

Abstract
Activity of glycogen synthase kinase-3β (GSK-3β) is required for long-term depression (LTD) via molecular mechanisms that are incompletely understood. Here, we report that PSD-95, a major scaffold protein of the postsynaptic density (PSD) that promotes synaptic strength, is phosphorylated on threonine-19 (T19) by GSK-3β. In cultured rat hippocampal neurons, phosphorylation of T19 increases rapidly with chemical LTD and is attenuated by pharmacologic or genetic suppression of GSK-3β. In organotypic rat hippocampal slices, we find that a nonphosphorylatable PSD-95 mutant (T19A) tagged with photoactivatable green fluorescent protein (PAGFP) shows enhanced stability in dendritic spines versus wild-type PSD-95, whereas the phosphomimetic mutant (PSD-95-T19D) is more readily dispersed. Further, overexpression of PSD-95-T19A, but not WT-PSD-95, impairs AMPA receptor internalization and the induction of LTD. These data indicate that phosphorylation on T19 by GSK-3β destabilizes PSD-95 within the PSD and is a critical step for AMPA receptor mobilization and LTD.
Introduction
PSD-95 is an abundant scaffold protein in the postsynaptic density (PSD) of excitatory synapses. Through its three PDZ domains, SH3 domain, and guanylate kinase-like domain, PSD-95 mediates interactions with structural and signaling proteins, including NMDA receptors and AMPA receptor/transmembrane AMPA receptor regulatory protein (TARP) complexes (Scannevin and Huganir, 2000; Funke et al., 2005; Peng et al., 2004; Sheng and Hoogenraad, 2007; Sheng and Kim, 2011). PSD-95 promotes synapse maturation and exerts a strong positive influence on synaptic strength (El-Husseini et al., 2000; Schnell et al., 2002; Stein et al., 2003; Ehrlich and Malinow, 2004; Elias et al., 2006; Futai et al., 2007). Recently PSD-95's essential role in synaptic structural organization was confirmed by electron microscopy tomography studies, where shRNA-mediated PSD-95 knockdown results in fragmentation of the PSD architecture (Chen et al., 2011).
Synaptic accumulation of PSD-95 is crucial for its ability to potentiate synaptic transmission. One factor enhancing synaptic accumulation of PSD-95 is JNK1-mediated phosphorylation of serine-295 (S295; Kim et al., 2007). Dephosphorylation of S295 of PSD-95, mediated by PP1/PP2A phosphatases, is required for AMPA receptor internalization and long-term depression (LTD), and S295 dephosphorylation occurs rapidly following LTD-like stimulation (Kim et al., 2007). It is not known whether S295 phosphorylation enhances synaptic transport or postsynaptic stability of PSD-95. Nevertheless, there is compelling evidence that the postsynaptic abundance of PSD-95 determines synaptic strength by controlling AMPA receptor trafficking (El-Husseini et al., 2000; Schnell et al., 2002; Stein et al., 2003; Ehrlich and Malinow, 2004; Nakagawa et al., 2004; Elias et al., 2006; Xu et al., 2008) and that post-translational modification of PSD-95 is involved in AMPA receptor mobilization and LTD induction.
The two mammalian isoforms of glycogen synthase kinase-3 (GSK-3α and GSK-3β) are ubiquitously expressed serine/threonine kinases impacting a variety of biological processes (for review, see Woodgett, 2001; Jope and Johnson, 2004). Both isoforms exhibit high enzymatic activity that is inhibited upon phosphorylation by Akt (on residue S9 of GSK-3β (Cross et al., 1995). In neurons, GSK-3 has been linked to many functions, including cell polarity and axon guidance (Hur and Zhou, 2010), antagonism of CREB (Grimes and Jope, 2001), modulation of monoamine signaling (Beaulieu et al., 2009), and phosphorylation of tau, which promotes the formation of neurofibrillary tangles observed in Alzheimer's Disease (Hanger et al., 1992; Mandelkow et al., 1992). GSK-3 is also a major target of Li+, a mainstay treatment for bipolar disorder, so any action of GSK-3β might be relevant to understanding this mental illness. With regard to synaptic plasticity, GSK-3β appears to be essential for LTD, and GSK-3β activity is suppressed during long-term potentiation (LTP; Peineau et al., 2007). However, the molecular basis of GSK-3β's role in the balance of LTP and LTD is unknown.
Here we report that GSK-3β can phosphorylate PSD-95 on threonine-19 (T19). T19 phosphorylation is induced by chemical LTD and inhibited by chemical LTP, and it is required for NMDA-mediated dispersal of PSD-95 clusters in cultured hippocampal neurons. We present evidence that phosphorylation of T19 destabilizes PSD-95 in spines, reduces membrane association of PSD-95, and is essential for AMPA receptor internalization and induction of LTD.
Materials and Methods
Antibodies and chemicals.
The following antibodies were used in this study: rabbit GSK-3β (Cell Signaling Technology), rabbit phospho-S9-GSK-3β (Cell Signaling Technology), rabbit pT19 PSD-95 (Abcam), rabbit GST (Santa Cruz Biotechnology), rabbit HA (Y11, Santa Cruz Biotechnology), mouse myc (9E10, Millipore), rabbit β-gal (ICN), mouse β-gal (Promega), mouse PSD-95 (clone K28/43, NeuroMab), rabbit GluA1-N (Millipore), mouse GluA2-N (Millipore), mouse pS-396 tau (Cell Signaling Technology), mouse tubulin (Sigma-Aldrich), and rabbit transferrin receptor (Sigma-Aldrich). Alexa-conjugated secondary antibodies were from Invitrogen. pS295 PSD-95 antibody was previously described (Kim et al., 2007). Roscovitine and SB216763 were from Tocris Bioscience. All other chemicals were purchased from Sigma-Aldrich unless otherwise stated.
Cell culture and transfection.
Hippocampal neurons were prepared from E18–E19 rats as described previously (Kim et al., 2007), and were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. COS-7 and HEK293 cells were maintained in DMEM supplemented with 10% fetal calf serum and transiently transfected with indicated plasmid constructs using Lipofectamine 2000 (Invitrogen).
DNA constructs.
WT-PSD-95 cDNA in pGW1 expression plasmid and WT-PSD-95-EGFP construct are previously described (Kim et al., 2007). pGW1-Myc-N-PDZ1 construct is previously described (Hsueh and Sheng, 1999). For electrophysiology experiments, PSD-95 constructs were subcloned into pCAG vector (chicken β-actin promoter with CMV-enhancer). PSD-95-EGFP was obtained in the pCAG and PSD-95-PAGFP was in the p16 expression vector. Rat GSK-3β cDNA was cloned from rat brain Match-Maker Yeast-two hybrid library (Clonetech) with PCR amplification, and subcloned into pGW1-HA vector. All PSD-95 and GSK-3β mutant constructs were generated with QuikChange II and QuikChange Multisite Site-Directed Mutagenesis Kits from Stratagene and verified by DNA sequencing. For the construction of GST-tagged fusion protein of PSD-95, the following two oligonucleotides were annealed and subcloned into BamH1/EcoR1 site of pGEX-4T1 vector (Pharmacia):
5′-GATCCCGCTACCAAGATGAAGACACGCCCCCTCTGGAACACAGCCCGGCCCACCTCTGA-3′
5′-AATTTCAGAGGTGGGCCGGGCTGTGTTCCAGAGGGGGCGTGTCTTCATCTTGGTAGCGG-3′.
For pSUPER-GSK-3β-RNAi construct, the following oligonucleotides were annealed and inserted into the HindIII/BglII sites of pSUPER vector (Brummelkamp et al., 2002):
GSK-3β-RNAi:
5′-GATCCCCGCTAGATCACTGTAACATATTCAAGAGATATGTTACAGTGATCTAGCTTTTTA-3′
5′-AGCTTAAAAAGCTAGATCACTGTAACATATCTCTTGAATATGTTACAGTGATCTAGCGGG-3′.
The pSUPER Luciferase-RNAi construct has been previously described (Zhang and Macara, 2006) and targets the sequence 5′-CGTACGCGGAATACTTCGA-3′.
In vitro GSK-3β kinase assay.
The PSD-95 N-terminal region (aa 13–29 of rat PSD-95) was GST-tagged and expressed in Escherichia coli BL21(DE3), and purified with GSH-agarose (Invitrogen). Purified GST-PSD-95 substrates (∼50 ng in 50 μl of kinase reaction volume) were incubated with 10 ng of purified GSK-3β (Millipore) in kinase buffer (25 mm Tris-HCl, pH 7.5, 5 mm β-glycerophosphate, 2 mm dithiothreitol (DTT), 0.1 mm Na3VO4, 10 mm MgCl2, 200 μm ATP) for 20 min at 30°C. After in vitro kinase reaction, protein samples were separated in 4–15% gradient SDS-PAGE gels (Bio-Rad) and analyzed with immunoblotting. Phosphorylation of T19 of PSD-95 was detected with immunoblotting with phospho-T19 PSD-95 antibody. After stripping the blot, GST-tagged fusion protein of PSD-95 were detected with GST antibody. GSK-3β kinase was detected with anti-GSK-3β antibody.
Chemical-LTD and chemical-LTP protocols.
All biochemical studies using hippocampal neurons were done with hippocampal neurons grown in 12-well plates at DIV 24–30. After the indicated drug treatments, hippocampal neurons were washed with ice-cold PBS twice, and lysed with 120 μl of DTT-containing Laemmli sample buffer per well. Protein samples were separated by SDS-PAGE and immunoblotted with the indicated antibodies. Chemical LTD was induced by bath application of 75 μm NMDA as previously described (Lee et al., 1998). For chemical LTP induction, cultured hippocampal neurons were incubated in ACSF (containing the following in mm: NaCl 168, KCl 2.6, HEPES 10, d-glucose 10, CaCl2 2, MgCl2 2, strychnine 0.001; pH 7.2) for 20 min at room temperature. Neurons were then incubated in ACSF without MgCl2 and with 200 μm glycine for 5 min as described previously (Lu et al., 2001; Man et al., 2003). After Gly/0 Mg2+stimulation, neurons were incubated in ACSF containing 2 mm MgCl2, and lysed with Laemmli sample buffer at indicated time points.
Immunostaining.
For endogenous PSD-95 staining, hippocampal neurons were fixed in methanol at −20°C for 15 min. After washing with PBS, hippocampal neurons were incubated with indicated antibodies in GDB buffer (30 mm phosphate buffer, pH 7.4, containing 0.1% gelatin, 0.3% Triton X-100, and 0.45 m NaCl). Alexa-488 and Alexa-568 secondary antibody (Invitrogen) were used for the visualization. Surface GluA1 labeling and NMDA-induced surface GluA2-internalization assay were performed as previously described (Kim et al., 2007). Primary mouse anti-PSD-95 antibody (clone K28/43) and Alexa-conjugated secondary antibodies were diluted in Tris-buffered saline (10 mm Tris, pH 7.4, 150 mm NaCl) containing 3% normal goat serum and 0.1% BSA.
Imaging of transfected hippocampal slices.
Organotypic slice cultures were prepared from p7 rat hippocampus and cultured as described previously (Nakagawa et al., 2004; Kim et al., 2007). Neurons were transfected by biolistic gene gun at DIV 3–5 (total 100 μg DNA; 25% PSD-95-EGFP construct, 25% DsRed as a transfection marker, 50% pCAG vector for EGFP imaging studies; 50% PSD-95 test construct, 50% DsRed for PAGFP imaging). PSD-95-EGFP images were acquired 24 h after transfection. For photoactivation experiments, PAGFP was activated by 405 nm laser pulses 48 h post-transfection and time-lapse images taken every 5 min over the course of 1 h at room temperature. Imaging was performed in oxygenated ACSF containing the following (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 26 NaHCO3, 1 NaH2PO4, and 11 glucose. All treatments were blinded to the observer for acquisition and quantification of images, and the confocal microscope settings were kept the same for all scans when fluorescence intensities were compared.
Image acquisition and quantification.
Confocal images from dissociated hippocampal cultures were obtained using a Zeiss inverted LSM 510 confocal microscope and 63× (NA 1.4) objective, whereas images from organotypic hippocampal slices were acquired on a Zeiss LSM 700 upright confocal microscope with a 40× (NA 1.4) objective. All treatments were blinded to the observer for acquisition and quantification, and the confocal microscope settings were kept the same for all scans when fluorescence intensities were compared. All measurements were performed using ImageJ (NIH) software unless otherwise indicated.
Membrane fractionation assay.
HEK293 cells were transfected with PSD-95-EGFP, T19A-PSD-95-EGFP or T19D-PSD-95-EGFP. Two days post-transfection, cells were harvested and prepared as previously described (Kim et al., 2007). Whole-cell, S2 cytosolic and P2 crude membrane fractions were collected and normalized for total protein. SDS-PAGE was used to resolve 5 μg of each fraction with immunoblots probed as indicated.
Coimmunoprecipitation assay.
HEK293 cells grown in 35 mm culture dishes were transfected with indicated DNAs using Lipofectamine 2000. Two days post-transfection, the cells were washed with cold PBS and lysed with cold RIPA buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 10 mm NaF, 1% NP40, 0.5% deoxycholate, and 0.1% SDS) containing protease inhibitors (PMSF, aprotinin, and leupeptin). After centrifugation at 4°C, cleared cell lysates were incubated with 1 μg of rabbit GFP antibody (Invitrogen) for 2–4 h at 4°C, then mixed with 20 ml of Protein A/G Plus agarose beads (Thermo) and washed four times with cold RIPA buffer. Immunoprecipitates were separated with a 4–15% gradient SDS-PAGE gel, and analyzed by immunoblotting using mouse PSD-95 antibody or mouse Myc antibody.
3H palmitate radiolabeling.
PSD-95 radiolabeling with 3H palmatic acid was performed as described previously (Fukata et al., 2006). Briefly, COS-7 cells in 35 mm dishes were transfected with wild-type or mutant PSD-95 constructs using Lipofectamine 2000. One day later, the cells were incubated in fresh DMEM with 10% fatty acid-free BSA for 30 min before the addition of 3H palmitate (PerkinElmer) at a concentration of 500 μCi/ml. After radiolabeling overnight, the cells were lysed in Laemmli sample buffer and separated by SDS-PAGE. The finished gels were stained and fixed with SimplyBlue colloidal Coomassie solution (Invitrogen), then soaked in Amplify fluorography solution (GE Healthcare) and dried. The dried gel was exposed to BioMax MS film (Carestream Health/Kodak) at −80°C and developed after 2 d.
Electrophysiology.
Electrophysiological recordings were performed on organotypic hippocampal slice cultures, prepared as described above. Neurons were transfected by biolistic gene gun at DIV 3–5 (total 100 μg DNA; 45% PSD-95 test construct; 45% empty pCAG vector; 10% 16 pl-EGFP marker, driven by β-actin promoter) and recorded 3 d after transfection. Recordings were performed in ACSF containing the following (in mm): 119 NaCl, 2.5 KCl, 4 CaCl2, 4 MgCl2, 26 NaHCO3, 1 NaH2PO4, 11 glucose, 0.1 picrotoxin, and 0.002–0.008 2-chloroadenosine, and bubbled continuously with 5% CO2/95% O2. Patch recording pipettes (2–4 MΩ) were filled with internal solution containing the following (in mm): 115 cesium methanesulfonate, 20 CsCl, 10 HEPES, 2.5 MgCl2, 4 ATP disodium salt, 0.4 GTP trisodium salt, 10 sodium phosphocreatine, and 0.6 EGTA, at pH 7.25. Simultaneous whole-cell recordings were obtained from a pair of transfected and neighboring untransfected CA1 pyramidal neurons during stimulation of Schaffer collateral pathways. For basal synaptic transmission experiments, presynaptic fibers were stimulated at 0.2 Hz. AMPA receptor EPSCs were recorded at −70 mV and NMDA receptor EPSCs at +40 mV in the presence of 0.01 mm NBQX. Each data point represents an average of 60 consecutive synaptic responses. For LTD experiments, presynaptic fibers were stimulated at 0.033 Hz during baseline recordings, and evoked AMPA EPSCs were measured at −70 mV. After at least 10 min of stable baseline, LTD was induced by 1 Hz stimulation (200 pulses) at −40 mV. Synaptic responses were then obtained under baseline conditions for another 30 min.
Statistical analysis.
Statistical significance was determined with Graphpad Prism software for all datasets, using the statistical analyses indicated in each figure legend. All values shown are the mean ± SEM.
Results
GSK-3 kinase activity mediates T19 phosphorylation of PSD-95
Previous work indicated that several N-terminal residues of PSD-95, including T19, are phosphorylated in cultured neurons (Morabito et al., 2004), and a recent study found elevated phosphorylation of PSD-95 on T19, as well as increased GSK-3β signaling, in TNiK knock-out mice (Coba et al., 2012). Given the importance of the N-terminal region of PSD-95 for targeting to dendritic spines and synaptic stability (Craven et al., 1999; El-Husseini Ael et al., 2002), we sought to identify the kinase that phosphorylates T19 of PSD-95 as a first step toward understanding the physiological significance of this post-translational modification. Using a PSD-95 T19-phosphospecific antibody, we found that cultured hippocampal neurons treated for ∼16 h with the GSK-3 inhibitors SB216763 (10 μm) or LiCl (10 mm) showed reduced phospho-T19 levels (∼65% decrease) without altering total PSD-95 (Fig. 1A). In contrast, roscovitine, an inhibitor of CDK5 that phosphorylates the S25 residue of PSD-95 (Morabito et al., 2004), had no effect on T19 phosphorylation. As expected, both SB216763 and LiCl also suppressed phosphorylation of Tau on S396, illustrating effective GSK-3 inhibition by these drugs (Fig. 1A). These data imply that GSK-3 activity is required for maintaining T19 phosphorylation of PSD-95 in neurons.
Figure 1.

Identification of GSK-3 as a PSD-95 T19 kinase. A, Cultured hippocampal neurons at DIV 25–28 were treated with DMSO, SB216763 (10 μm), LiCl (10 μm), or roscovitine (5 or 10 μm) for ∼16 h, and then immunoblotted with pT19 antibody. After stripping, the blot was reprobed with mouse total PSD-95 antibody and pS396 Tau antibody. Graph shows pT19 immunoblot intensity (corrected for total PSD-95 intensity) normalized to vehicle control. Statistical analysis was performed by one-way ANOVA, followed by Dunnett's test using DMSO treatment as control (n = 3, **p < 0.01). B, Phosphorylation of PSD-95 on T19 by GSK-3β in COS-7 cells. Wild-type PSD-95 was cotransfected with vector control (pGW1), HA-WT-GSK-3β, KD HA-KD-GSK-3β, or HA-R96A-GSK-3β. Thirty hours later, transfected COS-7 cell lysates were immunoblotted with pT19 antibody or HA antibody. After stripping, the blot was reprobed with mouse total PSD-95 antibody. C, In vitro phosphorylation of PSD-95 on T19 by recombinant purified GSK-3β. Purified GST-fused N-terminal peptide of PSD-95 (aa 13–29 of rat PSD-95) was mixed with recombinant GSK-3β. After the in vitro kinase reaction, protein samples were immunoblotted as indicated. Schematic diagram of GST-fused N-terminal peptide of PSD-95 is shown at bottom.
In heterologous cells, overexpression of wild-type GSK-3β or constitutively active S9A-GSK-3β, but not the K85M/K86I kinase-dead (KD) mutant, strongly promoted T19 phosphorylation of cotransfected PSD-95 (Fig. 1B). We also tested R96A-GSK-3β, a point mutant of GSK-3β which cannot phosphorylate substrates that require an initial “priming” by other kinases (Doble and Woodgett, 2003), and observed R96A-GSK-3β is able to phosphorylate T19 as efficiently as wt-GSK-3β and S9A-GSK-3β (Fig. 1B). We note that the +4 amino acid (Glu-23) is already negatively charged, so PSD-95 may mimic a constitutively primed GSK-3β substrate. Finally, we show that a GST fusion protein of the N terminus of PSD-95 was phosphorylated in vitro by purified GSK-3β, as measured by immunoblotting with phospho-T19 antibody (Fig. 1C). Thus GSK-3β is sufficient to phosphorylate PSD-95 on T19. Together, these data suggest that GSK-3β is a major kinase mediating PSD-95-T19 phosphorylation in hippocampal neurons.
Next, we studied the regulation of the phospho-T19 modification by synaptic activity. Treatment of cultured hippocampal neurons (DIV 24–28) with bath-applied NMDA (75 μm; “chemical LTD”; Lee et al., 1998) increased phosphorylation of T19-PSD-95 by >2-fold at 10 min, without affecting total PSD-95 levels (Fig. 2A). Conversely, a “chemical LTP” protocol (200 μm glycine/0 Mg2+; Lu et al., 2001; Man et al., 2003) led to a modest, but significant decrease in T19 phosphorylation (∼35% reduction by 45 min; Fig. 2B). Notably, this bidirectional regulation of T19 phosphorylation by chem-LTP and chem-LTD is opposite to that seen for S295 phosphorylation, which is reduced by chem-LTD and enhanced by chem-LTP (Fig. 2A,B; Kim et al., 2007). Additionally, we confirmed that chem-LTD also results in the activation of GSK-3β (Peineau et al., 2007), as judged by a decrease in the level of inhibitory S9 phosphorylation on GSK-3β (Fig. 2A). Thus, the phosphorylation of T19 in neurons is enhanced by NMDA stimulation that should induce AMPA receptor internalization and synapse weakening, coinciding with GSK-3β activation.
Figure 2.

Bidirectional regulation of phospho-T19 levels by chemical LTP and LTD treatments. A, Cultured hippocampal neurons at DIV 24–27 were stimulated with NMDA (75 μm, chem-LTD) for indicated times, and immunoblotted as indicated. After stripping, the phospho-T19 blot was reprobed with mouse total PSD-95 antibody. Graphs show the time course of pT19 band intensity relative to untreated control (normalized to total PSD-95) after NMDA stimulation. Statistical analysis was performed by one-way ANOVA, followed by Dunnett's test (n = at least 3 replicates per time point, ***p < 0.001, compared with untreated control). B, Cultured hippocampal neurons at DIV 24–28 were treated with 200 μm glycine in ACSF lacking MgCl2 (chem-LTP). After 5 min, neurons were replaced in regular ACSF and then lysed at indicated times, and immunoblotted as indicated. Statistical analysis was performed by one-way ANOVA, followed by Dunnett's test (n = 3 replicates per time point, *p < 0.05, compared with untreated control).
T19 phosphomutants exhibit altered stability in spines
What is the effect of T19 phosphorylation on PSD-95 in neurons? We compared the subcellular localization of wild-type PSD-95 and its T19 phosphomutants (phospho-null T19A, and phosphomimetic T19D) biolistically transfected into cultured hippocampal slices. The PSD-95 constructs were visualized via a C-terminal EGFP tag, whereas the transfected neuron was “filled” with cotransfected DsRed (Fig. 3A–C). We also tested mutants of PSD-95-S295, where we have previously shown phosphorylation by JNK1 promotes synaptic stability (Kim et al., 2007), as well as T19/S295 double mutants (Fig. 3D–I). We observed no differences in average spine density, spine length, or spine volume of neurons transfected with WT versus mutant PSD-95 (Fig. 3J–L). Further, no PSD-95 variants were significantly different from wild-type in their degree of localization in dendritic spines (as assayed by the ratio of EGFP intensity in spine heads relative to EGFP intensity in the adjacent dendrite (Fig. 3M)). These data indicate that, when overexpressed, the T19 and S295 mutants of PSD-95 have unaltered ability to concentrate in spine heads.
Figure 3.

T19 and S295 mutants of PSD-95 accumulate in spines and do not affect spine size or density. A–I, Sample images of dendrites of CA1 pyramidal neurons in rat cultured hippocampal slices transfected with DsRed and EGFP-tagged constructs of PSD-95, either wild-type or phosphomutant, as indicated. Confocal image stacks were quantified for (J) spine density, (K) spine length, and (L) spine volume. Amino acids at residue 19 and 295 are indicated in black (wild-type) or in color (amino acid substitutions). M, The ratio of EGFP intensity in spines versus adjacent dendritic shaft was quantified as a measure of PSD-95 enrichment at synapses. Statistical analysis was performed by one-way ANOVA, followed by Dunnett's test (n = 14, 9, 9, 5, 9, 6, 6, 5, and 8 neurons from left to right).
We next assessed the stability of PSD-95 at synapses using time-lapse imaging of PSD-95 tagged with photoactivatable-GFP (PAGFP; Patterson and Lippincott-Schwartz, 2002). Rat cultured hippocampal slices were biolistically transfected with PAGFP-tagged PSD-95 (WT, T19A, or T19D mutants), as well as DsRed to visualize neuron morphology. PAGFP was photoactivated specifically within the dendritic spine head (Fig. 4A–C), and the decay of PAGFP fluorescence (which should reflect exchange of fluorescent PSD-95-PAGFP in the PSD with nonfluorescent PSD-95 from elsewhere) was measured over time. The fluorescence of photoactivated wild-type PSD-95-PAGFP declined to 51% ±4.4% (SEM) of the initial activated intensity over the 55 min experimental time course (Fig. 4D,F), consistent with previous publications using this approach (Steiner et al., 2008; Sturgill et al., 2009). By comparison, the phospho-null PSD-95-T19A-PAGFP decayed more slowly, retaining 76 ± 4.8% of the PAGFP fluorescence in spines at 55 min, whereas the phosphomimetic T19D mutant decayed more rapidly, to an intensity of 23 ± 3.3% (Fig. 4D,F). At the end of monitoring, the fluorescence of all PAGFP constructs could be fully reactivated by a second set of 405 nm laser pulses, indicating that the loss of PAGFP signal was from dynamic exchange of PSD-95 molecules, not photobleaching of the fluorophore. These data suggest that phosphorylation of T19 accelerates PSD-95 turnover and promotes instability of PSD-95 in spine heads.
Figure 4.

Phospho-null and phosphomimic mutations of T19 alter PSD-95 stability in dendritic spines. A–C, Sample images of dendritic spines from CA1 pyramidal neurons expressing DsRed and (A) WT-PSD-95, (B) T19A-PSD-95, or (C) T19D-PSD-95 fused to photoactivatable GFP. Vertical dashed lines indicate initial activation laser and postexperimental reactivation with the 405 nm laser. D, Graph shows time course of PAGFP fluorescence (mean ± SEM) from individual spines of neurons expressing PSD-95-PAGFP constructs as indicated. Statistical significance for the T19A and T19D mutants was determined by two-way ANOVA with a Bonferroni post hoc test with comparisons to wild-type PSD-95-PAGFP at each time point (*p < 0.05, **p < 0.01, ***p < 0.001). Wild-type PSD-95-PAGFP, n = 18 spines; T19A-PSD-95-PAGFP, n = 13 spines; T19D-PSD-95-PAGFP, n = 14 spines. E, Time courses of PAGFP fluorescence for the PSD-95-T19/S295-PAGFP double phosphomutants. Data for wild-type PSD-95-PAGFP is replotted from D for reference. F, PAGFP intensities at the 55 min time point for the T19, S295, and T19/S295 mutants of PSD-95, compared with wild-type PSD-95 (gray bar). Statistical analysis was performed by one-way ANOVA, followed by a Bonferroni analysis comparing all treatments with wt-PSD-95-PAGFP (n = 18, 13, 14, 11, 12, 14, 11, 15, and 13 spines from left to right; *p < 0.05, **p < 0.01, ***p < 0.001).
For additional comparison, we examined the dynamic stability of PSD-95 mutants that are altered at the S295 site. The fluorescence of photoactivated phospho-null mutant S295A-PSD-95-PAGFP decayed more rapidly than wild-type PSD-95 (31 ± 4.3% fluorescence remaining at 55 min), whereas the phosphomimic mutant S295D-PSD-95 was more stable than wild-type (66 ± 2.4% at 55 min; Fig. 4D,F). In essence, S295A behaved like T19D, and S295D behaved like T19A, in this assay of PSD-95 stability at postsynaptic sites (Fig. 4D,F). We then investigated whether a functional interaction exists between T19 and S295 phosphorylation sites by testing T19/S295 double mutants of PSD-95. Interestingly, the double mutant T19A/S295A-PSD-95 with the stabilizing T19A and destabilizing S295A mutations showed a more rapid loss of PAGFP fluorescence than wild-type (33 ± 3.2% at 55 min; Fig. 4E,F). Conversely, the T19D/S295D double mutant showed a high stability in spines (63 ± 2.4% fluorescence remaining at 55 min), which is significantly higher than wild-type and much greater than the destabilizing T19D mutation alone (Fig. 4E, F). We also found the T19A/S295D and T19D/S295A double mutant combinations are not additive with regard to effect on PSD-95 synaptic stability (Fig. 4E,F). Together, these data indicate that the phosphorylation status of S295 is “dominant” over that of T19 with respect to the stability of PSD-95 in synapses, and dephosphorylation of S295 is required for phosphorylation of T19 to promote removal PSD-95 from synapses.
Constitutively active GSK-3β disperses PSD-95 clusters and reduces surface AMPAR subunits
The imaging studies of T19A- and T19D-PSD-95-PAGFP mutants suggest that phosphorylation of T19 promotes instability and mobilization of PSD-95 at postsynaptic sites. To investigate whether GSK-3β, which can phosphorylate T19, affects endogenous PSD-95 distribution, we transfected either wild-type GSK-3β or constitutively active (S9A) or KD (K85M/K86I) mutants of GSK-3β in cultured hippocampal neurons and imaged the cells 2 d later (DIV 16 + 2). Overexpression of constitutively active GSK-3β-S9A caused a ∼35% drop in the total intensity of PSD-95 staining in dendrites (Fig. 5A,B) as well as a decrease in surface GluA1 staining by ∼50% compared with neighboring untransfected neurons (Fig. 5C, D). KD-GSK-3β had no effect on dendritic PSD-95 staining intensity or surface GluA1 (Fig. 5A–D). We found that overexpressing wild-type GSK-3β did not significantly affect PSD-95 or surface GluA1 staining (Fig. 5A,B), likely due to GSK-3β activity being suppressed by S9 phosphorylation under basal culture conditions (Fig. 2). These data are consistent with GSK-3β phosphorylation reducing synaptic stability of PSD-95, leading to lower levels of PSD-95 in PSDs and reduced surface AMPA receptors.
Figure 5.

Overexpression of constitutively active S9A-GSK-3β reduces dendritic PSD-95 and surface GluA1. A, Cultured hippocampal neurons at DIV 16 were transfected with HA-WT-GSK-3β, constitutively active HA-S9A-GSK-3β, or KD HA-KD-GSK-3β. Two days later, transfected neurons were double-stained for HA and PSD-95. B, Bar graph showing dendritic immunostaining intensity of PSD-95, normalized to untransfected cells (t test, ***p < 0.001, compared with untransfected neurons; n = 15 neurons for each). C, Cultured hippocampal neurons at DIV 16 were transfected with HA-WT-GSK-3β, constitutively active HA-S9A-GSK-3β or KD HA-KD-GSK-3β. Two days later, transfected neurons were double-stained for HA and surface GluA1. D, Bar graph showing dendritic immunostaining intensity of surface GluA1, normalized to untransfected cells (t test, ***p < 0.001, compared with untransfected neurons; n = 17, 16, 17, and 16 neurons, left to right).
Are the effects of S9A-GSK-3β overexpression on PSD-95 and GluA1 specifically due to phosphorylation of T19? To address this question, we tested whether the effects of S9A-GSK-3β can be blocked by overexpression of T19A-PSD-95 (Fig. 6A,B). In control neurons transfected with WT-PSD-95-EGFP, S9A-GSK-3β overexpression caused a slight but significant decrease of EGFP intensity in spines (∼20%; Fig. 6C) and a ∼40% reduction in surface GluA1 immunostaining (Fig. 6D). In neurons cotransfected with mutant T19A-PSD-95-EGFP, overexpression of S9A-GSK-3β had no significant effect on PSD-95 or surface GluA1 (Fig. 6C,D). This ability of the T19A mutant overexpression to protect against GSK-3β-induced reduction of dendritic PSD-95 and surface GluA1 supports the hypothesis that the effects of GSK-3β on PSD-95 are mainly through phosphorylation of the T19 residue.
Figure 6.

Overexpression of T19A-PSD-95 prevents S9A-GSK-3β-induced loss of PSD-95 and surface GluA1. Rat cultured hippocampal neurons (DIV 16 + 3) were transfected as indicated, with empty vector or S9A-GSK-3β, and additionally cotransfected with either (A) WT-PSD-95-EGFP or (B) T19A-PSD-95-EGFP. Representative images of intrinsic EGFP fluorescence, surface GluA1 immunostaining, and GSK-3β immunostaining are shown. Signal intensity in the dendrites of transfected neurons was quantified for (C) PSD-95-EGFP (WT or T19A) and (D) surface GluA1. Statistical significance was determined by t test, comparing each treatment with and without S9A-GSK-3β (n = 18, 21, 15, and 22 neurons from left to right; *p < 0.05, **p < 0.01).
GSK-3β and T19 phosphorylation are required for NMDA-induced PSD-95 and AMPAR loss from synapses
NMDA receptor activation triggers the dispersal of a subset of PSD-95 from dendritic spines (Sturgill et al., 2009). Because NMDA stimulates GSK-3β and T19 phosphorylation of PSD-95 (Fig. 2A), we asked whether endogenous GSK-3β is required for NMDA-induced dispersal of synaptic PSD-95. We designed a GSK-3β shRNA that suppressed protein expression of cotransfected rat GSK-3β in COS cells (Fig. 7A). In cultured rat hippocampal neurons, transfection of the GSK-3β shRNA (DIV 15 + 3) reduced levels of endogenous GSK-3β protein by ∼70%, as measured by immunostaining with a GSK-3β antibody, whereas transfection of luciferase shRNA had no effect on GSK-3β immunoreactivity (Fig. 7B,C).
Figure 7.
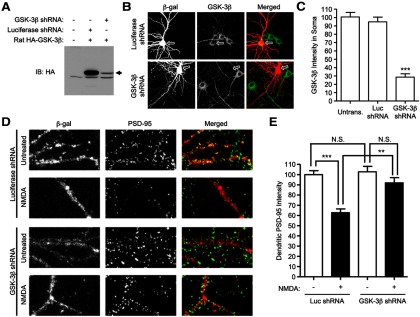
RNAi knockdown of GSK-3β reduces NMDA-induced loss of synaptic PSD-95. A, COS-7 cells were cotransfected with indicated expression vectors, and immunoblotted 60 h later for HA-GSK-3β. B, Cultured hippocampal neurons at DIV 15 were cotransfected with luciferase- or GSK-3β-shRNA constructs plus β-gal marker (4:1 shRNA: marker). Three days later, neurons were fixed and double-stained with mouse β-gal and rabbit GSK-3β antibody. Arrows point to soma of transfected neurons. C, Bar graph showing soma staining intensity of GSK-3β, normalized to untransfected cells (n = 12, 12, and 13 from left to right; t test, ***p < 0.001 compared with untransfected neurons or neurons transfected with luciferase shRNA). D, Cultured hippocampal neurons at DIV 15–16 were cotransfected with luciferase-shRNA, or GSK-3β-shRNA, plus marker β-gal. Three days later, transfected neurons were either untreated or treated with 75 μm NMDA for 10 min, and then double-labeled for β-gal and PSD-95. E, Bar graph showing dendritic immunostaining intensity of PSD-95, normalized to untransfected cells. Statistical analysis was performed by one-way ANOVA with a Bonferroni post hoc test for the indicated comparisons (n = 11 neurons for each condition; **p < 0.01, ***p < 0.001).
In neurons (DIV 16 + 3) transfected with GSK-3β shRNA and β-gal marker, there was no significant effect on mean intensity of PSD-95 staining under basal conditions, compared with luciferase shRNA-transfected control neurons. Treatment with NMDA (75 μm, 10 min) resulted in ∼35% reduction in mean immunofluorescence staining intensity of dendritic PSD-95 in control neurons transfected with Luciferase shRNA (Fig. 7D,E). Notably, total PSD-95 protein levels were unaffected by NMDA treatment, as assayed by Western blotting (Fig. 2A). However, in neurons transfected with GSK-3β shRNA, the NMDA-induced reduction of dendritic PSD-95 staining intensity was significantly curtailed (Fig. 7E), indicating that GSK-3β is required for the loss of PSD-95 from synapses following NMDA stimulation.
Given that the T19A mutation prevents the loss of PSD-95 from spines when cotransfected with S9A-GSK-3β (Fig. 6) and that GSK-3β knockdown prevents NMDA-induced PSD-95 dispersal (Fig. 7), we directly tested whether the T19A-PSD-95 mutant is resistant to synaptic release following NMDA treatment. Rat cultured hippocampal slices were transfected with WT-PSD-95-EGFP (Fig. 8A, left) or T19A-PSD-95-EGFP (Fig. 8A, right) and treated with NMDA [75 μm, 3 min, stopping the stimulation with the addition of APV (100 μm)]. By time-lapse imaging of spines of CA1 pyramidal neurons, we observed that the EGFP signal intensity for WT-PSD-95-EGFP decayed ∼40% over the 12 min following NMDA stimulation, similar to previous reports (Steiner et al., 2008; Sturgill et al., 2009), whereas the T19A mutant signal diminished by only ∼8% (Fig. 8B). These data strengthen the evidence that T19 phosphorylation is important for the declustering of synaptic PSD-95 following NMDA stimulation.
Figure 8.

NMDA-induced declustering of PSD-95 is blocked by overexpression of T19A-PSD-95. A, CA1 pyramidal neurons in rat cultured hippocampal slices were transfected with either WT-PSD-95-EGFP (left) or T19A-PSD-95-EGFP (right). One day later, transfected slices were treated with NMDA (75 μm, 3 min; stimulation stopped by addition of the NMDAR antagonist APV; 100 μm). Images were acquired before and at the indicated time points after NMDA application. B, Time course of WT-PSD-95-EGFP intensity (black) and T19A-PSD-95-EGFP intensity (blue) in spines following NMDA stimulation. Statistical significance was determined by two-way ANOVA with a Bonferroni post hoc test for each time point (n = 5 neurons each; *p < 0.05, **p < 0.01, ***p < 0.001).
Bath NMDA treatment is well known to stimulate AMPA receptor internalization (Lee et al., 1998; Malenka and Bear, 2004; Brown et al., 2005) and we have shown that this treatment stimulates GSK-3β and phosphorylation of PSD-95 on T19 (Fig. 1). Because PSD-95 is important for immobilizing AMPA receptors at postsynaptic sites (Bats et al., 2007), we hypothesized that T19 phosphorylation and ensuing destabilization of PSD-95 is mechanistically involved in NMDA-induced AMPA receptor internalization and LTD. Overexpression of the nonphosphorylatable mutant T19A-PSD-95 in cultured hippocampal neurons largely eliminated the internalization of GluA2 induced by NMDA, as measured by an antibody feeding internalization assay (Lee et al., 2002; Fig. 9A,B). In contrast, overexpression of wild-type PSD-95 did not significantly inhibit NMDA-induced GluA2 internalization under these conditions. Together, these data indicate that phosphorylation of PSD-95 T19 is important for NMDA-induced internalization of AMPA receptors, which is explicable by T19 phosphorylation causing the destabilization of PSD-95 in the PSD.
Figure 9.

Overexpression of PSD-95-T19A inhibits NMDA-induced GluA2 internalization. A, Cultured hippocampal neurons were transfected with EGFP, WT-PSD-95-EGFP, or T19A-PSD-95-EGFP at DIV 16. Three days later (DIV 19), surface GluA2 was live-labeled with GluA2-N antibody, washed, and then left untreated or treated with 75 μm NMDA for 10 min at 37°C. Internalized GluA2 receptors and remaining surface GluA2 receptors were visualized with Alexa-568 (red), and Alexa-647 (blue) secondary antibody, respectively (see Materials and Methods). Transfected neurons were identified with GFP-channel (green). Arrows point to the cell bodies of transfected neurons. B, Bar graph shows mean ± SEM. of integrated intensity of internalized GluA2 receptor. Statistical analysis was performed by one-way ANOVA with a Bonferroni post hoc test for the indicated comparisons (n = 14, 43, 9, 20, 8, and 20 from left to right; *p < 0.05, **p < 0.01, ***p < 0.001).
T19 phosphorylation of PSD-95 reduces membrane association
How does T19 phosphorylation lead to the destabilization of PSD-95 and its mobilization from the PSD? Clustering of PSD-95 at postsynaptic sites requires the N-terminal region, which contains determinants that are critical for head-to-head multimerization as well as palmitoylation of PSD-95 (Hsueh et al., 1997; Craven et al., 1999; El-Husseini et al., 2000; Christopherson et al., 2003). Given the effects of T19 phosphorylation on synaptic stability of PSD-95, and the location of T19 in the N-terminal region, we investigated whether T19 phosphorylation might affect palmitoylation or multimerization of PSD-95.
We first questioned whether T19 phosphorylation might affect the ability of PSD-95 to associate with membranes by assaying membrane versus cytosolic fractions of HEK293 cells transfected with wild-type PSD-95-EGFP, PSD-95-T19A-EGFP, or PSD-95-T19D-EGFP. The ratio of PSD-95 in the P2 membrane fraction versus the S2 supernatant fraction was used as a measure of membrane association (Fig. 10A,B). T19A-PSD-95 exhibited significantly greater membrane association relative to wild-type, whereas the T19D mutant showed significantly less than wild-type (Fig. 10A,B). As internal controls for our fractionation, we observed transferrin receptor was preferentially enriched in the P2 pellet, whereas tubulin was enriched in the soluble S2 fraction. These findings suggest that phosphorylation of T19 inhibits the association of PSD-95 with cell membranes, which might be a mechanism contributing to the destabilization and dispersal of PSD-95 from synapses.
Figure 10.

T19D phosphomimetic mutation reduces PSD-95 membrane association. A, Subcellular fractionation of HEK293 cells transfected as indicated. Whole-cell lysate, P2 membrane fraction, and S2 soluble fraction were collected as described previously (Kim et al., 2007), normalized for total protein and immunoblotted for PSD-95, tubulin, and transferrin receptor, as shown. B, Ratio of PSD-95 intensity (measured by densitometry) in P2/S2 fractions. Statistical analysis was performed by one-way ANOVA with a Bonferroni post hoc test for comparisons shown (n = 7 independent replicates; *p < 0.05, **p < 0.01, ***p < 0.001). C, COS-7 cells were transfected with EGFP-tagged PSD-95 constructs as indicated and metabolically labeled with 3H palmitate. Representative 3H fluorograph (top) and PSD-95 immunoblot (bottom) are shown with arrows indicating the position of PSD-95-EGFP. D, Average 3H fluorograph band densitometry for wild-type and mutant PSD-95-EGFP. Statistical significance was determined by one-way ANOVA with a Dunnett's post hoc test, comparing all mutants versus wild-type PSD-95-EGFP (n = 3 independent replicates; ***p < 0.001). E, HEK293 cells cotransfected with full-length PSD-95-EGFP and Myc-tagged PSD-95 truncation (N-Myc-PDZ1/2), with wild-type or T19 mutant residues as indicated. Representative immunoblots of HEK cell lysates (top), EGFP immunoprecipitates (middle), and coimmunoprecipitated Myc-tagged proteins (bottom) are shown. Filled arrows indicate the position of full-length PSD-95-EGFP constructs, whereas open arrows mark the N-Myc-PDZ1/2 fragments. F, Average densitometry measurements of coimmunoprecipitated N-Myc-PDZ1/2 protein, normalized to the amount of PSD-95-EGFP pulled down for each reaction. Statistical analysis was done by one-way ANOVA, with Dunnett's test comparing T19 mutants with wild-type (n = 5 independent experiments).
Palmitoylation of N-terminal residues cysteine-3 and cysteine-5 is known to promote interaction of PSD-95 with membranes and is required for PSD-95 accumulation in spines (Craven et al., 1999; El-Husseini et al., 2002). To test whether T19 phosphorylation affects the palmitoylation of PSD-95, COS-7 cells were transfected with wild-type, T19A-, T19D-, or the double C3, 5S-mutants of PSD-95 and incubated with 3H-palmitate overnight. After SDS-PAGE and 3H fluorography, we observed no significant difference in the incorporation of radioactive palmitate into wild-type, T19A-, and T19D-PSD-95, whereas the C3, 5S mutant showed a greatly reduced signal (not discernable above background; Fig. 10C,D).
Next, we examined PSD-95 multimerization. Consistent with previous studies (Hsueh et al., 1997; Christopherson et al., 2003), when coexpressed in HEK293 cells, full-length EGFP-tagged wild-type PSD-95 readily coimmunoprecipitated together with a Myc-tagged fragment containing the N-terminal region and the first two PDZ domains of PSD-95 (Myc-N-PDZ1/2; Fig. 10E,F). However, there was no difference in the amount of Myc-PDZ1/2 coimmunoprecipitated with T19A- or T19D-PSD-95 mutants (Fig. 10E,F). Similar results were obtained when precipitating the Myc-tagged fragment and blotting for coimmunoprecipitated EGFP-tagged PSD-95 constructs (data not shown). Taking these experiments together, T19 phosphorylation seems to negatively correlate with membrane association, but the mechanism underlying this effect is unclear and cannot be easily attributed to either the palmitoylation or multimerization state of PSD-95.
Phosphorylation of T19 is required for induction of LTD
Based on our finding that phosphorylation of T19 promotes PSD-95 mobilization and AMPA receptor internalization, we examined the effects of wild-type PSD-95 and T19A and T19D mutants on excitatory synaptic transmission in CA1 pyramidal neurons (Fig. 11). Cultured hippocampal slices at DIV 3–5 were biolistically transfected with PSD-95, T19A-PSD-95, or T19D-PSD-95, plus EGFP to mark the transfected cells. Simultaneous paired recordings of EPSCs were obtained 3 d post-transfection from neighboring untransfected and transfected CA1 pyramidal neurons. Wild-type PSD-95 strongly enhanced AMPA-EPSCs (∼4-fold) as previously reported (Schnell et al., 2002; Stein et al., 2003; Ehrlich and Malinow, 2004; Nakagawa et al., 2004; Elias et al., 2006; Futai et al., 2007; Kim et al., 2007; Xu et al., 2008), and overexpression of the phospho-null PSD-95-T19A and phosphomimetic PSD-95-T19D caused similarly large increases in AMPA-EPSC (Fig. 11A–C). All PSD-95 constructs also significantly enhanced NMDA-EPSCs though, consistent with earlier reports (Futai et al., 2007), the magnitude of this potentiation is much smaller than that observed for AMPA-EPSCs (Fig. 11A–C).
Figure 11.

Overexpression of PSD-95-T19A in CA1 pyramidal neurons blocks induction of LTD. A, Sample traces of evoked AMPA and NMDA receptor EPSCs recorded from transfected CA1 pyramidal neurons (identified by cotransfected GFP) versus untransfected controls. B, AMPA receptor (left) and NMDA receptor (right) EPSCs from transfected neurons versus neighboring untransfected controls. Average EPSC ±SEM for each treatment is indicated in red. PSD-95-T19A, n = 24 (AMPA), 22 (NMDA) pairs; wt-PSD-95, n = 33, 33 pairs; PSD-95-T19D, n = 23, 22 pairs. C, Summary of PSD-95-T19 mutant overexpression on AMPA-R-EPSCs (top) and NMDA-R-EPSCs (bottom), as indicated by the ratio of EPSC amplitudes between transfected and neighboring untransfected cells. Statistical significance was determined by paired t test (*p < 0.05). D, CA1 neurons were transfected with wild-type or mutant PSD-95 as indicated and recorded together with untransfected neighboring cells in double whole-cell patch-clamp mode. LTD was induced by a pairing protocol (see Materials and Methods). Statistical significance was determined by two-tailed t test on mean normalized EPSC (averaged over the last 10 min of postinduction recordings) for LTD experiments (n = 10, 8, and 9 pairs from left to right; **p < 0.01).
Because GSK3-β activity is primarily implicated in synaptic depression (Peineau et al., 2007), we examined LTD induced at Schaffer collateral-CA1 synapses by a pairing protocol. The normalized magnitude of LTD in neurons transfected with wild-type PSD-95 or PSD-95-T19D was similar to neighboring untransfected cells (∼50%; Fig. 11D). However, LTD induction was inhibited in cells overexpressing the mutant PSD-95-T19A, with the magnitude of depression only reaching ∼20% of untransfected cells (Fig. 11D). These results suggest that T19 phosphorylation of PSD-95 is necessary for the normal induction of LTD.
Discussion
Our findings offer a molecular basis for the involvement of GSK-3β in LTD: GSK-3β phosphorylates PSD-95 on T19, destabilizing PSD-95 in the PSD, which facilitates AMPA receptor mobilization and internalization from the synapse. This model is supported by several lines of evidence: First, T19 phosphorylation is induced by NMDA within minutes, in keeping with the time-frame of LTD. Second, the protein kinase we identify as responsible for T19 phosphorylation is GSK-3β, an enzyme that is activated during LTD and required for LTD induction (Peineau et al., 2007). Third, T19 phosphorylation reduces the association of PSD-95 with membranes and enhances turnover of PSD-95 in spines. Fourth, active GSK-3β causes a loss of PSD-95 from synapses and depletes surface AMPA receptors, both of which are blocked by overexpression of the nonphosphorylatable T19A-PSD-95 mutant. Finally, overexpression of T19A-PSD-95, but not wild-type PSD-95 or phosphomimetic T19D mutant, inhibits the induction of LTD.
N-terminal post-translational modifications of PSD-95 and synaptic stability
A number of post-translational modifications have been reported for the N-terminal region of PSD-95, which can affect the membrane targeting or synaptic stability of the protein (Craven et al., 1999; El-Husseini Ael et al., 2002; Morabito et al., 2004; Ho et al., 2011). We find the T19A-PSD-95 mutant is more strongly associated with plasma membranes than wild-type PSD-95, which in turn was more membrane-associated than T19D-PSD-95 (Fig. 7A,B). However, neither PSD-95 multimerization nor palmitoylation were noticeably affected in the T19 mutant constructs. Conceivably, electrostatic interference introduced by T19 phosphorylation may lower the affinity of PSD-95 for the plasma membrane.
In an elegant molecular replacement study, Xu et al. (2008) showed that PSD-95 mutants with small deletions or substitutions in the N-terminal region potentiated synaptic transmission but blocked LTD, a result similar to our findings with the PSD-95-T19A mutant. Thus, it is possible that some of the N-terminal mutations of Xu et al. (2008) could also be affecting T19 phosphorylation. Another study showed that overexpression of a PSD-95 mutant lacking the N-terminal PEST [proline (P), glutamic acid (E), serine (S), and threonine (T)-rich] motif impaired NMDA-induced AMPA receptor internalization (Colledge et al., 2003). This result was interpreted as the PEST sequence being important for ubiquitination and degradation of PSD-95 (but see Bingol and Schuman, 2004). Because T19 lies within the deleted PEST motif, the aforementioned results are also concordant with our conclusion that T19 phosphorylation is required for mobilization of PSD-95 from synapses, and consequently, AMPA receptor internalization. We did not see a reduction in total PSD-95 protein following NMDA/chemical LTD (Fig. 2A), arguing against the idea that degradation of PSD-95 is important for AMPA receptor endocytosis and LTD.
GSK-3-mediated destabilization of PSD-95 and other synaptic proteins during LTD
PSD-95 is a major scaffold protein of the PSD that promotes maturation and strength of excitatory synapses. According to our model, GSK-3β phosphorylation of PSD-95 T19 leads to weakened synapses. Our data show that T19 phosphorylation of PSD-95 is required for promoting AMPA receptor internalization and LTD. We hypothesize that mobilization of PSD-95 from the PSD, triggered by T19 phosphorylation, is a critical step that allows AMPA receptor/TARP complexes to “escape” more easily from the PSD (Bats et al., 2007). The untethered AMPA receptors could then diffuse more freely from the synapse to endocytic zones lateral to the PSD (Blanpied et al., 2002). In addition to activity-dependent modification and loosening of PSD-95, the level of AMPA receptors at synapses is also modulated by the phosphorylation status of the cytoplasmic tails of GluA and associated subunits, which affects their interaction with PSD-95 and other postsynaptic scaffolds (Lee et al., 2003; Malenka and Bear, 2004; Bats et al., 2007; Elias and Nicoll, 2007; Shepherd and Huganir, 2007; Newpher and Ehlers, 2008).
With overexpression of the T19A- or T19D-mutants of PSD-95 in cultured hippocampal slices, there were no differences in spine density, spine volume, or concentration of PSD-95 in spine heads compared with WT-PSD-95 (Fig. 3). There was also no significant difference between WT-, T19A-, or T19D-PSD-95 with regard to potentiation of synaptic transmission (Fig. 11A–C). We interpret these data to mean that the T19 phosphomutants traffic into spines and accumulate in the PSD to similar levels as wild-type PSD-95. However, unlike spine PSD-95-EGFP fluorescence and EPSC potentiation, which reflect steady-state levels of PSD-95 in postsynaptic sites, our PAGFP experiments measure the dynamic turnover of PSD-95 molecules in the spine head and they reveal that T19D is less stable than wild-type PSD-95, whereas T19A is more stable.
Why do the differences in postsynaptic stability of T19A versus WT versus T19D not manifest as differences in postsynaptic abundance of these PSD-95 variants? We can reconcile this apparent discrepancy by making the following assumption: spines have a limited capacity for PSD-95 at postsynaptic sites, and in neurons overexpressing PSD-95, this limited capacity is saturated no matter which variant is overexpressed. Thus the same ceiling level is reached in terms of spine accumulation and EPSC potentiation for WT, T19A, and T19D. However, WT-, T19A- and T19D-PSD-95 in the PSD do exchange at different rates with the nonsynaptic pool (as indicated by the PAGFP experiments). When one PSD-95 molecule leaves the PSD, it is quickly replaced by another PSD-95 molecule from the overexpressed nonsynaptic pool, thereby maintaining the saturated steady-state level of PSD-95 in the spine. This can explain why WT-, T19A-, and T19D-PSD-95 have different stabilities, but still show similar abundance in spines when overexpressed. However, we note that under non-steady-state conditions, such as S9A-GSK-3β overexpression (Figs. 5, 6) or NMDA stimulation (Figs. 7, 8), we do observe a net decrease in the amount of PSD-95 in spine heads and these effects depend on T19 phosphorylation.
The ability of GSK-3β to destabilize synaptic scaffolds may extend to inhibitory as well as excitatory synapses. It was recently reported that GSK-3β can phosphorylate gephyrin, the major scaffolding protein of GABAergic synapses, thereby inhibiting clustering of gephyrin and reducing inhibitory synaptic transmission (Tyagarajan et al., 2011). This raises the possibility that GSK-3β can regulate the function of distinct synapse types through phosphorylation of different scaffolding proteins.
PSD-95: a postsynaptic target of GSK-3β during LTD induction
We previously reported that phosphorylation of PSD-95 on S295 by JNK1 promotes the synaptic accumulation of PSD-95 (Kim et al., 2007). NMDA treatment induces rapid dephosphorylation of S295 mediated by PP1/PP2A phosphatase(s), and the phosphomimetic mutant PSD-95-S295D blocks AMPA receptor internalization and LTD (Kim et al., 2007). GSK-3β is activated by dephosphorylation of S9 by PP1/PP2A (Welsh and Proud, 1993; Zhang et al., 2003; Lin et al., 2007). It is widely believed that PP1 activity, and perhaps PP1 recruitment to stimulated synapses, are required for LTD (Mulkey et al., 1994; Morishita et al., 2001), though the exact molecular mechanisms of phosphatase action in LTD has been unclear. Our data show that PSD-95 could be an important convergent target of phosphatase signaling in LTD. By direct dephosphorylation of S295 and by indirect enhancement of T19 phosphorylation via GSK-3β, the concerted actions of PP1/PP2A can destabilize PSD-95 and promote its release from the PSD.
Moreover, our data with T19/S295 double mutants indicate that, with respect to dynamic stability of PSD-95 in synapses, phosphorylation of the S295 site is functionally “dominant” over phosphorylation of T19 (Fig. 4E,F). Most notably, T19D/S295D double mutant is relatively stable in synapses, implying that dephosphorylation of S295 is required before T19 phosphorylation can induce PSD-95 mobilization. On the basis of these results, we propose a model for PSD-95 dephosphorylation/phosphorylation in LTD (Fig. 12). Under basal conditions, JNK1 (which exhibits high basal activity in neurons) maintains phosphorylation of the S295 residue and promotes synaptic stability and retention of PSD-95 in the PSD. During LTD-inducing stimulation, PP1/PP2A phosphatases are engaged, leading to dephosphorylation of S295 as well as phosphorylation of T19 via activation of GSK-3β. This promotes removal from synapses of PSD-95 molecules that are both dephosphorylated on S295 and phosphorylated on T19. After the LTD stimulus wanes, the constitutively active JNK1 can rephosphorylate S295 and restore PSD-95 stability and accumulation at a reduced steady-state level. Requiring multiple concerted post-translational modifications for PSD-95 removal would allow synaptic activity to fine-tune the PSD (and thereby synaptic glutamate receptors) in a highly coordinated manner.
Figure 12.

Model for PSD-95 Phosphorylation Events During LTD. A, Under basal conditions in neurons, JNK1 promotes PSD-95 S295 phosphorylation stabilizing the molecule at the synapse, while GSK-3β activity is suppressed by Akt phosphorylation of S9. B, Weak or intermittent stimulation induces LTD, activating PP1 and PP2a phosphatases, which dephosphorylate PSD-95 S295 and GSK-3β. C, The newly activated GSK-3β phosphorylates PSD-95 on T19. D, PSD-95 molecules that have been dephosphorylated on the S295 residue and phosphorylated on T19 become destabilized and leave the PSD. E, After the LTD stimulus wanes, JNK1 can rephosphorylate the remaining PSD-95 molecules, stabilizing the spine at a new steady-state, depressed from the starting level of transmission.
Footnotes
We thank Dr Jin-Wu Tsai for custom MatLab algorithms used for spine analysis, Dr Yuanyuan Wang for experimental assistance with rat hippocampal slices, Dr Huaye Zhang for providing pSUPER Luciferase-RNAi construct, Dr Robby Weimer for PSD-95-EGFP, and Dr Baris Bingol for PSD-95-PAGFP. We also thank Dr Bingol for advice on image acquisition and analysis, and Dr Pascal Steiner for assistance with PAGFP imaging and for comments on this paper.
The authors declare no competing financial interests.
References
- Bats C, Groc L, Choquet D. The interaction between stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 2007;53:719–734. doi: 10.1016/j.neuron.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–347. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. A proteasome-sensitive connection between PSD-95 and GluR1 endocytosis. Neuropharmacology. 2004;47:755–763. doi: 10.1016/j.neuropharm.2004.07.028. [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Scott DB, Ehlers MD. Dynamics and regulation of clathrin coats at specialized endocytic zones of dendrites and spines. Neuron. 2002;36:435–449. doi: 10.1016/S0896-6273(02)00979-0. [DOI] [PubMed] [Google Scholar]
- Brown TC, Tran IC, Backos DS, Esteban JA. NMDA receptor-dependent activation of the small GTPase Rab5 drives the removal of synaptic AMPA receptors during hippocampal LTD. Neuron. 2005;45:81–94. doi: 10.1016/j.neuron.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Chen X, Nelson CD, Li X, Winters CA, Azzam R, Sousa AA, Leapman RD, Gainer H, Sheng M, Reese TS. PSD-95 is required to sustain the molecular organization of the postsynaptic density. J Neurosci. 2011;31:6329–6338. doi: 10.1523/JNEUROSCI.5968-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Sweeney NT, Craven SE, Kang R, El-Husseini Ael-D, Bredt DS. Lipid- and protein-mediated multimerization of PSD-95: implications for receptor clustering and assembly of synaptic protein networks. J Cell Sci. 2003;116:3213–3219. doi: 10.1242/jcs.00617. [DOI] [PubMed] [Google Scholar]
- Coba MP, Komiyama NH, Nithianantharajah J, Kopanitsa MV, Indersmitten T, Skene NG, Tuck EJ, Fricker DG, Elsegood KA, Stanford LE, Afinowi NO, Saksida LM, Bussey TJ, O'Dell TJ, Grant SG. TNiK is required for postsynaptic and nuclear signaling pathways and cognitive function. J Neurosci. 2012;32:13987–13999. doi: 10.1523/JNEUROSCI.2433-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/S0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven SE, El-Husseini AE, Bredt DS. Synaptic targeting of the postsynaptic density protein PSD-95 mediated by lipid and protein motifs. Neuron. 1999;22:497–509. doi: 10.1016/S0896-6273(00)80705-9. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini Ael-D, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS. Synaptic strength regulated by palmitate cycling on PSD-95. Cell. 2002;108:849–863. doi: 10.1016/S0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Elias GM, Nicoll RA. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol. 2007;17:343–352. doi: 10.1016/j.tcb.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Bredt DS, Fukata M. Protein palmitoylation by DHHC protein family. In: Kittler JT, Moss SJ, editors. the dynamic synapse: molecular methods in ionotropic receptor biology. Boca Raton, FL: CRC; 2006. [PubMed] [Google Scholar]
- Funke L, Dakoji S, Bredt DS. Membrane-associated guanylate kinases regulate adhesion and plasticity at cell junctions. Annu Rev Biochem. 2005;74:219–245. doi: 10.1146/annurev.biochem.74.082803.133339. [DOI] [PubMed] [Google Scholar]
- Futai K, Kim MJ, Hashikawa T, Scheiffele P, Sheng M, Hayashi Y. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95-neuroligin. Nat Neurosci. 2007;10:186–195. doi: 10.1038/nn1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J Neurochem. 2001;78:1219–1232. doi: 10.1046/j.1471-4159.2001.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- Ho GP, Selvakumar B, Mukai J, Hester LD, Wang Y, Gogos JA, Snyder SH. S-nitrosylation and S-palmitoylation reciprocally regulate synaptic targeting of PSD-95. Neuron. 2011;71:131–141. doi: 10.1016/j.neuron.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh YP, Sheng M. Requirement of N-terminal cysteines of PSD-95 for PSD-95 multimerization and ternary complex formation, but not for binding to potassium channel Kv1.4. J Biol Chem. 1999;274:532–536. doi: 10.1074/jbc.274.1.532. [DOI] [PubMed] [Google Scholar]
- Hsueh YP, Kim E, Sheng M. Disulfide-linked head-to-head multimerization in the mechanism of ion channel clustering by PSD-95. Neuron. 1997;18:803–814. doi: 10.1016/S0896-6273(00)80319-0. [DOI] [PubMed] [Google Scholar]
- Hur EM, Zhou FQ. GSK3 signaling in neural development. Nat Rev Neurosci. 2010;11:539–551. doi: 10.1038/nrm2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Futai K, Jo J, Hayashi Y, Cho K, Sheng M. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron. 2007;56:488–502. doi: 10.1016/j.neuron.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/S0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/S0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36:661–674. doi: 10.1016/S0896-6273(02)01024-3. [DOI] [PubMed] [Google Scholar]
- Lin CF, Chen CL, Chiang CW, Jan MS, Huang WC, Lin YS. GSK-3beta acts downstream of PP2A and the PI 3-kinase-akt pathway, and upstream of caspase-2 in ceramide-induced mitochondrial apoptosis. J Cell Sci. 2007;120:2935–2943. doi: 10.1242/jcs.03473. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/S0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, D'Souza S, Wong TP, Taghibiglou C, Lu J, Becker LE, Pei L, Liu F, Wymann MP, MacDonald JF, Wang YT. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38:611–624. doi: 10.1016/S0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, Vandenheede JR, Mandelkow E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–321. doi: 10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- Morabito MA, Sheng M, Tsai LH. Cyclin-dependent kinase 5 phosphorylates the N-terminal domain of the postsynaptic density protein PSD-95 in neurons. J Neurosci. 2004;24:865–876. doi: 10.1523/JNEUROSCI.4582-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Connor JH, Xia H, Quinlan EM, Shenolikar S, Malenka RC. Regulation of synaptic strength by protein phosphatase 1. Neuron. 2001;32:1133–1148. doi: 10.1016/S0896-6273(01)00554-2. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Futai K, Lashuel HA, Lo I, Okamoto K, Walz T, Hayashi Y, Sheng M. Quaternary structure, protein dynamics, and synaptic function of SAP97 controlled by L27 domain interactions. Neuron. 2004;44:453–467. doi: 10.1016/j.neuron.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Newpher TM, Ehlers MD. Glutamate receptor dynamics in dendritic microdomains. Neuron. 2008;58:472–497. doi: 10.1016/j.neuron.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Peng J, Kim MJ, Cheng D, Duong DM, Gygi SP, Sheng M. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J Biol Chem. 2004;279:21003–21011. doi: 10.1074/jbc.M400103200. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Huganir RL. Postsynaptic organization and regulation of excitatory synapses. Nat Rev Neurosci. 2000;1:133–141. doi: 10.1038/35039075. [DOI] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci U S A. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim E. The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol. 2011;3:a005678. doi: 10.1101/cshperspect.a005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, Nicoll RA. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci. 2003;23:5503–5506. doi: 10.1523/JNEUROSCI.23-13-05503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60:788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill JF, Steiner P, Czervionke BL, Sabatini BL. Distinct domains within PSD-95 mediate synaptic incorporation, stabilization, and activity-dependent trafficking. J Neurosci. 2009;29:12845–12854. doi: 10.1523/JNEUROSCI.1841-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagarajan SK, Ghosh H, Yévenes GE, Nikonenko I, Ebeling C, Schwerdel C, Sidler C, Zeilhofer HU, Gerrits B, Muller D, Fritschy JM. Regulation of GABAergic synapse formation and plasticity by GSK3beta-dependent phosphorylation of gephyrin. Proc Natl Acad Sci U S A. 2011;108:379–384. doi: 10.1073/pnas.1011824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh GI, Proud CG. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem J. 1993;294:625–629. doi: 10.1042/bj2940625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR. Judging a protein by more than its name: GSK-3. Sci STKE. 2001;2001:re12. doi: 10.1126/stke.2001.100.re12. [DOI] [PubMed] [Google Scholar]
- Xu W, Schlüter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium.: evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–33077. doi: 10.1074/jbc.M212635200. [DOI] [PubMed] [Google Scholar]
- Zhang H, Macara IG. The polarity protein PAR-3 and TIAM1 cooperate in dendritic spine morphogenesis. Nat Cell Biol. 2006;8:227–237. doi: 10.1038/ncb1368. [DOI] [PubMed] [Google Scholar]