

Abstract
Smoking is a major risk factor for diabetes, cardiovascular disease, and nonalcoholic fatty liver disease. The health risk associated with smoking can be exaggerated by obesity. We hypothesize that nicotine when combined with a high-fat diet (HFD) can also cause ectopic lipid accumulation in skeletal muscle, similar to recently observed hepatic steatosis. Adult C57BL6 male mice were fed a normal chow diet or HFD and received twice-daily ip injections of nicotine (0.75 mg/kg body weight) or saline for 10 weeks. Transmission electron microscopy of the gastrocnemius muscle revealed substantial intramyocellular lipid accumulation in close association with intramyofibrillar mitochondria along with intramyofibrillar mitochondrial swelling and vacuolization in nicotine-treated mice on an HFD compared with mice on an HFD treated with saline. These abnormalities were reversed by acipimox, an inhibitor of lipolysis. Mechanistically, the detrimental effect of nicotine plus HFD on skeletal muscle was associated with significantly increased oxidative stress, plasma free fatty acid, and muscle triglyceride levels coupled with inactivation of AMP-activated protein kinase and activation of its downstream target, acetyl-coenzyme A-carboxylase. We conclude that 1) greater oxidative stress together with inactivation of AMP-activated protein kinase mediates the effect of nicotine on skeletal muscle abnormalities in diet-induced obesity and 2) adipose tissue lipolysis is an important contributor of muscle steatosis and mitochondrial abnormalities.
Cigarette smoking is the leading preventable cause of death and disability worldwide (1). Smoking is a major risk factor for cardiovascular disease, chronic obstructive pulmonary disease, and lung cancer (2–4). There is increasing evidence that smoking also contributes to nonalcoholic fatty liver disease (5, 6) and has direct adverse effects on muscle function (7, 8). Of further importance, the health risk associated with smoking, whether passive or active, is exaggerated by obesity, and smoking and obesity are the leading causes of morbidity and mortality worldwide (9, 10). The life expectancy of an obese smoker is 13 years less than that of a normal-weight nonsmoker (10). Furthermore, smoking lowers the body weight and body mass index, which make many people reluctant to quit smoking (10).
In a recent study, we demonstrated that twice-daily ip injections of nicotine (0.75 mg/kg body weight) significantly reduced body weight in mice on a high-fat diet (HFD) relative to mice fed HFD alone (11). Nicotine treatment also led to a reduction in body fat on mice on an HFD as determined by both dual-energy x-ray absorptiometry (DXA) and computed tomography scans (11). The effect of nicotine on weight loss in mice on an HFD was completely blocked by mecamylamine, suggesting a direct role of nicotine in preventing HFD-induced weight gain and abdominal fat accumulation in mice (11). This, at first glance, suggests that nicotine may prevent HFD-induced obesity. However, adipose tissue has the unique function of storing triglycerides in lipid droplets and, upon lipolysis, to provide free fatty acid (FFA) to other organs during a time of energy shortage (12). In obesity and other conditions where cellular lipid homeostasis is perturbed, lipolysis can contribute to ectopic lipid accumulation (13). Accordingly, increased adipose tissue lipolysis triggered by combined treatment with nicotine and an HFD could be expected to result in ectopic lipid accumulation in tissues such as liver and muscle. Consistent with this notion, we recently demonstrated that combined treatment with nicotine and an HFD triggers greater oxidative stress, activates hepatocellular apoptosis, and exacerbates HFD-induced hepatic steatosis (14). In the current studies, we tested the hypothesis that nicotine plus an HFD can also cause intramyocellular lipid (IMCL) accumulation and skeletal muscle abnormalities through increased adipose tissue lipolysis in male C57BL/6J mice fed an HFD with 34.9% fat (11), a commonly used model of diet-induced obesity (15–18).
Materials and Methods
Animals
Male C57BL/6 mice weighing 22 to 24 g (Taconic Farms) were used in all experiments. Mice were housed under controlled temperature (22°C) and photoperiod (12-hour light, 12-hour dark cycle) with free access to water and food. Mice were fed either a normal chow diet (NCD) with 5% fat (8.5 kJ/g; laboratory rodent diet 5001; Lab Diet) or HFD consisting of 26.2% protein, 26.3% carbohydrate, and 34.9% fat (21.9 kJ/g; D12492; Research Diets) for 10 weeks. Mice on either diet received twice-daily ip injections of nicotine (0.75 mg/kg body weight) or saline for 10 weeks. Nicotine liquid was purchased from Sigma Life Science and was covered with aluminum foil during storage to prevent light exposure. To determine whether nicotine-induced adipose tissue lipolysis mediated the ectopic fat deposition in the muscle, additional groups of mice on an HFD were given nicotine or saline in the presence or absence of acipimox (0.05% in the drinking water).
Animals were weighed weekly. The amount of food consumed per mouse was determined daily. Before killing, mice underwent DXA scanning as described previously (11). Mice were fasted overnight before euthanasia with a lethal injection of sodium pentobarbital (200 mg/kg body weight). Blood samples were collected from each animal by cardiac puncture immediately after death, and plasma was separated and stored for subsequent measurements of FFA. Gastrocnemius muscles were removed and weighed. Muscle pieces were snap-frozen in liquid N2 and stored frozen for subsequent measurements of triglycerides, oxidative stress, and changes in protein expression. The remaining portions of muscles were fixed in 2.5% glutaraldehyde for transmission electron microscopy (EM). Animal handling and experimentation were in accordance with the recommendation of the American Veterinary Medical Association and were approved by the Charles R. Drew University School of Medicine and Science Institutional Animal Care and Use Committee.
Measurements of plasma FFA, muscle triglyceride, and oxidative stress
Plasma FFA levels were measured by using Abcam's quantitation kit according to the manufacturer's protocol. Muscle triglyceride levels were measured by using Abcam's triglyceride quantitation kit according to the manufacturer's protocol. The muscle reduced glutathione (GSH) to oxidized glutathione (GSSG) ratio was measured using a commercial kit (BIOXYTECH GSH/GSSG-412 assay kit; OXISResearch, a division of Oxis Health Products, Inc), as described previously (14, 19). The GSH to GSSG ratio is inversely related to reactive oxygen species (ROS) levels.
Muscle ultrastructure
Gastrocnemius muscle morphology was evaluated by transmission EM. Portions of glutaraldehyde-fixed muscles were further diced into small pieces, postfixed in 1% osmium tetroxide, and embedded in Epon 812 (Polysciences). Thin sections from selected tissue blocks were sectioned with an LKB ultramicrotome, stained with uranyl acetate, and examined with an Hitachi 600 EM. An observer who was unaware of the treatment assignment took and analyzed the electron micrographs. From each treatment group, 80 micrographs (20 micrographs per mouse) were selected for ultrastructural analysis. The point-counting method (20, 21) was used to estimate the volume density (Vv) (the volume of a given cellular component per unit cell volume) of IMCL and abnormal mitochondria, such as vacuolated or swelled mitochondria with broken cristae, by superimposing a transparent overlay bearing a double-lattice grid on electron micrographs of muscle fibers. The Vv was obtained by dividing the points on IMCL and abnormal mitochondria by the total number of points counted over the muscle fiber. Values are expressed as percentages of the myofibrillar volume (percent Vv), obtained by multiplying Vv values by 100.
Numerical density of IMCL droplets and abnormal mitochondria (number per unit area) were determined according to an unbiased 2-dimensional rule (20, 21) by counting the number of lipid droplets or the abnormal mitochondria within a reference area encompassing 250 μm2 of muscle fiber cytoplasm after superimposing a transparent overlay bearing a lattice grid onto electron micrographs of the muscle fiber. Only the profiles intersected by inclusion edges (upper and right borders) and within the frame were counted. The mean diameter of the lipid droplets was obtained by direct measurements of its largest cross-sectional profiles in the electron micrograph.
Western blotting
Western blotting was performed using muscle lysates as described previously (19, 22–24). In brief, proteins (50–80 μg) were separated on a 4% to 12% SDS-polyacrylamide gel with 2-(N-morpholino)ethanesulfonic acid buffer purchased from Invitrogen at 200 V. The gel was transferred onto an Immunoblot polyvinylidene difluoride Membrane (Bio-Rad) overnight at 4°C. Membranes were blocked in blocking solution (0.3% Tween 20 in Tris-buffered saline [TBS] and 10% nonfat dry milk) for 1 hour at room temperature and then probed using rabbit monoclonal phospho–AMP-activated protein kinase (p-AMPK) (1:300), total AMPK (1:300), phospho–acetyl-coenzyme A-carboxylase (p-ACC (1:200), and total ACC (1:200) antibodies from Cell Signaling Technology for 1 hour at room temperature or overnight at 4°C with constant shaking. After three 10-minute washes in TBS-T buffer, membranes were then incubated in antirabbit IgG secondary antibody (Amersham Biosciences) at a 1:2000 dilution. All antibodies were diluted in blocking buffer. For immunodetection, membranes were washed three times in TBS/Tween 20 wash buffer, incubated with enhanced chemiluminescence solutions per the manufacturer's specifications (Amersham Biosciences), and exposed to Hyperfilm ECL. Band intensities were determined using Quantity One software from Bio-Rad. Data for p-AMPK and p-ACC levels were normalized to total AMPK and total ACC, respectively.
Statistical analysis
Statistical analyses were performed using the SigmaStat version 2.0 Program (Jandel Corporation). Data are presented as mean ± SEM. We used one-way ANOVA to compare group differences. If overall ANOVA revealed significant differences, post hoc (pairwise) comparisons were performed using Tukey's test. Differences were considered significant if P < .05.
Results
Effects of nicotine plus HFD on body weight, food intake, plasma FFA, and muscle oxidative stress and triglyceride levels
There was no difference in body weight in mice on an NCD with or without nicotine (Figure 1A). Mice fed an HFD exhibited a progressive increase in body weight relative to mice fed an NCD in the absence or presence of nicotine (Figure 1A). As expected from our previous studies (11, 14), nicotine significantly (P < .05) reduced body weight in mice on an HFD compared with mice on an HFD plus saline (Figure 1A). By 10 weeks of combined treatment with nicotine and HFD, mean body weight was reduced by 17.6% relative to mice fed the HFD alone. This reduction in body weight in the combined treatment group was further associated with reduced food intake (Figure 1B). With 10 weeks of treatment, a DXA scan showed a significant (P < .05) decrease in the total amount of fat after combined treatments with nicotine and HFD (8.0 ± 1.2 g) compared with mice on an HFD alone (14.8 ± 1.2 g). There was no difference in total fat content in mice on an NCD in the absence (5.5 ± 0.6 g) or presence (5.2 ± 0.05 g) of nicotine.
Figure 1.

Panel A, Time course of body weight measured over 10 weeks of nicotine treatment in mice fed an NCD or HFD. By 10 weeks of combined treatment with nicotine and HFD, mean body weight was significantly (P < .05) reduced relative to mice fed an HFD and saline. Panel B, Average food intake in mice fed an NCD or HFD in the absence or presence of nicotine. Panel C, Plasma FFA levels in various treatment groups. Panels D and E, Addition of nicotine to HFD also caused a significant increase in muscle triglyceride levels (panel D) and muscle oxidative stress as indicated by a low GSH to GSSG ratio (panel E). Values are given as mean ± SE of 5–6 mice per group. Means with different letters are significantly (P < .05) different from each other. Means with the same letter are not different.
Compared with mice fed with NCD in the presence or absence of nicotine, mice on an HFD exhibited a significant (P < .05) increase in plasma FFA levels (Figure 1C). Combining HFD with nicotine caused a further significant (P < .05) increase in plasma FFA levels (Figure 1C). There were no differences in muscle triglyceride levels in mice fed with either NCD with or without nicotine or HFD alone (Figure 1D). However, the HFD when combined with nicotine caused a significant (P < .05) increase in muscle triglyceride levels (Figure 1D). To test whether nicotine plus HFD causes greater oxidative stress, we measured the muscle GSH to GSSG ratio, which is inversely related to ROS levels. As shown in Figure 1E, compared with mice fed an NCD with or without nicotine or HFD, combined treatment with nicotine and HFD resulted in a significant (P < .05) increase in muscle oxidative stress as indicated by a low GSH to GSSG ratio.
Nicotine plus an HFD causes IMCL accumulation and mitochondrial abnormalities in obese mice
We performed transmission EM to evaluate myofibrillar architecture in various treatment groups (Figure 2). Gastrocnemius muscle from mice fed NCD in the absence (Figure 2A) or presence of nicotine (Figure 2B) exhibited normal myofibrillar architecture and sarcomeric pattern with abundant normal looking mitochondria and no IMCL accumulation. Although HFD plus saline did not affect myofibrillar architecture (Figure 2C), it induced varying degrees of muscle abnormalities when combined with nicotine. These included IMCL accumulation, usually found in close association with intramyofibrillar (IMF) mitochondria (Figure 2, D and E), IMF mitochondrial vacuolization (Figure 2F), and swelling with broken cristae (Figure 2G). IMCL accumulation was not seen in the subsarcolemmal (SS) region, and SS mitochondria were not affected by HFD and nicotine treatment. Treatment with acipimox, a lipolysis inhibitor, prevented skeletal muscle changes in response to nicotine plus HFD, as assessed by comparable myofibrillar architecture between mice fed an HFD in the absence (Figure 3A) or presence of acipimox (Figure 3B). It also fully attenuated nicotine plus HFD-induced IMCL accumulation and mitochondrial abnormalities (Figure 3, D vs C).
Figure 2.

A and B, Representative transmission electron micrographs of gastrocnemius muscle from mice fed an NCD without (A) or with (B) nicotine exhibit normal myofibrillar architecture and sarcomeric pattern with abundant normal-appearing IMF mitochondria and no IMCL accumulation. C, Appearance of muscle fiber in mice fed an HFD is similar to that seen in mice on an NCD in the absence (A) or presence (B) of nicotine. D, Representative electron micrograph shows IMCL accumulation in close association with IMF mitochondria (arrow). E, A higher-magnification view of the area marked in D showing IMCL accumulation (asterisk). F and G, Nicotine plus HFD also causes mitochondrial vacuolization (F; arrow) and mitochondrial swelling with broken cristae (G; asterisk). Scale bar, 1 μm (A–D, F, and G) and 0.4 μm (E). Data are representative of 4 mice in each group.
Figure 3.

A and B, Representative transmission electron micrographs of gastrocnemius muscles from mice fed an HFD (A) and mice receiving acipimox plus HFD (B) show normal ultrastructural myofibrillar architecture, with normal morphology of both SS and IMF mitochondria. C, As expected, nicotine plus HFD treatment shows IMF mitochondrial abnormalities, including IMCL accumulation. D, Acipimox treatment fully prevents nicotine plus HFD-induced IMCL accumulation and mitochondrial abnormalities. Scale bar, 1 μm. Data are representative of 4 mice in each group.
Ultrastructural analysis of gastrocnemius muscles from mice in various treatment groups is summarized in Table 1. There were no differences in percent Vv of IMCL and abnormal mitochondria in mice fed with NCD in the presence or absence of nicotine and mice fed HFD. However, a significant (P < .01) increase (4.1-fold) in the percent Vv of IMCL was noted after combined treatment with HFD and nicotine compared with mice on HFD and saline. Likewise, the percent Vv of abnormal mitochondria was increased significantly (P < .01) in nicotine-treated HFD-fed mice compared with mice fed HFD alone. Although HFD alone resulted in a significant (P < .002) increase in the average diameter of the IMCL droplet relative to NCD-fed mice in the absence or presence of nicotine, combined treatment with HFD and nicotine caused a further significant (P < .001) increase in IMCL droplet diameter. We also noted a significant increase in the number of lipid droplets (P < .05) as well as in the number of abnormal mitochondria (P < .002) after combined treatment with nicotine and HFD compared with mice fed NCD in the absence or presence of nicotine or HFD alone.
Table 1.
Morphometric Data on Gastrocnemius Muscles in Mice on an NCD or HFD in the Absence or Presence of Nicotinea
| Treatment | NCD plus saline | NCD plus nicotine | HFD plus saline | HFD plus nicotine |
|---|---|---|---|---|
| IMCL (percent Vv) | 0.65 ± 0.2b | 0.81 ± 0.1b | 0.92 ± 0.2b | 3.81 ± 0.3c |
| IMCL (droplets/250 μm2) | 1.70 ± 0.66b | 1.66 ± 0.33b | 2.13 ± 0.95b | 5.00 ± 0.86c |
| IMCL diameter (nm) | 252 ± 22b | 218 ± 54b | 416 ± 72c | 802 ± 71d |
| Abnormal mitochondria (percent Vv) | 0.17 ± 0.1b | 0.18 ± 0.1b | 0.25 ± 0.1b | 2.25 ± 0.5c |
| Abnormal mitochondria number (organelles/250 μm2) | 1.90 ± 0.21b | 2.13 ± 0.07b | 2.63 ± 0.75b | 7.10 ± 2.64c |
Percent Vv is the volume density expressed as a percentage of the myofibrillar volume. Abnormal mitochondria include vacuolated mitochondria and swelled mitochondria with broken cristae. Values are given as mean ± SE of 4 mice per group.
In each row, means with different superscript letters are significantly (P < 0.01) different from each other.
Nicotine plus an HFD inactivates muscle AMPK
AMPK, a central regulator of cellular energy homeostasis, plays an important role in fatty acid metabolism through its ability to regulate key fatty acid biosynthetic pathways (25, 26). To investigate whether nicotine plus HFD-induced IMCL accumulation was associated with inactivation of AMPK, we carried out Western blot analysis of p-AMPK and total AMPK in muscle lysates (Figure 4A). Compared with mice on an NCD with or without nicotine, mice fed an HFD had decreased levels of phospho-AMPK (Figure 4A). Notably, addition of nicotine to the HFD led to complete dephosphorylation (inhibition) of AMPK. AMPK phosphorylates and inactivates ACC, the rate-limiting enzyme in fatty acid biosynthesis (25, 26). Consistent with the finding of inactivation of AMPK in HFD- and HFD plus nicotine-treated groups, no p-ACC was detected in muscle lysates in these groups (Figure 4A). Densitometric analysis further revealed a significant (P < .05) decrease in p-AMPK in muscle lysates by 70.9% in mice on an HFD compared with that of mice on an NCD. A further significant decrease (65.2%) in p-AMPK levels was detected in muscle lysates of the combined treatment group compared with that of mice on an HFD. We also observed a complete (∼99%) reduction in p-ACC levels in muscles in mice fed an HFD with or without nicotine. There were no significant differences in total AMPK or total ACC levels among various groups.
Figure 4.
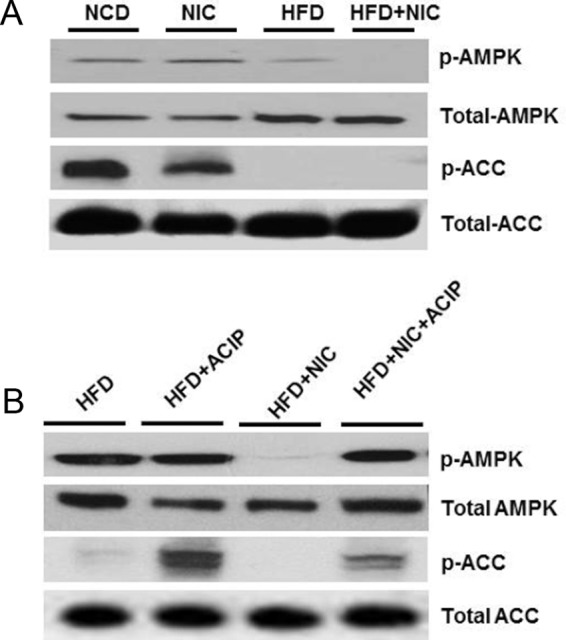
A, Western blot analysis shows that mice fed an HFD have decreased p-AMPK and p-ACC levels compared with mice on an NCD with or without nicotine treatment. Addition of nicotine to HFD leads to complete dephosphorylation of AMPK and ACC. B, Treatment with acipimox significantly attenuates the HFD plus nicotine-induced decrease in p-AMPK and p-ACC levels.
We next assessed the effect of acipimox on HFD plus nicotine-induced inactivation of AMPK and its downstream target ACC (Figure 4B). As expected, nicotine plus HFD resulted in a marked reduction in p-AMPK (70.7%) and almost complete reduction in p-ACC (98.5%) levels compared with mice fed HFD. Treatment with acipimox significantly attenuated HFD plus nicotine-induced decrease in p-AMPK and p-ACC levels.
Discussion
In this study, we used the model of diet-induced obesity in C57BL6J mice to examine the underlying mechanisms of the detrimental effects of 2 common lifestyle factors, nicotine and HFD, on skeletal muscle. We elected to use nicotine as opposed to first- or secondhand smoke to have a single drug determining the molecular basis of the combined effects of nicotine and HFD. To make effects specific to nicotine, we purposely used a shorter (10 weeks) duration to examine whether there are any detrimental effects of nicotine on muscle in mice on an HFD, because longer exposure to HFD alone results in insulin resistance and systemic inflammation (27, 28), which may cause greater muscular abnormalities (29). The results of the present study provide evidence, for the first time, that 1) nicotine combined with an HFD causes IMCL accumulation and IMF mitochondrial abnormalities in obese mice and 2) abdominal lipolysis is an important contributor to nicotine-induced muscle abnormalities in mice on an HFD. We have also shown that the detrimental effects of the combined treatment of nicotine and HFD on muscle abnormalities are associated with increased oxidative stress, plasma FFA, and muscle triglyceride levels together with inactivation (dephosphorylation) of AMPK and activation of its downstream target ACC. These results together with our recent findings that nicotine when combined with HFD also causes hepatic steatosis (14) suggest an association between hepatic and muscle steatosis in nicotine-treated mice on an HFD.
In skeletal muscle, mitochondria are divided into SS and IMF mitochondria based on their spatial locations in muscle fiber. SS mitochondria are found immediately underneath the sarcolemmal membrane, and IMF mitochondria are intermingled within the myofibers. It is worth noting that these mitochondrial subpopulations respond differently to various physiological perturbations. For example, SS mitochondria are more malleable in response to exercise (30, 31), whereas IMF mitochondria have higher metabolic enzymes (32) and are essential for muscular contraction (32). IMF mitochondria are also more sensitive to oxidative stress (33). We are intrigued by the observation that combined treatment of nicotine and HFD results in IMCL accumulation juxtaposed with IMF mitochondria and IMF mitochondrial damage as opposed to SS mitochondria compared with either insult alone. The mechanism responsible for the selective response of an HFD and nicotine on IMF mitochondria remains unknown but may be mediated by a greater degree of sensitivity of IMF mitochondria to oxidative stress (33). Thus, one could assume that nicotine when combined with an HFD may cause IMF mitochondrial dysfunction that would have detrimental functional consequences on muscle metabolism.
Because of the low affinity for nicotine at the muscle nicotinic acetylcholine receptor levels (34), it is likely that nicotine may not have a direct role in causing IMCL accumulation and IMF mitochondrial abnormalities in mice on an HFD. Thus, one possibility is that nicotine could trigger adipose tissue lipolysis and, in turn, muscle abnormalities in part by augmenting catecholamine release and stimulation of adrenoreceptors in the fat cells and in part by directly activating the nicotinic acetylcholine receptors in the adipose tissue (35–38). Indeed, consistent with a role of adipose tissue-mediated lipolysis contributing to muscle abnormalities, we found that lipolysis inhibitor acipimox fully attenuates nicotine plus HFD-induced IMCL accumulation and IMF mitochondrial abnormalities. There have been studies in murine models indicating that a high-fat high-sucrose cafeteria diet or streptozotocin treatment can also mimic the results observed on muscle after combined treatments with nicotine and HFD (29, 39). This, at first glance, suggests a nonspecific effect of nicotine on muscle in mice on an HFD. However, this seems unlikely, because HFD alone, within the study paradigm, did not lead to IMCL accumulation and IMF mitochondrial abnormalities but caused these changes only when combined with nicotine.
Increased oxidative stress has been implicated in IMCL accumulation and mitochondrial damage in the skeletal muscle (40, 41). Furthermore, Bonnard and colleagues (29) have demonstrated that increased generation of oxidative stress triggered by a high-fat high-sucrose diet or streptozotocin treatment led to a marked alterations in mitochondrial structure, including swelling, vacuolization, and broken cristae. Notably, these alterations were blocked by attenuation of oxidative stress either through normalization of glycemia or by antioxidant treatment (29). Elevated circulating FFA levels associated with adipose tissue lipolysis play a major role in developing IMCL accumulation and generation of oxidative stress (42, 43). Indeed, we found significantly elevated plasma FFA levels and higher oxidative stress in the combined treatment group compared with either treatment alone. Thus, it is conceivable that nicotine plus HFD could induce IMCL accumulation and cause mitochondrial abnormalities through generation of oxidative stress. This is consistent with earlier reports indicating that nicotine in the presence of a FFA enhances the production of ROS in C2C12 skeletal myocytes (44). This concept is further supported by our recent data showing that combined treatment of HFD and nicotine triggers a higher degree of oxidative stress in the liver compared with either insult alone (14).
Consistent with a pivotal role for AMPK in lipid homeostasis (26, 45, 46), here we show that combined treatments with nicotine and HFD that caused muscle abnormalities also caused inhibition of AMPK. The net effect of AMPK inactivation is decreased phosphorylation and activation of ACC, leading to increased IMCL accumulation, possibly through decreased fatty acid oxidation (26). This is consistent with our recent report indicating that nicotine when combined with an HFD causes inhibition of AMPK in the liver (14). Additional support of this notion came from the findings that mice lacking both AMPKβ1 and -2 isoforms in skeletal muscle had drastically reduced exercise capacity and IMF mitochondrial number with no effect on SS mitochondria (47). In this context, it is interesting to note that we have also found a 2.7-fold increase in the number of abnormal IMF mitochondria after combined treatments with nicotine and HFD compared with that of mice on an HFD alone. Of further interest, emerging evidence suggests that AMPK inhibition elevates oxidative stress in a variety of cell systems (46, 48, 49). Given that oxidative stress can cause IMCL accumulation and mitochondrial abnormalities (29), nicotine plus HFD may induce these abnormalities through AMPK inactivation, which in turn contributes to oxidative stress.
In summary, we have provided insights into the molecular mechanisms by which nicotine plus HFD induces IMCL accumulation and mitochondrial abnormalities in obese mice. We conclude that 1) greater oxidative stress coupled with inactivation of AMPK may be critical for the detrimental effect of nicotine and HFD on skeletal muscle in obese mice and 2) lipolysis of abdominal fat is an important contributor to IMCL accumulation and mitochondrial abnormalities. We surmise that nicotine plus HFD is likely to be a very toxic combination in patients with detrimental effects on both muscle and liver.
Acknowledgments
This work was supported by the Minority Institution Drug Abuse Research Program (MIDARP) from the National Institutes of Health (R24DA017298).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ACC
- acetyl-coenzyme A-carboxylase
- DXA
- dual-energy x-ray absorptiometry
- EM
- electron microscopy
- FFA
- free fatty acid
- GSH
- reduced glutathione
- GSSG
- oxidized glutathione
- HFD
- high-fat diet
- IMCL
- intramyocellular lipid
- IMF
- intramyofibrillar
- NCD
- normal chow diet
- p-AMPK
- phospho-AMP–activated protein kinase
- ROS
- reactive oxygen species
- SS
- subsarcolemmal
- TBS
- Tris-buffered saline
- Vv
- volume density.
References
- 1. He J, Gu D, Wu X, et al. Major causes of death among men and women in China. N Engl J Med. 2005;353:1124–1134 [DOI] [PubMed] [Google Scholar]
- 2. Barnes PJ. New concepts in chronic obstructive pulmonary disease. Annu Rev Med. 2003;54:113–129 [DOI] [PubMed] [Google Scholar]
- 3. Zaher C, Halbert R, Dubios R, George D, Nonikov D. Smoking-related diseases: the importance of COPD. Int J Tuberc Lung Dis. 2004;8:1423–1428 [PubMed] [Google Scholar]
- 4. Hudson NL, Mannino DM. Tobacco use: a chronic illness? J Community Health. 2010;35:549–553 [DOI] [PubMed] [Google Scholar]
- 5. Hamabe A, Uto H, Imamura Y, et al. Impact of cigarette smoking on onset of nonalcoholic fatty liver disease over a 10-year period. J Gastroenterol. 2011;46:769–778 [DOI] [PubMed] [Google Scholar]
- 6. Zein CO, Unalp A, Colvin R, Liu YC, McCullough AJ. Smoking and severity of hepatic fibrosis in nonalcoholic fatty liver disease. J Hepatol. 2011;54:753–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Petersen AM, Magkos F, Atherton P, et al. Smoking impairs muscle protein synthesis and increases the expression of myostatin and MAFbx in muscle. Am J Physiol Endocrinol Metab. 2007;293:E843–E848 [DOI] [PubMed] [Google Scholar]
- 8. Tang K, Wagner PD, Breen EC. TNF-α-mediated reduction in PGC-1α may impair skeletal muscle function after cigarette smoke exposure. J Cell Physiol. 2010;222:320–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haslam DW, James WP. Obesity. Lancet. 2005;366:1197–1209 [DOI] [PubMed] [Google Scholar]
- 10. Chiolero A, Faeh D, Paccaud F, Cornuz J. Consequences of smoking for body weight, body fat distribution, and insulin resistance. Am J Clin Nutr. 2008;87:801–809 [DOI] [PubMed] [Google Scholar]
- 11. Mangubat M, Lutfy K, Lee ML, et al. Effect of nicotine on body composition in mice. J Endocrinol. 2012;212:317–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ahmadian M, Duncan RE, Sul HS. The skinny on fat: lipolysis and fatty acid utilization in adipocytes. Trends Endocrinol Metab. 2009;20:424–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DeFronzo RA. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture 2009. Diabetologia. 2010;53:1270–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Friedman TC, Sinha-Hikim I, Parveen M, et al. Additive effects of nicotine and high-fat diet on hepatic steatosis in male mice. Endocrinology. 153:5809–5820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collins S, Martin TL, Surwit RS, Robidoux J. Genetic vulnerability to diet-induced obesity in the C57BL/6J mouse: physiological and molecular characteristics. Physiol Behav. 2004;81:243–248 [DOI] [PubMed] [Google Scholar]
- 16. Behan JW, Avramis VI, Yun JP, Louie SG, Mittelman SD. Diet-induced obesity alters vincristine pharmacokinetics in blood and tissues of mice. Pharmacol Res. 2010;61:385–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Meijer VE, Le HD, Meisel JA, et al. Dietary fat intake promotes the development of hepatic steatosis independently from excess caloric consumption in a murine model. Metabolism. 2010;59:1092–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patsch JM, Kiefer FW, Varga P, et al. Increased bone resorption and impaired bone microarchitecture in short-term and extended high-fat diet-induced obesity. Metabolism. 2011;60:243–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sinha-Hikim I, Sinha-Hikim AP, Parveen M, et al. Long-term supplementation with a cystine-based antioxidant delays loss of muscle mass in aging. J Gerontol A Biol Sci Med Sci. 2013;68:749–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cruz-Orive LM, Weibel ER. Recent stereological methods for cell biology: a brief survey. Am J Physiol. 1990;258:L148–L156 [DOI] [PubMed] [Google Scholar]
- 21. Mahapatra N, O'Connor DV, Vaingankar SM, et al. Hypertension from targeted ablation of chromogranin A can be rescued by the human ortholog. J Clin Invest. 2005;115:1942–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sinha-Hikim I, Braga M, Shen R, Sinha Hikim AP. Involvement of c-Jun NH2-terminal kinase and nitric oxide-mediated mitochondria-dependent intrinsic pathway signaling in cardiotoxin-induced muscle cell death: role of testosterone. Apoptosis. 2007;12:1965–1978 [DOI] [PubMed] [Google Scholar]
- 23. Braga M, Sinha Hikim AP, et al. Involvement of oxidative stress and caspase 2-mediated intrinsic pathway signaling in age-related increase in muscle cell apoptosis in mice. Apoptosis. 2008;13:822–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kovacheva EL, Hikim AP, Shen R, Sinha I, Sinha-Hikim I. Testosterone supplementation reverses sarcopenia in aging through regulation of myostatin, c-Jun NH2-terminal kinase, Notch, and Akt signaling pathways. Endocrinology. 2010;151:628–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118:829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9:407–416 [DOI] [PubMed] [Google Scholar]
- 27. Bernardi S, Zauli G, Tikellis C, et al. TNF-related apoptosis-inducing ligand significantly attenuates metabolic abnormalities in high-fat-fed mice reducing adiposity and systemic inflammation. Clin Sci. 2012;123:547–555 [DOI] [PubMed] [Google Scholar]
- 28. He HJ, Wang GY, Gao Y, Ling WH, Yu ZW, Jin TR. Curcumin attenuates Nrf2 signaling defect, oxidative stress in muscle and glucose intolerance in high fat diet-fed mice. World J Diabetes. 2012;15:94–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bonnard C, Durand A, Peyrol S, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krieger DA, Tate CA, McMillin-Wood J, Booth FW. Populations of rat skeletal muscle mitochondria after exercise and immobilization. J Appl Physiol. 1980;48:23–28 [DOI] [PubMed] [Google Scholar]
- 31. Ljubicic V, Joseph AM, Saleem A, et al. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: effects of exercise and aging. Biochem Biophys Acta. 2010;1800:223–234 [DOI] [PubMed] [Google Scholar]
- 32. Ferreira R, Vitorino R, Alves RM, et al. Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specialization in skeletal muscle. Proteomics. 2010;10:3142–3154 [DOI] [PubMed] [Google Scholar]
- 33. Adhihetty PJ, Ljubicic V, Menzies KJ, Hood DA. Differential susceptibility of subsarcolemmal and intermyofibrillar mitochondria to apoptotic stimuli. Am J Physiol Cell Physiol. 2005;289:C994–C1001 [DOI] [PubMed] [Google Scholar]
- 34. Xiu X, Puskar NL, Shanata JA, Lester HA, Dougherty DA. Nicotine binding to brain receptors requires a strong cation-π interaction. Nature. 2009;458:534–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Andersson K, Arner P. Systemic nicotine stimulates human adipose tissue lipolysis through local cholinergic and catecholaminergic receptors. Int J Obes Relat Metab Disord. 2001;25:1225–1232 [DOI] [PubMed] [Google Scholar]
- 36. Liu RH, Mizuta M, Matsukura S. The expression and functional role of nicotinic acetylcholine receptors in rat adipocytes. J Pharmacol Exp Ther. 2004;310:52–58 [DOI] [PubMed] [Google Scholar]
- 37. An Z, Wang H, Song P, Zhang M, Geng X, Zou MH. Nicotine-induced activation of AMP-activated protein kinase inhibits fatty acid synthase in 3T3L1 adipocytes: a role for oxidant stress. J Biol Chem. 2007;282:26793–26801 [DOI] [PubMed] [Google Scholar]
- 38. Kolditz CI, Langin D. Adipose tissue lipolysis. Curr Opin Clin Nutr Metab Care. 2010;13:377–381 [DOI] [PubMed] [Google Scholar]
- 39. Cabot C, Pouillot K, Roy S, et al. Effect of cafeteria diet feeding on soleus intramyocellular lipid of Wistar rats. J Endocrinol Metab. 2012;2:21–25 [Google Scholar]
- 40. Crane JD, Devries MC, Safdar A, Hamadeh MJ, Tarnopolsky MA. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J Gerontol A Biol Sci Med Sci. 2010;65:119–128 [DOI] [PubMed] [Google Scholar]
- 41. Martin C, Dubouchaud H, Mosoni L, et al. Abnormalities of mitochondrial functioning can partly explain the metabolic disorders encountered in sarcopenic gastrocnemius. Aging Cell. 2007;6:165–177 [DOI] [PubMed] [Google Scholar]
- 42. Coen PM, Goodpaster BH. Role of intramyocellular lipids in human health. Trends Endocrinol Metab. 2012;23:391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Blaak E. Metabolic fluxes in skeletal muscle in relation to obesity and insulin resistance. Best Pract Res Clin Endocrinol Metab. 2005;19:391–403 [DOI] [PubMed] [Google Scholar]
- 44. Tatebe J, Morita T. Enhancement of TNF-α expression and inhibition of glucose uptake by nicotine in the presence of a free fatty acid in C2C12 skeletal myocytes. Horm Metab Res. 2011;43:11–16 [DOI] [PubMed] [Google Scholar]
- 45. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Park CS, Bang BR, Kwon HS, et al. Metformin reduces airway inflammation and remodeling via activation of AMP-activated protein kinase. Biochem Pharmacol. 2012;84:1660–1670 [DOI] [PubMed] [Google Scholar]
- 47. O'Neill HM, Maarbjerg SJ, Crane JD, et al. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role of AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A. 2011;108:16092–16097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jia F, Wu C, Chen Z, Lu G. AMP-activated protein kinase inhibits homocysteine-induced dysfunction and apoptosis in endothelial progenitor cells. Cardiovasc Drugs Ther. 2011;25:21–29 [DOI] [PubMed] [Google Scholar]
- 49. Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665 [DOI] [PMC free article] [PubMed] [Google Scholar]