERRα expression after injury (Fig. 2). Previous studies demonstrated that ERRα transcript and protein are upregulated early in differentiation in cultured myocytes (22, 24). In agreement, ERRα transcript peaked 3 d after CTX injury at the onset of myogenesis in vivo (9) and returned to baseline by d 15 in the M-ERRαWT muscles (Fig. 2B). IHC analysis in regenerating muscles revealed that ERRα protein was present in the nucleus and cytosol of the M-ERRαWT TA cells at d 7, possibly because of transient accumulation of ERRα protein in nascent myofibers (Fig. 2A). By d 15, ERRα was detected only in the nucleus, reflecting the localization observed in uninjured TA. As expected, no ERRα protein was detected in the M-ERRα−/− animals, confirming that antibody detection was specific for ERRα and that no genetic reversion of the Esrra allele had occurred in the new myofibers.
Figure 2.
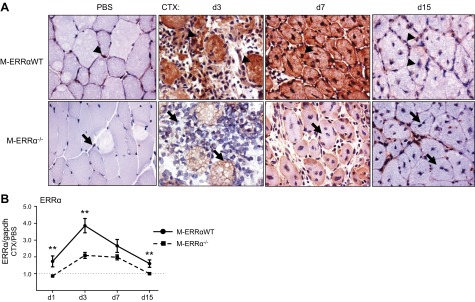
ERRα protein and transcript expression in regenerating M-ERRαWT and M-ERRα−/− TA. A) ERRα protein expression and localization was assessed in PBS-injected control or CTX-injected regenerating TA of M-ERRαWT and M-ERRα−/− mice by immunostaining on the indicated day after CTX injury (×400). B) ERRα transcript was analyzed in M-ERRαWT and M-ERRα−/− TA during regeneration by real-time PCR and corrected to Gapdh (control transcript) expression. Values in CTX-injected TA were normalized to expression in the PBS-injected TA from the same mouse. Data are expressed as the means ± sem (n=4 mice/time point/genotype). **P ≤ 0.01.
After injury, macrophages enter the injury site to remove tissue debris and stimulate satellite cell proliferation. To rule out differences between the M-ERRαWT and M-ERRα−/− mice in these essential early stages, we assessed markers of satellite cells and activated macrophages (Supplemental Fig. S2). We detected no difference between the M-ERRα−/− and WT muscles in the expression of the satellite cell transcription factor Pax7 (3), by IHC or real-time PCR (Supplemental Fig. S2A, B). Analysis of the macrophage marker CD11b by IHC revealed identical peak stain intensity at d 3 after CTX injury and return to baseline by d 7 in the M-ERRαWT and the M-ERRα−/− muscles (Supplemental Fig. S2C). These results suggest that inflammatory and satellite cell activation phases of the injury-regeneration program were unaltered in ERRα-deficient muscles; therefore, differences between WT and M-ERRα−/− myofiber recovery most likely arose during myogenesis.
Impaired recovery of mitochondrial activity in M-ERRα−/− muscles during regeneration
Mitochondrial biogenesis is an essential program for myocyte differentiation that may be altered in ERRα-deficient muscles (7, 9). Since regeneration was delayed in the M-ERRα−/− mice, we examined the time course of recovery of mitochondrial biogenesis and enzyme activity. At baseline, the M-ERRαWT muscles had modestly higher COX activity than did M-ERRα−/− muscles (Fig. 3A). In response to CTX injury, COX activity declined during postinjury d 1 and 3 and began to recover similarly by d 7 in both groups, concomitant with early myofiber regeneration (Fig. 3A). In contrast, the ERRα-deficient muscles had significantly lower COX activity than did the M-ERRαWT muscles in the d 15 myofibers. We observed the same differential time course in d 15 recovery using SDH as a marker of mitochondrial activity (Supplemental Fig. S3). To quantitatively assess mitochondrial recovery, we measured the CS activity in muscle extracts (Fig. 3B) and found that it declined to its lowest point at d 3 and began to recover by d 7 similarly in the regenerating WT and M-ERRα−/− muscles. In agreement with histochemical data, CS activity at d 15 was significantly lower in the M-ERRα−/− than in the M-ERRαWT TA.
Figure 3.
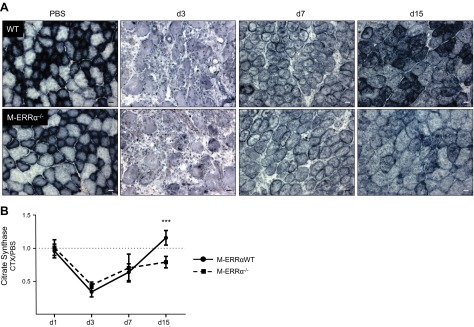
Mitochondrial activity was reduced in regenerated myofibers of M-ERRα−/− muscle. A) COX activity in PBS-injected control (left panels) or CTX-injected regenerating TA of M-ERRαWT and M-ERRα−/− mice at the indicated day after CTX injury (×200). Scale bars = 20 μm. B) CS activity rates in CTX-treated regenerating M-ERRαWT (circles) and M-ERRα−/− (squares) TA. Means ± sem were calculated from the ratio of CS activity in CTX-treated to that of PBS-treated TA (n=4/time point/genotype), ***P ≤ 0.001.
We then visualized changes in sarcomeric structure and mitochondria during regeneration at d 7 and 15 at higher resolution using transmission electron microscopy (Fig. 4). In the M-ERRαWT muscles, the mitochondria were localized between discontinuous sarcomeres of newly forming myofibers. By d 15, the subsarcolemmal mitochondrial population was evident, and the intermyofibrillar mitochondria were well-organized between the ordered sarcomeres, similar to the noninjured muscles. In contrast, at d 7 and 15, the regenerating M-ERRα−/− muscles had fewer mitochondria, and the intermyofibrillar populations were not organized between the sarcomeres, as in the d 15 M-ERRαWT muscles. The sarcomeres were irregularly assembled, resulting in myofibrils that appeared widely spaced and out of register in the d 15 M-ERRα−/− muscles. On the collective basis of the biochemical and structural analyses, M-ERRα−/− muscles exhibit impaired recovery of mitochondrial metabolic activity and number during skeletal muscle regeneration after acute injury.
Figure 4.
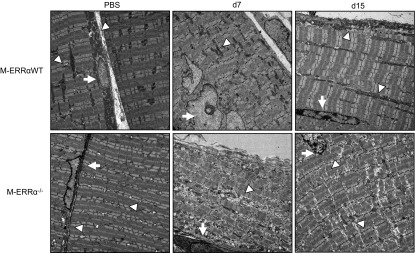
Regenerating M-ERRα−/− muscles had reduced and disorganized mitochondria. Ultrastructural analysis by TEM of PBS- or CTX-treated regenerating TA from M-ERRαWT (WT) and M-ERRα−/− on d 7 and 15 after injury. Arrows indicate nuclei; arrowheads define sites of mitochondria or disrupted mitochondria. At least 3 samples were examined per condition; images are representative (×1100).
Mitochondrial biogenesis-related factors were downregulated in M-ERRα−/− regenerating muscles
Given the evidence of reduced mitochondrial activity, we predicted that mitochondrial biogenesis gene programs may be dysregulated in M-ERRα−/− muscles during regeneration. The expression values in PBS-treated TA, reported in Table 1, reveal no significant differences in basal transcript levels between the M-ERRαWT and M-ERRα−/− TA. We examined regeneration-associated changes in ERRα pathway-related factors (Fig. 5). Unlike the pattern seen for ERRα, both PGC-1α and ERRγ transcript levels were decreased throughout regeneration of the M-ERRαWT muscles. Deletion of ERRα had no effect on ERRγ expression. In contrast, PGC-1α transcript was higher at postinjury d 1 in the M-ERRα−/− TA than in M-ERRαWT TA, but was downregulated at subsequent time points. The related PGC-1 coactivators, PGC-1β and PGC-1-related coactivator (PRC), were increased on d 3 and returned to baseline by d 7 in the regenerating M-ERRαWT muscles, suggesting that they may be involved in activating mitochondrial biogenesis factors during regeneration. In the M-ERRα−/− muscles, PGC-1β expression was significantly reduced at d 3 but continued to rise during regeneration, resulting in elevated expression at d 7 relative to the M-ERRαWT muscles, then returned to baseline by d 15 (Fig. 5). In contrast, PRC expression in the M-ERRα−/− TA followed the same expression pattern as the M-ERRαWT TA during regeneration, with only modest up-regulation seen at d 1 and 7.
Table 1.
Transcript expression levels in PBS-injected M-ERRαWT and M-ERRα−/− TA
| Transcript | Genotype | Average | P |
|---|---|---|---|
| ERRγ | WT | 1.19 ± 0.17 | 0.01 |
| MKO | 1.89 ± 0.19 | ||
| Gabpa | WT | 1.21 ± 0.25 | 0.48 |
| MKO | 1.00 ± 0.15 | ||
| TFAM | WT | 1.22 ± 0.42 | 0.52 |
| MKO | 1.55 ± 0.30 | ||
| Mfn1 | WT | 1.74 ± 0.40 | 0.26 |
| MKO | 1.25 ± 0.15 | ||
| Mfn2 | WT | 1.40 ± 0.15 | 0.70 |
| MKO | 1.32 ± 0.14 | ||
| PDK4 | WT | 0.98 ± 0.13 | 0.08 |
| MKO | 1.45 ± 0.23 | ||
| PGC-1α | WT | 2.69 ± 0.69 | 0.18 |
| MKO | 1.51 ± 0.49 | ||
| PGC-1β | WT | 1.42 ± 0.50 | 0.96 |
| MKO | 1.40 ± 0.12 | ||
| PRC | WT | 0.81 ± 0.16 | 0.21 |
| MKO | 0.58 ± 0.08 | ||
| AMPKγ3 | WT | 1.28 ± 0.33 | 0.35 |
| MKO | 1.96 ± 0.64 | ||
| AMPKα1 | WT | 0.91 ± 0.21 | 0.09 |
| MKO | 0.52 ± 0.06 | ||
| AMPKα2 | WT | 1.16 ± 0.14 | 0.002 |
| MKO | 2.35 ± 0.33 | ||
| AMPKβ1 | WT | 0.84 ± 0.25 | 0.79 |
| MKO | 0.76 ± 0.09 | ||
| AMPKβ2 | WT | 1.39 ± 0.12 | 0.02 |
| MKO | 1.86 ± 0.14 | ||
| STK11/LKB1 | WT | 0.84 ± 0.14 | 0.34 |
| MKO | 0.69 ± 0.07 | ||
| Erk | WT | 0.85 ± 0.21 | 0.47 |
| MKO | 1.03 ± 0.14 | ||
| Dusp1 | WT | 0.75 ± 0.15 | 0.64 |
| MKO | 0.83 ± 0.09 |
Values are means ± sem normalized to Gapdh; n = 16/genotype.
Figure 5.
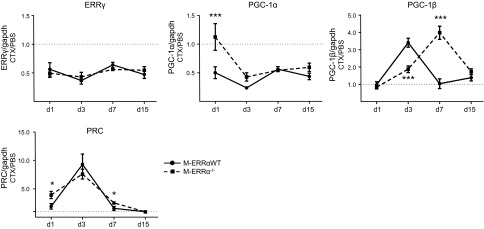
Expression of ERRγ and PGC isoforms during TA muscle regeneration in M-ERRαWT and M-ERRα−/− mice. Quantitative real-time PCR analysis was performed to analyze ERRγ, PGC-1α, PGC-1β, and PRC expression in regenerating TA muscles of M-ERRαWT (circles) and M-ERRα−/− (squares) mice at the indicated time points. Transcripts were corrected to Gapdh (control transcript) expression, and values in CTX-injected TA were normalized to expression in the PBS-injected TA from the same mouse. Data are expressed as means ± sem (n=4 mice/time point/genotype). *P ≤ 0.05, ***P ≤ 0.001.
We next examined the expression of ERRα target genes involved in mitochondrial biogenesis, mitochondrial fusion, and metabolism. Gabpa, also known as NRF-2a, is the DNA-binding subunit of the NRF-2 complex, which regulates numerous mitochondrial electron transport chain (ETC)/oxidative phosphorylation enzymes, and mitochondrial transcription factors (35). Another important target is Tfam, which regulates mtDNA maintenance and expression (35). NRF-2a and Tfam transcripts were significantly decreased in the M-ERRα−/− regenerating muscles at d 3, which corresponds to their peak expression in the M-ERRαWT muscles (Fig. 6A). At subsequent time points, NRF-2a and Tfam expression returned to baseline in both groups. The transcripts encoding the Mfn isoforms 1 and 2, which were coordinately upregulated on d 3 in M-ERRαWT during regeneration, were increased in M-ERRα−/− TA at d 3 and 7, respectively. Despite the observed dysregulation of mitochondrial biogenesis–related factors, expression of pyruvate dehydrogenase kinase (PDK)-4 was unchanged in the regenerating M-ERRα−/− muscles (Fig. 6A). At the protein level, Mfn2 protein expression was not different in the M-ERRα−/− muscles compared to that in the M-ERRαWT muscles (Fig. 6B). However, Tfam and Gabpa protein levels were dramatically decreased at d 7, consistent with transcript down-regulation in the M-ERRα−/− regenerating muscles. Thus, impaired regeneration in M-ERRα−/− is accompanied by dysregulation of key factors involved in the mitochondrial biogenesis program.
Figure 6.
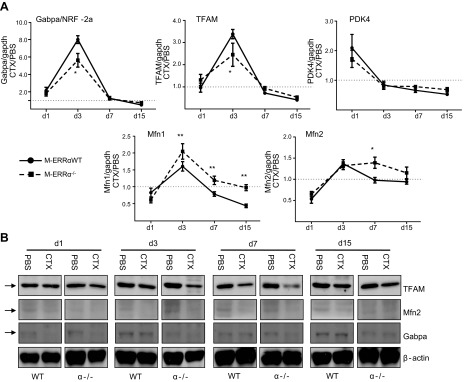
Expression of genes involved in mitochondrial biogenesis and metabolism during TA muscle regeneration. A) Real-time PCR was used to quantify transcripts encoding metabolic target genes from regenerating TA muscles of M-ERRαWT (circles) and M-ERRα−/− (squares) at the indicated time points. Transcripts were corrected to Gapdh expression, and values in CTX-injected TA were normalized to expression in the PBS-injected TA from the same mouse. Data are expressed as means ± sem (n=4 mice/time point/genotype). *P ≤ 0.05, **P ≤ 0.01. B) Western blot analysis of TFAM, Mfn2, Gabpa, and β-actin protein expression in CTX- or PBS-treated TA of M-ERRαWT and M-ERRα −/− mice (n=3–4 mice/time point/genotype) at the indicated postinjection days.
Dysregulation of the AMPK pathway in regenerating M-ERRα−/− muscles
Our previous studies in ERRα−/− primary myocytes revealed that early differentiation is slowed by ERK/MAP kinase activation caused by down-regulation of the ERRα target gene, dual specificity phosphatase-1 (Dusp1; ref. 22). Early ERK activation in the regenerating M-ERRαWT muscles coincided with down-regulation of Dusp1, as observed in myogenesis in vitro (Supplemental Fig. S4). In contrast to the results in ERRα−/− myocytes, Dusp1 and ERK regulation appeared normal in the M-ERRα−/− muscles during regeneration.
Given the impaired recovery of mitochondrial activity in the M-ERRα−/− muscles, we then explored the time course of AMPK pathway activity during regeneration. AMPK is activated in response to a rise in the intracellular AMP/ATP (or ADP/ATP) ratio, associated with impaired ATP synthesis or increased demand, and is an important regulator of mitochondrial biogenesis and respiration in skeletal muscles (36). AMPK activation also increases during differentiation and myotube formation in cultured myocytes (37). During regeneration in the CTX injury model, AMPK was activated similarly at d 3 in the M-ERRαWT and M-ERRα−/− muscles (Fig. 7A). However, although AMPK phosphorylation declined by d 7 in the WT muscles, (Fig. 7A, B) it remained elevated in the M-ERRα−/− muscles, resulting in a 2-fold higher AMPK activation over the M-ERRαWT (Fig. 7B). Phosphorylation of the AMPK target ACC was also elevated at d 7 in the regenerating M-ERRα−/− TA compared to that in the M-ERRαWT TA (Fig. 7A, B). By d 15, AMPK activity was equivalent to PBS-treated muscles in regenerating TA of both genotypes. We then measured the AMPK isoform-specific activity in muscles at d 7 of regeneration (Fig. 7C). A similar modest increase in AMPKα1 activity was observed in the M-ERRαWT and M-ERRα−/− TA. In contrast, AMPKα2 activity was selectively elevated in the M-ERRα−/− d 7 regenerating muscles compared to that in the uninjured limbs, whereas, it was reduced in the WT muscles. Therefore, AMPKα2 accounts for the elevated AMPK activity observed in M-ERRα−/− regenerating muscles.
Figure 7.

Activation of the AMPK pathway was elevated during TA muscle regeneration in M-ERRα−/− mice. A) Western blot analysis of whole protein lysate PBS- or CTX-treated TA isolated from M-ERRαWT and M-ERRα−/− mice at the indicated time points. Antibodies used were phospho-Thr172 AMPKα, total AMPKα, phospho-Ser79 ACC, and total ACC. B) Left panel: densitometry analysis of band intensity from the Western blot analysis performed in A with Image J software. Ratio of phospho-Thr172 AMPK/tAMPK in muscle extracts from M-ERRαWT (circles) and M-ERRα−/− (squares) at the indicated time points. Right panel: d 7 percentage increase calculated from A in phospho-Thr172 AMPK (left) or phospho-Ser79ACC (right) for M-ERRαWT (solid) or M-ERRα−/− (hatched) mice. C) AMPK activity of AMPKα1 or AMPKα2 catalytic complexes immunoprecipitated from d 7 regenerating or PBS-treated muscle. Activity was calculated as picomoles/minute per milligram protein and reported as the ratio of specific isoform activity in CTX- or PBS-treated TA of the same mouse (mean±se; n=4). *P ≤ 0.05. D) Top panel: total ATP levels in M-ERRαWT and M-ERRα−/− muscle extracts from CTX-treated TA on the indicated posttreatment days, determined by luciferase assay. Values are reported as means ± sem. *P ≤ 0.05. Bottom panel: analysis of AMP and ATP levels in M-ERRαWT and M-ERRα−/− muscle extracts from d 7 and 15 CTX-treated TA by UV-HPLC. E) Quantitative real-time PCR analysis of transcripts encoding AMPK catalytic and regulatory isoforms in regenerating TA muscles of M-ERRαWT (solid) and M-ERRα−/− (dashed) mice at the indicated time points. Transcripts were corrected to Gapdh and values in CTX-injected TA were normalized to expression in PBS-injected TA from the same mouse. Data are expressed as means ± sem (n=4 mice/time point/genotype). *P ≤ 0.05, ***P ≤ 0.001.
Because AMPK is stimulated in response to energy depletion, we measured ATP content in muscle extracts (Fig. 7D). Consistent with the pattern of AMPK activation, the M-ERRα−/− regenerating muscles had significantly lower ATP content only at the d 7 time point, whereas no changes were observed at d 1, 3, or 15 compared to WT. No differences were seen between the PBS-injected muscles of the M-ERRαWT and M-ERRα−/− mice (data not shown). Using HPLC analysis we confirmed that ATP was reduced only at d 7 in the M-ERRα−/− muscles, whereas AMP content was unchanged (Fig. 7D). No differences in AMP or ATP were observed between the WT and M-ERRα−/− in d 15 muscles. Thus, enhanced AMPK activation was associated with an energy deficit in the M-ERRα−/− TA during regeneration.
AMPK is a heterotrimeric complex comprised of catalytic (AMPKα) and regulatory (AMPKβ and AMPKγ) subunits that have distinct expression patterns during myogenesis and in adult muscle fiber types (37–39). Subunit composition influences sensitivity of the AMPK complex to allosteric regulation (36). We assessed whether AMPK dysregulation in regenerating M-ERRα−/− muscles is associated with changes in subunit expression. The catalytic subunits α1 and α2 showed distinct expression patterns during regeneration, with α1 showing peak expression at d 3, followed by α2 up-regulation at d 7 in the M-ERRαWT muscles (Fig. 8E). In contrast, α2 expression was blunted in the M-ERRα−/− muscles at the d 7 time point. The AMPKβ1 subunit was transiently increased at d 3 of regeneration, whereas AMPKβ2 was downregulated throughout the regeneration. No difference was observed between the WT and M-ERRα−/− muscles for either transcript. The AMPKγ3 regulatory subunit was upregulated by d 7 and remained high in the M-ERRαWT muscles, whereas in the M-ERRα−/− muscles, γ3 showed enhanced peak expression at d 7, then returned to control levels. In uninjured TA, only the AMPKα2 and β2 subunits were moderately upregulated in M-ERRα−/− muscle (Table 1). Thus, basal expression most likely had a minimal effect on the changes observed during regeneration. Finally, the upstream AMPK activating kinase LKB1 was induced at d 3 of regeneration but was not differentially regulated in the M-ERRαWT and M-ERRα−/− muscles. Thus, although AMPK subunits are dynamically regulated during muscle recovery, we found little evidence that altered AMPK signaling in ERRα-deficient skeletal muscle is due to changes in subunit expression.
Figure 8.
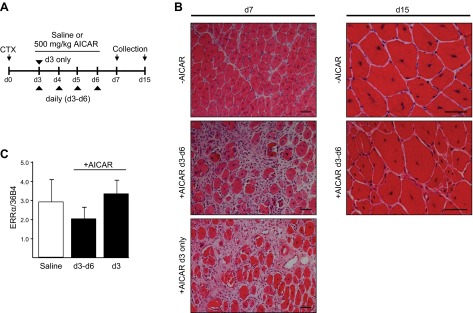
Activation of AMPK with systemic AICAR administration after CTX injury led to impaired muscle regeneration. A) Experimental design for testing effects of pharmacologic AMPK activation on muscle regeneration in WT mice in response to CTX injury. AICAR was administered (500 mg/kg, i.p.) once daily beginning on d 3 after CTX injection on a single day (d 3 only) or on multiple consecutive days (d 3–6). B) Histologic (H&E) examination of regenerating skeletal muscle in the saline or AICAR treatment groups (±AICAR) at the specified time points. C) Real-time PCR quantitation of ERRα transcript expression in d 7 vastus in saline- and AICAR-treated mice. Values were corrected to 36B4 (control transcript) and are reported as means ± sem (n=3 mice/time point/treatment).
AMPK activation by AICAR impaired skeletal muscle regeneration after acute injury
Delayed myofiber recovery in the M-ERRα−/− muscles was associated with AMPK activation. AMPK activation has been shown to impair myocyte differentiation and protein synthesis in muscles (40, 41). However, the role of AMPK in regulating muscle regeneration after acute injury has not been studied. To investigate, we subjected WT mice to acute CTX-induced injury and subsequently treated them with the AMP analogue AICAR or saline on d 3 alone or on 4 consecutive days (d 3–6) after CTX injury (Fig. 8A). AMPK activation in response to AICAR administration was confirmed by using blood glucose monitoring (reduced 30 min after treatment) and by AMPK activity assays (data not shown). Regeneration in CTX-treated TA was dramatically impaired at d 7 and 15 in mice treated with AICAR on d 3–6, which is a critical period of rapid myofiber formation (Fig. 8B). More remarkable, however, even a single administration of AICAR to activate AMPK on d 3 alone was sufficient to impair myofiber recovery at d 7. The regenerating muscles of the AICAR-treated mice from both dose groups had increased the nonmyofiber area and showed fewer myofibers at d 7 than did the saline-treated control. By d 15, the TA of mice treated on d 3–6 of regeneration with AICAR had greater heterogeneity of myofiber size, with an increased proportion of small myofibers compared to the saline-treated muscles. ERRα transcript levels in the muscles were not changed in either AICAR treatment group. Thus, the effects of AMPK activation on skeletal muscle regeneration are not due to altered ERRα expression.
AMPK stimulated ERRα transcript expression through Esrra promoter activation
AMPK is known to regulate mitochondrial oxidative metabolism in skeletal muscle through activation of transcription factors (42, 43). Based on our observations that AMPK activation occurred early in muscle regeneration and coincided with ERRα up-regulation, we sought to determine whether AMPK can regulate ERRα expression. WT mice treated systemically for 7 d with AICAR had increased ERRα transcript expression 2- and 1.9-fold in TA and white vastus, respectively, compared to expression in the saline-treated controls (Fig. 9A). By contrast, PGC-1α expression was not changed in response to AICAR. Similarly, C2C12 myotubes treated with AICAR for 2 d showed up-regulation of ERRα transcript expression (Fig. 9B), with no effect of AMPK stimulation on endogenous PGC-1α transcript levels.
Figure 9.
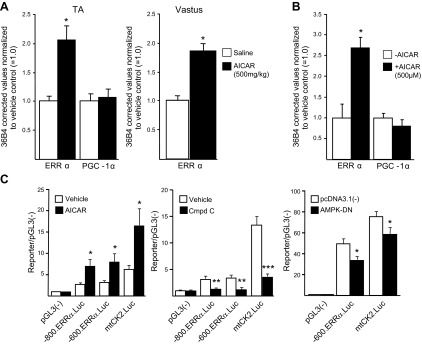
Pharmacologic activation of AMPK up-regulated endogenous ERRα transcript expression in skeletal muscle. A) Real-time PCR quantitation of ERRα and PGC-1α transcript expression in TA or white vastus muscles of mice given 500 mg/kg/d AICAR or saline for 1 wk. Values were corrected to 36B4 transcript normalized to expression in saline-treated control and are reported as means ± sem. *P ≤ 0.05. B) ERRα and PGC-1α transcripts were measured in C2C12 myotubes treated with PBS (−AICAR) or 500 μM AICAR (+AICAR) for 48 h by real-time PCR and corrected to 36B4 (control transcript) levels. Values in AICAR-treated myocytes were normalized to PBS-treated control values and expressed as means ± sem (3 independent trials). *P ≤ 0.05. C) Left and middle panels: differentiated C2C12 myotubes transfected with pGL3(−); the Esrra gene promoter-reporters −800.ERRα.Luc and −600.ERRα.Luc; or the mitochondrial creatine kinase gene promoter-reporter, −1.27.mtCK2.Luc, followed by treatment with AICAR (left panel) or compound C (middle panel). Right panel: C2C12 myoblasts were transiently cotransfected with the promoter–reporter constructs pGL3(−), −600.ERRα.Luc, or −1.27.mtCK2.Luc and the expression vector pcDNA3.1(−) or pcDNA3.1-AMPK-DN. Data are expressed as means ± sem (4 independent trials). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
We next tested whether AMPK activation of ERRα transcript expression might involve direct genetic regulation of the Esrra gene. Using a reporter construct containing the Esrra proximal promoter, we first examined the response to AMPK activation (Fig. 9C). AICAR treatment of C2C12 myotubes activated both the −800.ERRa.Luc and −600.ERRa.Luc constructs by ∼3-fold. The −1.27.mtCK2.Luc mitochondrial creatine kinase control promoter-reporter was similarly induced. Conversely, Esrra promoter activity was significantly blunted by treating cells with the AMPK inhibitor compound C. Likewise, cotransfection with a dominant-negative AMPKα2 mutant (AMPK-DN) inhibited the Esrra promoter–reporter activity. Taken together, the results show that AMPK modulates ERRα expression via transcriptional regulation of the Esrra promoter.
DISCUSSION
In the current study, ERRα controlled mitochondrial biogenesis and recovery of oxidative function during muscle regeneration and was essential for normal myofiber regrowth. Several novel findings of the study support these conclusions. First, in response to injury, ERRα-deficient muscles had delayed regeneration, resulting in smaller myofibers with centralized nuclei. Second, impaired mitochondrial oxidative recovery in the M-ERRα−/− muscles was associated with down-regulation of mitochondrial biogenesis-related factors. Third, the AMPK pathway was activated during normal muscle regeneration and up-regulated ERRα expression through a transcriptional mechanism. However, elevated AMPK activation early in regeneration was sufficient to impair myofiber growth. Thus, delayed myofiber recovery in the M-ERRα−/− muscles may be due to elevated and sustained AMPK activation resulting from impaired mitochondrial activity. Taken together, our results demonstrate that the ERRα pathway, which is stimulated downstream of AMPK, regulates the mitochondrial biogenesis program required to support muscle regeneration.
Skeletal muscle regeneration involves several overlapping stages, including necrosis and inflammation, satellite cell activation/proliferation, myogenesis and fusion, and myofiber remodeling (3). We used a muscle-specific ERRα-knockout model, thereby precluding functional alterations in nonmyocyte cell types involved in injury-stimulated regeneration, such as macrophages and satellite cells. As expected, CTX treatment caused similar necrotic damage in the WT and M-ERRα−/− muscles, as well as equivalent macrophage infiltration and satellite cell proliferation in response to injury. These data support the notion that ERRα specifically regulates the myogenesis stage of muscle regeneration.
An integral metabolic component of myocyte differentiation involves mitochondrial biogenesis and increased mitochondrial enzyme activity, which is detectable within 24 h after the onset of myoblast differentiation (5, 10). Likewise, in skeletal muscle injury models, recovery of mitochondrial enzyme expression and activity generally begins at d 3 of regeneration, consistent with the results of this study (8, 9). Mitochondrial biogenesis requires coordinated expression of mitochondrial structural, transport proteins, and metabolic enzymes; replication and transcription of mtDNA; and mitochondrial fission and fusion (35). Most mitochondrial proteins are nuclear encoded, although several ETC subunits as well as tRNAs and rRNAs, involved in translation of mitochondrial transcripts, are encoded by mtDNA. Thus, mechanisms regulating mitochondrial biogenesis necessitate communication between the nuclear and mitochondrial genomes. Key factors regulating mitochondrial function in skeletal muscle include the coactivator PGC-1, the corepressors RIP140 and NCoR, and the transcription factors PPARs, NRF-1 and 2, and TFAM (11, 35, 44, 45). In addition, our previous studies showed that ERRα is necessary for maximum mitochondrial oxidative activity and for normal differentiation in primary myotubes, suggesting that ERRα controls metabolic programs during myogenesis in vivo (15, 22).
In the regenerating WT muscles, ERRα, PGC-1β, and PRC were upregulated in parallel with the onset of myogenesis and expression of mitochondrial enzyme genes (9), whereas, unexpectedly, PGC-1α and ERRγ were downregulated. Previous studies have yielded conflicting results regarding PGC-1α expression during regeneration. PGC-1α was induced at d 10 postinjury in one model, but was downregulated throughout regeneration in another study, in agreement with our findings (8, 9). In differentiating C2C12 myocytes, ERRα expression increases early in parallel with PGC-1α and PGC-1β and to a lesser magnitude PRC (22, 23). The variable PGC-1α expression patterns reported may be related to the different skeletal muscle types examined in various injury models. ERRγ expression during muscle regeneration has not been evaluated, but it is highly expressed in mature myotubes. Our previous studies showed that oxidative stress inhibits myogenesis in ERRγ−/− myocytes (24, 34). However, in our study, ERRγ was downregulated throughout regeneration in the WT and M-ERRα−/− muscles, suggesting that it may function in later stages of muscle remodeling. Meanwhile, PGC-1β and PRC may regulate metabolic recovery with ERRα, in that they were coordinately upregulated with NRFs, TFAM, and other mitochondrial biogenesis-related genes during regeneration (Figs. 2 and 6 and ref. 9). Moreover, in M-ERRα−/− muscles, PGC-1β expression was reduced at the critical d 3 time point, with its peak expression shifted to d 7 when myofiber recovery was clearly impaired. Thus, PGC-1β expression may be partially dependent on ERRα or on another upstream factor that is dysregulated in M-ERRα−/− muscle. Of particular note, PGC-1β was downregulated with Gabpa/NRF-2a and TFAM in M-ERRα−/− muscles. NRF-2 and NRF-1 regulate many mitochondrial transcription factors, such as TFAM, along with mitochondrial ETC subunits and protein import complexes (35). ERRα directly regulates Gabpa/NRF-2a expression via binding to the Gabpa gene promoter (15, 21). In addition, ERRα was necessary for maximum TFAM and NRF-1 induction by PGC-1α in cell-based studies, but it is unclear whether ERRα directly binds to regulatory elements in the Tfam or Nrf1 genes (13, 15). Furthermore, ERRα and NRF-1 binding sites are clustered on many mitochondrial genes, suggesting regulatory coordination between the NRFs and ERRα (21). Indeed, ERRα and NRF-1 cooperate with PGC-1β to activate Gabpa/NRF-2a and TFAM expression and to stimulate mitochondrial biogenesis during myogenesis (23). Collectively, the evidence supports an essential role for ERRα with the PGC-1β coactivator in activating mitochondrial biogenesis-related genes and downstream mitochondrial activity during muscle regeneration.
Biochemical analysis of enzyme activity and ultrastructural examination of new myofibers revealed that recovery of mitochondrial function and content was impaired in the regenerating M-ERRα−/− muscles. Furthermore, tissue ATP content was reduced specifically at d 7, when regeneration was lagging, suggesting that the M-ERRα−/− muscles had insufficient energetic capacity to support normal myofiber formation. Consistent with this notion, AMPK activation, associated with higher AMPKα2 isoform activity, remained elevated in ERRα-deficient muscles after it had returned to basal levels in WT muscles. The AMPK pathway is a homeostatic energy sensor that balances the myocyte energetic state with nutrient availability, energetic demand, and hypertrophy and growth (36). During C2C12 myogenesis AMPK is activated and subunit expression is dynamically regulated during the transition from proliferating myoblasts to mature myotubes (37). In our study, in vivo evidence showed that AMPK functioned as a regulatory signal during skeletal muscle regeneration. Indeed, AMPK was activated at the onset of regeneration (d 3) followed by a rapid return to basal levels by d 7. Our results suggest that a precise regulation of AMPK activity is necessary for normal muscle regrowth (discussed below). In skeletal muscle, AMPK plays an important role in regulating mitochondrial biogenesis and oxidative capacity in response to physiological stimuli that increase ATP demand (36). On the basis of the observed dynamics, AMPK is most likely activated by the increased energy demand associated with growth (e.g., protein synthesis) and stimulates the transcriptional pathways involved in mitochondrial biogenesis and energy generation.
AMPK has been shown to regulate mitochondrial oxidative gene expression through the activation of PGC-1α and NRFs (42, 43, 46). However, our data show that PGC-1β and PRC were dynamically upregulated in parallel with ERRα, coincident with maximum mitochondrial recovery, whereas PGC-1α expression was reduced throughout the regeneration time course. Consistent with a role for AMPK in regulating ERRα and downstream mitochondrial biogenesis pathways during regeneration, we showed that ERRα was upregulated in skeletal muscles in response to 1 wk of AICAR treatment, which is a sufficient duration to stimulate β-oxidation (36). Regulation of endogenous ERRα expression by the AMPK pathway may involve transcriptional activation of the Esrra gene promoter. Although the mechanism of Esrra gene activation is not clear, the region of the Esrra promoter shown to be AMPK responsive in our trials and in cardiac myocytes (47) contains clusters of sites for Sp1, which has been shown to interact and cooperate with Gabpa/NRF-2a (47, 48). Future studies will investigate the dependence of Esrra induction by AMPK-regulated transcription factors, including PGC-1α and NRF-1 and NRF-2 and potentially ERRα itself. Thus, ERRα may respond to AMPK signaling and regulate metabolic gene programs during regeneration.
In addition to stimulating mitochondrial oxidative capacity AMPK pathway activation also inhibits ATP-consuming processes. Sustained AMPK activation has been demonstrated to inhibit growth or differentiation in various models. In C2C12 myocytes, low glucose conditions, AICAR treatment, or expression of constitutively-active AMPK mutants can inhibit myogenesis, whereas AMPK inhibition advances myotube formation (41, 49). Of note, AICAR inhibition of myogenesis was associated with PGC-1α-dependent Foxo3a repression (49). One mechanism by which AMPK blocks myocyte differentiation and hypertrophy is by inhibiting mTOR pathway-regulated protein synthesis (40, 50). AMPK signaling may serve as a modulator that coordinates the establishment of energy-generating and -consuming specializations of the adult myofiber (51). In the ERRα-deficient muscles AMPK activation was elevated and sustained well beyond the time at which activity had returned to basal levels in the WT regenerating muscles. Indeed, we showed that activation of AMPK by a single dose of AICAR in the WT mice was sufficient to impair muscle regeneration and to replicate the M-ERRα−/− phenotype. Thus, appropriate AMPK signaling is essential for coordination of metabolism and growth during muscle regeneration, and its dysregulation most likely contributes to delayed recovery in M-ERRα−/− muscles in response to injury.
In summary, in our study, ERRα was required for normal skeletal muscle repair in response to injury. M-ERRα−/− mice displayed impaired muscle regeneration that coincided with blunted mitochondrial biogenesis and AMPK pathway activation. The ERRα pathway may promote maintenance of skeletal muscle mass and contractile function in the context of exercise-induced injury and aging. More broadly, the current study highlights the interrelatedness of metabolic regulation and muscle growth. Thus, ERRα and related metabolic regulatory pathways may present important therapeutic targets to promote maintenance of muscle mass in the context of skeletal muscle dystrophies and other disease-associated muscle loss.
Supplementary Material
Acknowledgments
The authors thank Dr. Brian Armstrong and Mariko Lee (Microscopy Core, City of Hope), Dr. Marcia Miller, Dr. Zhou Li, and Ricardo Zerda (Electron Microscopy Core, City of Hope; TEM-Office of Naval Research (ONR) grant N00014-02–10958), and the Animal Resources Center (Department of Comparative Medicine, City of Hope) for assistance; and Dr. Timothy Synold for his contribution to the HPLC assay methodology.
This work was supported by Public Health Service grant R01 DK074700 (to J.H.) from the National Institute of Diabetes and Digestive and Kidney Diseases, U.S. National Institutes of Health (NIH; Bethesda MD, USA) and by the Beckman Research Institute (City of Hope) and the Morgan and Helen Chu Graduate Student Fellowship (to S.L.). J.A. is the Nestlé Chair in Energy Metabolism, and his laboratory is supported by grants from the École Polytechnique Fédérale de Lausanne, the EU Ideas program (AdG-231138), the Swiss National Science Foundation (31003A-140780), and NIH (R01AG043930). The Developmental Studies Hybridoma Bank was established under the auspices of the National Institute of Child Health and Human Development (NICHD), NIH, and is maintained by The University of Iowa Department of Biology (Iowa City, IA, USA). The Analytical Pharmacology Core is supported by grant P30CA33572 from the National Cancer Institute, NIH.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- ACC
- acyl-CoA carboxylase
- AICAR
- 5-aminoimidazole-4-carboxamide-1-β-d-ribosyl monophosphate
- AMPK
- AMP-activated protein kinase
- AMPK-DN
- dominant-negative AMP-activated protein kinase
- BAT
- brown adipose tissue
- CN
- central nuclei
- COX
- cytochrome c oxidase
- CS
- citrate synthase
- CSA
- cross-sectional area
- CTX
- cardiotoxin
- DN
- dominant negative
- DTNB
- 5,5′-dithiosbis(2-nitrobenzoic acid)
- ETC
- electron transport chain
- ERRα
- estrogen-related receptor α
- Gabpa
- GA-binding protein transcription factor, α subunit
- H&E
- hematoxylin and eosin
- IHC
- immunohistochemistry
- MCK
- muscle creatine kinase
- M-ERRα−/−
- muscle-specific ERRα deletion
- M-ERRαWT
- muscle-specific ERRα wild type
- Mfn
- mitofusin
- mtDNA
- mitochondrial DNA
- NRF
- nuclear respiratory factor
- PGC-1
- PPARγ coactivator 1
- PDK
- pyruvate dehydrogenase kinase
- PPAR
- peroxisome proliferator-activated receptor
- PRC
- PGC-1-related coactivator
- SDH
- succinate dehydrogenase
- TA
- tibialis anterior
- TCA
- tricarboxylic acid
- TEM
- transmission electron microscope
- TFAM
- mitochondrial transcription factor A
- WT
- wild-type
REFERENCES
- 1. Wallace G. Q., McNally E. M. (2009) Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu. Rev. Physiol. 71, 37–57 [DOI] [PubMed] [Google Scholar]
- 2. Vettor R., Milan G., Franzin C., Sanna M., De Coppi P., Rizzuto R., Federspil G. (2009) The origin of intermuscular adipose tissue and its pathophysiological implications. Am. J. Physiol. Endocrinol. Metab. 297, E987–E998 [DOI] [PubMed] [Google Scholar]
- 3. Ciciliot S., Schiaffino S. (2010) Regeneration of mammalian skeletal muscle: basic mechanisms and clinical implications. Curr. Pharm. Des. 16, 906–914 [DOI] [PubMed] [Google Scholar]
- 4. Bentzinger C. F., Wang Y. X., Rudnicki M. A. (2012) Building muscle: molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 4, pii: a008342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lyons C. N., Leary S. C., Moyes C. D. (2004) Bioenergetic remodeling during cellular differentiation: changes in cytochrome c oxidase regulation do not affect the metabolic phenotype. Biochem. Cell Biol. 82, 391–399 [DOI] [PubMed] [Google Scholar]
- 6. Hamai N., Nakamura M., Asano A. (1997) Inhibition of mitochondrial protein synthesis impaired C2C12 myoblast differentiation. Cell Struct. Funct. 22, 421–431 [DOI] [PubMed] [Google Scholar]
- 7. Vignaud A., Hourde C., Torres S., Caruelle J. P., Martelly I., Keller A., Ferry A. (2005) Functional, cellular and molecular aspects of skeletal muscle recovery after injury induced by snake venom from Notechis scutatus scutatus. Toxicon 45, 789–801 [DOI] [PubMed] [Google Scholar]
- 8. Duguez S., Feasson L., Denis C., Freyssenet D. (2002) Mitochondrial biogenesis during skeletal muscle regeneration. Am. J. Physiol. Endocrinol. Metab. 282, E802–E809 [DOI] [PubMed] [Google Scholar]
- 9. Wagatsuma A., Kotake N., Yamada S. (2011) Muscle regeneration occurs to coincide with mitochondrial biogenesis. Mol. Cell. Biochem. 349, 139–147 [DOI] [PubMed] [Google Scholar]
- 10. Remels A. H., Langen R. C., Schrauwen P., Schaart G., Schols A. M., Gosker H. R. (2010) Regulation of mitochondrial biogenesis during myogenesis. Mol. Cell. Endocrinol. 315, 113–120 [DOI] [PubMed] [Google Scholar]
- 11. Giguere V. (2008) Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr. Rev. 29, 677–696 [DOI] [PubMed] [Google Scholar]
- 12. Huss J. M., Torra I. P., Staels B., Giguere V., Kelly D. P. (2004) Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Biol. 24, 9079–9091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schreiber S. N., Emter R., Hock M. B., Knutti D., Cardenas J., Podvinec M., Oakeley E. J., Kralli A. (2004) The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. U. S. A. 101, 6472–6477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wende A. R., Huss J. M., Schaeffer P. J., Giguere V., Kelly D. P. (2005) PGC-1alpha coactivates PDK4 gene expression via the orphan nuclear receptor ERRalpha: a mechanism for transcriptional control of muscle glucose metabolism. Mol. Cell. Biol. 25, 10684–10694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mootha V. K., Handschin C., Arlow D., Xie X., St Pierre J., Sihag S., Yang W., Altshuler D., Puigserver P., Patterson N., Willy P. J., Schulman I. G., Heyman R. A., Lander E. S., Spiegelman B. M. (2004) Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl. Acad. Sci. U. S. A. 101, 6570–6575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huss J. M., Kopp R. P., Kelly D. P. (2002) Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J. Biol. Chem. 277, 40265–40274 [DOI] [PubMed] [Google Scholar]
- 17. Schreiber S. N., Knutti D., Brogli K., Uhlmann T., Kralli A. (2003) The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha). J. Biol. Chem. 278, 9013–9018 [DOI] [PubMed] [Google Scholar]
- 18. Luo J., Sladek R., Carrier J., Bader J. A., Richard D., Giguere V. (2003) Reduced fat mass in mice lacking orphan nuclear receptor estrogen-related receptor alpha. Mol. Cell. Biol. 23, 7947–7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Villena J. A., Hock M. B., Chang W. Y., Barcas J. E., Giguere V., Kralli A. (2007) Orphan nuclear receptor estrogen-related receptor alpha is essential for adaptive thermogenesis. Proc. Natl. Acad. Sci. U. S. A. 104, 1418–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huss J. M., Imahashi K., Dufour C. R., Weinheimer C. J., Courtois M., Kovacs A., Giguere V., Murphy E., Kelly D. P. (2007) The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 6, 25–37 [DOI] [PubMed] [Google Scholar]
- 21. Dufour C. R., Wilson B. J., Huss J. M., Kelly D. P., Alaynick W. A., Downes M., Evans R. M., Blanchette M., Giguere V. (2007) Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 5, 345–356 [DOI] [PubMed] [Google Scholar]
- 22. Murray J., Huss J. M. (2011) Estrogen-related receptor alpha regulates skeletal myocyte differentiation via modulation of the ERK MAP kinase pathway. Am. J. Physiol. Cell Physiol. 301, C630–C645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shao D., Liu Y., Liu X., Zhu L., Cui Y., Cui A., Qiao A., Kong X., Chen Q., Gupta N., Fang F., Chang Y. (2010) PGC-1 beta-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERR alpha. Mitochondrion 10, 516–527 [DOI] [PubMed] [Google Scholar]
- 24. Wang S. C., Myers S., Dooms C., Capon R., Muscat G. E. (2010) An ERRbeta/gamma agonist modulates GRalpha expression, and glucocorticoid responsive gene expression in skeletal muscle cells. Mol. Cell. Endocrinol. 315, 146–152 [DOI] [PubMed] [Google Scholar]
- 25. Rangwala S. M., Li X., Lindsley L., Wang X., Shaughnessy S., Daniels T. G., Szustakowski J., Nirmala N. R., Wu Z., Stevenson S. C. (2007) Estrogen-related receptor alpha is essential for the expression of antioxidant protection genes and mitochondrial function. Biochem. Biophys. Res. Commun. 357, 231–236 [DOI] [PubMed] [Google Scholar]
- 26. Sonoda J., Laganiere J., Mehl I. R., Barish G. D., Chong L. W., Li X., Scheffler I. E., Mock D. C., Bataille A. R., Robert F., Lee C. H., Giguere V., Evans R. M. (2007) Nuclear receptor ERR alpha and coactivator PGC-1 beta are effectors of IFN-gamma-induced host defense. Genes Dev. 21, 1909–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Naya F. J., Mercer B., Shelton J., Richardson J. A., Williams R. S., Olson E. N. (2000) Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. J. Biol. Chem. 275, 4545–4548 [DOI] [PubMed] [Google Scholar]
- 28. Liu D., Zhang Z., Gladwell W., Teng C. T. (2003) Estrogen stimulates estrogen-related receptor alpha gene expression through conserved hormone response elements. Endocrinology 144, 4894–4904 [DOI] [PubMed] [Google Scholar]
- 29. Bancroft J. D., Gamble M. (2002) Theory and Practice of Histological Techniques. Churchill Livingstone, London/New York [Google Scholar]
- 30. Mascorro J. A., Bozzola J. J. (2007) Processing biological tissues for ultrastructural study. Methods Mol. Biol. 369, 19–34 [DOI] [PubMed] [Google Scholar]
- 31. Srere P. A. (1969) Citrate synthase: [EC 4.1.3.7. citrate oxaloacetate-lyase (CoA-acetylating)]. Methods Enzymol. 13, 3–11 [Google Scholar]
- 32. Figarola J. L., Rahbar S. (2013) Smallmolecule COH-SR4 inhibits adipocyte differentiation via AMPK activation. Int. J. Mol. Med. 31, 1166–1176 [DOI] [PubMed] [Google Scholar]
- 33. Raney M. A., Turcotte L. P. (2008) Evidence for the involvement of CaMKII and AMPK in Ca2+-dependent signaling pathways regulating FA uptake and oxidation in contracting rodent muscle. J. Appl. Physiol. 104, 1366–1373 [DOI] [PubMed] [Google Scholar]
- 34. Murray J., Auwerx J., Huss J. M. (2013) Impaired myogenesis in estrogen-related receptor gamma (ERRgamma)-deficient skeletal myocytes due to oxidative stress. FASEB J. 27, 135–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scarpulla R. C. (2012) Nucleus-encoded regulators of mitochondrial function: integration of respiratory chain expression, nutrient sensing and metabolic stress. Biochim. Biophys. Acta. 1819, 1088–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Witczak C. A., Sharoff C. G., Goodyear L. J. (2008) AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell. Mol. Life. Sci. 65, 3737–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Niesler C. U., Myburgh K. H., Moore F. (2007) The changing AMPK expression profile in differentiating mouse skeletal muscle myoblast cells helps confer increasing resistance to apoptosis. Exp. Physiol. 92, 207–217 [DOI] [PubMed] [Google Scholar]
- 38. Durante P. E., Mustard K. J., Park S. H., Winder W. W., Hardie D. G. (2002) Effects of endurance training on activity and expression of AMP-activated protein kinase isoforms in rat muscles. Am. J. Physiol. Endocrinol. Metab. 283, E178–E186 [DOI] [PubMed] [Google Scholar]
- 39. Mahlapuu M., Johansson C., Lindgren K., Hjalm G., Barnes B. R., Krook A., Zierath J. R., Andersson L., Marklund S. (2004) Expression profiling of the gamma-subunit isoforms of AMP-activated protein kinase suggests a major role for gamma3 in white skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 286, E194–E200 [DOI] [PubMed] [Google Scholar]
- 40. Chan A. Y., Soltys C. L., Young M. E., Proud C. G., Dyck J. R. (2004) Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J. Biol. Chem. 279, 32771–32779 [DOI] [PubMed] [Google Scholar]
- 41. Fulco M., Cen Y., Zhao P., Hoffman E. P., McBurney M. W., Sauve A. A., Sartorelli V. (2008) Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 14, 661–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bergeron R., Ren J. M., Cadman K. S., Moore I. K., Perret P., Pypaert M., Young L. H., Semenkovich C. F., Shulman G. I. (2001) Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 281, E1340–E1346 [DOI] [PubMed] [Google Scholar]
- 43. Jager S., Handschin C., St-Pierre J., Spiegelman B. M. (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. U. S. A. 104, 12017–12022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Seth A., Steel J. H., Nichol D., Pocock V., Kumaran M. K., Fritah A., Mobberley M., Ryder T. A., Rowlerson A., Scott J., Poutanen M., White R., Parker M. (2007) The transcriptional corepressor RIP140 regulates oxidative metabolism in skeletal muscle. Cell Metab. 6, 236–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamamoto H., Williams E. G., Mouchiroud L., Canto C., Fan W., Downes M., Heligon C., Barish G. D., Desvergne B., Evans R. M., Schoonjans K., Auwerx J. (2011) NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 147, 827–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Canto C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., Elliott P. J., Puigserver P., Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu X., Xu X., Lu Z., Zhang P., Fassett J., Zhang Y., Xin Y., Hall J. L., Viollet B., Bache R. J., Huang Y., Chen Y. (2011) AMP activated protein kinase-alpha2 regulates expression of estrogen-related receptor-alpha, a metabolic transcription factor related to heart failure development. Hypertension 58, 696–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Galvagni F., Capo S., Oliviero S. (2001) Sp1 and Sp3 physically interact and co-operate with GABP for the activation of the utrophin promoter. J. Mol. Biol. 306, 985–996 [DOI] [PubMed] [Google Scholar]
- 49. Williamson D. L., Butler D. C., Alway S. E. (2009) AMPK inhibits myoblast differentiation through a PGC-1alpha-dependent mechanism. Am. J. Physiol. Endocrinol. Metab. 297, E304–E314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bolster D. R., Crozier S. J., Kimball S. R., Jefferson L. S. (2002) AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 277, 23977–23980 [DOI] [PubMed] [Google Scholar]
- 51. Mounier R., Lantier L., Leclerc J., Sotiropoulos A., Pende M., Daegelen D., Sakamoto K., Foretz M., Viollet B. (2009) Important role for AMPKalpha1 in limiting skeletal muscle cell hypertrophy. FASEB J. 23, 2264–2273 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.