

Abstract
Lipid metabolism is tightly regulated by nuclear receptors, transcription factors, and cellular enzymes. In this study, we demonstrated that the liver-enriched transcription factor CREBH (cAMP-responsive element binding protein, hepatocyte specific) and peroxisome proliferator-activated receptor α (PPARα) function as binary transcriptional activators to regulate lipid metabolism by activating fibroblast growth factor 21 (FGF21), a hepatic hormone that regulates whole-body energy homeostasis. Gain- and loss-of-function studies indicated that CREBH regulates triglyceride and fatty acid metabolism in animals under fasting or on an atherogenic high-fat (AHF) diet. CREBH and PPARα act as interactive trans-activators that regulate each other for their expression. Activated CREBH protein interacts with PPARα to form a functional complex upon fasting or the AHF diet, and both factors are required for induction of the metabolic hormone FGF21. The CREBH-PPARα complex was found to bind to integrated CRE-PPAR-responsive element-binding motifs in the FGF21 gene promoter. Whereas CREBH and PPARα function in synergy to activate FGF21 gene expression, PPARα relies on CREBH to exert its trans-activation effect on FGF21. Supporting the key role of CREBH in regulating FGF21, infusion of recombinant FGF21 protein can reverse hypertriglyceridemia and hypoketonemia and partially rescue nonalcoholic steatohepatitis developed in the CREBH-null mice after the AHF diet. Our study demonstrated a transcriptional regulatory axis of CREBH-PPARα-FGF21 in maintaining lipid homeostasis under metabolic stress. The functional relationship between CREBH and PPARα in regulating FGF21 may represent an important transcriptional coactivation mechanism that orchestrates the processes of energy supply upon metabolic alteration.
Hepatic lipid metabolism is tightly regulated by nuclear receptors, transcription factors, and cellular enzymes in response to nutritional, hormonal, and stress signals (1). Previously, we identified an endoplasmic reticulum (ER)-localized, liver-enriched transcription factor CREBH (cAMP-responsive element-binding protein, hepatocyte specific), which is activated by ER stress, inflammatory stimuli, and metabolic signals (2). Proinflammatory cytokines TNFα, IL6, and IL1β, bacterial endotoxin lipopolysaccharide, insulin signal, saturated fatty acids, nutrient starvation, or atherogenic high-fat (AHF) feeding, can all induce expression and/or activation of CREBH in the liver (2, 3). CREBH is activated through regulated intramembrane proteolysis mediated by Golgi-resident site-1 protease and site-2 protease, the same enzymes that process sterol response element-binding proteins for cholesterol biosynthesis (2, 4). Activated CREBH functions as a potent transcription factor that induces expression of the genes involved in hepatic acute-phase response, lipogenesis, fatty acid (FA) oxidation, and lipolysis (2, 3, 5). Supporting the role of CREBH in regulating lipid homeostasis, CREBH-knockout mice display profound nonalcoholic steatohepatitis (NASH) and hypertriglyceridemia along with diminished abdominal fat upon AHF feeding (3). In human population, patients with hypertriglyceridemia exhibit high rates of functional mutations of the CREBH gene (5). Moreover, CREBH was shown to regulate hepatic gluconeogenesis under fasting or diet-induced obese conditions by activating expression of phosphoenol pyruvate carboxykinase and glucose-6-phosphatase (6).
Fibroblast growth factor 21 (FGF21) is a potent metabolic hormone predominantly produced by the liver (7). It regulates multiple pathways in lipid and glucose metabolism through pleiotropic actions (8–10). In the liver, fasting stress increases peroxisome proliferator-activated receptor (PPAR)α-dependent expression of FGF21 to stimulate FA oxidation and ketogenesis during the adaptive response to starvation (9, 10). In the fed state, FGF21 regulates other important metabolic processes, including lipolysis and glucose uptake in the liver or other peripheral tissues, such as white adipose tissue in which FGF21 also plays a role in browning upon cold exposure (11). In human patients, serum FGF21 concentrations correlate with hepatic steatosis, insulin resistance, hypertriglyceridemia, pericardial fat accumulation, and type 2 diabetes (12–14). It has been shown that infusion of exogenous FGF21 protein elicits therapeutic effects on metabolic symptoms associated with lipid and/or glucose dysregulation in animal models. In obese mice or diabetic monkeys, administration of FGF21 can decrease fasting serum glucose, triglycerides (TGs), and insulin concentrations and cause small but significant weight loss (8, 15–17). Because of its multiple metabolic benefits, FGF21 has been targeted as a potential therapeutic agent for treatment of metabolic syndrome.
In this study, we demonstrated that CREBH functions as a key transcriptional regulator of FGF21 expression by interacting with the nuclear receptor PPARα under metabolic stress. CREBH and PPARα rely on each other for their expression and function in synergy to regulate expression of FGF21. The activated CREBH protein interacts with PPARα to bind to an integrated CRE-PPAR-responsive element (PPRE) binding consensus sequence in the FGF21 gene promoter upon fasting or AHF feeding. We demonstrated that administration of recombinant FGF21 protein can reverse hypertriglyceridemia, hypoketonemia, and hepatic steatosis developed in the CREBH-null mice under the AHF diet.
Materials and Methods
Materials
Synthetic oligonucleotides were purchased from Integrated DNA Technologies, Inc. Antibodies against flag and β-actin were from Sigma. Mouse monoclonal anti-PPARα antibody was purchased from Millipore Corp. Mouse monoclonal anti-FGF21 antibody and FGF21 ELISA kit were from R&D Systems, Inc. Mouse monoclonal anti-CREB antibody was from Santa Cruz Biotechnology. Polyclonal anti-CREBH antibody was raised by immunizing rabbits with a mouse CREBH protein fragment spanning N-terminal amino acids 75–250 of mouse CREBH protein. The crude blood serum antibody was purified through affinity purification. Kits for measuring TG, FA, and ketone body 3-hydroxybutyric acid were from BioAssay System. Purified, bioactive recombinant human (rh) FGF21 protein was purchased from ProSpec-Tany TechnoGene Ltd.
Mouse experiments
The CREBH-null mice with exons 4–7 of the CrebH gene deleted were previously described (18). The PPARα-null mice were purchased from The Jackson Laboratory. CREBH-null, PPARα-null, and wild-type control mice on a C57Bl/6J background of approximately 3-month-old were used for the experiments. For the diet study, the AHF diet (16% fat, 41% carbohydrate, supplied with 0.5% sodium cholate by weight) was from Harlan Laboratories. All the animal experiments were approved by the Wayne State University IACUC committee and carried out under the institutional guidelines for ethical animal use. For the FGF21 rescue animal experiment, daily injections of purified, bioactive rh FGF21 protein and control injections of vehicle (sterile water) were carried out for 8 days. CREBH-null and wild-type control male mice after the AHF diet for 6 months were injected ip with 0.1 μg FGF21 protein per gram body weight or vehicle. On the fifth day of injection, mice were fasted for 12 hours. Following fasting, on the sixth day of injection, glucose tolerance tests were performed. Mice were allowed to recover for 2 days with ongoing injections before they were euthanized and tissues were harvested.
Protein interaction assay, immunoprecipitation (IP), and Western blot analysis
The interaction between CREBH and PPARα was determined by IP-Western blot assay. For preparation of the whole lysates, liver tissues were homogenized and lysed with Nonidet P-40 lysis buffer containing protease inhibitor cocktail as previously described (19). For IP, 200 μg of the whole lysate were incubated with 3 μg of specific protein antibody for overnight at 4°C. Immunoprecipitated proteins were resolved by SDS-PAGE followed by immunoblotting using an anti-CREBH or anti-PPARα antibody.
Nuclear extracts (NEs) and EMSA
To perform the EMSA, liver NEs from wild-type, CREBH-null, or PPARα-null mice were prepared by Subcellular Protein Fractionation kit (Thermo Scientific). EMSA was performed with Lightshift Chemiluminescent EMSA kit (Thermo Scientific). Liver NE (2.5 μg) was incubated with biotin-labeled probe (2.6 ng) in binding buffer containing 20 mM HEPES, 100 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, 15% (vol/vol) glycerol, and 1 μg polydeoxyinosinic deoxycytidylic acid for 30 minutes at room temperature. The reaction mixtures were separated in a 5% nondenaturing polyacrylamide gel. The gel was transferred to nylon membrane and followed by Chemiluminescent Nucleic Acid Detection Module (Thermo Scientific). The sequence information for the EMSA probes is described in Supplemental Figure 10 published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org.
For a full description of materials and methods used in this work, see Supplemental Data.
Results
CREBH-knockout mice display hypertriglyceridemia, hypoketonemia, and FGF21 deficiency upon fasting or under an AHF diet
We recently demonstrated that CREBH plays a key role in maintaining lipid homeostasis by regulating hepatic lipogenesis, FA oxidation, and lipolysis (3). However, the molecular mechanism through which CREBH exerts its pleiotropic effects on lipid metabolism remains elusive. The involvement of CREBH in lipid-associated pathophysiology also needs to be further delineated. To address these issues, we subjected CREBH-null and wild-type control mice to nutrient starvation or fed them with an AHF diet. The AHF diet (also called Paigen diet) is known to induce atherosclerosis and nonalcoholic fatty liver disease in murine models (20, 21). Supporting the role of CREBH in lipolysis (3), CREBH-null mice exhibited hypertriglyceridemia, reflected by significantly elevated TG levels in the blood sera or liver tissues, after 14-hour fasting or under the AHF diet for 6 months (Figure 1A and Supplemental Figure 1). Moreover, CREBH-null mice produced increased serum-free FA but decreased ketone body 3-hydroxybutyric acid (BOH) upon fasting or after the AHF diet (Figure 1, B and C), thus confirming the requirement of CREBH for FA oxidation and ketogenesis under metabolic stress, as suggested by our previous gene expression analysis (3). Furthermore, we expressed an activated form of CREBH in the liver of mice using an adenovirus-based overexpression system. Overexpression of the activated CREBH, but not green fluorescent protein (GFP) control, significantly decreased levels of serum TG and FA in the mice (Figure 1D), thus validating the role of CREBH in lipolysis and FA oxidation. Additionally, we overexpressed the activated CREBH or GFP control in high-fat diet-induced obese animals. Consistent with the roles of CREBH in lipolysis and de novo lipogenesis as we previously revealed (3), adenoviral expression of the activated CREBH significantly decreased serum TG but increased hepatic TG levels in the obese mice (Supplemental Figure 2, A and B). One week after the adenoviral injection, the obese animals overexpressing CREBH displayed slight body weight loss, compared with those overexpressing GFP (Supplemental Figure 2C).
Figure 1.

CREBH-knockout (KO) mice display hypertriglyceridemia, hypoketonemia, and FGF21-deficiency upon metabolic stress. A–C, Levels of serum TG (A), free FA (B), and ketone body BOH (C) in the CREBH-null (KO) and wild-type (WT) control mice under the feeding condition (feed), after 14-hour fasting (fast), or after 6 months of the AHF diet. Each bar denotes the mean ± SEM (n = 8 WT or KO mice under feeding or fasting condition; n = 6 WT or KO mice under the AHF diet). *, P < .05; **, P < .01. D, Levels of serum TG and free FA in the mice overexpressing GFP or the activated form of CREBH. WT male mice 3 months of age were injected with adenovirus expressing GFP or the activated form of CREBH via tail vein injection. One week after injection, blood samples were collected to determine serum levels of TG and FA. Each bar denotes the mean ± SEM (n = 3 mice per group). **, P < .01. Levels of the activated CREBH protein in the livers of mice infected with adenovirus expressing GFP or the activated form of CREBH were determined by Western blot analysis (left panel). E, Levels of plasma FGF21 in the CREBH-null and WT control mice after 14-hour fasting. Each bar denotes the mean ± SEM. *, P < .05. F, Levels of serum FGF21 in the mice overexpressing GFP or the activated form of CREBH. WT mice were infected with adenovirus expressing GFP or the activated form of CREBH as described in panel D. Each bar denotes the mean ± SEM (n = 3 mice per group). **, P < .01.
Next, we sought to understand the mechanism by which CREBH simultaneously regulates lipolysis, FA oxidation, and ketogenesis under metabolic conditions. Recently, we performed liver gene expression analysis and identified several groups of potential CREBH-regulated genes, including FGF21 (3). FGF21, a fasting hormone mainly produced by the liver, is known to exert pleiotropic effects on FA oxidation, ketogenesis, and lipolysis upon prolonged fasting or feeding with a methonine- and choline-deficient diet (9, 10). We wondered whether CREBH regulates FGF21 production in mice under metabolic stress. Supporting this scenario, levels of serum FGF21 in the CREBH-null mice were significantly reduced, compared with those of the control mice, upon 14-hour fasting (Figure 1E). Furthermore, overexpression of the activated form of CREBH in mouse liver dramatically increased serum levels of FGF21 in the mice after the fasting (Figure 1F). These results suggested the role of CREBH as a proactive regulator in driving production of FGF21 in the animals upon fasting.
Metabolic stress induces a regulatory axis of CREBH-PPARα-FGF21 in the liver
It was shown that the liver nuclear receptor PPARα is required for hepatic expression of FGF21 upon prolonged fasting (9, 10, 22). Given the requirement of CREBH for FGF21 production, we asked whether CREBH functions as a transcriptional regulator of FGF21 expression associated with or independent of PPARα. To address this question, we analyzed expression of CREBH, PPARα, and FGF21 in CREBH- or PPARα-knockout mice. Expression of the PPARα mRNA was significantly decreased in the liver of CREBH-null mice, compared with that in wild-type control mice, under feeding conditions (Figure 2A). Upon 14-hour fasting, expression levels of the PPARα gene were increased in the wild-type mice, but not the CREBH-null mice (Figure 2A). Levels of the FGF21 mRNA were significantly decreased in the liver of CREBH-null mice under the feeding or fasting condition (Figure 2A). Western blot analysis showed that PPARα and FGF21 proteins were not detectable in the liver of CREBH-null mice under feeding conditions (Figure 2C). Upon fasting, PPARα and FGF21 proteins were detectable in the CREBH-null liver, but the levels were lower than those in the wild-type control mice (Figure 2C). These results suggest that CREBH is required for expression of PPARα and FGF21 in the liver. There are discrepancies between the PPARα or FGF21 protein and mRNA levels in mouse livers or blood sera upon fasting. These discrepancies may be partially due to feedback regulation and protein maturation processes observed with hormones or protein regulators involved in lipid metabolism and stress response (23, 24).
Figure 2.

Fasting stress induces an interdependent regulatory axis of CREBH-PPARα-FGF21 in the liver. A and B, Expression levels of CREBH, PPARα, and FGF21 mRNAs in the livers of wild-type (WT) control, CREBH-null (A), and PPARα-null (B) mice under feeding or fasting condition. Total RNAs were isolated from liver tissues of WT, CREBH-null, and PPARα-null mice under the normal chow diet (feeding) or after 14 hours of fasting. These RNAs were subjected to quantitative real-time RT-PCR analysis. Expression values were normalized to β-actin mRNA levels. Fold changes of mRNA levels are shown by comparison to that in one of the WT control mice under the feeding condition. Each bar denotes the mean ± SEM (n = 3). *, P < .05; **, P < .01.WT, wild-type; KO, knockout. C and D, Levels of the CREBH, PPARα, and FGF21 proteins in the livers of WT control, CREBH-null (C), and PPARα-null (D) mice under feeding or fasting condition. Liver whole lysates were prepared from the WT, CREBH-null, or PPARα-null mice under the normal chow (feeding) or after 14 hours of fasting. Equal amounts of the proteins were separated on SDS-PAGE gels and immunoblotted using the antibody against CREBH, PPARα, FGF21, or β-actin. The values below the gels represent the PPARα, FGF21, or activated form of CREBH protein signal intensities that were quantified using NIH ImageJ software and normalized to β-actin signal intensities.
Next, we analyzed CREBH and FGF21 expression in the PPARα-null mice. Levels of the CREBH and FGF21 mRNAs in the livers of the PPARα-null mice were significantly decreased under either feeding or after 14-hour fasting (Figure 2B). Levels of CREBH protein, both the precursor and activated forms, in the livers of the PPARα-null mice were lower than those in the control mice under the fasting condition (Figure 2D). Levels of FGF21 protein in the PPARα-null liver were detectable, but lower than those in the control mice, under the feeding or fasting condition (Figure 2D). Together, the results obtained with the CREBH- and PPARα-null mice suggested that CREBH and PPARα regulate each other for their expression, and both factors are required for optimal expression of FGF21 in the liver.
Regarding the regulatory relationship between FGF21, CREBH, and PPARα, it is important to note that expression of FGF21 in the CREBH-null liver was barely detectable under the feeding condition; in contrast, expression of FGF21 in the PPARα-null liver was detectable, albeit with reduced levels (Figure 2, C and D). Under the fasting condition, the reduction in FGF21 expression in the CREBH-null liver was more profound than that in the PPARα-null liver (Figure 2, A–D). This led us to speculate that CREBH may play a more dominant role in activating FGF21 expression than PPARα. To further test this hypothesis, we examined expression of FGF21 in PPARα-null mice expressing exogenous CREBH and in CREBH-null mice expressing exogenous PPARα. Adenoviral expression of exogenous CREBH or PPARα in the liver of PPARα-null or CREBH-null mice was first confirmed by immunoblotting analyses (Supplemental Figure 3). Supporting the role of PPARα or CREBH in FGF21 expression, levels of both hepatic FGF21 mRNA and serum FGF21 protein were reduced in PPARα-null or CREBH-null mice, compared with those in the control mice, under feeding or fasting condition (Figure 3, A–D). Importantly, expression of exogenous CREBH, but not GFP, increased production of FGF21, at both mRNA and protein levels, in the PPARα-null mice upon fasting (Figure 3, A and B). In contrast, expression of exogenous PPARα failed to rescue diminished FGF21 expression in the CREBH-null mice upon fasting (Figure 3, C and D). These results confirm that CREBH plays a determinant role in driving FGF21 expression and that PPARα likely relies on CREBH to exert its trans-activation effect on FGF21.
Figure 3.
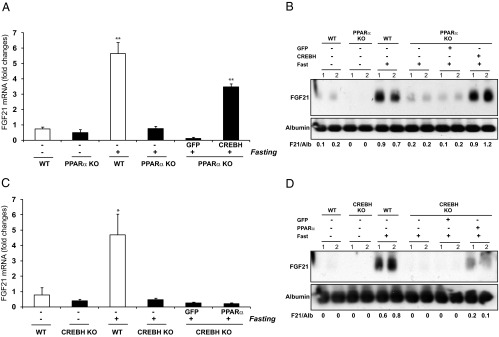
CREBH plays a determinant role in activating FGF21 expression, and PPARα relies on CREBH to exert its effect on transcriptional activation of FGF21. PPARα-null mice were infected with adenovirus expressing GFP or the activated form of CREBH, whereas CREBH-null mice were infected with adenovirus expressing GFP or PPARα via tail vein injection. Total liver RNAs and blood serum samples were prepared from the wild-type (WT) control mice, PPARα-null mice, CREBH-null mice, PPARα-null mice expressing exogenous GFP or CREBH, or CREBH-null mice expressing exogenous GFP or PPARα under feeding or 14-hour fasting condition. A, Levels of the FGF21 mRNA in livers of the WT control mice, PPARα-null mice, or PPARα-null mice infected with adenovirus expressing GFP or the activated form of CREBH. B, Levels of the serum FGF21 protein in the WT mice, PPARα-null mice, or PPARα-null mice infected with adenovirus expressing GFP or the activated form of CREBH. C, Levels of the FGF21 mRNA in livers of the WT control mice, CREBH-null mice, or CREBH-null mice infected with adenovirus expressing GFP or PPARα. D, Levels of the serum FGF21 protein in the WT control mice, CREBH-null mice, or CREBH-null mice infected with adenovirus expressing GFP or PPARα. For panels A and C, the mRNA expression values were determined by quantitative real-time RT-PCR analysis. Expression values were normalized to β-actin mRNA levels. Fold changes of mRNA levels are shown by comparison with that of one of the WT control mice under the feeding condition. Each bar denotes the mean ± SEM (n = 3). *, P < .05; **, P < .01. For panels B and D, levels of serum FGF21 protein were determined by Western blot analysis. Levels of albumin in the serum were determined as loading controls. The values below gel images represent quantification of FGF21 protein signal intensities after normalization to those of albumin. The experiments were repeated at least 3 times and the representative images were shown.
Activated CREBH interacts with PPARα and binds to the FGF21 gene promoter in the liver upon metabolic challenges
To understand the molecular mechanism underlying the regulation of FGF21 by CREBH and PPARα, we studied cis-regulatory elements in the promoter region of the FGF21 gene. A highly conserved DNA sequence was identified in the promoter regions spanning nucleotides −62 to −93 of the FGF21 genes of mammalian species ranging from mouse to human (Figure 4A). This conserved sequence in mice contains a cAMP response element (CRE)-binding motif (TGACG) and 2 hexameric PPREs, PPRE1 (TGATAT) and PPRE2 (TGACAC) (Figure 4A and Supplemental Figure 4) (25). This finding led us to hypothesize that CREBH and PPARα, as transcriptional activators, may interact with each other and bind to the FGF21 gene promoter. To test this hypothesis, we examined binding activities of CREBH and PPARα to the promoter of human FGF21 gene by chromatin immunoprecipitation (ChIP) analysis with the human hepatocellular cell line Huh-7 expressing exogenous PPARα and/or CREBH. Binding activity of CREBH or PPARα to the conserved FGF21 promoter region containing the CRE-PPRE motif can be detected in the Huh-7 cells expressing CREBH or PPARα, but not GFP control (Figure 4B). Importantly, binding activity of CREBH or PPARα to the FGF21 gene promoter was significantly increased in the cells expressing both CREBH and PPARα, compared with that in the cells expressing either CREBH or PPARα alone (Figure 4B). These results suggest that both CREBH and PPARα are recruited to the FGF21 gene promoter, and both factors are likely interactive to enhance their binding activities. Interestingly, no binding activity of CREB to the FGF21promoter region containing the conserved CRE-PPRE sequence was detected in the ChIP assay with the Huh-7 cells expressing CREB, whereas strong binding activity to the same FGF21 promoter region was detected with the cells expressing CREBH (Figure 4C). As a control, strong binding activity to the known CREB-target gene PCK1 promoter was detected with the cells expressing CREB (Figure 4C) (26). Further, gene reporter analysis showed that overexpression of CREB failed to activate the FGF21 promoter whereas overexpression of CREBH can significantly increase expression of the FGF21 promoter reporter (Supplemental Figure 5). These results suggest that CREBH, but not CREB, binds to the conserved CRE-PPRE motif to activate the FGF21 gene promoter.
Figure 4.
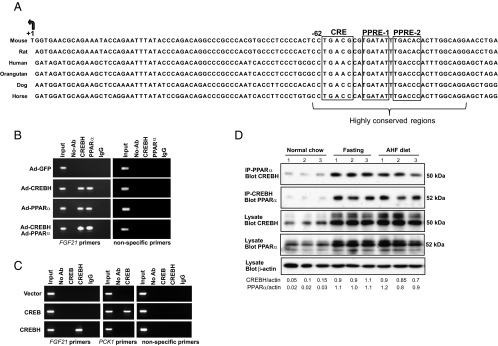
CREBH interacts with PPARα to bind to the FGF21 gene promoter. A, Alignment of FGF21 gene promoter sequences in mammalian species including mouse, rat, orangutan, dog, horse, and human. The arrow indicates transcription start site of the FGF21 gene, and the boxes highlight the predicted binding motifs for CREBH and PPARα. B, ChIP analysis of CREBH and PPARα binding activities to the FGF21 gene promoter. Huh-7 cells were infected with adenovirus expressing GFP, the activated CREBH, PPARα, or the same titer of combination of CREBH-expressing and PPARα-expressing adenovirus. An anti-CREBH or an anti-PPARα antibody was used to pull down the exogenously expressed activated CREBH or PPARα binding complex from Huh-7 cells. PCR was performed to determine the binding activities of CREBH and PPARα to the FGF21 promoter using the primers amplifying the promoter region containing the conserved CRE-PPRE motif (Supplemental Figure 4). Mock ChIP (no-antibody) and antibody control ChIP (IgG as an IP antibody) were included as negative controls. The PCRs with the genomic DNA isolated from sonicated cell lysates were included as positive controls (Input). C, ChIP analysis of CREBH and CREB binding activities to the FGF21 gene promoter. Huh-7 cells were transfected with plasmid vector expressing the activated CREBH or CREB. An anti-CREBH or anti-CREB antibody was used to pull down the CREBH- or CREB-binding complex from Huh-7 cells. PCR was performed to determine the binding activities of CREBH and CREB to the FGF21 promoter. As a control, the binding activity of CREB to its known target, PCK1 gene promoter, was determined with the same DNAs pulled down by the CREB antibody. D, IP-Western blot analysis of CREBH and PPARα interaction in the livers of mice under normal chow, after 14-hour fasting, or after 6 months of AHF diet. Whole-protein lysates were prepared from mouse liver tissues and subjected to IP to pull down the endogenous PPARα or CREBH protein complex using an anti-PPARα or anti-CREBH antibody. The precipitates were analyzed by immunoblotting with the antibody against CREBH or PPARα. Whole-liver protein lysates were subjected to Western blot analysis to determine the levels of CREBH, PPARα, and β-actin as the controls. The values below the gels represent the interactive CREBH (with PPARα) and PPARα (with CREBH) protein signal intensities after normalization to β-actin signal intensities. Ab, antibody; Ad, adenovirus.
Further, we tested whether CREBH and PPARα interact with each other in the liver upon metabolic challenges that stimulate lipolysis, FA oxidation, and ketogenesis, such as fasting and AHF diet. To detect potential interaction between CREBH and PPARα, wild-type mice were subjected to fasting for 14 hours or AHF diet feeding for 6 months. IP-Western blot analysis was performed with mouse liver protein lysates using an anti-PPARα or anti-CREBH antibody to pull down endogenous PPARα or CREBH and an anti-CREBH or PPARα antibody to detect the interaction between endogenous PPARα and CREBH. Through this approach, we detected increased amounts of the activated form of CREBH (50 kDa) that were associated with PPARα in the liver of the mice after fasting or AHF diet (Figure 4D), thus demonstrating the interaction between the activated CREBH and PPARα in the liver under metabolic stress that stimulates lipolysis, FA oxidation, and ketogenesis.
CREBH-PPARα complex binds to integrated CREB-PPRE motifs in the FGF21 gene promoter
The interaction between CREBH and PPARα and their binding activities to the FGF21 gene promoter suggest that CREBH and PPARα may form a functional complex and bind to the integrated CRE-PPRE motifs to activate transcription of the FGF21 gene under metabolic stress (Figure 4A). To test this hypothesis, we performed EMSA with an oligo containing the CRE-PPRE consensus sequence (nucleotide −58 to −98) and the liver nuclear extract (NE) from mice expressing CREBH, PPARα, or GFP control. Binding activities to the CRE-PPRE consensus sequence were detected with the liver NE from the mice expressing GFP, CREBH, or PPARα (Figure 5A, lanes 2–18). When either CREBH or PPARα, but not GFP, was overexpressed, shifted binding signals, which reflected the binding activities of homo- or heterodimerized CREBH or PPARα, were detected (Figure 5A, lanes 8–18).
Figure 5.
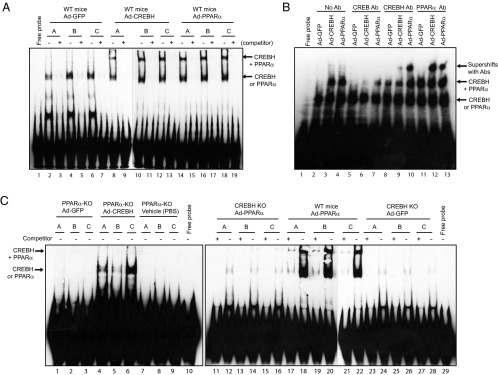
CERBH, PPARα, and CREBH-PPARα complex bind to the integrated CRE-PPRE motif in the FGF21 gene promoter. A, Binding activities of liver NE expressing GFP, CREBH, or PPARα to the CRE-PPRE binding motif. EMSA was performed using the liver NE from wild-type (WT) mice infected with adenovirus expressing GFP, CREBH, or PPARα and the FGF21 gene probe containing the CRE-PPRE motif. Competition assays were performed by using excessive concentrations (400-fold) of unlabeled probe. Free probe indicates the control reaction with the CRE-PPRE probe in the absence of NE. B, Antibody gel supershift assay for the recruitments of CREB, CREBH, and PPARα binding complexes to the FGF21 promoter. Gel supershift assay was performed using liver NEs from WT mice infected with adenovirus expressing GFP, CREBH, or PPARα and the FGF21 gene probe containing the conserved CRE-PPRE binding motifs in the presence of an anti-CREB, anti-CREBH, or anti-PPARα antibody. The protein-DNA binding complexes were visualized by shifted signals and highlighted by arrows. C, Binding activities of liver NE from wild-type, PPARα-null, or CREBH-null mice expressing GFP, CREBH, or PPARα to the CRE-PPRE binding motif. EMSA was performed with liver NE from WT, PPARα-null, or CREBH-null mice infected with adenovirus expressing GFP, CREBH, or PPARα and the FGF21 gene probe containing the CRE-PPRE binding motif. Competition assays were performed by using excessive concentrations of the unlabeled probe.
To confirm the binding activities of CREBH and/or PPARα to the CRE-PPRE motif, we performed antibody supershift gel assay. When the anti-CREBH or anti-PPARα antibody was included in the binding reaction, supershifted binding complexes were detected in the liver NEs expressing CREBH or PPARα, but not from those expressing GFP (Figure 5B, lanes 8–13). As a control, when the anti-CREB antibody was included in the binding reaction, no supershifted signal was detected in the liver NEs expressing CREBH, PPARα, or GFP (Figure 5B, lanes 5–7). This is consistent with the ChIP analysis demonstrating that CREBH, but not CREB, binds to the FGF21 gene promoter (Figure 4C). Further, we performed EMSA with liver NEs from PPARα-null, CREBH-null, and wild-type mice expressing PPARα, CREBH, or GFP. Contrast to the liver NE from the wild-type mice expressing GFP (Figure 5A, lanes 2–7), the liver NE from the PPARα-null or CREBH-null mice expressing GFP did not exhibit any binding activity to the CRE-PPRE motifs (Figure 5C, lanes 1–3; References 23–28). When exogenous CREBH was expressed in the liver of PAPRα-null mice, binding activities to the CRE-PPRE motifs were detected with the liver NE (Figure 5C, lanes 4–6). However, when PPARα was expressed in the liver of CREBH-null mice, liver NE binding activities to the CRE-PPRE motifs were diminished (Figure 5C, lanes 11–16). In contrast, when exogenous PPARα was expressed in the liver of the wild-type mice, shifted signals that indicate the binding activities of CREBH- or PPARα-homodimer or heterodimer complexes were detectable (Figure 5C, lanes 17–22). These results not only confirmed the binding activities of CREBH and PPARα to the integrated CRE-PPRE motifs, but also implicated that CREBH recruitment to the CRE-PPRE motif is a prerequisite for PPARα binding to the FGF21 gene promoter.
PPARα acts as a transcriptional coactivator of CREBH and functions in synergy with CREBH to activate expression of the FGF21 gene
As described above, although both CREBH and PPARα are required for stress-induced FGF21 expression, PPARα relies on CREBH for its recruitment to the FGF21 promoter and transcriptional activation of the FGF21 gene (Figures 2, 3, and 5C). Therefore, PPARα likely functions as a transcriptional coactivator of CREBH in activating transcription of the FGF21 gene. To further validate this scenario, we evaluated the binding ability of CREBH or PPARα to a mutant form of the CRE-PPRE motif in which 2 key CRE-binding nucleotides, AC, were changed to TG (Figure 6A). EMSAs were performed with the FGF21 promoter oligo containing the conserved CRE-PPRE binding sequence or its mutant version and liver NE from wild-type or PPARα-knockout mice. Binding activities of CREBH and/or PPARα to the wild-type oligo were detected with the liver NE from wild-type mice expressing exogenous CREBH or PPARα (Figure 6B, lanes 2–4). In contrast, the binding activities of CREBH or PPARα were disrupted by the mutation in the CRE-binding site (Figure 6B, lanes 5–7). In the absence of PPARα, the liver NE expressing exogenous CREBH or GFP displayed binding activities to the CRE-PPRE motif, but not its CRE-mutant version, albeit no shifted binding signals for CREBH-PPARα complex were detected (Figure 6B, lanes 10–14). These results again suggest that CREBH binding to the CRE motif is the prerequisite for the recruitment of PPARα or PPARα-CREBH complex to the FGF21 gene promoter. Moreover, to discern the PPARα-binding site within the highly conserved CRE-PPRE consensus sequence, EMSA was performed with the CRE-PPRE probe containing a mutation in PPRE1 or PPRE2 (Figure 6A). Whereas binding activities of CREBH or PPARα to the wild-type, PPRE1 mutant, or PPRE2-mutant oligo can all be detected with the liver NEs, the shifted CREBH-PPARα binding complex signals can only be detected with the wild-type or PPRE1-mutant oligo, but not the PPRE2-mutant oligo (Figure 6C). These results suggest that PPRE2, but not PPRE1, is required for the recruitment of CREBH-PPARα complex to the CRE-PPRE consensus sequence in the FGF21 gene promoter by functioning as the PPARα binding site. Additionally, we verified the binding activities of CREBH and PPARα to the CRE-PPRE motif by EMSA with in vitro translated CREBH and PPARα proteins. Individual CREBH and PPARα, as well as CREBH-PPARα complex, can all bind to the CRE-PPRE motif (Supplemental Figure 6). Consistent with EMSAs with liver NEs, the CRE- and PPRE-binding site within the CRE-PPRE motif are essential for the binding of CREBH-PPARα complex to the motif.
Figure 6.
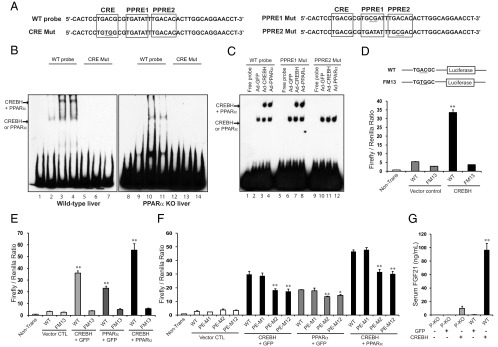
PPARα relies on CREBH to bind to the CRE-PPRE motif and functions in synergy with CREBH to activate expression of the FGF21 gene. A, Wild-typ (WT) and mutant oligo probes used in EMSA. B, Binding activities of CREBH or PPARα to the WT and CRE mutant (CRE Mut) probes in the presence or absence of PPARα. EMSA was performed using the WT and mutant probes and the liver NE from WT or PPARα-null mice infected with adenovirus expressing GFP, CREBH, or PPARα. C, Binding activities of CREBH or PPARα to the WT, PPRE1-mutant, and PPRE2-mutant probes. EMSA was performed using the WT and mutant probes and the liver NEs from WT mice infected with adenovirus expressing GFP, CREBH, or PPARα. D, Luciferase reporter analysis of trans-activation activities of the human FGF21 gene promoter or its mutant form by CREBH. Mouse hepatoma cell line Hepa1–6 was transiently transduced with the expression vectors containing the WT or CRE-mutant (FM13) FGF21 promoter and adenovirus expressing the activated form of CREBH. Renilla reporter plasmid was included in the cotransfection for normalization of luciferase reporter activities. E, Reporter analysis of trans-activation activities of the FGF21 promoter (WT) and its mutant form (FM13) by PPARα and/or CREBH. Hepa1–6 cells were cotransfected with the expression vectors containing the WT or mutant FGF21 promoter and adenovirus expressing GFP, PPARα, or activated CREBH. The same amounts of adenovirus titers were used for individual infections. F, Reporter analysis of trans-activation activities of the human FGF21 gene promoter or its mutant forms by CREBH and/or PPARα. Hepa1–6 cells were transduced with the expression vectors containing the wild-type, PPRE1-mutant (PE-M1), PPRE2-mutant (PE-M2), or both PPRE1- and PPRE2-mutant (PE-M12) FGF21 promoter and adenovirus expressing GFP, the activated CREBH, and/or PPARα. For panels D–F, each bar denotes the mean ± SEM (n = 3 biological repeats). **, P < .01. G, Serum levels of FGF21 in the WT and PPARα-knockout (P-KO) mice infected with adenovirus expressing GFP or CREBH. Blood serum samples were collected from the mice after 14-hour fasting, and levels of serum FGF21 were determined by ELISA. Each bar denotes the mean ± SEM (n = 3 biological repeats). **, P < .01. CTL, control.
Next, we performed gene expression reporter analysis with the human FGF21 gene promoter or its mutant versions. The mutation in the CRE-binding site significantly decreased transcriptional activation of the FGF21 gene promoter by CREBH (Figure 6D). Supporting the conclusion that CREBH binding to the CRE-PPRE motif is a prerequisite for PPARα to exert its trans-activation effect on FGF21, expression of either PPARα alone or both PPARα and CREBH failed to activate the FGF21 promoter with the mutation in the CRE-binding site (Figure 6E). Moreover, the mutation in PPRE2, but not PPRE1, significantly decreased transcriptional activation of the FGF21 gene promoter by CREBH or PPARα (Figure 6F). Mutations in both PPRE1 and PPRE2 did not further decrease transcriptional activation of the FGF21 promoter, compared with the PPRE2 mutation alone, thus confirming that PPRE2, but not PPRE1, is involved in the transcriptional activation of the FGF21 gene by CREBH or PPARα. Although the mutation in PPRE2 repressed transcriptional activation of the FGF21gene promoter by CREBH and/or PPARα, CREBH can still activate the FGF21 promoter in the presence of the PPRE2 mutation (Figure 6F), suggesting that PPRE2 likely serves as a coactivator binding site that optimizes the transcriptional activity. It is important to note that, in comparison to expression of PPARα or CREBH alone, coexpression of PPARα and CREBH exerted synergistic effect on activation of the wild-type FGF21 promoter (Figure 6E). At the protein level, whereas expression of the activated CREBH significantly increased serum FGF21 levels in the PPARα null mice, expression of the activated CREBH increased production of serum FGF21 in the wild-type mice more dramatically than in the PPARα-null mice (Figure 6G). Together, these results suggest that PPARα likely acts as a coactivator of CREBH and functions with CREBH to synergistically activate the FGF21 gene promoter.
Infusion of recombinant FGF21 protein reverses hypertriglyceridemia, hypoketonemia, and hepatic steatosis, and partially rescues NASH phenotype in CREBH-null mice after the AHF diet
Previously, we established that deletion of CREBH leads to a severe NASH phenotype in mice under the AHF diet (3). To determine whether FGF21 is the major player targeted by CREBH in regulating lipid metabolism and to evaluate the effect of FGF21 on reversing NASH phenotype resulting from CREBH deletion, we fed the CREBH-null animals with the AHF diet. After 6 months on the AHF diet, CREBH-null mice developed a profound NASH phenotype, characterized by hepatocyte ballooning, lobular and portal inflammation, steatosis, and fibrosis (Figure 7) (3). Along with the NASH state, the CREBH-null mice displayed hypertriglyceridemia, hypoketonemia, and impaired FA metabolism, as reflected by much higher levels of serum TG and FA but lower levels of the ketone body BOH, compared with the wild-type mice under the AHF diet (Figure 7, A–C). The CREBH-null animals under the NASH state were administrated with rh FGF21 protein or vehicle once a day, for 8 days. After the FGF21 treatment, the symptoms of hypertriglyceridemia and hypoketonemia in the AHF-fed CREBH-null mice were reversed, as the levels of serum TG and FA were reduced whereas the levels of serum BOH were increased, compared with wild-type and CREBH-null mice treated with the vehicle (Figure 7, A–C). Consistent with the previous studies (27), the average body weight of the animals after FGF21 injection was lower than that of the control animals (Supplemental Figure 7). Together, these results confirm that FGF21 is the major downstream factor targeted by CREBH in regulating TG lipolysis, FA oxidation, and ketogenesis.
Figure 7.
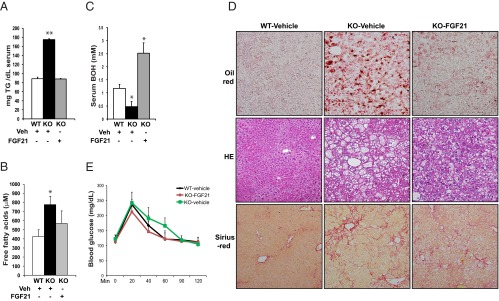
Infusion of recombinant FGF21 protein reverses hypertriglyceridemia, hypoketonemia, and hepatic steatosis, and partially rescues NASH phenotype in CREBH-null mice after the AHF diet. CREBH-null and wild-type (WT) control mice were fed with AHF diet for 6 months before they were injected ip with purified rh FGF21protein (0.1 μg per gram body weight) or vehicle once a day for 8 days. On the sixth day of injection, mice were subjected to fasting blood sample collection and glucose tolerance test after 12-hour fasting. Mice were allowed to recover for 2 days with ongoing injections before they were euthanized and tissues were harvested. A–C, Levels of serum TG, ketone body BOH, and FA in the WT and CREBH-null mice administrated with recombinant FGF21 protein or vehicle (Veh). The blood samples were collected from the mice after 12-hour fasting on the sixth day of administration of FGF21 protein or vehicle. Each bar denotes the mean ± SEM (n = 5 WT or KO mice treated with vehicle and 4 KO mice treated with FGF21). *, P < .05; **, P < .01. D, Histologic staining analysis of liver tissue sections from the wild-type and CREBH-null mice after 8 days of FGF21 protein or vehicle treatment. The upper panel shows the oil-red O staining of hepatic lipid droplets; the middle panel shows hematoxylin and eosin (HE) staining of liver tissue sections; and the lower panel shows Sirius red staining of hepatic collagens. Magnification: ×200. E, Glucose tolerance tests of the CREBH-null and wild-type control mice after 6 days of administration of the FGF21 protein or vehicle. Animals were fasted for 12 hours before being infused with 1 mg d-glucose per gram body weight. Each bar denotes the mean ± SEM (n = 5 WT or KO mice treated with vehicle and 4 KO mice treated with FGF21). KO, CREBH knockout.
We further investigated whether FGF21 protein treatment can rescue the NASH phenotype in the CREBH-null mice after the AHF diet. Histologic analysis indicated that hepatic steatosis, was significantly relieved in the CREBH-null mice receiving the recombinant FGF21 protein, compared with that in the CREBH-null mice treated with vehicle (Figure 7D, upper panel). The effect of FGF21 treatment on reducing hepatic lipid accumulation was confirmed by significantly reduced levels of hepatic TG in the CREBH-null mice receiving FGF21 protein (Supplemental Figure 8A). We examined the impact of FGF21 treatment in hepatic inflammation and fibrosis in the CREBH-null mice after the AHF diet. The extent of hepatocyte ballooning was reduced in the CREBH-null mice after the FGF21 treatment (Figure 7D, middle panel). This was further evidenced by the significant reduction in the sizes of the hepatocytes in the livers of the FGF21-treated CREBH-null mice (Supplemental Figure 8B). Only insignificant change in lobular and portal inflammation was observed in the CREBH-null mice upon FGF21 treatment (Figure 7D, middle panel). Similarly, an insignificant reduction in hepatic fibrosis in the FGF21-treated mice was evidenced by collagen staining of liver tissue sections (Figure 7D, lower panel). The limited effects of the FGF21 treatment on the liver tissue pathology may be due to irreversible morphologic changes caused by NASH after 6 months of AHF diet. Because NASH activities are associated with glucose intolerance and insulin resistance (28), we assessed whether the FGF21 treatment exerts any effect on glucose homeostasis in the CREBH-null mice under NASH state. The glucose tolerance test indicated that the CREBH-null animals treated with FGF21 exhibited improved ability in clearance of blood glucose, compared with the CREBH-null animals treated with vehicle (Figure 7E).
Discussion
In this study, we revealed the molecular mechanism through which 2 liver-specific transcriptional regulators, CREBH and PPARα, interact to activate expression of a key metabolic hormone, FGF21 (Supplemental Figure 9). We also demonstrated the functional significance of this regulatory mechanism in maintaining lipid homeostasis and its associated metabolic syndrome. Our major findings include: 1) CREBH is required for lipolysis, FA oxidation, and ketogenesis upon metabolic stress by regulating FGF21 production; 2) CREBH and PPARα function as stress-inducible binary transcriptional activators that regulate each other for their expression; 3) CREBH interacts with PPARα to synergistically activate expression of the FGF21 gene; 4) CREBH and PPARα form a transcriptional complex that binds to integrated CRE-PPRE binding motifs in the FGF21 gene promoter; 5) PPARα relies on CREBH for its recruitment to the FGF21 promoter and transcriptional activation of the FGF21 gene; 6) FGF21 protein treatment can reverse hypertriglyceridemia, hypoketonemia, and NASH state developed in CREBH null mice after the metabolic diet. These findings provide novel insights into the key transcriptional programs that control lipid homeostasis and have important implications in the understanding and treatment of metabolic disease.
As a newly-identified metabolic regulator, CREBH appears to play crucial roles in maintaining lipid homeostasis by regulating multiple lipid-associated pathways, including lipogenesis, lipolysis, FA oxidation, and ketogenesis (Figure 1) (3). However, it remains elusive how CREBH regulates perplex metabolic programs involved in multiple lipid-catalyzing pathways under metabolic stress conditions. The finding that FGF21 is the direct CREBH target gene provides an answer to this question. At the molecular level, CREBH coordinates with PPARα to activate FGF21 expression through a multiple-layer, fine-tuned regulatory mechanism. CREBH and PPARα regulate each other for their expression in the liver under fasting or AHF diet. Metabolic stress-induced expression of FGF21 requires both CREBH and PPARα, but PPARα relies on CREBH to exert its effect on driving FGF21 expression (Figures 2 and 3). The coordination between CREBH and PPARα in regulating FGF21 expression also occurs through protein-protein interaction upon fasting or the AHF diet (Figure 4). Through this interactive mechanism, CREBH and PPARα bind to the integrated CRE-PPRE motifs in the FGF21 promoter and function in synergy to activate expression of the FGF21 gene (Figures 5 and 6). Apparently, CREBH and PPARα act as binary transactivators that regulate each other for their gene expression and interact with each other for their function. The functional relationship between CREBH and PPARα may represent an important transcriptional coactivation mechanism that facilitates energy supplies under metabolic stress.
Previous studies showed that hepatic expression of FGF21 is regulated by PPARα (8–10). Gene reporter analysis suggested that expression of CREBH may be regulated by PPARα (29). Our work is consistent with the previous studies, but it does indicate an unexpected conclusion that CREBH plays a more determinant role in transcriptional activation of the FGF21 gene. In the absence of CREBH, PPARα binding activity to the CRE-PPRE motifs was diminished, and no CREBH-PPARα complex was formed in the FGF21 promoter (Figures 5C and 6B). Our results from DNA-protein interaction and gene reporter analyses suggest that PPARα acts as a coactivator of CREBH and functions with CREBH to synergistically activate expression of the FGF21 gene (Figures 5C and 6, B–G). Interestingly, our previous study showed that CREBH regulates many target genes in TG and FA metabolism that are also regulated by PPARα (3, 9, 10). Whether CREBH functions with PPARα to activate all their target genes is an intriguing question to be investigated in the future.
An important implication of this work is the therapeutic potential of FGF21 in NASH and its related symptoms. CREBH-null mice develop profound hypertriglyceridemia, hypoketonemia, and NASH under the AHF diet (Figure 7) (3). Therefore, the AHF-fed CREBH-null animal model is a perfect model system for evaluating therapeutic potential of FGF21 protein treatment in metabolic symptoms associated with impaired lipid metabolism. Our study demonstrated that administration of rh FGF21 protein can simultaneously reverse hypertriglyceridemia, hypoketonemia, and hepatic steatosis, and partially rescue hepatic inflammation and fibrosis in the CREBH-null mice after the AHF diet (Figure 7). The therapeutic effect of FGF21 on NASH was also evidenced by the observation that FGF21 protein treatment improved glucose tolerance in the AHF-fed CREBH-null mice under the NASH state (Figure 7E). These results confirm the pleiotropic effects of exogenous FGF21 administration on lipid metabolism and its promising therapeutic potential in metabolic disorders.
In summary, we revealed an unprecedented regulatory mechanism through which CREBH and PPARα interact to activate expression of the metabolic hormone FGF21 in the liver in response to metabolic stress (Supplemental Figure 9). This finding extends the relationship between the nuclear receptors and ER-associated trans-activators in regulating energy metabolism. The interdependency and synergism between PPARα and CREBH provide regulatory mechanisms for nuclear receptors or stress-inducible bZIP transcription factors to extend their biological actions and thereby coordinate adaptive physiological responses.
Acknowledgments
We thank Dr Eleftheria Maratos-Flier for her inspiring idea and constructive comments on this work. We also thank Dr Gregory Kapatos for providing the CREB expression vector.
This work was supported, in part, by National Institutes of Health (NIH) Grants DK090313 and ES017829 and American Heart Association Grants 0635423Z and 09GRNT2280479 (to K.Z.). R.M. is supported by NIH Training Grant R25GM058905.
Disclosure Summary: H.K., R.M., Z.Z., L.C., J.C., R.Z., and K.Z. have nothing to declare.
Footnotes
- AHF
- atherogenic high-fat
- BOH
- 3-hydroxybutyric acid
- ChIP
- chromatin immunoprecipitation
- CRE
- cAMP response element
- CREBH
- cAMP-response element-binding protein, hepatic specific
- ER
- endoplasmic reticulum
- FA
- fatty acid
- FGF21
- fibroblast growth factor 21
- GFP
- green fluorescent protein
- IP
- immunoprecipitation
- PPARα
- peroxisome proliferator-activated receptor α
- NASH
- nonalcoholic steatohepatitis
- NE
- nuclear extract
- PPRE
- PPAR response element
- TG
- triglyceride.
References
- 1. Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog Lipid Res. 2009;48(1):1–26 [DOI] [PubMed] [Google Scholar]
- 2. Zhang K, Shen X, Wu J, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124(3):587–599 [DOI] [PubMed] [Google Scholar]
- 3. Zhang C, Wang G, Zheng Z, et al. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology. 2012;55(4):1070–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100(4):391–398 [DOI] [PubMed] [Google Scholar]
- 5. Lee JH, Giannikopoulos P, Duncan SA, et al. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat Med. 2011;17(7):812–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee MW, Chanda D, Yang J, et al. Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab. 2010;11(4):331–339 [DOI] [PubMed] [Google Scholar]
- 7. Nishimura T, Nakatake Y, Konishi M, Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta. 2000;1492(1):203–206 [DOI] [PubMed] [Google Scholar]
- 8. Kharitonenkov A, Shiyanova TL, Koester A, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115(6):1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5(6):426–437 [DOI] [PubMed] [Google Scholar]
- 10. Inagaki T, Dutchak P, Zhao G, et al. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5(6):415–425 [DOI] [PubMed] [Google Scholar]
- 11. Fisher FM, Kleiner S, Douris N, et al. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012;26(3):271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dushay J, Chui PC, Gopalakrishnan GS, et al. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology. 2010;139(2):456–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chavez AO, Molina-Carrion M, Abdul-Ghani MA, Folli F, Defronzo RA, Tripathy D. Circulating fibroblast growth factor-21 is elevated in impaired glucose tolerance and type 2 diabetes and correlates with muscle and hepatic insulin resistance. Diabetes Care. 2009;32(8):1542–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li H, Dong K, Fang Q, et al. High serum level of fibroblast growth factor 21 is an independent predictor of non-alcoholic fatty liver disease: A 3-year prospective study in China. J Hepatol. 2013;58:557–563 [DOI] [PubMed] [Google Scholar]
- 15. Kharitonenkov A, Wroblewski VJ, Koester A, et al. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology. 2007;148(2):774–781 [DOI] [PubMed] [Google Scholar]
- 16. Fisher FM, Estall JL, Adams AC, et al. Integrated regulation of hepatic metabolism by fibroblast growth factor 21 (FGF21) in vivo. Endocrinology. 2011;152(8):2996–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu J, Stanislaus S, Chinookoswong N, et al. Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models–association with liver and adipose tissue effects. Am J Physiol Endocrinol Metab. 2009;297(5):E1105–E1114 [DOI] [PubMed] [Google Scholar]
- 18. Luebke-Wheeler J, Zhang K, Battle M, et al. Hepatocyte nuclear factor 4α is implicated in endoplasmic reticulum stress-induced acute phase response by regulating expression of cyclic adenosine monophosphate responsive element binding protein H. Hepatology. 2008;48(4):1242–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zheng Z, Zhang C, Zhang K. Measurement of ER stress response and inflammation in the mouse model of nonalcoholic fatty liver disease. Methods Enzymol. 2011;489:329–348 [DOI] [PubMed] [Google Scholar]
- 20. Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68(3):231–240 [DOI] [PubMed] [Google Scholar]
- 21. Matsuzawa N, Takamura T, Kurita S, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. 2007;46(5):1392–1403 [DOI] [PubMed] [Google Scholar]
- 22. Lundåsen T, Hunt MC, Nilsson LM, et al. PPARα is a key regulator of hepatic FGF21. Biochem Biophys Res Commun. 2007;360(2):437–440 [DOI] [PubMed] [Google Scholar]
- 23. Tian Q, Stepaniants SB, Mao M, et al. Integrated genomic and proteomic analyses of gene expression in mammalian cells. Mol Cell Proteomics. 2004;3(10):960–969 [DOI] [PubMed] [Google Scholar]
- 24. Lee JS, Zheng Z, Mendez R, Ha SW, Xie Y, Zhang K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol Lett. 2012;211(1):29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boergesen M, Pedersen TÅ, Gross B, et al. Genome-wide profiling of liver X receptor, retinoid X receptor, and peroxisome proliferator-activated receptor α in mouse liver reveals extensive sharing of binding sites. Mol Cell Biol. 2012;32(4):852–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quinn PG, Wong TW, Magnuson MA, Shabb JB, Granner DK. Identification of basal and cyclic AMP regulatory elements in the promoter of the phosphoenolpyruvate carboxykinase gene. Mol Cell Biol. 1988;8(8):3467–3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coskun T, Bina HA, Schneider MA, et al. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149(12):6018–6027 [DOI] [PubMed] [Google Scholar]
- 28. Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology. 2010;51(2):679–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Danno H, Ishii KA, Nakagawa Y, et al. The liver-enriched transcription factor CREBH is nutritionally regulated and activated by fatty acids and PPARα. Biochem Biophys Res Commun. 2010;391(2):1222–1227 [DOI] [PubMed] [Google Scholar]