

Abstract
Acromegaly is associated with an increased incidence of cardiovascular disease. Transgenic mice expressing bovine GH (bGH) gene have previously been used to examine the effects of chronic GH stimulation on cardiovascular function. Results concerning systolic blood pressure (SBP) in bGH mice are conflicting. We hypothesized that these discrepancies may be the result of the various ages of the mice used in previous studies. In the current study, SBP was assessed monthly in male bGH mice from 3–12 months of age. Factors known to alter blood pressure were assessed during this time and included: levels of brain natriuretic peptide (BNP) and glucose homeostasis markers, and renal levels of angiotensin-converting enzyme 2 and endothelial nitric oxide synthase. Beginning at 6 months of age bGH had increased SBP compared with wild-type controls, which remained elevated through 12 months of age. Despite having increased blood pressure and cardiac BNP mRNA, bGH mice had decreased circulating levels of BNP. Additionally, bGH mice had an age-dependent decline in insulin levels. For example, they were hyperinsulinemic at 3 months, but by 11 months of age were hypoinsulinemic relative to wild-type controls. This decrease in insulin was accompanied by improved glucose tolerance at 11 months. Finally, both angiotensin-converting enzyme 2 and endothelial nitric oxide synthase expression were severely depressed in kidneys of 11-month-old bGH mice. These results indicate that elevated SBP in bGH mice is dependent on age, independent of insulin resistance, and related to alterations in both the natriuretic peptide and renin-angiotensin systems.
Acromegaly is a disease caused by increased GH secretion often secondary to a pituitary adenoma and results in profound metabolic and cardiovascular complications. The development of the clinically recognized disease is slow, and most patients are not diagnosed until the fourth decade of life (1). Metabolically, patients with acromegaly have altered body composition, with increased lean mass and decreased fat mass, and often develop insulin resistance and subsequent diabetes mellitus (1). In the cardiovascular system, acromegaly leads to left ventricular hypertrophy with subsequent cardiomyopathy and a predisposition to developing hypertension and atherosclerosis (2, 3). Left untreated, the chronic exposure to high GH levels causes the development of a unique cardiomyopathy characterized by concentric hypertrophy of both ventricles and eventual congestive failure (4, 5). The evolution of cardiovascular disease in patients with acromegaly is unclear. The overarching question has been: are the resulting cardiovascular deficits due to direct actions of GH/IGF-I on cardiomyocytes or are they secondary to the chronic changes in the vasculature and renal systems?
Bovine GH (bGH) transgenic mice are commonly used as an animal model of acromegaly because they exhibit gigantism, with increased body length and mass, and have elevated serum levels of GH and IGF-I (6–8). bGH mice also exhibit altered body composition with increased lean mass, decreased fat mass, increased fluid mass (9, 10), altered metabolism including the development of insulin resistance (11), endothelial dysfunction (12), altered blood pressure (BP) (13–17), and decreased life span (18). Previous studies concerning the cardiovascular function of bGH mice have been mixed. Reports concerning BP in bGH mice have shown both normal (13, 14, 16) and elevated BP (15, 17). Salt challenge does not affect BP in bGH mice, which suggests a structural or hormonal deficit underlying the change in BP (15). There is evidence from studies of bGH mice that chronic GH may lead to endothelial dysfunction and structural changes of mesenteric vasculature (12, 15). Hearts of bGH mice are enlarged, show deteriorating function, and exhibit increased hypertrophy and fibrosis (13, 14). In contrast, isolated bGH cardiomyocytes show no change in function and demonstrate an increased sensitivity to calcium (19) and an inability to respond to acute insulin stimulation (19), implicating a multisystem mechanism for the cardiovascular dysfunction found in bGH mice that may be linked to insulin resistance.
In addition to the heart, the kidneys of bGH mice exhibit glomerular hypertrophy with severe mesangial sclerosis, which correlate with an increasing albumin to creatinine ratio (20). Counterintuitively, this renal phenotype has been associated with an increased glomerular filtration rate (GFR) (15). Despite this report of increased kidney function, increased GH action has been implicated with alterations in the renin-angiotensin system (RAS). Indeed, circulating aldosterone levels are known to be increased in both patients with acromegaly and bGH mice; however, the increased levels appear to be dependent on GH and independent of renin and IGF-I (21). Interestingly, long-lived GH receptor (GHR−/−) mice (which lack GH signaling) show a shift in the RAS system away from the angiotensin-converting enzyme (ACE)2/angiotensin II (AngII) arm to the favor the ACE2/angiotensin 1–7 (Ang(1–7))/Mas pathway (22). The ACE2/Ang(1–7)/Mas pathway is thought to be protective and antagonizes the development of renal sclerosis and cardiac fibrosis (23). Unfortunately, no studies have been conducted that examine this alternate pathway in bGH mice.
Despite several previous studies describing the nature of cardiovascular complications in bGH mice, there are discrepancies that may be due to various ages of the mice examined. Given the shortened life span of bGH mice, the time course of the development of cardiovascular complications is important in understanding the progression of acromegaly and the chronic effects of GH. Here we present data describing an age-dependent elevation in systolic blood pressure (SBP), which develops independent of body weight. Further, we challenge the traditional belief that bGH mice are insulin resistant throughout life and show that older bGH mice are more glucose tolerant and have lower circulating insulin levels than controls. Finally, we examine several factors that may influence BP in bGH mice with a focus on brain natriuretic peptide (BNP) and renal expression of the ACE2/Ang(1–7)/Mas pathway.
Materials and Methods
Animals
Male bGH transgenic mice, generated as previously described (9), and wild-type (WT) littermate controls were used for this study. Mice were housed at a maximum density of 4 mice per cage in the temperature-controlled (23°C) vivarium and exposed to 14-hour light/14-hour dark cycle. All mice were allowed ab libitum access to water and food (ProLab RMH 3000; PMI Nutrition International). All procedures performed with the mice were approved by the Institutional Animal Care and Use Committee at Ohio University and are in accordance with all standards set forth by federal, state, and local authorities.
Body composition measurements
Body composition was measured in bGH (n = 9) and WT littermates (n = 7) monthly from 3–12 months of age. Measurements were collected using a desktop NMR Bruker LF50 Minispec as previously described (8).
Noninvasive BP measurement
SBP was measured monthly from 3–12 months of age in bGH (n = 9) and WT littermates (n = 7). SBP measurements were made using a noninvasive BP tail-cuff system (catalog no. IN125/M, ADInstruments) connected to a PowerLab system (catalog no. PL3508, ADInstruments). For each measurement, mice were placed in a mouse restrainer and maintained at approximately 37°C using a heating pad throughout all training periods and measurements. To ensure acclimation to the procedure, each mouse underwent 4 days of mock measurements prior to data collection. Each training day consisted of 5 SBP measurements. For SBP readings, at least 5 technical replicates were performed for each time point on each mouse. The first measurement was always discarded to avoid variation from stress due to handling. As suggested by the manufacturer, we identified SBP as the cuff pressure at which the tail pulse first reappeared after occlusion. All data were analyzed using Lab Chart v7.4 (ADInstruments).
Histologic preparation and measurements
Whole hearts and kidneys were dissected from 3-, 6-, and 12-month bGH and WT littermates (n = 2) and fixed in 15% (vol/vol) neutral buffered formalin for 24 hours. Samples were then transferred to 70% ethanol and shipped to AML Laboratories for paraffin embedding and processing. Longitudinal sections (5 μm) of whole heart were stained with Masson Trichrome to highlight collagen deposits. Longitudinal sections (5 μm) of whole kidney were stained with periodic acid Schiff stain without diastase digestion to highlight areas of increased matrix expansion. All micrographs were collected using Nikon Elements imaging software. All images were processed using ImageJ (24) software. Kidney glomerular area was collected in 10 nonoverlapping fields (×400) from each mouse. Heart collagen content was determined by collecting 20 nonoverlapping fields (×200) from each mouse. An automated threshold procedure was used to separate collagen from other cellular components (see Supplemental Materials and Methods published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org for description).
Real-time quantitative PCR (RTqPCR)
Two-step RTqPCR was performed using total RNA isolated from heart ventricular samples of bGH (n = 5) and WT (n = 9) mice. Reverse transcription was performed using the Maxima First Strand cDNA Synthesis Kit (catalog no. K1642, Thermo Scientific). RTqPCR was performed in a Bio-Rad iCycler machine using the Maxima SYBR Green/Fluorescein qPCR kit (catalog no. K0242, Thermo Scientific). All data were analyzed using qBase Plus v2.4 (Biogazelle) and are in compliance with MIQE (25) standards. Details can be found in the Supplemental Materials and Methods. Primer sequences are listed in Supplemental Table 1.
Plasma measurements
Whole blood was collected from the tail tips of bGH and WT littermates after a 12-hour overnight fast by clipping approximately 1 mm of the tail tip and collecting about 250 μL of blood using heparinized capillary tubes. Plasma was collected after centrifugation for 10 minutes at 7000 × g, 4°C. Circulating levels of BNP were determined using the Mouse BNP EIA (catalog no. EIA-BNP-1; RayBiotech) following the manufacturer's directions. Levels of insulin, C-peptide, monocyte chemotactic protein 1 (MCP-1), and IL-6 were measured using a Milliplex Mouse Metabolic Panel (catalog no. MMHMAG-44K; Millipore Corp.) according to the manufacturer's instructions. Fasting blood glucose levels were collected every 3 months from tail vein bleeding using OneTouch Ultra test strips and glucometers (Lifescan).
Intraperitoneal insulin/glucose tolerance testing (ITT/GTT)
For the GTT, bGH (n = 8) and WT littermates (n = 14) were fasted for 12 hours prior to measurements. Each mouse received an ip injection of 10% glucose at 0.01 mL/g body weight. For the ITT, bGH (n = 8) and WT littermates (n = 14) were nonfasted. Each mouse received an ip injection of 1 U/kg body weight insulin (Humilin R; Lilly). For both GTT and ITT, blood glucose measurements were collected using OneTouch Ultra test strips and glucometers before glucose or insulin injection and at 15, 30, 45, 60, 90, 120, and 150 minutes after injection.
Immunoblots
Protein was isolated from homogenized sections of flash-frozen kidney tissue (∼30 mg) from bGH (n = 4) and WT (n = 3). Protein samples (20 μg) were separated on 10% SDS-polyacrylamide gels before being transferred to polyvinylidene difluoride membranes (catalog no. RPN2020LFP; GE Healthcare). Subsequently, membranes were blocked and probed with primary antibodies: ACE (catalog no. SC-20791; 1:200), ACE2 (catalog no. SC-20998, 1:200), endothelial nitric oxide synthase (eNOS) (catalog no. SC-654; 1:200), (Santa Cruz Biotechnology); glyceraldehyde-3 phosphate dehydrogenase (catalog no. 2118; 1:1000), (Cell Signaling Technology). Finally, membranes were incubated with Cy5-conjugated secondary antibody (1:2500) (catalog no. PA45011; GE Healthcare) and scanned using a Pharos FX laser scanner (Bio-Rad Laboratories). Full details can be found in the Supplemental Materials and Methods.
Statistics
All values are reported as mean ± SEM. Statistics were performed using SPSS v 17.0 (IBM). All time-dependent analysis was performed using two-way ANOVA with repeated measures, age, and genotype as factors. Maulchy's test of sphericity was performed on all repeated-measures data. The Green-House Geisser correction was applied if Maulchy's test was found to be significant. For single time point measurements, equality of variance was tested using Levene's test, after which group means were then compared using an independent Student's t test or a Welch's t test for unequal variances. For all tests, statistical significance was determined when P < .05.
Results
Body weight and body composition
There was a significant interaction between genotype and age for body weight, fat, and lean and fluid mass (P < .05) (Figure 1A–D). At every time point from 3–12 months of age, bGH mice had significantly increased body weight (P < .05) (Figure 1A). bGH mice showed a trend of failure to gain fat mass beginning at 4 months of age that became significant at 6 months of age (P < .05) (Figure 1B). This failure to gain fat mass was accompanied by an increase in lean mass percentage (Figure 1C), which also became significant at 6 months of age (P < .05). Fluid mass showed a similar trend, with increased fluid mass in bGH mice from 6–8 months of age and from 10–12 months of age (P < .05).
Figure 1.

Body composition, length, and tissue masses. A, Body weight in grams, (B) % fat mass, (C) % lean mass, and (D) % fluid mass from 3–12 months of age in bGH (dot-dash line) and WT littermates (solid line). E, Length (millimeters) in 12-month mice from tip of nose to anus and (F) tissue mass normalized to body weight and then to WT of 10 dissected tissues. All values are presented as the average within genotype ± SEM. *, Significant difference between genotype (P < .05); †, significant interaction between age and genotype (P < .05); ‡, significant difference with age (P < .05). BAT, brown adipose tissue; Ing, inguinal white adipose; Epi, epididymal white adipose; Retro, retroperitoneal white adipose; Mes, mesenteric white adipose.
Body length and tissue mass
Concomitant with increased body weight, bGH mice demonstrated an increased body length at dissection (Figure 1E) with a mean length of 110.2 ± 1.3 mm vs 95.1 ± 0.6 mm in controls (P < .05). Additionally, 12-month-old tissue masses were significantly different in bGH vs control mice (P < .05). bGH mice had significant decreases in relative weights of white adipose tissue depot mass (72%–87%), interscapular brown adipose tissue mass (19%), and in whole brain size (14%) compared with controls. Tissues significantly increased in bGH mice included liver (112%), kidney (43%), heart (60%), and lung (25%). In terms of absolute mass, the hearts of 12-month-old bGH mice were 0.248 ± 0.011 g vs 0.122 ± 0.003 g in WT. The mass of the kidneys of 12-month-old bGH mice were 0.604 ± 0.019 g vs 0.334 ± 0.010 g in WT.
Cardiac histology and fibrosis
In order to determine the effects of chronic GH on cardiac fibrosis, cardiac tissue from 3-, 6-, and 12-month-old bGH and control mice were evaluated for the level of collagen deposition. Figure 2A shows representative sections from bGH mice and WT littermates at each time point with the light blue staining representing collagen against the purple and red muscle fibers. The bGH mice had significant increases in percent collagen area at 3 (WT, 7.1 ± 0.6%; bGH, 9.2 ± 0.4%) and 12 (WT, 11.6 ± 0.4%; bGH, 15.9 ± 1.1%) months of age (P < .05). No difference between bGH and control mice was observed at 6 months of age.
Figure 2.
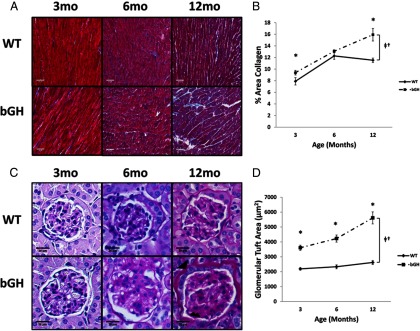
Heart and kidney histology. A, Sections of heart stained with Masson's Trichrome from 3-, 6-, and 12-month-old bGH and WT littermates (n = 2). B, Collagen content calculated from 10–20 nonoverlapping sections of LV cardiac tissue at each time point. C, Sections of glomeruli stained with Periodic Acid Schiff stain from 3-, 6-, and 12-month-old bGH and WT littermates (n = 2). D, Glomerular tuft area calculated from at least 10 glomeruli from at least 5 nonoverlapping sections at each time point. All values are presented as average ± SEM. Scale bars in panel A are 50 μm and in panel C are 20 μm. *, Significant difference between genotype (P < .05); †, significant interaction between age and genotype (P < .05); ‡, significant difference with age (P < .05). mo, months.
Glomerular histology
Kidneys were examined from 3-, 6-, and 12-month-old bGH mice and littermate controls to assess glomerular size and structure (Figure 2, C and D). There was a significant interaction between age and genotype in glomerular tuft size in bGH mice from 3–12 months of age (P < .05). Visible membrane thickening and mesangial sclerosis were also apparent in kidneys of bGH mice (Figure 2C).
SBP
In order to establish longitudinal SBP, measurements were taken each month from 3–12 months of age. Changes in SBP demonstrated a significant interaction between age and genotype (P < .05). Specifically, from 3–5 months of age, bGH and littermate controls had similar SBP ranging between 102 and 107 mm Hg (Figure 3A). At 6 months of age, however, bGH mice had significantly elevated SBP relative to controls (113.6 ± 1.2 mm Hg vs WT: 107.5 ± 0.9 mm Hg, P < .05). From 7–12 months of age, bGH mice continued to exhibit higher SBP than controls. The bGH mice had a peak SBP of 119.6 ± 0.7 mm Hg (10 months), and the controls had a peak SBP of 111.5 ± 0.6 mm Hg (12 months).
Figure 3.

SBP, BNP, and heart calcium channels. A, SBP measured in unanesthetized male mice (n = 7–9) from 3–12 months of age. Cardiac LV BNP RNA levels were measured at 6 (B) and 12 (C) months of age (n = 5–9). Circulating BNP levels were measured by ELISA at 5 (D) and 7 (E) months of age (n = 7–9). RNA levels of the major calcium channels in heart were measured at 6 months (F) and 12 months (G) of age. All values are presented as average ± SEM. *, Significant difference between genotype (P < .05); †, significant interaction between age and genotype (P < .05); ‡, significant difference with age (P < .05); SERCA2, sarcoplasmic endoreticulum calcium exchanger 2; LTCC, L-type calcium channel, NCX1, sodium calcium exchanger 1; RYR2, ryanodine receptor 2.
BNP levels in serum and cardiac tissue
Circulating levels of BNP were measured at 5 and 7 months of age to assess levels of natriuretic peptides around the age of increased SBP in bGH mice. Levels of circulating BNP tended to be decreased at 5 months of age; however, levels did not reach statistical significance (59.7 ± 2.1 pg/mL vs WT: 108.3 ± 27.2 pg/mL; P = .12). At 7 months of age, bGH mice had a significant and more substantial decrease in circulating BNP levels (29.0 ± 6.2 pg/mL vs 116.7 ± 25.5 pg/mL; P < .05). In cardiac tissue, BNP mRNA levels showed a nearly significant (P = .054) increase at 6 months of age and were significantly increased by more than 3-fold at 12 months of age (P < .05).
Cardiac calcium channel gene expression
Results from both 6- and 12-month-old bGH and WT littermates (Figure 3, F and G) showed that there was no difference in mRNA expression of any of the 4 cardiac calcium channels at either age between genotypes.
Glucose homeostasis and insulin levels
Both fasting plasma glucose (FPG) and plasma insulin levels showed a significant interaction between age and genotype (P < .05). FPG levels (Figure 4A) at 3 months of age in bGH and WT were similar (WT: 112.3 ± 6.8 mg/dL vs bGH: 111.8 ± 9.6 mg/dL; P = .968). By 6 months of age, bGH mice tended (P = .07) to have decreased FPG (WT: 163.9 ± 14.2 mg/dL vs bGH: 132.2 ± 3.9 mg/dL). At 9 and 12 months of age, bGH mice had significantly (P < .05) decreased FPG levels relative to littermate controls (9 months WT: 166.2 ± 10.4 mg/dL vs bGH: 125.0 ± 4.3 mg/dL; 12 months WT: 170.6 ± 11.8 mg/dL vs bGH: 92.7 ± 3.8 mg/dL).
Figure 4.

Glucose homeostasis. Fasting blood glucose (A), fasting plasma insulin (B), and fasting plasma c-peptide (C) levels at 3, 9, and 11 months of age (n = 7–10). D, Intraperitoneal ITT was performed in 11-month-old nonfasted mice by injecting mice (n = 8–10) with 1U/kg body weight insulin and subsequent sampling of blood glucose. E, Intraperitoneal GTT was performed in 11-month-old mice (n = 8–10) following a 12-hour fast by injecting mice with a 10% glucose solution at 0.01 mL/g bodyweight and sampling glucose before injection at 15, 30, 45, 60, 90, 120, and 150 minutes after injection. F, Area under the curve (AUC) of the GTT was calculated for both groups of mice. *, Significant difference between genotype (P < .05); †, significant interaction between age and genotype (P < .05); ‡, significant difference with age (P < .05).
Plasma insulin levels (Figure 4B) showed a significant interaction between age and genotype (P < .05). At 3 months of age bGH mice were hyperinsulinemic relative to controls (WT: 707 ± 124 pg/mL vs bGH: 1352 ± 166 pg/mL; P < .05). Insulin levels were similar at 6 months of age, but by 12 months of age bGH mice were hypoinsulinemic compared with controls (WT: 1192 ± 144 pg/mL vs bGH: 765 ± 76 pg/mL). C-peptide levels were also measured at 3, 9, and 11 months of age but did not reach statistical significance (Figure 4C). The trend of C-peptide levels did match that of plasma insulin.
ITT/GTTs were conducted using 11-month-old bGH and WT controls. bGH mice showed no difference at any time point in response to the ITT (Figure 4D). In contrast, the GTT (Figure 4, E and F) demonstrated that bGH mice had significantly improved glucose clearance with a calculated area under the curve (AUC) of 29 378 ± 1549 mg/dL*min−1 vs 42 399 ± 5499 mg/dL*min−1 (P < .05) in littermate controls.
Circulating inflammatory cytokines
Levels of circulating MCP-1 and IL-6 were measured in 2-, 8-, and 11-month-old bGH and WT controls (Figure 5). IL-6 levels showed an increase throughout age regardless of genotype whereas both IL-6 and MCP-1 levels showed an interaction with age and genotype. In young 2-month-old mice, bGH had similar levels of IL-6 but increased levels of MCP-1. By 8 months of age, bGH had significantly elevated circulating levels of both IL-6 and MCP-1. By 11 months of age, levels of both cytokines were significantly elevated in bGH mice with IL-6 levels nearly triple (WT: 46.3 ± 8.6 pg/mL; bGH: 117.3 ± 33.2 pg/mL) and MCP-1 levels almost double (WT: 98.4 ± 25.1 pg/mL; bGH: 171.6 ± 22.9 pg/mL) that of littermate controls (P < .05).
Figure 5.

Circulating inflammatory cytokines. Plasma levels of IL-6 (A) and MCP-1 (B) at 2, 8, and 11 months of age (n = 7–10). *, Significant difference between genotype (P < .05); †, significant interaction between age and genotype (P < .05); ‡, significant difference with age (P < .05);.
ACE, ACE2, and eNOS expression in 12-month-old bGH kidney
Renal levels of ACE, ACE2, and eNOS protein were measured in 12-month-old mice. Expression of ACE (Figure 6A) tended to be higher in bGH mice vs controls (P = .08), and the levels of ACE2 (Figure 6B) and eNOS (Figure 6C) were severely and significantly depressed (P < .05) in bGH mice by approximately 82% and 75%, respectively.
Figure 6.

Expression of ACE, ACE2, and eNOS in kidney. Immunoblot analysis of 12-month-old ACE (A), ACE2 (B), and eNOS (C) levels normalized to glyceraldehde 3-phosphate dehydrogenase (GAPDH) in kidney (n = 3–4). *, Significant difference between genotype (P < .05). A.U., arbitrary units.
Discussion
The goal of our study was to follow the development of elevated SBP in bGH mice and simultaneously consider several contributory factors. The key findings from our study were that bGH mice developed elevated SBP throughout the first year of life, and the time period between 5 and 7 months of age marked a significant change in both body composition and BP. Our longitudinal histologic examination of kidney and heart indicated that renal changes preceded the development of cardiac fibrosis, suggesting that changes in the kidney may precipitate elevated BP in bGH mice. However, the maintenance of the elevated BP is likely due to derangements in multiple organs. The bGH mice showed a dramatic increase in renal ACE:ACE2 ratio indicating a shift in the RAS toward the proinflammatory, profibrotic AngII pathway and away from the cardioprotective Ang(1–7) pathway. In the heart of bGH mice, BNP mRNA expression was increased, as expected, but the circulating levels of active BNP were severely depressed, indicating the inability to produce an adequate natriuretic peptide response. Finally, the initial hyperinsulinemia in bGH mice was not sustained because the bGH mice became hypoinsulinemic by 11 months of age. This hypoinsulinemia was accompanied by improved glucose tolerance and improved fasting blood glucose levels. Overall, elevated SBP in bGH mice is age dependent and associated with hormonal changes in kidney and heart, but not related to insulin resistance or hyperinsulinemia in old age.
To date, 5 studies have assessed BP in bGH mice. Our results of normal SBP in early life between 3–5 months and increased SBP from 6–12 months of age in bGH mice are in agreement with most of these studies (15–17) but are inconsistent with 2 studies (13, 14). Early studies of bGH mice at 3 (16), 7 (13), and 8 months (14) of age describe no difference in SBP vs WT controls. More recent studies, however, find that 5- (15) and 9- (17) month-old bGH mice have elevated SBP relative to WT controls. Both Bollano et al (14) and Dilley and Schwartz (13) report extremely low pressures for WT mice of 84.0 ± 4 mm Hg at 7 months of age and 88.0 ± 3.7 mm Hg at 8 months of age, respectively. These pressures are well below what we and others (15, 17) measure for WT mice. This difference may reflect an environmental or methodological variable that was not accounted for in the earlier studies.
Insulin resistance is thought to be a contributory factor to hypertension (26). Indeed, a 2004 analysis of more than 1000 mixed-ethnicity people found that insulin resistance, but not circulating insulin levels, is positively correlated with BP (27). Chronic GH action is commonly accepted to exert an overall anti-insulin effect, an effect underscored by the increased incidence of hyperinsulinemia and diabetes in acromegalic individuals (1). In our current study, bGH mice had hyperinsulinemia in early life but improved glucose homeostasis and hypoinsulinemia in later life. Our data argue against insulin resistance being a significant factor in the chronic maintenance of elevated SBP in bGH mice. Nevertheless, we cannot rule out the possibility that early hyperinsulinemia (before 9 months of age) played a role in the establishment of increased SBP. Our laboratory has previously reported this trend in insulin levels in bGH mice (28). The drastic decline in insulin levels throughout life may point to a defect in pancreatic β-cells. Although young, 3-month-old bGH mice are reported to have increased islet cell mass (29), to date there has not been a study addressing the function or structure of the pancreas in bGH mice from birth to death.
To our knowledge, only one prior study has commented on the disconnect between the presence of improved glucose homeostasis in the face of the chronic GH stimulation in bGH mice (30). In that study, bGH mice have improved glucose tolerance at 3, 6, and 9 months of age regardless of sex. Together with the improved glucose tolerance, the bGH mice also have improved glucose-stimulated insulin secretion but a decreased response to pyruvate tolerance testing, indicating a deficit in gluconeogenesis (30). Although the exact mechanism causing this disconnect is not known, bGH mice have elevated IGF-I (6–8), which may play a role. IGF-I is known to decrease gluconeogenesis through inhibition of hepatic phosphoenol pyruvate carboxykinase (31, 32) and has been shown to stimulate glucose uptake in the absence of functional insulin receptor (31). Another intriguing possibility may be that bGH mice demonstrate tissue-specific insulin resistance that may not be reflected in the results of ITT and GTTs. It was recently demonstrated that bGH mice hearts are resistant to acute insulin stimulation but, at baseline, actually exhibit an increase in insulin-signaling activation (33).
Glomerulosclerosis is a well-documented age-dependent feature in bGH mice (34–37). The mesangial sclerosis is thought to be a direct result of increased GH action and not IGF-I (36). Our histologic analysis of the kidney supports previous studies (35), showing a disproportionate increase in mesangial scarring and glomerular capsule thickening throughout age in bGH mice vs WT controls. Further, our findings showed that renal histologic changes occurred early in the life of the bGH mouse, by 3 months, and before the onset of increased SBP at 6 months with progressive enlargement and mesangial scarring throughout age. Given these renal histologic alterations, it would be expected that function is compromised. In one study examining GFR in 5- to 7-month-old bGH mice, GFR is not altered when normalized to body weight (15). However, in a more recent study, bGH mice have an increased albumin-to-creatinine ratio at both 5 and 12 weeks of age (20). The role of GH in renal function is supported by clinical studies of acromegalic patients showing a positive correlation between GH, hypercalciuria, nephrolithiasis, microalbuminuria, and decreased sodium secretion (38). Although the exact mechanism underlying this pathology is not known, a partial explanation may be that, in vitro, GH can induce the production of reactive oxygen species and promote reorganization of the actin cytoskeleton in glomerular podocytes (39). The precipitating event may be an increase in lipid accumulation within the mesangium (37).
The kidney plays a critical role in BP homeostasis through ion balance and through the production of vasoactive substances such as renin, AngII, and angiotensin (1–7) (Ang(1–7)). The canonical renin-angiotensin system (RAS) relies on an enzymatic cascade resulting in the formation of the principal mediator AngII through action of the ACE. The discovery of another arm of the RAS by which AngI and AngII can be converted into Ang(1–7) via the action of ACE2, expressed primarily in kidney, heart, and testis (40), has added complexity to the system. As opposed to AngII, Ang(1–7) counteracts, through interaction with its receptor Mas, the classical actions of the RAS exerting an eNOS-mediated vasodilatory and antiproliferative effect (23). The ACE:ACE2 ratio, therefore, gives insight into which arm of the pathway is most activated. Recently, long-lived GHR knockout mice were shown to have decreased ACE:ACE2 ratios in both heart and kidney with a concurrent rise in eNOS expression in both tissues (22). Our results showed that unlike GHR knockout mice, bGH mice have a drastically increased ACE:ACE2 ratio and decreased eNOS expression in kidney at 12 months of age vs WT. This implies that local levels of AngII may be elevated in the kidney of bGH mice and may contribute to the observed glomerulosclerosis which, in turn, contributes to the elevated SBP. The cardiac fibrosis that we observed in the bGH mice may also be a consequence of the imbalance in ACE/ACE2 expression.
The leading cause of death in acromegalic individuals is cardiovascular disease (41). Untreated, these patients develop a unique form of cardiomyopathy characterized initially by a hyperkinetic syndrome with improved cardiac contractility and output. This evolves to concentric left ventricular hypertrophy, diastolic dysfunction due to a stiffened ventricle, and eventual systolic failure (5). bGH mice have been used in several studies to consider cardiac function with varying results. Using echocardiography in 8-month-old bGH mice, Bollano et al (14) showed a dramatic decrease in systolic ejection fraction and fractional shortening. A report from the same laboratory 1 year later, however, showed no change in cardiac contractility using direct ventricular cannulation (15). In vitro work using isolated ventricular myocytes from bGH mice showed that the isolated cells have increased cell shortening, increased maximal shortening and relengthening velocities, and increased peak calcium transients, supporting an overall enhanced contractile ability (19). Our measurement of the 4 main cardiac calcium channel mRNAs revealed no difference between bGH and WT. Therefore, factors such as cardiac fibrosis or altered blood volume may play a more prominent role in the pathophysiology of chronic GH excess. Indeed, our results demonstrated that cardiac fibrosis was a distinguishing feature in old bGH mice, a finding that agrees with previous studies (14), but was not prominent until 12 months of age.
Upon being stretched, cells of the atrium and ventricle release natriuretic peptides (NP). These proteins are cardioprotective and work to counteract the effects of the RAS by promoting vasorelaxation and urinary sodium excretion (42). The 2 main natriuretic peptides are atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) and are released primarily from the heart atrium and ventricle, respectively (43). The NPs act primarily through the G-coupled NP receptor A and are cleared through the NP receptor C or via proteolysis (44). Disruption in the action of these hormones can have a direct effect on systemic fluid homeostasis. Our results showed disconnect between levels of BNP transcript in the heart and circulating BNP, indicating that GH may be inhibiting BNP posttranscriptionally at the level of translation, release, or by accelerating clearance. An inhibitory effect of GH on ANP (45), BNP (46), and N-terminal pro-BNP (which is released in 1:1 quantities after cleavage of BNP) (47) has been described in humans with GH disturbances. A study conducted using bGH mice demonstrated that circulating levels of active ANP were increased at 7 but not 27 weeks of age (48). This decrease in circulating active ANP mirrored an increase in the prohormone at 27 weeks in the bGH mice, pointing to an inability to properly cleave ANP to its active form (48). Our measurements of BNP are in line with these observations concerning ANP.
Attenuation of the GH/IGF-I axis is associated with a decrease in inflammatory markers and an improvement in aging (49). Thus, the opposite may be true in chronic GH excess, which may promote a deleterious proinflammatory environment. Inflammation in bGH kidney (50) and adipose tissue (51) is thought to contribute to the overall dysregulated physiology of the bGH mouse. It was recently shown that adipocytes cultured from patients with acromegaly show an increased production of monocyte chemotactic protein 1 (MCP-1) and several other proinflammtory cytokines (52). Our data demonstrated that the level of circulating MCP-1 was higher throughout age in bGH mice whereas the level of IL-6 increased from 2 to 8 months and remained elevated at 11 months of age. MCP-1 is a potent macrophage chemoattractant that, via its receptor CCR2, is thought to play an important role in early macrophage infiltration during the pathogenesis of atherosclerosis (53). Further, CCR2−/− mice are known to develop less kidney damage in models of AngII-induced hypertension (54). IL-6 is an important mediator of the acute phase inflammatory response and is necessary for the survival and proliferation of T and B cell populations (55). Human studies associate increased IL-6 levels with an increased risk of all-cause mortality (56, 57). Despite this relationship, IL-6 is a major driver of cardiac development and hypertrophy (58) with IL-6−/− mice demonstrating severe ventricular dilation and fibrosis (59). Interestingly, AngII can increase expression of IL-6 in vascular smooth muscle of rats (60). Thus the increased MCP-1 levels that we observed may be promoting damage in the kidneys, whereas the increased IL-6 may be a compensatory mechanism driving cardiac hypertrophy to deal with the increased BP in bGH mice.
There are several limitations to our study. The first is that we used only male mice. We know that female bGH mice have differences in body composition (8) and that estrogen is known to directly inhibit GH signaling (61); thus, it is likely there are sex differences in BP. Second, bGH mice have been reported to have increased spontaneous locomotor activity in response to new environments (62, 63). This may play a role in the measurement of our early SBP, as mice acclimate to the procedure. Third, the tail-cuff system we used for SBP measurement does not permit measuring diastolic BP, which may show earlier changes than SBP (64). Finally, the increased SBP in bGH mice may be the result of altered levels of vasoactive substances or may be related to vasculature dysfunction, which was not explored in this study but has been previously evaluated in other studies (12, 13, 15). Previous studies using bGH mice show that the small-resistance vasculature (such as mesenteric vessels) has a decreased average diameter (15) and an increased wall-to-lumen ratio (13). There is also evidence that increased oxidative stress plays a role in the vascular function of 2- to 3-month-old bGH mice (12). In humans, acromegalic patients exhibit vascular abnormalities with increased intima medial thickness and decreased flow-mediated dilatation (5). Based on these data, GH likely plays a role in both endothelial function and vessel structure.
In conclusion, bGH mice developed elevated SBP with advancing age. This change was accompanied by increased cardiac fibrosis, glomerular hypertrophy, and mesangial sclerosis. A disconnect was observed between increased cardiac BNP mRNA and decreased circulating BNP levels. In the kidney, bGH mice showed a dramatic decrease in both ACE2 and eNOS protein expression levels, which may indicate loss of the ability to form antiproliferative and cardioprotective Ang(1–7). Finally, systemic insulin resistance and hyperinsulinemia were not contributing factors to the maintenance of higher SBP in older bGH mice. Our current study identified several contributory pathways, which may play a role in cardiovascular function of bGH mice. In the future it will be important to 1) examine the effects of GH on BNP with a focus on BNP production, release, and cleavage in the presence of acute and chronic GH stimulation; 2) further investigate the status of the ACE2/Ang(1–7)/Mas pathway in animal models of GH disruption including measurements of circulating and tissue-specific AngII and Ang(1–7), and 3) solidify our understanding of the interaction between chronic GH action and insulin signaling by examining both systemic and tissue level insulin sensitivity and performing an in-depth evaluation of pancreatic structure and function throughout age in bGH mice.
Acknowledgments
We thank Diana Cruz-Topete (National Institutes of Health/National Institue of Environmental Health Sciences) for her help in performing the Milliplex MAP experiment and Lauren Volpe (Ohio University Patton College of Education) for her careful editing of the manuscript.
This work was supported, in part, by the Gates Millennium Scholars (GMS) Graduate Fellowship program; the State of Ohio's Eminent Scholar Program that includes a gift from Milton and Lawrence Goll; National Institutes of Health Grants P01AG031736, AG032290, DK58259, and DK083729; the American Veterans (AMVETS) Organization and the Provost Undergraduate Research Fund and the Diabetes Institute at Ohio University.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ACE2
- angiotensin-converting enzyme 2
- Ang(1–7)
- angiotensin 1–7
- AngII
- angiotensin II
- ANP
- atrial natriuretic peptide
- bGH
- bovine GH
- BNP
- brain natriuretic peptide
- BP
- blood pressure
- eNOS
- endothelial nitric oxide synthase
- FPG
- fasting plasma glucose
- GFR
- glomerular filtration rate
- GHR
- GH receptor
- GTT
- glucose tolerance testing
- ITT
- insulin tolerance testing
- MCP-1
- monocyte chemotactic protein 1
- NP
- natriuretic peptide
- RAS
- renin-angiotensin system
- RTqPCR
- real-time quantitative PCR
- SBP
- systolic blood pressure
- WT
- wild type.
References
- 1. Chanson P, Salenave S. Acromegaly. Orphanet J Rare Dis. 2008;3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boysan SN, Kantarci F, Celik O, Mihmanli I, Gazioglu N, Kadioglu P. Atherosclerotic risk factors and premature atherosclerosis in acromegaly before and after 48 months of octreotide-LAR treatment. Angiology. 2012;63:522–527 [DOI] [PubMed] [Google Scholar]
- 3. Colao A, Ferone D, Marzullo P, Lombardi G. Systemic complications of acromegaly: epidemiology, pathogenesis, and management. Endocr Rev. 2004;25:102–152 [DOI] [PubMed] [Google Scholar]
- 4. Schwarz ER, Jammula P, Gupta R, Rosanio S. A case and review of acromegaly-induced cardiomyopathy and the relationship between growth hormone and heart failure: cause or cure or neither or both? J Cardiovasc Pharmacol Ther. 2006;11:232–244 [DOI] [PubMed] [Google Scholar]
- 5. Colao A. Improvement of cardiac parameters in patients with acromegaly treated with medical therapies. Pituitary. 2012;15:50–58 [DOI] [PubMed] [Google Scholar]
- 6. Chen WY, White ME, Wagner TE, Kopchick JJ. Functional antagonism between endogenous mouse growth hormone (GH) and a GH analog results in dwarf transgenic mice. Endocrinology. 1991;129:1402–1408 [DOI] [PubMed] [Google Scholar]
- 7. Naar EM, Bartke A, Majumdar SS, Buonomo FC, Yun JS, Wagner TE. Fertility of transgenic female mice expressing bovine growth hormone or human growth hormone variant genes. Biol Reprod. 1991;45:178–187 [DOI] [PubMed] [Google Scholar]
- 8. Palmer AJ, Chung MY, List EO, et al. Age-related changes in body composition of bovine growth hormone transgenic mice. Endocrinology. 2009;150:1353–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berryman DE, List EO, Coschigano KT, Behar K, Kim JK, Kopchick JJ. Comparing adiposity profiles in three mouse models with altered GH signaling. Growth Horm IGF Res. 2004;14:309–318 [DOI] [PubMed] [Google Scholar]
- 10. Ding J, Berryman DE, Kopchick JJ. Plasma proteomic profiles of bovine growth hormone transgenic mice as they age. Transgenic Res. 2011;20:1305–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dominici FP, Balbis A, Bartke A, Turyn D. Role of hyperinsulinemia on hepatic insulin receptor concentration and autophosphorylation in the presence of high growth hormone levels in transgenic mice overexpressing growth hormone gene. J Endocrinol. 1998;159:15–25 [DOI] [PubMed] [Google Scholar]
- 12. Andersson IJ, Johansson ME, Wickman A, et al. Endothelial dysfunction in growth hormone transgenic mice. Clin Sci (Lond). 2006;110:217–225 [DOI] [PubMed] [Google Scholar]
- 13. Dilley RJ, Schwartz SM. Vascular remodeling in the growth hormone transgenic mouse. Circ Res. 1989;65:1233–1240 [DOI] [PubMed] [Google Scholar]
- 14. Bollano E, Omerovic E, Bohlooly-y M, et al. Impairment of cardiac function and bioenergetics in adult transgenic mice overexpressing the bovine growth hormone gene. Endocrinology. 2000;141:2229–2235 [DOI] [PubMed] [Google Scholar]
- 15. Bohlooly-Y M, Carlson L, Olsson B, et al. Vascular function and blood pressure in GH transgenic mice. Endocrinology. 2001;142:3317–3323 [DOI] [PubMed] [Google Scholar]
- 16. Peten EP, Striker LJ, Fogo A, Ichikawa I, Patel A, Striker GE. The molecular basis of increased glomerulosclerosis after blockade of the renin angiotensin system in growth hormone transgenic mice. Mol Med. 1994;1:104–115 [PMC free article] [PubMed] [Google Scholar]
- 17. Izzard AS, Emerson M, Prehar S, et al. The cardiovascular phenotype of a mouse model of acromegaly. Growth Horm IGF Res. 2009;19:413–419 [DOI] [PubMed] [Google Scholar]
- 18. Bartke A, Chandrashekar V, Bailey B, Zaczek D, Turyn D. Consequences of growth hormone (GH) overexpression and GH resistance. Neuropeptides. 2002;36:201–208 [DOI] [PubMed] [Google Scholar]
- 19. Colligan PB, Brown-Borg HM, Duan J, Ren BH, Ren J. Cardiac contractile function is enhanced in isolated ventricular myocytes from growth hormone transgenic mice. J Endocrinol. 2002;173:257–264 [DOI] [PubMed] [Google Scholar]
- 20. Doi SQ, Chilakamarri GC, Mendonca MC, et al. Increased class A scavenger receptor and glomerular lipid precede mesangial matrix expansion in the bGH mouse model. Growth Horm IGF Res. 2010;20:326–332 [DOI] [PubMed] [Google Scholar]
- 21. Bielohuby M, Roemmler J, Manolopoulou J, et al. Chronic growth hormone excess is associated with increased aldosterone: a study in patients with acromegaly and in growth hormone transgenic mice. Exp Biol Med (Maywood). 2009;234:1002–1009 [DOI] [PubMed] [Google Scholar]
- 22. Giani JF, Miquet JG, Muñoz MC, et al. Upregulation of the angiotensin-converting enzyme 2/angiotensin-(1–7)/Mas receptor axis in the heart and the kidney of growth hormone receptor knock-out mice. Growth Horm IGF Res. 2012;22:224–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Santos RA, Ferreira AJ, Simões E Silva AC. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1–7)-Mas axis. Exp Physiol. 2008;93:519–527 [DOI] [PubMed] [Google Scholar]
- 24. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bustin SA, Benes V, Garson JA, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–622 [DOI] [PubMed] [Google Scholar]
- 26. Ferrannini E, Haffner SM, Stern MP. Essential hypertension: an insulin-resistant state. J Cardiovasc Pharmacol. 1990;15(Suppl 5):S18–S25 [PubMed] [Google Scholar]
- 27. Saad MF, Rewers M, Selby J, et al. Insulin resistance and hypertension: the Insulin Resistance Atherosclerosis study. Hypertension. 2004;43:1324–1331 [DOI] [PubMed] [Google Scholar]
- 28. Ding J, List EO, Okada S, Kopchick JJ. Perspective: proteomic approach to detect biomarkers of human growth hormone. Growth Horm IGF Res. 2009;19:399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parsons JA, Bartke A, Sorenson RL. Number and size of islets of Langerhans in pregnant, human growth hormone-expressing transgenic, and pituitary dwarf mice: effect of lactogenic hormones. Endocrinology. 1995;136:2013–2021 [DOI] [PubMed] [Google Scholar]
- 30. Boparai RK, Arum O, Khardori R, Bartke A. Glucose homeostasis and insulin sensitivity in growth hormone-transgenic mice: a cross-sectional analysis. Biol Chem. 2010;391:1149–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Di Cola G, Cool MH, Accili D. Hypoglycemic effect of insulin-like growth factor-1 in mice lacking insulin receptors. J Clin Invest. 1997;99:2538–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J, Zhu X, Chen C, et al. Effect of insulin-like growth factor-1 (IGF-1) on the gluconeogenesis in calf hepatocytes cultured in vitro. Mol Cell Biochem. 2012;362:87–91 [DOI] [PubMed] [Google Scholar]
- 33. Miquet JG, Giani JF, Martinez CS, et al. Prolonged exposure to GH impairs insulin signaling in the heart. J Mol Endocrinol. 2011;47:167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quaife CJ, Mathews LS, Pinkert CA, Hammer RE, Brinster RL, Palmiter RD. Histopathology associated with elevated levels of growth hormone and insulin-like growth factor I in transgenic mice. Endocrinology. 1989;124:40–48 [DOI] [PubMed] [Google Scholar]
- 35. Doi T, Striker LJ, Quaife C, et al. Progressive glomerulosclerosis develops in transgenic mice chronically expressing growth hormone and growth hormone releasing factor but not in those expressing insulin like growth factor-1. Am J Pathol. 1988;131:398–403 [PMC free article] [PubMed] [Google Scholar]
- 36. Doi T, Striker LJ, Gibson CC, Agodoa LY, Brinster RL, Striker GE. Glomerular lesions in mice transgenic for growth hormone and insulinlike growth factor-I. I. Relationship between increased glomerular size and mesangial sclerosis. Am J Pathol. 1990;137:541–552 [PMC free article] [PubMed] [Google Scholar]
- 37. Machado MO, Hirata RD, Sellitti DF, et al. Growth hormone promotes glomerular lipid accumulation in bGH mice. Kidney Int. 2005;68:2019–2028 [DOI] [PubMed] [Google Scholar]
- 38. Auriemma RS, Galdiero M, De Martino MC, et al. The kidney in acromegaly: renal structure and function in patients with acromegaly during active disease and 1 year after disease remission. Eur J Endocrinol. 2010;162:1035–1042 [DOI] [PubMed] [Google Scholar]
- 39. Reddy GR, Pushpanathan MJ, Ransom RF, et al. Identification of the glomerular podocyte as a target for growth hormone action. Endocrinology. 2007;148:2045–2055 [DOI] [PubMed] [Google Scholar]
- 40. Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87:E1–E9 [DOI] [PubMed] [Google Scholar]
- 41. Colao A, Marzullo P, Di Somma C, Lombardi G. Growth hormone and the heart. Clin Endocrinol (Oxf). 2001;54:137–154 [DOI] [PubMed] [Google Scholar]
- 42. Nishikimi T, Maeda N, Matsuoka H. The role of natriuretic peptides in cardioprotection. Cardiovasc Res. 2006;69:318–328 [DOI] [PubMed] [Google Scholar]
- 43. Nishikimi T, Kuwahara K, Nakao K. Current biochemistry, molecular biology, and clinical relevance of natriuretic peptides. J Cardiol. 2011;57:131–140 [DOI] [PubMed] [Google Scholar]
- 44. Potter LR. Natriuretic peptide metabolism, clearance and degradation. FEBS J. 2011;278:1808–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McKnight J, McCance D, Hadden D, et al. Basal and saline-stimulated levels of plasma atrial natriuretic factor in acromegaly. Clin Endocrinol (Oxf). 1989;31:431–438 [DOI] [PubMed] [Google Scholar]
- 46. Lazúrová I, Pura M, Wagnerová H, et al. Effect of growth hormone replacement therapy on plasma brain natriuretic peptide concentration, cardiac morphology and function in adults with growth hormone deficiency. Exp Clin Endocrinol Diabetes. 2010;118:172–176 [DOI] [PubMed] [Google Scholar]
- 47. Andreassen M, Faber J, Vestergaard H, Kistorp C, Kristensen LO. N-terminal pro-B-type natriuretic peptide in patients with growth hormone disturbances. Clin Endocrinol (Oxf). 2007;66:619–625 [DOI] [PubMed] [Google Scholar]
- 48. Dirsch V, Wolf E, Wanke R, Schulz R, Hermanns W, Vollmar A Effect of chronic GH overproduction on cardiac ANP expression and circulating ANP levels. Mol Cell Endocrinol. 1998;144:109. [DOI] [PubMed] [Google Scholar]
- 49. Masternak MM, Bartke A. Growth hormone, inflammation and aging. Pathobiol Aging Age Relat Dis. 2012;2:17293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Coschigano KT, Wetzel AN, Obichere N, et al. Identification of differentially expressed genes in the kidneys of growth hormone transgenic mice. Growth Horm IGF Res. 2010;20:345–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Z, Masternak MM, Al-Regaiey KA, Bartke A. Adipocytokines and the regulation of lipid metabolism in growth hormone transgenic and calorie-restricted mice. Endocrinology. 2007;148:2845–2853 [DOI] [PubMed] [Google Scholar]
- 52. Olarescu NC, Ueland T, Godang K, Lindberg-Larsen R, Jorgensen JO, Bollerslev J. Inflammatory adipokines contribute to insulin resistance in active acromegaly and respond differently to different treatment modalities. Eur J Endocrinol. 2013;170:39–48 [DOI] [PubMed] [Google Scholar]
- 53. Wan W, Murphy PM. Regulation of atherogenesis by chemokines and chemokine receptors. Arch Immunol Ther Exp (Warsz). 2013;61:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liao TD, Yang XP, Liu YH, et al. Role of inflammation in the development of renal damage and dysfunction in angiotensin II-induced hypertension. Hypertension. 2008;52:256–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. German AJ, Ryan VH, German AC, Wood IS, Trayhurn P. Obesity, its associated disorders and the role of inflammatory adipokines in companion animals. Vet J. 2010;185:4–9 [DOI] [PubMed] [Google Scholar]
- 56. Wassel CL, Barrett-Connor E, Laughlin GA. Association of circulating C-Reactive protein and interleukin-6 with ongevity into the 80s and 90s: The Rancho Bernardo Study. J Clin Endocrinol Metab. 2010;95:4748–4755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Giovannini S, Onder G, Liperoti R, et al. Interleukin-6, C-reactive protein, and tumor necrosis factor-α as predictors of mortality in frail, community-living elderly individuals. J Am Geriatr Soc. 2011;59:1679–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci USA. 1995;92:4862–4866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Banerjee I, Fuseler JW, Intwala AR, Baudino TA. IL-6 loss causes ventricular dysfunction, fibrosis, reduced capillary density, and dramatically alters the cell populations of the developing and adult heart. Am J Physiol Heart Circ Physiol. 2009;296:H1694–H704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Funakoshi Y, Ichiki T, Ito K, Takeshita A. Induction of interleukin-6 expression by angiotensin II in rat vascular smooth muscle cells. Hypertension. 1999;34:118–125 [DOI] [PubMed] [Google Scholar]
- 61. Leung KC, Johannsson G, Leong GM, Ho KK. Estrogen regulation of growth hormone action. Endocr Rev. 2004;25:693–721 [DOI] [PubMed] [Google Scholar]
- 62. Söderpalm B, Ericson M, Bohlooly M, Engel JA, Törnell J. Bovine growth hormone transgenic mice display alterations in locomotor activity and brain monoamine neurochemistry. Endocrinology. 1999;140:5619–5625 [DOI] [PubMed] [Google Scholar]
- 63. Bohlooly-Y M, Olsson B, Gritli-Linde A, et al. Enhanced spontaneous locomotor activity in bovine GH transgenic mice involves peripheral mechanisms. Endocrinology. 2001;142:4560–4567 [DOI] [PubMed] [Google Scholar]
- 64. Vitale G, Pivonello R, Auriemma RS, et al. Hypertension in acromegaly and in the normal population: prevalence and determinants. Clin Endocrinol (Oxf). 2005;63:470–476 [DOI] [PubMed] [Google Scholar]