

Abstract
For several decades antibodies raised against specific proteins, peptides, or peptide epitopes have proven to be versatile and very powerful tools to demonstrate molecular identity in cells and tissues. New techniques of immunohistochemistry and immunofluorescence have improved both the optical resolution of such protein identification as well as its sensitivity, particularly through the use of amplification methodology. However, this improved sensitivity has also increased the risks of false-positive and false-negative staining and thereby raised the necessity for proper and adequate controls. In this review, the authors draw on many years of experience to illuminate many of the more common errors and problematic issues in immunohistochemistry, and how these may be avoided. A key factor in all of this is that techniques need to be properly documented and especially antibodies and procedures must be adequately described. Antibodies are a valuable and shared resource within the scientific community; it is essential therefore that mistakes involving antibodies and their controls are not perpetuated through inadequate reporting in the literature.
Antibodies, particularly for use in immunohistochemistry, represent one of the most powerful tools in modern biological science. They combine extremely high precision of identification at the protein level, with high sensitivity, and also localization at a cellular or even a subcellular scale. Although the technique of immunocytochemistry has been around for some 50 years (1), the methodology itself is still relatively crude, and our understanding of what factors influence specificity and sensitivity is often rudimentary. In the postgenomic era of the Internet we are inundated by information from companies offering large numbers of antibodies, mostly against peptides or recombinant proteins, all of which postulate very high specificity combined with rigorous controls. But how much of this should we believe, and what are the minimal controls that still need to be carried out to ensure adequate scientific rigor in our experiments? Referees and journal editors are becoming alarmed by the often superficial way in which antibody specificity is dealt with (2–4). This guide briefly discusses how antibodies are produced, how they function in the context of immunohistochemistry, and what controls and documentation are essential if a result is to be believed. Unfortunately, the scientific literature is pervaded by examples of erroneous results using antibodies, particularly in immunohistochemistry. Here we hope to alert the investigator and potential referees to the possible pitfalls that can be encountered.
What Are Antibodies? What Types Are There? How Are They Generated?
This article cannot hope to summarize the vast amount of very detailed literature concerning antibodies, their generation, and their functionality. There are numerous excellent reviews and books covering these topics (eg, Refs. 5–9). Here we intend to introduce only sufficient knowledge of antibodies to explain the issues that contribute to the way they work in our experimental systems and, consequently, also their limitations and potential artifacts. In general, antibodies are produced by B cells (or plasma cells) within the body as part of the humoral response to infection. Antibodies circulate in the blood or in peritoneal fluid, or may be attached to the surface of lymphocytes, and serve to interact specifically with foreign antigens, causing these to be ingested by phagocytosis.
Antibodies are of several types, the most common being IgG, which possesses 2 larger “heavy” chains, each linked to shorter chains by disulfide bridges (Figure 1). The ends of the chains form a hypervariable paratope (Figure 1), which can specifically recognize a small 3-dimensional differentially charged surface (the immunological epitope) of its cognate immunogen (the protein used for immunization), which we refer to as the “antigen” (meaning “antibody-generating” molecule). Upon infection or immunization, specific IgG molecules, and the cells producing them, are clonally selected, and variability can be amplified by recombination and site-specific mutation within these cells. Immunization of a living mammal with a large immunogen gives rise to so-called polyclonal antisera, because many different IgGs are generated, each recognizing a different 3-dimensional epitope within the same immunizing protein. For comprehensive details of immunization procedures, see Harlow and Lane (6, 7). Antisera are the serum or sometimes plasma fractions from the blood of immunized animals. The IgG fraction within the antiserum may be further purified crudely, for example, using the IgG-specific binding properties of Protein A or Protein G, or by differential ammonium sulfate precipitation. Alternatively, an antiserum can be purified quite specifically by using affinity chromatography binding to the original immunizing antigen. Such affinity-purified antisera, although highly enriched in the specific IgG, may have lost concentration (titer) because the highest affinity antibodies do not elute well from the columns, and/or may be structurally damaged by the exposure to the very acidic pH needed to elute the antibodies from the affinity column. In general, species are chosen for immunization, which are evolutionarily distant from either the species of the immunizing antigen, and/or the species in which the antibodies are to be applied. Most commonly, polyclonal antibodies are raised in rabbits, guinea pigs, donkeys, goats, or sheep, although other species (eg, rats or chicken) may also be used.
Figure 1.

Diagrammatic representation of an IgG molecule to indicate terms used in the text. Fab fragments are those generated by papain cleavage and comprise only a single antigen-combining site, whereas F(ab)2 fragments are generated by pepsin cleavage and have 2 antigen-combining sites.
Monoclonal antibodies are created in much the same way as polyclonal antibodies by the immunization of living mice (or sometimes rats). However, once a sufficient titer of the specific polyclonal antibody is attained, the animals are killed, their spleens are removed, and the individual antibody-producing lymphocytes (each producing a different individual but specific IgG) from the spleen are immortalized by fusion with tumor cells, to produce clonal cell lines (hybridomas) capable of making each specific IgG. Such clones are cultured in vitro to secrete into the culture medium only one (monoclonal) antibody that will recognize only a single immunogenic epitope. Whereas most hybridoma cells are used to make IgG in vitro by secretion into culture media, in the past, monoclonal antibodies could also be produced in vivo as ascites fluid often with advantageously high titers, although this procedure is ethically problematic (10).
In addition to IgGs, other Ig types may be generated. For example, the mucosal immune system of the gut or uterine lining generates preferentially IgA molecules in response to a mucosal surface immunogen. There are also IgD, IgE, and IgM types of antibody al though these are not relevant in the context of immunohistochemistry. Chicken have proven very useful, because they can make IgY molecules in response to immunization, which are transported to the egg yolk in large amounts, such that specific antibodies can be produced simply by collecting eggs, with more than 1 mg of pure IgY in every egg (11).
Properties of Different Antibodies and Immunogens
Individual monoclonal antibodies, or single IgG molecules, or the variable moieties of these, the so-called Fab fragments (fragment antibody-binding; peptidase-cleaved IgG variable regions; Figure 1), all bind to their single immunogenic epitope with high specificity but often only modest affinity (ca. 10−5 to 10−7 M). This means that they need to be applied in a relatively concentrated form (cell supernatant dilutions of the order of 1:1000 or 1:100, or even less are usual here), and washing procedures cannot be too stringent because the specific binding could be ruptured. Because of the opportunity for clone selection in the generation of monoclonal antibodies, it is possible also to obtain higher affinity antibodies, although these are less common. Polyclonal antisera, on the other hand, bind as polyvalent entities, usually recognizing multiple immunogenic epitopes on a single target protein. Antibody detection relies on secondary antibodies or similar molecular bridges, which build molecular networks between different IgG molecules binding to the same target, resulting in cooperativity and binding affinities that are much greater (eg, 10−8 to 10−12 M). This, in turn, allows for antibodies to be applied at higher dilutions (eg, 1:1000 to 1:100 000 or greater) and also permits much more stringent washing. Antibodies with high affinity, as in general most polyclonal antibodies, bind larger amounts of antigen with a greater stability in a shorter time than those with low affinity and are therefore preferred in immunohistochemistry (12).
In between these two extremes are many of the antibodies that we use today, because they can be generated quickly from just a DNA sequence. Such antibodies make use of a small peptide sequence generated by translation of a DNA or RNA sequence, followed by chemical synthesis. Usually such peptides are about 10–14 amino acids long and without much secondary structure. They often represent an unconstrained (flexible) region of a protein that is the equivalent of a single antigenic epitope (often a group of 4–6 amino acids with charged or hydrophobic side chains). Because they represent only single immunogenic epitopes, these antibodies, although polyclonal in terms of their production, are little different from monoclonal antibodies and are referred to as monotypic antibodies. Like many monoclonal antibodies, they often have only modest binding affinity and cannot be rigorously washed. A brief summary of the main kinds of antibody and their various advantages and disadvantages is given in Table 1.
Table 1.
Attributes of Different Common Antibody Types
| Antibody type | Production | Advantages | Disadvantages |
|---|---|---|---|
| Conventional polyclonal | Production time short and inexpensive. Require larger pure proteins as immunogens. | Recognize multiple epitopes on one antigen and can thus help amplify a signal from a target protein with low expression level. Comprises heterogeneous mixture of different IgGs; hence, higher affinity through cooperativity, and thus more resistant to rigorous washing. More tolerant of antigen variation; preferred choice for detection of denatured proteins. More stable over a broad pH and salt concentration. | Greater likelihood of cross-reactivity, high background, and immune mimicry. Batch-to-batch variability. |
| Monotypic polyclonal | Production time short. Uses small peptides as immunogens; hence, easy to start from DNA sequence. | Simple production procedure. Can tailor antibodies to specific peptide epitopes. | Single epitope recognition means often lower affinity, and high background and cross-reactivity, also due to hapten-recognizing IgGs. |
| Monoclonal | Production time long and expensive, requiring substantial technical skill. Procedure allows for selection of optimal antibodies. | A hybridoma is a constant, renewable source of identical antibodies; hence, increased reproducibility. Procedure allows for selection for optimal characteristics. Single specific IgG means lower background. | Often low concentration and modest affinity; hence, less rigorous washing possible. High epitope specificity means less tolerance of antigen variation or damage; thus highly susceptible to changes in pH and salt concentration. |
It must be appreciated that short peptide sequences alone are not strongly antigenic. The way such antipeptide antibodies are made is to covalently link the small peptides to a larger hapten protein (or other molecule), which itself may be immunogenic, such as keyhole limpet hemocyanin (KLH), BSA, or human thyroglobulin. Many hundreds of identical small peptides covalently linked by either their N- or C-termini can bind to one KLH molecule. In special examples, more than one peptide from the same DNA/RNA sequence (or protein) can be used, thus effectively recreating a true polyclonal situation, but this is not common. What needs to be remembered for all such antibodies, is that they are mostly monotypic (having relatively low affinity) and also that many IgG molecules are present in the resulting antiserum that recognize the hapten molecule (eg, KLH). These are deliberately chosen for this purpose, because haptens act as a kind of adjuvant, inducing a greater immune response. Where the antigen is very small, for example, for some neurotransmitters or amino acids, the chemistry by which the antigen molecule is attached to the hapten becomes important and may need to be taken into account during tissue fixation for immunohistochemistry (see Figure 2).
Figure 2.

Importance of fixation for small molecule immunohistochemistry. A and B, To generate specific antibodies, the immunogen was linked to a larger protein by carbodiimide. Only when carbodiimide is used with the fixative (4% formaldehyde) (panel B), will the antigen be presented to the antibody in the “right” form. C, Serotonin is not fixed well with strong complexing fixatives such as acrolein (compare panel D, where the mild fixative 4% paraformaldehyde was used). In contrast, the small peptide TRH is only fixed by rapid strong fixation, here using acrolein (cf. panels E and F).
Sometimes it is convenient to use a recombinant protein made in Escherichia coli or in a eukaryotic cell line as immunogen. These can produce very good antibodies, but again there are aspects that require consideration. First, such proteins may contain additional peptide sequences used for their purification (eg, GST; glutathione-S-transferase), which may not subsequently be completely removed and may contribute to the polyclonal pool of IgG. Second, as with the peptide sequences above, such recombinant proteins are often denatured because of the way they are purified, eg, using 8 M urea, and consequently the immunogenic epitopes may only be presented by denatured proteins and not by proteins in their natural in vivo conformation. This may not matter if tissue sections are automatically denatured, eg, by using an antigen-retrieval procedure (13). But in many fixed or native preparations, the natural protein may simply not exhibit any of the epitopes against which the antibodies are directed. Occasionally, we have also observed that antibodies may be present in such preparations that recognize contaminating E. coli proteins, giving rise to a nonspecific signal, which, however, can be effectively blocked by additionally using a crude preparation of E. coli protein to preadsorb the antibodies.
A clever variant of these immunogen methodologies, which have been used to obtain a good effect in the localization of neurotransmitters in the brain, is to take account of the postfixation chemistry by using reagents such as formaldehyde, carbodiimide, or glutaraldehyde, in the coupling of the immunogen to its hapten. In this way the antibodies generated are able to recognize precisely the molecular epitope present in the brain tissue that results from fixation if the same agent is used (14, 15). Small neurotransmitters, like many small molecules, are notoriously difficult to localize in tissue sections, because they are quickly washed away unless there is rapid fixation or conversion to a more easily complexed product.
Adjuvants (eg, Freund's adjuvant) are complex molecular mixtures of known high immunogenicity (ie, they provoke a strong immune reaction, for example, because they contain pertussis toxin), which are added to the immunogen in order to stimulate the B cells and ensure the production of antibodies. Although in themselves they generally do not interfere with the use of antibodies for purposes such as immunohistochemistry, it is important to appreciate that all such antisera will also contain many IgG molecules specific for the adjuvant or similar entities. A more worrying aspect consequent to the boosting of the animal's immune system arises if the animal had been previously exposed to a different antigen. One striking case of this was observed when the first specific antibody titer had fallen to insignificantly low levels, and a different antigen was injected followed by Freund's adjuvant; this new serum contained significant amounts of both specific antibodies (16).
Finally, in this section, the increasing manufacture of antibodies by so-called genetic immunization needs mention. In this procedure, rats or rabbits are immunized by either sc or im injection, or by biolistic delivery (“gene-gun”), of a DNA expression construct which for some days at least will be transcribed and translated in vivo to yield a novel immunogenic protein, against which antibodies are generated (17). This method does not involve the use of adjuvants or haptens, although sequences may be incorporated into the DNA construct having a comparable effect, and most importantly, there is a very high likelihood that the immunogen is presented in its native conformation, and not denatured as is typical of most recombinant proteins. We used this approach successfully to produce antibodies recognizing a fetal antigen, against which we had failed to develop antibodies by more conventional procedures (17).
Validating Antibodies
At the outset it needs to be made clear that there is no single or perfect way to validate the specificity of antibodies used for immunohistochemistry. Multiple approaches need to be taken and judged by the “weight of evidence.” The following is a critical appraisal of the many methods that have been used to support the specificity of antibodies.
Omission of the primary antiserum
One of the most common controls used in immunohistochemistry is simply to leave out the primary antibody and otherwise to complete the protocol identically to sections in which the primary antibody is included. Although informative when high background staining is noted along with more specific staining, this is probably the least useful control for specificity, because it cannot distinguish any false-positive (see later) results. It is a control for possible nonspecific binding of the secondary antibody and says little about the specificity of the primary antibody. If this control is used, it is important to substitute the primary antibody by an equivalent amount of a preimmune serum, ideally from the same animal as was subsequently immunized, or failing that, at least normal serum from the same species. Nonetheless, this will not establish specificity of the generated staining: some other approach must be used. The reason for using a preimmune serum from the same animal is that for the sake of economy some antibody-producing companies reuse animals for antibody production, which may not have generated a good immune response to immunogens previously, although may in fact have moderate antibody titers against specific proteins in their blood or could generate these (see above). Nor is it acceptable to replace the primary antiserum simply with PBS solution. If preimmune serum is not available, replacement of the primary antibody by an IgG of the same class as the primary antibody may be an option. In this way one can determine whether the primary antibody IgG sticks to the section.
Preadsorption of antibodies using the immunizing antigen or equivalent
Here, usually prior to the application of the primary antibody solution to a tissue section, the solution is preincubated with the immunizing antigen (peptide or protein, although usually not covalently linked to a hapten) in considerable molar excess in order to quench any available specific binding sites of the IgG for the target protein in the section. The high molar excess is required usually because of the relatively low affinity that particularly monotypic antibodies have for their target, which will subsequently be in equilibrium with the immunogen during the usually long (eg, overnight) primary antibody incubation. Although mostly a convincing control for antibody specificity, this control will not exclude the possibility that antibodies are recognizing a so-called immune-mimetic epitope in the target tissue. This is a 3-dimensional molecular structure, which, although not of identical sequence to the immunogen, has a similar molecular “shape,” which would be equally recognized by the antibodies. Moreover, the higher the concentration of antibodies used (see below), the more likely that some immune-mimetic sequences may be detected. It must be recognized, however, that if not applied, the possibility of false-positive results greatly increases. It is better first to show that staining is blocked and then to investigate the possibility for immune-mimicry.
Western blotting
Western blots are frequently used (particularly in company catalogues) to prove the specificity of antibodies. Here it is important to note, that it is essential to show the whole molecular size range of the blot to exclude any unspecific binding to proteins which from their size could not be the specific antigen. If there is a trail of nonspecifically interacting proteins, then these will also bind antibodies in tissue sections. It is not appropriate to perform Western blots with a single purified recombinant protein, because with only mild washing any Coomassie stainable band (ie, in sufficient quantity) could bind nonspecifically and at low affinity, suggesting a specific immune reaction. For the same reason, it is important also to indicate just where Coomassie-stainable proteins are running on Western blots, because coincidence of the correct molecular size is not evidence of molecular identity, and any protein if in high enough concentration on a Western blot can bind antibodies, albeit with only modest affinity. Good controls using Western blots are those using, for example, extracts of cells transfected or not with appropriate gene constructs; or possibly a panel of cells or tissues with known positive and negative expression profile, yielding known specifically sized immunopositive bands, and no others. Even this may not be sufficient, as a recent article has shown (18), where protein sequencing was subsequently carried out to show that a band of the correct size on a Western blot was not in fact the expected band, but an immune-mimetic artifact. An excellent example of how antibodies can be evaluated using a Western blotting approach is described by Panjwani et al (19). It is worth noting that it is not always possible to obtain reliable Western blot results using all antibodies: some antibodies demand a native conformation of the target protein not present on electrophoresed and blotted proteins; some molecules are too small for Western blotting; and sometimes antigenic epitopes are occluded during the blotting procedure. Ironically, when controls for Western blots are used, preadsorption with purified antigens is usually applied.
Immunocytochemistry of transfected cells
One of the simplest and clearest of controls is to carry out transient transfections of cell lines using various expression constructs and then to apply cytospin immunocytochemistry for the transfected antigen (20) (Figure 3). The advantage of using transient transfection is that there are both expressing and nonexpressing cells for comparison within the same field of view. In Figure 3 we, in fact, used 3 different expression constructs, one for the target protein, a G protein-coupled receptor called RXFP1, and two for other more or less closely related G protein-coupled receptors (all immunocytochemically negative). Note also that here we have used immune sera from quite different rabbits (thus having a different background pool of polyclonal antibodies in addition to the specific monotypic antibodies raised against the common immunogen). This technique can also easily be adapted to quantitative flow cytometry, although there is a caveat here in that many commercial and other antisera contain sodium azide as a stabilizer; this substance is toxic to cells even at low concentration, and hence would kill living cells treated with such antibodies.
Figure 3.

Cytospin preparations of human embryonic kidney293T cells transfected with different expression plasmids encoding the human G protein-coupled receptors RXFP1, RXFP2, or HE6 (as indicated), and then subjected to immunocytochemistry using the double-peroxidase anti-peroxidase technique and polyclonal antibodies raised against the peptide antigen L7–2 (amino acids 278–292 of the human RXFP1 sequence), coupled to KLH. In panels A and B antibodies derived from 2 independent rabbits immunized with the same L7–2 antigen were used. Note in these panels using transiently transfected cells that there are both labeled (dark stain) and unlabeled (pale cell outlines) cells. Panels C and D represent negative controls with cells transfected with plasmids for non-cognate receptors. See Ivell et al (20) for more details.
Immunohistochemistry of tissues from gene-deleted animals
An ideal control, where feasible, is to make use of tissues in which the target gene product of interest has been genetically deleted. This is obviously only relevant for those species, such as mice, in which an appropriate knockout animal is available, or where there is a natural mutation. Because all other aspects of the tissue sections are identical, this indeed offers a perfect control situation. The use of knockout tissues is also ideal for validation of Western blot approaches (19), especially to ensure that erroneous secondary bands are not generated.
Parallel reproduction of results using alternative antibodies and/or techniques
Another very good control situation is to make use of 2 or more different antibodies recognizing different epitopes within the same target protein, and which thus should show coincidence of the cell type-specific signal. An example is shown in Figure 4, in which 3 different antibodies (L7–1, L7–2, and L7–3), each recognizing a different peptide sequence within the human RXFP1 receptor, indicate that the same cell types within the endometrium are being recognized (cf Figure 4, A, D, and G). Although ideally, one should use serial sections for such analyses, it is possible to use nonserial sections, as here, if the cell types are clearly identifiable, and the tissues used are structurally and hormonally equivalent and have been prepared and treated identically (21). Similarly, panels of monoclonal antibodies recognizing different epitopes within the same protein immunogen can be very effectively used in such a context. Here it is important to thoroughly check that antibodies do indeed recognize discrete peptide epitopes. In various company catalogues the same antisera may be marketed under quite different catalogue numbers. Also, it pays to check the peptide sequences given in such catalogues, that when cross-reactivity is suggested for several species, a BLAST search of the GenBank database will quickly show whether the antigenic peptide sequence is indeed present in the species sequence of interest.
Figure 4.

Sections (8 μm) of formaldehyde-fixed and paraffin-embedded endometrial tissue from a nonpregnant macaque monkey of reproductive age, stained immunohistochemically using the double-peroxidase anti-peroxidase technique and 3 different polyclonal antibodies raised against 3 different peptide epitopes within the human RXFP1 protein sequence (panels A–C, L7–1: amino acids 145–159; panels D–F. L7–2: amino acids 278–292; panels G–I. L7–3: amino acids 609–624). Using the immune sera (panels A, D, and G), significant cytoplasmic staining is observed in endometrial stromal cells. However, there is also staining in the nuclei of epithelial cells (arrowheads) using all 3 antibodies. All staining was eliminated when the primary antisera were replaced by the corresponding preimmune sera from the same rabbits (panels B, E, and H). However, only the epithelial nuclear staining disappeared when antibodies were preincubated with an excess of KLH (panels C, F, and I), showing that this quasi-specific staining was due to the presence of antibodies raised against the hapten KLH, which had been used in the generation of all 3 primary antisera. For more details; see Ivell et al (20).
A further very convincing variant of this approach is to make use of a quite different technique, such as in situ mRNA hybridization (Figure 5) or laser capture RT-PCR, to verify the cellular localization of the target protein of interest and its absence in other cell types. Also such techniques can effectively target different regions within a transcript (22), offering the added advantage of checking whether or not the signal represents a functional full-length transcript. A variant of this approach, most easily carried out in mice, is to take advantage of transgenically introduced unique proteins, such as ß-galactosidase, green fluorescent protein, or Cre-recombinase, the tissue-specific expression of which is driven by a promoter from an endogenously expressed gene of interest (23–25). Sometimes, one can also take advantage of the older techniques of enzyme-histochemistry (for the localization of specific substrate-metabolizing enzymes), or ligand binding, where a specific protein-protein interaction (such as hormone-receptor binding) can be demonstrated in a tissue section by hybridization with radioactively or fluorescently labeled ligands (26–28).
Figure 5.
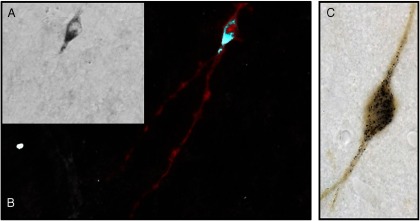
Double labeling of mRNA and protein in the same cell. A, GnRH. The image on the left shows GnRH mRNA using a biotinylated riboprobe stained with immunoperoxidase detection of the biotin in black. B, Inversion converts the black product to white, and staining using immunofluorescence in red shows that the same cell that possessed mRNA also possesses the product of that mRNA, GnRH peptide. C, Tyrosine hydroxylase. As in panels A and B, but the cell in this image has its mRNA stained black and the protein tyrosine hydroxylase in brown.
Antibody Titration, Signal Amplification, and Effects on Specificity
As already discussed, the ideal primary antibody for immunocytochemistry is one binding its specific antigen with very high affinity. This means that it can be applied at high dilution, such that any nonspecific interactions, which are generally low-affinity interactions, are obviated. Theoretically, for long incubation times, the number of antibody molecules binding the specific antigen in a section should only depend on the number of target molecules in the tissue and not on the antibody dilution. In practice, however, where incubation times are finite, very high dilution may also lead to signal attenuation. A way to overcome this is to make use of antibody amplification systems, such as the avidin-biotin-peroxidase complex method (29), the double-peroxidase anti-peroxidase/avidin-biotin-peroxidase complex method (30), or more recently tyramine amplification (reviewed in detail by Hoffman et al in Reference 31). Signal amplification allows a higher dilution of the primary antibody (and hence higher specificity), although it should be remembered, that amplification systems could also amplify mimicry with equal efficacy if this is not eliminated.
Homologous Immunohistochemistry
Often it is necessary to use mouse monoclonal antibodies on mouse tissues (Figure 6A), a procedure that is inevitably complicated by high levels of background staining when indirect immunohistochemical detection methods are used. This background staining is caused, in general, by binding of the secondary antimouse antibody to endogenous mouse tissue IgGs (Figure 6, C and F; in this case omission of the primary antibody is an appropriate and necessary control). Different approaches have been suggested to resolve this problem. In Figure 6, an example is illustrated whereby the primary mouse IgG has been purified and directly biotinylated, allowing the use of a secondary goat antibiotin antibody, now giving comparable staining specificity (Figure 6, D and G) to the equivalent heterologous situation (Figure 6, B and E). Another solution for this problem is to use monovalent IgG Fab fragments that recognize both the Fc and F(ab)2 regions of IgG as a preincubation step (Figure 7). This step does not affect the antigen specificity of the primary antibody. Instead of the normally used biotin-labeled goat-antimouse secondary antibody, a biotin-conjugated F(ab)2 goat-antimouse antibody is then applied to the tissue (Figure 7C and inset). This leads to almost complete elimination of the background staining (cf Figure 7A) (32).
Figure 6.
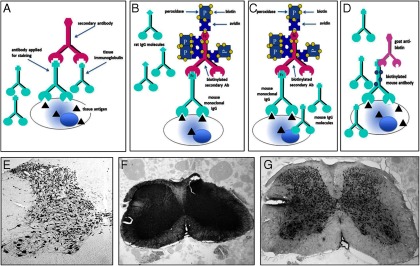
In conventional heterologous immunohistochemistry (panel A), there is little risk that the secondary antibody will cross-react with endogenous IgG in the tissue sections (here mouse-on-rat). Thus a biotin-peroxidase approach using a biotinylated second antibody (panel B) will give rise to good specific staining (panel E). However, for homologous immunohistochemistry (here mouse-on-mouse; panel C), such secondary antibodies will also react with the endogenous IgG in the tissue, giving rise to high unspecific background staining (panel F). One way that this problem can be overcome is to use a primary antibody that has been purified and directly biotinylated (panel D), combined with a heterologous goat-antibiotin antibody as secondary antibody. This would then be detected using a conventional conjugated antigoat tertiary antibody, to give rise to an image (panel G) little different from the heterologous situation (cf. panel E). Ab, antibody.
Figure 7.

Immunohistochemical labeling of mouse testis tissue with a mouse monoclonal antibody against the protein p62 (brown staining), a marker for autophagy. Examples of labeled spermatogonia and primary spermatocytes are indicated by arrowheads. A, Conventional immunohistochemical staining procedure, using normal goat serum and a biotinylated goat-antimouse secondary antibody. Note that except for the cytoplasm of the elongating spermatids all cells seem to stain. B, Omission of the primary p62 antibody, otherwise using the same procedure as in panel A; the staining in spermatogonia and spermatocytes has more or less disappeared, although background staining in most cells remains present. C, Normal goat serum has been replaced by a preincubation with goat-antimouse Fab fragments, and the biotinylated secondary goat-antimouse antibody has been replaced by a F(ab)2-labeled biotinylated goat-antimouse antibody; now there is specific brown staining in the spermatogonia and primary spermatocytes only and an absence of any background staining. The inset in panel C shows a detail of this. D, As negative control, the p62 antibody has been replaced by IgG of the same class as the primary antibody; otherwise the procedure is exactly as in panel C. Scale bars: panel A, 42 μm; panel D, 10.5 μm.
Specific Issues Relating to Immunofluorescence
The use of fluorescently tagged antibody systems, especially combined with high-resolution microscopy, such as laser confocal scanning microscopy, represents a revolution in morphological imaging. The great advantage of using such fluorescence-based systems is that they capture very narrow depths of field, and filters can be applied that eliminate signals from other wavelengths, making detection highly selective for relatively narrow wavelength ranges (ie, specific antibody signals). Data from multiple wavelength ranges can then be overlaid in the computer to assess colocalization. However, exactly the same problems regarding specificity and controls apply for immunofluorescence as for conventional immunohistochemistry. Additionally, however, immunofluorescence is accompanied by some problems of its own, the most common of which relates to quality and choice of suitable fluorophores, and corresponding excitation and emission filters (Figure 8). It is essential to control individually for each fluorophore being used, to ensure that the chosen wavelengths for the excitation and emission filters are indeed mutually discrete, especially when double labeling is planned. Figure 8 illustrates a typical artifact where the tail of emission from a bright green fluorophore falls within the range of emission from a red fluorophore. This could easily be confused with the specific signal coming from a red fluorophore (note: in the example illustrated in Figure 8, this slide had only one fluorescent molecule that had peak emission in the green range; no second fluorophore was applied, and yet there is evidently a marked signal corresponding to the red fluorophore). The most obvious giveaway for such an artifact is that the immunofluorescence for what should be the second primary antibody appears so similar to that for the first antigen that it is simply “too good to be true” in terms of its cellular colocalization and ranges of intensity within each structure. Biologically, it would be hard to imagine 2 different proteins to be always present in exactly the same amounts in exactly the same cells. Placement of a single-labeled specimen under both sets of filters controls for this “bleed-through” phenomenon (33). Often if the weaker of the 2 antigens is used with the green fluorophore in double-labeling with red and green, the problem can be avoided.
Figure 8.

Relative intensity and separation of fluorophores. Low-sensitivity methods for immunofluorescence detection using a Cy2-tagged secondary antibody usually only gives a signal in the green channel. However, when such signals are amplified, for example using a tyramine-based procedure, not only are strong signals evident in the green emission channel (panel A), there is often sufficient “bleed-through” fluorescence in the tail of the emission range of the green fluorophore now to give a signal that is detected also as red (panel B, here using very specific Texas Red filters), and the image appears as if it is double-labeled, even though in this case only a single, Cy2-tagged antibody has been used. A further control for this is to reverse the order of the red and green fluorophores, using the weaker antigen as green, and the stronger one as red. Note the highly suspicious morphologic identity of the 2 images.
False Negatives and False Positives
A false negative is where, on the basis of other information, one would expect to see an immunohistochemical signal, but this is not evident. Clearly, the antibodies themselves may not be specific or have the appropriate titer. But sometimes a false-negative situation may occur where the antigenic sequence of the target protein is in some way hidden from the antibody or presented in a different way because of fixation chemistry. One solution may be to use an antigen or epitope retrieval procedure that applies a strong chemical denaturation step or heat that exposes subcellular and cellular structures not otherwise accessible in conventional sections. For example, this approach has worked well for transcription factors and nuclear receptors located within the nuclei of cells. One caveat to this approach is that it will only work where the immunogenic epitope is equivalently denatured, ie, for small peptide immunogens, or for predenatured recombinant proteins. An alternative source of false-negative results is when the target antigen is relatively small and becomes washed out from thin sections during processing. A simple solution here can be to use different fixation chemistry, or possibly to employ thicker sections (despite loss of resolution).
Just as different fixation procedures can give rise to differing results, so also can antibodies behave differently applied to fixed and paraffin-embedded tissues, compared with frozen sections, or to cell culture preparations. Although the latter are like frozen sections in some ways, generally such cells require specialist permeabilization protocols (eg, using mild nonionic detergents, or saponin), which allow for entry of an antibody into the cell (for intracellular target antigens), without loss of cellular structural integrity. Insufficient or excessive permeabilization or inadequate subsequent washing steps can easily lead to false-positive or false-negative results, or even to false subcellular localization (34). Otherwise, the kinds of controls required are similar to those for conventional tissue sections.
A false positive occurs, where, despite apparently good and reproducible validation (see above), an antibody-specific signal is observed in cells in which no such signal is expected, or where other techniques (eg, in situ mRNA hybridization; immunohistochemistry of knockout animals) suggest that no signal should be present. The most obvious cause is an impure antigen or the presence of an immune-mimetic epitope (as described above). The former requires knowledge of the source of the immunogen; the latter is common when the epitope is identical to or conformationally similar to sequences in other unrelated proteins. Controlling mimicry can be difficult and in this regard can be minimized through use of polyclonal rather than lower affinity monotypic or monoclonal antibodieş where stringent washing is often not feasible. A further cause of false positives can be that antibodies are present that have been generated against moieties in the immunogen unrelated to the target antigen. For example, we had observed what looked like a very specific signal in cell nuclei of the macaque uterine epithelium (Figure 4 and Reference 20). We had used a peptide-specific antibody generated using KLH as hapten. However, this signal occurred only in sections from the macaque monkey, and not in human, and could be completely eliminated by preincubation of the antibodies with pure KLH (Figure 4, C, F, and I). Similar examples are known in which antibodies have been raised against recombinant proteins retaining a GST moiety in the immunogen, and which then recognize GST wherever this is expressed, eg, in the male reproductive tract. This may also occur when thyroglobulin or BSA has been used in preparation of the immunogen. Such issues may be resolved by using a panel of monoclonal antibodies, if available, recognizing different epitopes within the same target molecule, and which, because of selection, cannot recognize the hapten.
Conclusions
The list of controls and possible artifacts encountered in immunohistochemistry summarized here cannot be exhaustive. Ultimately, it comes down to the “weight of evidence” in favor of specificity or against it. But in publishing immunohistochemical results it is essential that this evidence is presented adequately, so that informed readers can make up their own minds. What is equally essential is that information on company websites is regarded cautiously and that companies begin to specify the methodology they have used for both staining and tissue preparation. In presentation of data using antibodies, authors should be aware that a reference to someone else's work where a different mode of tissue acquisition and/or a different method of detection in a different location is used, may not provide adequate evidence of control for their experiment. Authors should ensure that the antibodies used are fully documented with proper identification (eg, catalogue and preferably lot or batch numbers, indicating bleed and/or animal identity). Newly created antibodies require a full description of the immunizing antigen (as now demanded in the current Instructions to Authors for Endocrinology and other journals). A failure to provide rigorous documentation on identity and specificity of antibodies represents grounds for rejecting scientific work.
Acknowledgments
We thank the many students, colleagues, and authors who have directly and indirectly taught us much about the quirks and trials of using antibodies. We also want to acknowledge particularly the contributions of Ms Marga Balvers (Hamburg) and Dr. Wei Wei Le, a long-time colleague and friend of Dr. Gloria Hoffman, who was instrumental in devising some of the strategies presented in this article. She died in June, 2013.
This work was supported by the following agencies: Deutsche Forschungsgemeinschaft, National Health and Medical Research Council of Australia, Australian Research Council, and US National Institutes of Health-National Institute of Neurological Disorders and Stroke.
Disclosure Summary: None of the authors has anything to declare.
Footnotes
- GST
- glutathione-S-transferase
- KLH
- keyhole limpet hemocyanin.
References
- 1. Coons AH. Fluorescent antibody methods. Gen Cytochem Methods. 1958;1:399–422 [PubMed] [Google Scholar]
- 2. Saper CB. An open letter to our readers on the use of antibodies. J Comp Neurol. 2005;493:477–478 [DOI] [PubMed] [Google Scholar]
- 3. Saper CB. A guide to the perplexed on the specificity of antibodies. J Histochem Cytochem. 2009;57:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gore A. Editorial: antibody validation requirements for articles published in Endocrinology. Endocrinology. 2013;154:579–580 [DOI] [PubMed] [Google Scholar]
- 5. Neuberger MS. Antibody diversification by somatic mutation: from Burnet onwards. Immunol Cell Biol. 2008;86:124–132 [DOI] [PubMed] [Google Scholar]
- 6. Harlow E, Lane D. Antibodies: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1988 [Google Scholar]
- 7. Harlow E, Lane D. Using antibodies: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999 [Google Scholar]
- 8. Goding JW. Monoclonal Antibodies, Principles and Practice. Amsterdam: Elsevier; 1996 [Google Scholar]
- 9. Howard GC, Kaser MR. Making and Using Antibodies. Boca Raton, FL: CRC Press; 2006 [Google Scholar]
- 10. Hendriksen CF. Replacement, reduction and refinement in the production and quality control of immunobiologicals. ALTEX. 2006;23:187–190 [PubMed] [Google Scholar]
- 11. Spillner E, Braren I, Greunke K, Seismann H, Blank S, du Plessis D. Avian IgY antibodies and their recombinant equivalents in research, diagnostics and therapy. Biologicals. 2012;40:313–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lipman NS, Jackson LR, Trudel LJ, Weis-Garcia F. Monoclonal versus polyclonal antibodies: distinguishing characteristics, applications, and information resources. ILAR J. 2005;46:258–268 [DOI] [PubMed] [Google Scholar]
- 13. Shi SR, Shi Y, Taylor CR. Antigen retrieval immunohistochemistry: review and future prospects in research and diagnosis over two decades. J Histochem Cytochem. 2011;59:13–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pow DV, Crook DK. Extremely high titre polyclonal antisera against small neurotransmitter molecules: rapid production, characterisation and use in light- and electron-microscopic immunocytochemistry. J Neurosci Methods. 1993;48:51–63 [DOI] [PubMed] [Google Scholar]
- 15. Pow DV, Crook DK. Rapid postmortem changes in the cellular localisation of amino acid transmitters in the retina as assessed by immunocytochemistry. Brain Res. 1994;653:199–209 [DOI] [PubMed] [Google Scholar]
- 16. Clayton CJ, Hoffman GE. Immunocytochemical evidence for anti-LHRH and anti-ACTH activity in the “F” antiserum. Am J Anat. 1979;155:139–145 [DOI] [PubMed] [Google Scholar]
- 17. McKinnell C, Sharpe RM, Mahood K, et al. Expression of insulin-like factor 3 protein in the rat testis during fetal and postnatal development and in relation to cryptorchidism induced by in utero exposure to di (n-Butyl) phthalate. Endocrinology. 2005;146:4536–4544 [DOI] [PubMed] [Google Scholar]
- 18. Warthemann R, Eildermann K, Debowski K, Behr R. False-positive antibody signals for the pluripotency factor OCT4A (POU5FI) in testis-derived cells may lead to erroneous data and misinterpretations. Mol Hum Reprod. 2012;18:605–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Panjwani N, Mulvihill EE, Longuet C, et al. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE−/− mice. Endocrinology. 2013;154:127–139 [DOI] [PubMed] [Google Scholar]
- 20. Ivell R, Balvers M, Pohnke Y, et al. Immunoexpression of the relaxin receptor LGR7 in breast and uterine tissues of humans and primates. Reprod Biol Endocrinol. 2003;1:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stubbs SA, Hardy K, Da Silva-Buttkus P, et al. Anti-Müllerian hormone protein expression is reduced during the initial stages of follicle development in human polycystic ovaries. J Clin Endocrinol Metab. 2005;90:5536–5543 [DOI] [PubMed] [Google Scholar]
- 22. Raj A, Tyagi S. Detection of individual endogenous RNA transcripts in situ using multiple singly labeled probes. Methods Enzymol. 2010;472:365–386 [DOI] [PubMed] [Google Scholar]
- 23. Spergel DJ, Krüth U, Hanley DF, Sprengel R, Seeburg PH. GABA- and glutamate-activated channels in green fluorescent protein-tagged gonadotropin-releasing hormone neurons in transgenic mice. J Neurosci. 1999;19:2037–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hakamata Y, Tahara K, Uchida H, et al. Green fluorescent protein-transgenic rat: a tool for organ transplantation research. Biochem Biophys Res Commun. 2001;286:779–785 [DOI] [PubMed] [Google Scholar]
- 25. Wolfe A, Divall S, Singh SP, et al. Temporal and spatial regulation of CRE recombinase expression in gonadotrophin-releasing hormone neurones in the mouse. J Neuroendocrinol. 2008;20:909–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kauffman FC, Pickel VM, Sims KL, Bloom FE. Localization of nicotinamide adenine dinucleotide phosphate-dependent dehydrogenases in catecholamine-containing neurons of rat brain. Studies on the nucleus locus ceruleus. J Histochem Cytochem. 1974;22:20–28 [DOI] [PubMed] [Google Scholar]
- 27. Einspanier A, Müller D, Lubberstedt J, et al. Characterization of relaxin binding in the uterus of the marmoset monkey. Mol Hum Reprod. 2001;7:963–970 [DOI] [PubMed] [Google Scholar]
- 28. Körner M, Stöckli M, Waser B, Reubi JC. GLP-1 receptor expression in human tumors and human normal tissues: potential for in vivo targeting. J Nucl Med. 2007;48:736–743 [DOI] [PubMed] [Google Scholar]
- 29. Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem. 1981;29:577–580 [DOI] [PubMed] [Google Scholar]
- 30. Davidoff M, Schulze W. Combination of the peroxidase anti-peroxidase (PAP)- and avidin-biotin-peroxidase complex (ABC)-techniques: an amplification alternative in immunocytochemical staining. Histochemistry. 1990;93:531–536 [DOI] [PubMed] [Google Scholar]
- 31. Hoffman GE, Le WW, Sita LV. The importance of titrating antibodies for immunocytochemical methods. Curr Protoc Neurosci. 2008;45:2.12.1–2.12.26 [DOI] [PubMed] [Google Scholar]
- 32. Brown JK, Pemberton AD, Wright SH, Miller HR. Primary antibody-Fab fragment complexes: a flexible alternative to traditional direct and indirect immunolabeling techniques. J Histochem Cytochem. 2004;52:1219–1230 [DOI] [PubMed] [Google Scholar]
- 33. Bacia K, Petrášek Z, Schwille P. Correcting for spectral cross-talk in dual-color fluorescence cross-correlation spectroscopy. Chemphyschem. 2012;13: 1221–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spector DL. Subcellular localization of genes and their products. In: Spector DL, Goldman RD, Leinwand LA, eds. Cells. A Laboratory Manual. Vol 3 Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1998:98.1–98.20 [Google Scholar]