

Abstract
N-acetylserotonin (NAS) is an immediate precursor of melatonin, which we have reported is neuroprotective against ischemic injury. Here we test whether NAS is a potential neuroprotective agent in experimental models of ischemic injury. We demonstrate that NAS inhibits cell death induced by oxygen–glucose deprivation or H2O2 in primary cerebrocortical neurons and primary hippocampal neurons in vitro, and organotypic hippocampal slice cultures ex vivo and reduces hypoxia/ischemia injury in the middle cerebral artery occlusion mouse model of cerebral ischemia in vivo. We find that NAS is neuroprotective by inhibiting the mitochondrial cell death pathway and the autophagic cell death pathway. The neuroprotective effects of NAS may result from the influence of mitochondrial permeability transition pore opening, mitochondrial fragmentation, and inhibition of the subsequent release of apoptogenic factors cytochrome c, Smac, and apoptosis-inducing factor from mitochondria to cytoplasm, and activation of caspase-3, -9, as well as the suppression of the activation of autophagy under stress conditions by increasing LC3-II and Beclin-1 levels and decreasing p62 level. However, NAS, unlike melatonin, does not provide neuroprotection through the activation of melatonin receptor 1A. We demonstrate that NAS reaches the brain subsequent to intraperitoneal injection using liquid chromatography/mass spectrometry analysis. Given that it occurs naturally and has low toxicity, NAS, like melatonin, has potential as a novel therapy for ischemic injury.
Introduction
Cerebral ischemia, caused by reduction in the supply of glucose and oxygen to the brain, leads to a complex cascade of cellular events, culminating in both acute and delayed neuronal death (Siesjö, 1992). Apoptosis, necrosis, inflammation, and oxidative stress contribute to the development of ischemic cascade (Friedlander, 2003; Pandya et al., 2011). Autophagy leads to degradation of proteins and removes harmful product after ischemia, especially dysfunctional mitochondria (Wen et al., 2008; Pan, 2013). Interestingly, autophagy not only has a protective effect on neuronal survival, but also promotes cell death, known as “autophagic cell death,” in a wide range of important situations including cerebral ischemia (Koike et al., 2008; Puyal et al., 2009; Nopparat et al., 2010; Gao et al., 2012; Sheng et al., 2012; Shi et al., 2012). Mitochondrial dysfunction is an important event in the middle cerebral artery occlusion (MCAO) animal model and cellular models of ischemic stroke. In cerebral ischemia, the translocation of apoptotic triggering proteins from mitochondria to cytoplasm, often as a consequence of the mitochondrial permeability transition (mPT), activates a cascade of caspases that eventually trigger cell death (Soane et al., 2011). Autophagy overactivation can induce autophagic neuron death and caspase-dependent apoptosis (Cui et al., 2012).
N-acetylserotonin (NAS) is a naturally occurring chemical intermediate that is produced from serotonin and is converted to melatonin, an agent that has been reported by us and other researchers to be neuroprotective in experimental models of ischemic stroke and other neurological diseases (Wang, 2009; Wang et al., 2009, 2011; Pandya et al., 2013; Zhang et al., 2013). Both NAS and melatonin belong to a library of 1040 FDA-approved compounds assembled by the National Institute of Neurological Disorders and Stroke (the NINDS Custom Collection). Although many of the biological effects of NAS (including antioxidant, anti-aging, anti-anxiety, and neuroprotection) are similar to those produced by melatonin, evidence indicates that NAS may play unique roles in the CNS beyond merely functioning as a precursor of melatonin: (1) NAS has a stronger antioxidant capacity (Wölfler et al., 1999) and its antioxidant activity may be independent of its conversion to melatonin (Oxenkrug et al., 2001), (2) NAS differs from melatonin in that it is found in some areas of the brain where melatonin is absent (Jang et al., 2010), (3) NAS is present in human serum in nanomolar concentrations, which is ∼10- to 100-fold higher than concentrations of melatonin (Wölfler et al., 1999), and (4) NAS, but not melatonin, acts as a potent TrkB receptor agonist (Jang et al., 2010).
It is unknown whether NAS, like melatonin, protects neuronal cells from hypoxia/ischemia-induced injury. Here we evaluate the neuroprotective effects of NAS in primary cerebrocortical neurons (PCNs), primary hippocampal neurons (PHNs), and organotypic hippocampal slice cultures (OHSCs) exposed to oxygen-glucose deprivation (OGD) and H2O2 cell death stimuli, as well as in vivo in MCAO cerebral ischemia. We test the molecular mechanisms of NAS in inhibiting mitochondrial death pathways and evaluate whether NAS could orchestrate the induction of neuronal autophagy.
Materials and Methods
Drugs.
NAS and monodansylcadaverine (MDC) were purchased from Sigma-Aldrich. Rhodamine 123 (Rh 123), Ca-Green-5N, and DAPI were purchased from Life Technologies.
Cell cultures, slice cultures, and induction of cell death.
PCNs and PHNs were isolated from E14 to E16 mice and subjected to OGD or H2O2 for 2 or 18 h as previously described (Wang et al., 2009). OHSCs were prepared from mouse pups 5- to 9-d-old as previously described (Stoppini et al., 1991; De Simoni and Yu, 2006) with slight modification. In brief, pups were decapitated and the brains rapidly removed and transversely cut into 400 μm slices using the Vibroslice (World Precision Instruments). Slices were transferred into ice-cold Hanks balanced salt solution supplemented with 5 mg/ml d-glucose and placed onto 30-mm-diameter, 0.4 μm pore, porous transparent membrane inserts (Millipore). The culture medium consisted of 50% Eagle minimal essential medium with Earle's salt, 25% Hanks balanced salt solution, 25% horse serum, 25 mmol/l HEPES, 1 mmol/l glutamine, and 1% antibiotic/antimycotic solution (penicillin G 10,000 U/ml, streptomycin sulfate 10,000 μg/ml, Fungizone 1%), supplemented with d-glucose to a final concentration of 6.5 mg/ml.
Primary cultures or OHSCs were preincubated for 2 h with NAS, luzindole, 3-methyladenine (3-MA), or cyclosporin A (CsA). Experiments on primary cultures and OHSCs lasted for ∼7 d and 12 to ∼14 d, respectively. OHSCs were treated by H2O2 for 18 h while OGD was induced (Pringle et al., 1997; Pellegrini-Giampietro et al., 1999) in an airtight chamber in a serum- and glucose-free balanced salt solution containing the following (in mmol/l): NaCl 124, KCl 5, MgCl 1.3, NaH2PO4 1.25, NaHCO3 26, CaCl2 2, pH 7.4. Dishes were sealed in an airtight chamber with anaerobic system envelopes with palladium catalyst (BD BBL GasPak Plus, Becton Dickinson) (Zhang et al., 2003; Wang et al., 2009). Following induction of hypoxia and hypoglycemia, OHSCs were transferred to normal serum-free medium (75% MEM, 25% Hanks balanced salt solution, 5 mg/ml glucose) with 0.25 mg/ml propidium iodide (PI) and replaced in the incubator under normoxic conditions.
Cell death induced by OGD or H2O2 in OHSCs was assessed using PI staining (Wang et al., 2004; Bai and Lipski, 2010). Slice cultures were examined using a Nikon ECLIPSE TE-200 fluorescence microscope and processed with IP LAB software. PI images were analyzed, and the intensity of PI fluorescence in the selected region of interest (CA1, CA3, and dentate gyrus) was used as an index of cell death. Cell death of PCNs, PHNs, and OHSCs was also quantitatively evaluated by the lactate dehydrogenase (LDH) assay as previously described according to the manufacturer's instructions (Roche; Wang et al., 2009, 2011).
Wild-type Atg5 (autophagy-competent) mouse embryonic fibroblasts (Atg5+/+ MEFs) and Atg5-deficient (autophagy-deficient) mouse embryonic fibroblasts (MEFs; Atg5−/−) were cultured as previously described (Kuma et al., 2004) and subjected to H2O2 for 18 h. Cell death was quantified by the MTS assay [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] (Roche; Wang et al., 2011).
MDC staining.
PCNs were subjected to H2O2 for 2 or 18 h and then stained with 20 μm MDC in PBS at 37°C for 1 h. After washing with PBS, cells were immediately analyzed by fluorescence microscopy.
Determination of mitochondrial transmembrane potential and Image-It Live mPTP assay.
For mitochondrial transmembrane potential (ΔΨm), PCNs and PHNs were treated as indicated with or without 10 μmol/L NAS. Living cells were stained with 2 μmol/L Rh 123 as previously described (Wang et al., 2009) for 5 min at room temperature. In digital images, reduced green Rh 123 fluorescence indicated dissipated ΔΨm. Mitochondrial transition pore (mPTP) assays were performed according to the manufacturer's instructions (Life Technologies). Briefly, PCNs were coincubated with NAS (10 μmol/L) or CsA (10 μmol/L), washed with modified HBSS buffer, and loaded with calcein AM and CoCl2 for 15–20 min; 1 μmol/L ionomycin was then added to test whether mPTP was activated. Digital images were taken.
Mitochondrial permeability transition assay in isolated brain mitochondria.
Nonsynaptosomal brain mitochondria were isolated from C57BL/6J mice. The mPTP assay was performed as described previously (Stavrovskaya et al., 2010).
Western blot.
Primary neuronal cultures or OHSCs were exposed to OGD or H2O2 with or without NAS. Cells and mouse brain samples were lysed (Wang et al., 2009). Antibody to Beclin 1, caspase-9, caspase-3, and active caspase-3 were purchased from Cell Signaling Technology, LC3 antibody from Novus Biologicals, p62 antibody from Santa Cruz Biotechnology, melatonin receptor 1A (MT1) antibody from Millipore, and β-actin antibody from Sigma-Aldrich.
Subcellular fractionation.
PCNs, OHSCs, and mouse brain cytosolic mitochondria fractionations were performed as described previously (Zhang et al., 2003; Wang et al., 2009). Released cyto. c (cytochrome c)/Smac/AIF (apoptosis-inducing factor) was analyzed by Western blot. Antibody to cyto. c was purchased from PharMingen, Smac/Diablo from Novus Biologicals, and AIF from Sigma-Aldrich.
Assays of caspase activity.
Cells or slices were extracted and enzyme assays were performed as previously described (Wang et al., 2001, 2009) with the ApoAlert Caspase 3 Assay Kit and ApoAlert Caspase 9 Assay Kit (Clontech). The lysates were incubated with caspase-3-like substrate Ac-DEVD-AFC and caspase-9-like substrate Ac-LEHD-AMC. Enzyme activity was determined using a Tecan GENios microplate reader (excitation at 400 nm and emission at 505 nm).
Immunocytochemistry.
PCNs were exposed to H2O2 with or without NAS. Cells were fixed in 4% paraformaldehyde for 15 min, 0.1 m glycine for 15 min, and 1% Triton X-100 for 30 min. After blocking in 5% BSA in PBS for 30 min, cells were incubated with anti-TOM20 or anti-active caspase-3. A fluorescent stain DAPI was used to stain nuclei. Digital images were taken with a fluorescence microscope. For the quantitative measurement of mitochondrial length, ImageJ (v. 1.43) software was used to set scale according to the picture pixel and size. Nano Measurers (v.1.2.5) software was used to track mitochondria and measure mitochondrial length. A minimum of 200 mitochondria for each picture was counted and percentage of different length of mitochondria in each picture was measured.
Permanent middle cerebral artery occlusion and drug treatment.
Focal cerebral ischemia was induced as described previously (Zhang et al., 2008, 2011; Wang et al., 2009). Briefly, anesthesia was induced in 5- to 7-week-old male C57BL/6J mice (body weight 20–25 g, The Jackson Laboratory) with 2% (v/v) isoflurane (70% N2O/30% O2) and maintained with a 1–0.5% concentration. Rectal temperature was maintained between 37.0 and 37.5°C with a heating pad (Harvard Apparatus). Focal cerebral ischemia was induced by an intraluminal 7-0 nylon thread with a silicone-blunted tip introduced into the right cervical internal carotid artery. The thread was inserted 9 ± 1.0 mm into the internal carotid artery up to the middle cerebral artery. A laser Doppler perfusion monitor (Perimed AB) was adhered to the right temporal aspect of the animal's skull and used to confirm middle cerebral artery flow disruption. NAS was dissolved in saline containing 1.2% ethanol and administered at a dose of 10 mg/kg by intraperitoneal injection; pretreatment took place 10 min before and 20 min after the operation and post-treatment was administered 30 min after the operation. Control animals, which underwent sham surgery consisting of anesthesia and carotid artery dissection, were injected with equivalent volumes of saline containing 1.2% ethanol. Animals were killed after surgery, brains were removed, and the cerebral hemisphere was retained for either evaluation of infarct volume (after 24 h of MCAO) or Western blot analysis (after 12 h of MCAO). Experiments were conducted in accordance with protocols approved by the Harvard Medical School Animal Care Committee.
Determination of neurological score and infarct volume.
MCAO was sustained for a period of 24 h, after which each animal was assigned a neurological score based on a previously reported scale (Zhang et al., 2011) and killed, while brains were rapidly removed for 2,3,5-triphenyltetrazolium chloride (TTC) staining, and infarct volume was measured as previously described (Zhang et al., 2008, 2011; Wang et al., 2009).
LC/MS assay.
Mice received intraperitoneal injections of NAS, and brain tissues were harvested 30 min after the final injection. Homogenate was obtained using a Dounce homogenizer with added ddH2O to reach a concentration of 400 mg (brain weight)/ml. Cold methanol was added four times to aliquots of brain homogenate followed by centrifugation at 14,000 rpm for 15 min to collect the supernatant, which was tested by liquid chromatography/mass spectrometry (LC/MS). Measured samples were quantified by standard curve to obtain the concentration of targeting NAS contained in the submitted samples.
Statistical analysis.
Densitometry was conducted using the Quantity One Program (Bio-Rad). Statistical significance was evaluated by t test. Error bars represent SEM; *p < 0.05; **p < 0. 01. Data on viability or the extent of cell death were compared by ANOVA or repeated measures of ANOVA and by nonpaired Student's t test. Drug analysis, including IC50 and maximum protection, were performed by GraphPad Prism program.
Results
NAS inhibits OGD- and H2O2-induced cell death in primary neurons in vitro
NAS exhibited neuroprotective effects against both glutamate-induced HT22 cell death and H2O2-induced SK-N-MC cell death (Moosmann and Behl, 1999) and rescued dopaminergic neurons from death in a model of 6-OHDA-induced lesion (Aguiar et al., 2005). We recently reported the protective effect of NAS against acute hepatic ischemia-reperfusion injury in mice (Yu et al., 2013). To determine whether NAS, like melatonin (Wang et al., 2009, 2011), protects a variety of neuronal cells from hypoxia/ischemia-induced injury, we tested it in models of cerebral ischemia in vitro. OGD is an ischemic-like in vitro experimental model. Exposing PCNs to OGD induces cyto. c release and caspase-3 activation, ultimately leading to cell death (Zhang et al., 2003; Wang et al., 2009). To provide more evidence of NAS protection against neuronal cell death, the OGD ischemic model was used. Indeed, incubation of PCNs with NAS (0.01 nm to 100 μm) resulted in statistically significant inhibition of OGD-mediated PCN cell death (Fig. 1A). In addition, exposing PCNs to H2O2 with NAS resulted in better dose-dependent inhibition of cell death than OGD-mediated one (Fig. 1B). Plotting the dependence between drug concentration and OGD- or H2O2-mediated PCN cell death as a semi-log plot revealed the IC50 to be 53 nm (Fig. 1A) and 4 nm (Fig. 1B), with maximum protection 43% (Fig. 1A) and 78% (Fig. 1B), respectively.
Figure 1.

Neuroprotective effects of NAS on the cell death of PCNs. Cell death was induced by 3 h exposure to OGD (A, B) or 18 h exposure to 1000 μmol/L H2O2 (C, D) with or without a series of concentrations of NAS. PCNs were preincubated with NAS for 2 h. Cell death was evaluated by LDH assay (A, C). Data from three independent experiments are presented, and statistically significant differences are indicated with *p < 0.05 and **p < 0.01. The resulting curves (plotted semilogarithmically) define the IC50 and maximum protection calculated by GraphPad Prism program (B, D).
Specific cell populations or brain regions show selective vulnerability in stroke. Next to the cerebral cortex, hippocampus are one of the types of nerve nuclei that are most vulnerable to stroke and excitotoxicity (Aguiar et al., 2005). To determine whether NAS-mediated neuroprotection could be extended to other neuronal types, PHNs were subjected to OGD or H2O2 with or without NAS in indicated doses. Similar to PCNs, the LDH assay demonstrated that NAS significantly inhibited PHN cell death, although its neuroprotective effects on cell death were weaker than in PCNs (Fig. 2A,B).
Figure 2.

NAS attenuates neuronal cell death in PHNs and OHSCs. Neuronal cell death of PHNs and OHSCs induced by 3 h OGD or 18 h H2O2 (1000 μmol/L for PHNs and 1500 μmol/L for OHSCs) with or without NAS was evaluated by the LDH assay (A–D). PHNs (A, B) and OHSCs (C, D) were preincubated with NAS for 2 h. Data from three independent experiments are graphed, and statistically significant differences are indicated with *p < 0.05 and **p < 0.01. NAS (15 μm) inhibited cell death in OHSCs exposed to OGD (E) and H2O2 (F), and PI fluorescence images were obtained. Hippocampal slices under normal control conditions displaying background PI fluorescence (E, F, left). Intense PI labeling in OHSCs exposed to OGD and H2O2 mainly occurred in the CA1, CA3 pyramidal cell field as well as dentate gyrus (E, F, middle). NAS significantly attenuated PI labeling, demonstrating neuroprotective effects (E, F, right). Scale bars: top lanes, 0.5 mm; middle and bottom lanes, 0.1 mm.
NAS inhibits OGD- and H2O2-induced cell death in organotypical hippocampal slice cultures ex vivo
Because the cytoarchitecture and neuronal circuit-based functional activities are well maintained, brain-slice models simulate the essential features of in vivo pathologies of neurological diseases (Cho et al., 2007). OHSC cultures can live for several weeks; they more closely resemble the cerebral microenvironment than primary neuronal cultures and provide many parallels with the MCAO animal model. In particularly, OGD-treated OHSC cultures mimic ischemic conditions in vivo and provide models of ischemic stroke ex vivo (Strasser and Fischer, 1995; Pringle et al., 1997) to test NAS. We thus demonstrated that NAS protected OHSCs from OGD- or H2O2-mediated cell death (Fig. 2C,D). Furthermore, the distribution of neuronal cell death induced by OGD and H2O2 in hippocampal slices and the protective effects of NAS to different types of hippocampal neurons were determined by analyzing PI uptake. The pyramidal and granule-cell layers of slice cultures 12- to 14-d-old could be distinguished under phase-contrast microscopy. PI fluorescence uptake in control slice cultures was at a very low background level. However, OGD or H2O2 significantly increased PI fluorescence uptake throughout the neuronal cell layers (Fig. 2E,F). Interestingly, the intensity of PI fluorescence in the CA1 subregion was significantly reduced by the administration of NAS, demonstrating the protective capability of NAS against OGD- or H2O2-induced cell death characterized by regional distribution of PI fluorescence uptake (Fig. 2E,F).
NAS does not activate melatonin receptor 1A in primary neurons in vitro
Our above results demonstrate the neuroprotective effectiveness of NAS against the OGD- and H2O2-induced cell death of PCNs, PHNs, and OHSCs. We then investigated the molecular mechanisms underlying NAS-mediated neuroprotection. Our previous studies showed that melatonin-mediated protection is dependent on the presence and activation of MT1 in chronic neurodegenerative diseases including Huntington's disease and Amyotrophic lateral sclerosis (Wang et al., 2011; Zhang et al., 2013). We further examined whether NAS, an immediate precursor of melatonin and an agonist of MT1, provides neuroprotection through the activation of MT1 in PCNs. The administration of NAS in a time course mode (0, 1, 2, 6, and 24 h) produced no significant activation of MT1 in these primary neurons, suggesting that NAS, unlike melatonin, does not provide neuroprotection through the activation of MT1 (Fig. 3A, left). We next directly evaluated whether the melatonin receptor antagonist luzindole counters NAS-mediated neuroprotection. Unlike melatonin (Wang et al., 2011), luzindole did not block NAS-mediated neuroprotection in PCNs exposed to H2O2, further confirming that melatonin receptor binding is not central to the mechanism of NAS protection against H2O2-induced stress (Fig. 3A, right).
Figure 3.

NAS does not activate MT1 but slows the dissipation of ΔΨm, influences the opening of mPTP, and reduces the release of mitochondrial apoptogenic factors during H2O2- and/or OGD-induced cell death. PCNs were treated with NAS for 0, 1, 2, 6, and 24 h. Whole cells were extracted and analyzed by Western blotting using antibody to MT1. β-Actin was used as loading control. This blot is representative of three independent experiments. Densitometry was performed to quantify the intensity of the bands from the three independent experiments (A, left). PCNs (A, right; B, top) and PHNs (B, bottom) were subjected to 1000 μmol/L H2O2 for 18 h with or without NAS (10 μmol/L; B) or luzindole (A, right). The supernatants were collected for LDH assay (A, right); **p < 0.01. The living cells were then stained with 2 μmol/L Rh 123 to determine the electrostatic charge of the mitochondria (B). PCNs were submitted for Image It Live mPTP assay (C). PCNs were coincubated with 10 μmol/L NAS or CsA and loaded with calcein AM and CoCl2 for 15 min, and 1 μmol/L ionomycin was then added. Tests of mPTP activation were conducted and digital images were taken (C). Cell death was induced in PCNs by 18 h exposure to 1000 μmol/L H2O2 with or without indicated concentrations of CsA and evaluated by LDH assay (D). Statistically significant differences (n = 6) are indicated with *p < 0.05 and **p < 0.01. The representative images of mitochondria stained for TOM20 (E, top) and matched DAPI images for nuclei (E, bottom) were shown and the quantitation of mitochondrial fragmentation was measured (E, right). The data represent the three independent experiments. *p < 0.05 and **p < 0.01. Brain mitochondria (0.25 mg/ml) energized with glutamate/malate 5 mmol/l were challenged with a series of Ca2+ additions (25 μm each) until they began to spontaneously release Ca2+ (F). Changes in ΔΨm (F, left) and absorbance (F, rightl), as indicators of mitochondrial swelling and induction of mPT, were monitored simultaneously. Pore-forming agent Alameticin (Ala) was added in the end of each sample. NAS (30 μm) was added 1 min before Ca2+ addition. Cell death was induced by subjecting PCNs to OGD for 3 h (G) or 1000 μmol/L H2O2 for 18 h (H) with or without 10 μmol/L NAS (G, H). Subsequently, cells were extracted, and either cytosolic components (G, H, left) or mitochondrial lysates (G, H, right) were obtained and analyzed by Western blotting using antibodies to cyto. c, Smac, and AIF. β-Actin and COX IV were used as cytosolic and mitochondrial component loading controls, respectively. This blot is representative of three independent experiments. Scale bars, 5 μm.
NAS slows dissipation of the mitochondrial potential gradient and influences mitochondrial permeability transition pore opening and mitochondrial fragmentation in intact cells in vitro
We previously reported that another neuroprotective agent, minocycline (also belonging to the NINDS Custom Collection), acts directly on the mitochondria to alter mPT-mediated cyto. c release (Zhu et al., 2002). We tested whether NAS acts on mitochondria. Given that the loss of ΔΨm is an important event associated with progression of mitochondrial dysfunction and leads to cell death of the host neuron (Chang and Johnson, 2002), we first measured the depolarization of ΔΨm of PCNs and PHNs by Rh 123 staining. The green fluorescence resulting from the accumulation of Rh 123 within negatively charged (functioning) mitochondria displays a punctuate pattern, as demonstrated in healthy control cells (Fig. 3B, left). Rh 123 staining became diffuse after H2O2 insult (Fig. 3B, middle), presumably due to the loss of ΔΨm mitochondrial depolarization. NAS significantly inhibited dissipation of ΔΨm induced by H2O2 in PCNs (Fig. 3B, top) and PHNs (Fig. 3B, bottom).
Loss of ΔΨm is observed subsequent to mPT pore activation. mPT results from the opening of a mPTP, a multimeric complex of proteins that opens under certain pathological conditions including ischemic stroke (Matsumoto et al., 1999). Given that the Image iT LIVE mPTP assay provides a more direct method by observing mitochondrial calcein signal in intact cells to measure mPTP than the Rh 123 assay (which relies on ΔΨm alone), we used it to test whether NAS plays a direct role in regulating mPTP in intact PCNs.
Mitochondrial calcein signal in control PCNs (the uniform cellular fluorescence from unquenched calcein; Fig. 3C, left) was lost through Ca2+-mediated pore opening resulting from ionomycin treatment (Fig. 3C, second left). However, the addition of CsA, a compound reported to prevent mPTP formation via direct interaction with mPTP constituents, maintained the mitochondrial calcein signal by preventing Ca2+-mediated mPTP formation (Fig. 3C, right). Interestingly, similar to CsA, NAS significantly inhibited Ca2+-mediated pore opening, suggesting its role in the regulation of mPTP formation in intact PCNs (Fig. 3C, second right). To further test whether CsA has an effect similar to NAS on cell survival, we tested its protective effects at different concentrations. The best protection was conferred by 0.5 μm CsA (Fig. 3D), consistent with the findings of others working with PCNs (Domañska-Janik et al., 2004).
The mitochondria are the key target organelles affected by apoptotic proteins, which may cause mitochondrial swelling through the formation of membrane pores or may increase the permeability of the mitochondrial membrane and cause apoptotic effectors to leak out (Cotran et al., 1999; Büki et al., 2000). Interestingly, mitochondrial fragmentation correlates with H2O2-induced primary neuronal death (Jahani-Asl et al., 2007; Young et al., 2010). We therefore tested whether NAS inhibits mitochondrial fragmentation/morphology alterations in intact PCNs by using TOM20, a mitochondrial marker, to track mitochondrial fragmentation (Jahani-Asl et al., 2007). Mitochondrial length varied in PCNs. Mitochondria were classified into different categories from a length ranging from <1, 1–2, 2–3, 3–4, 4–5, to >5 μm. In control healthy PCNs, ∼95% of mitochondria (green) had a length of >1 μm (Fig. 3E, right, white bars). Of these 16% ranged within 1–2 μm; 27% within 2–3 μm, 27% within 3–4 μm, 15% within 4–5 μm, and 10% within >5 μm. The treatment of H2O2 caused the mitochondria of majority of PCNs clearly fragmented/broken and swollen (Fig. 3E, right, black bars) along with morphologic features indicative of apoptosis (i.e., chromatin condensation and cell shrinkage; blue; Fig. 3E, left bottom) which made 49% of mitochondria became <1 μm and 39% of mitochondria ranged within 1–2 μm, whereas only 11% of mitochondria had a length within 2–3 μm, 1% within 3–4 μm and <1% within >5 μm. Our observation found that these alterations of mitochondrial fragmentation/morphology were significantly reduced by the administration of NAS (Fig. 3E, right, gray bars). Our data showed that ∼87% of mitochondria had a length of >1 μm in PCNs incubated by NAS combined with H2O2. Of these 28% ranged within 1–2 μm; 32% within 2–3 μm, 18% within 3–4 μm, 7% within 4–5 μm, and 2% within >5 μm. We therefore concluded that NAS-mediated neuroprotective effects in primary neurons might not only preserve ΔΨm for appropriate cellular energetics, but also influence mPTP opening and mitochondrial fragmentation/morphology in intact cells in vitro.
NAS is not a mPT inhibitor in isolated brain mitochondria
To test whether mitochondria are also direct targets of NAS in the brain, purified brain mitochondria were used to investigate the effects of NAS on mPT and other mitochondrial physiological parameters. Studies were done using an adapted fluorescence system (Baranov et al., 2008) that allows simultaneous measurement of membrane potential (by following TMRM fluorescence), Ca2+ flux (using Calcium Green 5N), NAD+/NADH redox status (using autofluorescence of the NAD+/NADH couple), and swelling (via light scatter). However, the addition of NAS in a concentration range from 1 to 30 μm (not toxic) to isolated brain mitochondria had no significant effect on ΔΨm, calcium transport, NAD+/NADH redox status, or swelling (Fig. 3F). This suggests that the action mechanisms of NAS in isolated mitochondria are similar to those we previously reported NINDS Custom Collection agents, e.g., melatonin, methazolamide (Wang et al., 2008), and dipyrone (Zhang et al., 2011), but dissimilar to those of minocycline (Zhu et al., 2002).
NAS inhibits mitochondrial cell death pathways
Release of apoptogenic factor cyto. c from mitochondria was observed subsequent to mPTP activation. Cyto. c/Smac/AIF, released into the cytoplasm from mitochondria, trigger both the caspase-dependent and -independent mitochondrial death pathways (Li et al., 1997; Green and Reed, 1998; Susin et al., 1999; Li et al., 2001). Hypoxic injury to primary neurons causes the mitochondria to release apoptogenic factors, which in turn activate caspases (Plesnila et al., 2001; Zhang et al., 2003; Wang et al., 2009). We hypothesized that NAS, like other agents neuroprotective against ischemic injury (including we previously reported melatonin, methazolamide, nortriptyline, and dipyrone), inhibits the release not only of cyto. c, but also of other mitochondrial apoptogenic factors Smac and/or AIF. Indeed, Western blot showed elevated levels of cyto. c/Smac/AIF in cytosol and concomitantly lower levels of cyto. c/Smac/AIF in the mitochondrion, suggesting that these apoptogenic factors were released into the cytoplasm from the mitochondria of primary neurons challenged with OGD (Fig. 3G) or H2O2 (Fig. 3H). Moreover, our data confirmed that NAS blocked release of these cell death mediators into the cytoplasm (Fig. 3G,H).
The release of cyto. c/Smac/AIF from mitochondria triggers activation of downstream caspases. Indeed, Western blot analysis demonstrated that caspase-9 and -3 were activated in primary neurons after H2O2 or OGD insult, and NAS effectively inhibited caspase-9 and -3 activation (Fig. 4A,C,E). In parallel, NAS-mediated inhibition of caspase-9 and -3 activities was confirmed by fluorogenic assay (Fig. 4B,D,F).
Figure 4.
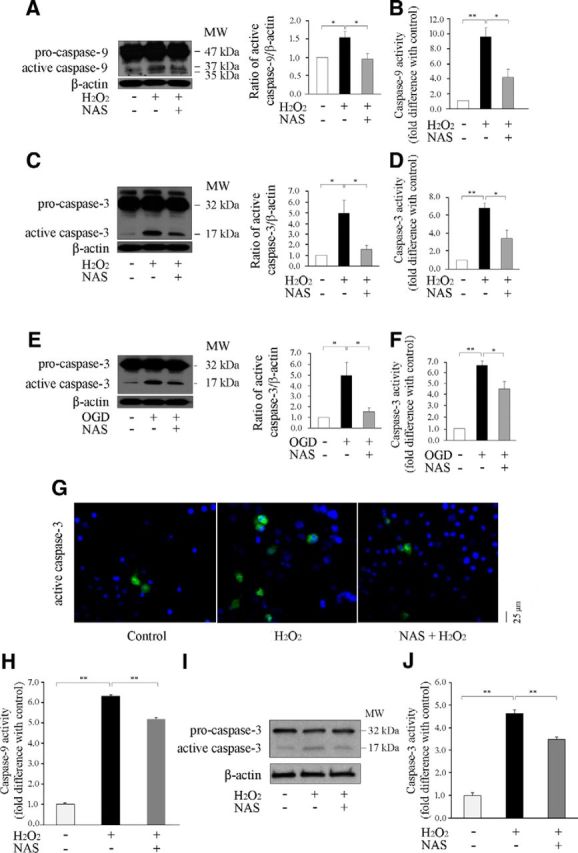
NAS inhibits activation of caspase-9 and -3 in PCNs and OHSCs. Cell death of PCNs and OHSCs was induced by subjecting PCNs to 1000 μmol/L H2O2 for 18 h (A–D, G) and subjecting OHSC to 1500 μmol/L H2O2 for 18 h (H–J), or 3 h OGD (E, F) with or without 10 μmol/L NAS. Whole-cell lysates were extracted, and samples were analyzed by Western blot (each of which contained 50 μg protein) using antibodies to caspase-9 and -3 (A, C, F, I). β-Actin was used as a loading control. The blots are representative of three independent experiments. Densitometry was performed to quantify the intensity of the bands from the three independent experiments. Caspase-3 and -9 activities were also quantified using a fluorogenic assay in lysed PCNs (B, D, F) and OHSCs (H, J). Results come from at least three independent experiments; *p < 0.05, **p < 0.01. Nuclei of PCNs were stained with Hoechst 33342, whereas immunostaining showed that the increased active caspase-3 in H2O2 insult was reduced by the administration of NAS (G).
Moreover, Western blot analysis (Fig. 4I) and fluorogenic assay (Fig. 4H,J) also revealed activation of both caspase-9 and -3 in H2O2-induced OHSCs, whereas NAS significantly inhibited the activation of caspase-9 and -3 in H2O2-mediated OHSC cell death. Together, our data suggest that the inhibition of the release of mitochondrial apoptogenic factors cyto. c/Smac/AIF and inhibitory activation of caspase-9 and -3 relate to the neuroprotective ability of NAS against cell death in both primary neurons and OHSCs.
NAS inhibits H2O2-mediated activation of autophagy
The concept that mitochondria are central to the cell death decision process has relevance to autophagic cell death, while crosstalk involving autophagy-related proteins (Atg5 or Atg7) and Beclin-1 has been reported between autophagy and the apoptotic pathway (Nagley et al., 2010). Among the Atg proteins, Atg5 is one of the key regulators of autophagy, whereas Beclin-1 (yeast Atg6) and LC3 (yeast Atg8) are two pacemakers in the autophagic cascade.
A number of reports have demonstrated that autophagic cell death in primary neurons and other cell lines upon insult by the apoptotic inducer H2O2 (Ha et al., 2012), OGD (Sheng et al., 2012) or staurosporine (Higgins et al., 2011) can be inhibited by protective agents, although increased autophagy is associated with oxidative damage to mitochondria in PCNs. To determine the role of H2O2 in autophagic cell death and whether the inhibition of autophagy recapitulates the protective role of NAS, Atg5−/− (autophagy-deficient) MEFs and Atg5+/+ (WT, autophagy-competent) MEFs were challenged with H2O2 (Fig. 5A) in the presence or absence of NAS. We demonstrate that H2O2 induces cell death and NAS inhibits cell death in both Atg5−/− and WT MEFs (Fig. 5A). Compared with WT cells, Atg5 knock-out rendered MEFs significantly more resistant to H2O2-mediated cell death (in other words, WT MEFs are more vulnerable to H2O2-mediated cell death), suggesting a possible contribution of autophagic cell death to under H2O2 stress, and that autophagy deficiency can recapitulate the protective role of NAS in the presence of H2O2 (Fig. 5A). Moreover, Atg5−/− MEFs seemed to be more receptive to the cell-death-repressive effects of NAS than WT MEFs. That is, NAS (1 μm) significantly reduced H2O2-mediated cell death in Atg5−/− MEFs, whereas it had no such effect in WT MEFs (Fig. 5A). Our data suggest that only partial of the rescue effects of NAS may be due to its modulation of autophagy, and the rest are due to other reasons including its suppression of the mitochondria cell-death pathways.
Figure 5.

NAS inhibits autophagic cell death in vitro. Atg5−/− MEFs and Atg5+/+ MEFs (A), and PCN (B) cell death were induced by 2 h or 18 h exposure as indicated to 1000 μmol/L H2O2, with or without a series of indicated concentrations of NAS as well as 20 μmol/L 3-MA. Cells were preincubated with NAS or 3-MA for 2 h. Cell viability was evaluated by MTS assay (A) and cell death was quantified by LDH assay (B). Statistically significant differences (n = 6–8) are indicated with *p < 0.05 and **p < 0.01. The data are presented as mean ± SEM. Autophagy induced by subjecting PCNs to 2 h (C, E) or 18 h (D, E) H2O2 (1000 μmol/L) with or without 10 μmol/L NAS. PCNs were treated by the administration of 10 μmol/L NAS for 2 and 18 h. Whole-cell lysates were extracted, and samples were analyzed by Western blot (each of which contained 50 μg protein) using antibodies to LC3, Beclin-1, and p62. β-Actin was used as a loading control. The blots are representative of three independent experiments (C, D). Densitometry was performed to quantify the intensity of the bands from the three independent experiments; *p < 0.05, **p < 0.01. Autophagy of PCNs was assessed by 20 μm MDC stain. Autophagic vacuoles were labeled by MDC staining (E).
Furthermore, we tested 3-MA, an autophagy inhibitor in PCNs, and found that like NAS, it inhibits PCN cell death (Fig. 5B). Our data therefore suggest that autophagic cell death contributes to H2O2-mediated toxicity and that inhibition of autophagy may recapitulate the protective role of NAS in PCNs.
The phosphatidylethanolamine-conjugated form of LC3, LC3-II, specifically associates with the autophagosome membrane. Levels of LC3-II directly correlate with the number of autophagosomes, and are therefore one of the most reliable markers with which to study autophagy (Klionsky et al., 2012). Beclin-1, a key protein involved in the regulation of autophagy, is essential for the recruitment of other autophagic proteins during the expansion of preautophagosomal membrane (Wen et al., 2008). Additionally, autophagic flux can be assessed by turnover of a specific substrate, p62 (SQSTM1), which is efficiently degraded by autophagy through direct interaction with LC3 (Pankiv et al., 2007). Inhibition of autophagy correlates with increased levels of p62 (Komatsu et al., 2007), suggesting that steady-state levels of this protein reflect autophagic status. It has been reported that autophagic cell death correlates with apoptotic signaling in OGD- or H2O2-induced neurotoxicity in PCNs through increasing LC3-II and/or Beclin-1 levels (Sheng et al., 2010; Higgins et al., 2011; Shi et al., 2012) and reducing p62 expression (Sheng et al., 2010), but it is not clear whether NAS can block autophagic cell death in PCNs.
We next investigated whether NAS exerts its neuroprotection by interfering autophagic cell death pathways. By testing the protective effect of NAS on H2O2-induced neurotoxicity of PCNs in the time course mode, we found a slight cell death upon 2 h H2O2 challenge and a significant cell death upon 18 h H2O2 insult by LDH measurement (Fig. 5B). In other words, our data indicate that PCNs were in the status of initial cell death upon 2 h H2O2 challenge and actually undergo dying upon 18 h H2O2 insult (Fig. 5B). Furthermore, we demonstrate that the H2O2-generated cell death is associated with increased autophagic phenotype. Levels of LC3-II in cultured cells were moderately increased upon 2 h (Fig. 5C) and greatly increased upon 18 h (Fig. 5D) insult. However, the administration of NAS significantly reduced the H2O2-mediated increase in LC3-II levels. Because LC3-II is specifically bound to the autophagosomes, we then stained PCNs with MDC, an autofluorescent compound used for labeling autophagic vacuoles/autophagosomes. Our observations from MDC staining further confirmed the immunoblotting data on LC3-II levels (Fig. 5E). The 2 h H2O2 challenge to PCNs caused some accumulation of autophagic vacuoles, whereas 18 h H2O2 challenge generated a massive accumulation of autophagic vacuoles (Fig. 5E). Interestingly, NAS was found to inhibit H2O2-induced autophagic vacuoles in both 2 h and 18 h challenge (Fig. 5E). In parallel, upregulation of Beclin-1 was observed in PCNs upon 2 h or 18 h insult with H2O2, whereas treatment with NAS for 2 h (Fig. 5C) or 18 h (Fig. 5D) resulted in a moderate or marked reduction of Beclin-1 protein levels (Fig. 5B), respectively.
Furthermore, we measured the endogenous levels of p62 to assess autophagic activity. We detected subtle downregulation of p62 in apoptotic PCNs upon H2O2 challenge for 2 h and pronounced regulation after 18 h, whereas NAS treatment significantly restored decreased p62 levels (Fig. 5C,D). Together, our data indicate that H2O2-induced PCN cell death is mediated, at least partly, by activating autophagy and autophagic cell death, whereas NAS exerts its neuroprotection by inhibiting both the mitochondrial cell death and autophagic cell death pathways.
NAS reduces cerebral ischemia-induced injury in vivo
Cerebral ischemia, through reducing perfusion to a brain region, results in permanent neurological deficits and/or death. Our previous reports indicated that the neuroprotective agents melatonin (Wang et al., 2009), methazolamide (Wang et al., 2009), nortriptyline (Zhang et al., 2008), and dipyrone (Zhang et al., 2011) not only rescue cultured primary neurons from a variety of cell death stimuli and inhibit the mitochondrial cell death pathway, but also decrease the volume of the infarct and improve neurological score in the MCAO mouse model of cerebral ischemia. Here we evaluated the ability of NAS to reduce ischemic damage in the MCAO model. Several doses of NAS were tested, and 10 mg/kg showed the best protection. The infarct volume was quantified by staining with TTC. Mice given NAS intraperitoneal injections both pretreatment and post-treatment had significantly smaller infarcts after MCAO than mice injected with vehicle alone (Fig. 6A,B). In addition, as rated on the neurological score, mice treated with NAS had significantly better postischemic behavior than vehicle-treated control mice by pretreatment and had the improved trend (but not significant difference) than vehicle-treated control mice by post-treatment (Fig. 6C).
Figure 6.

NAS diminishes damage from cerebral ischemia. Lesion size (A, B) and neurological scores (C) were determined for mice injected with saline (controls) and NAS. NAS (10 mg/kg) was administered by intraperitoneal injection 10 min before and 20 min after the onset of MCAO (pretreatment) or 30 min after the onset of MCAO (post-treatment). Brains were quickly removed after 24 h of ischemia, cut into coronal sections, and stained with 2% TTC, and neurological scores were rated (C). The data are presented as mean ± SEM for the saline (n = 11 for pretreatment, n = 7 for post-treatment) and NAS groups (n = 7 for pretreatment, n = 8 for post-treatment); *p < 0.05, **p < 0.01. Male mice received pretreatment or post-treatment of 10 mg/kg NAS or vehicle (D–F). Brain tissues were harvested 30 min after the injection. The processed homogenate samples from NAS-treated or vehicle-treated mice were tested by LC/MS to record the relative absorbance (D). After 12 h of MCAO, the brains were removed and the ischemic territory was dissected; either cytosolic fractions were analyzed by Western blotting with antibodies against cyto. c/Smac/AIF (E, top), or whole-cell lysates were analyzed with antibodies to caspase-3 (E, bottom) or Beclin-1, LC3, and p62 (F) and reprobed with anti-β-actin. Densitometric scans of these gels quantified the intensity of the bands (each of which contained 50 μg proteins); *p < 0.05, **p < 0.01.
NAS determination in brains of mice with ischemic injury
To determine whether NAS can easily reach the brain and increase the concentration of NAS in the brain tissue of male C57BL/6J mice receiving acute stroke treatment, we used LC/MS to investigate whether NAS can penetrate the blood–brain barrier (BBB) of MCAO mice. As shown in Figure 6D, there was endogenous NAS secretion in the brains of MCAO mice (counts 4232) comparing with blank (counts 7) and standard control as 200 fg/μl NAS (counts 18268), whereas a remarked NAS peak was detected in the brain tissue of MCAO mice treated with NAS (counts 366593). Our findings regarding the high brain level of NAS provide direct evidence that NAS is absorbed, penetrates the BBB, and reaches the CNS.
NAS inhibits mitochondrial cell death pathways and autophagy activation following ischemic injury
We have demonstrated that NAS inhibited the release of mitochondrial apoptogenic factors and the activation of caspases in primary neurons in vitro and OHSCs ex vivo challenged with OGD or H2O2 (Figs. 3, 4). Because cyto. c release and caspase-3 activation are found in the brains of mice that have suffered ischemic damage (Zhang et al., 2003, 2008, 2011; Wang et al., 2009), whereas NAS inhibits caspase-3 activation in acute hepatic ischemia-reperfusion injury in mice (Yu et al., 2013), we next investigated whether NAS inhibited cyto. c release and caspase-3 activation in ischemic brain regions in MCAO mice in vivo. Our data demonstrate that NAS reduced cyto. c release and diminished caspase-3 activation (Fig. 6E) in the brain tissue of ischemic mice in vivo. NAS inhibits autophagy activation in environmentally stressed PCNs (Fig. 5). To determine the influence of NAS on neuronal autophagy in cerebral ischemia, we further tested the effects of NAS on autophagy in vivo. We examined the levels of autophagy markers Beclin-1 and LC3-II as well as p62 in MCAO mice. The levels of Beclin-1 and LC3-II were greatly increased in vehicle-treated control MCAO mice, whereas the administration of NAS significantly reduced their levels (Fig. 6F). Furthermore, we detected a marked decrease in p62 levels after hypoxic-ischemic injury in mice, whereas NAS treatment significantly this downregulation (Fig. 6F). Our findings that NAS inhibits mitochondrial death pathways and autophagic activation following ischemic injury in MCAO mice and in environmentally stressed primary neurons and OHSCs systems are consistent.
Discussion
Despite great efforts to identify drugs protective against cerebral ischemia, effective agents remain elusive. To our knowledge, this is the first demonstration that NAS offers neuroprotection in models of hypoxic-ischemic brain injury, identifying it as a novel potential drug candidate for ischemic stroke.
Blocking of the mitochondrial cell death pathway and autophagic cell death pathway have become very attractive targets for pharmacotherapy. Atg5 has been shown to translocate from the cytosol to mitochondria, where it associates with Bcl-XL, triggering cyto. c redistribution and downstream caspase activation (Yousefi et al., 2006). Beclin-1 is negatively regulated by interacting with the anti-apoptotic Bcl-2 or Bcl-XL protein (Nagley et al., 2010), and knockdown of Beclin-1 and Atg5 reduces hypoxia-induced cell death (Azad et al., 2008). In the present study, H2O2 induced both the induction of autophagy and the release of proapoptotic mitochondrial proteins. It is possible that there is a mutually promoting relationship between the induction of autophagy and the release of proapoptotic mitochondrial proteins.
In recent years, autophagy has become an attractive topic in the study of neuronal death in cerebral ischemia. A deeper understanding of autophagic cell death in cerebral ischemia will provide novel insights into the pathogenic mechanisms and help to develop effective therapeutics. We observed autophagy stimulation not only in apoptotic PCNs under H2O2 stress in vitro, but also in ischemic injury in vivo associated with upregulation of LC3-II and Beclin-1 and downregulation of p62, confirming autophagy activation in focal cerebral ischemia. It has been reported that the levels of LC3-II and Beclin-1 were downregulated, whereas p62 was upregulated following cerebral ischemia, consistent with an activation of autophagy (Gao et al., 2012). More recently, Cui et al. (2012) have further proved the evidence that inhibiting autophagy activation can reduce ischemia injury and improve cell survival. Administration of NAS inhibited the increase in LC3-II and Beclin-1 levels and reversed the reduction of p62 both 12 h postischemia in vivo and after 2 h and 18 h H2O2 treatment in vitro, implying that NAS promotes neuronal survival through suppressing the autophagic pathway. Collectively, our data indicate that inhibition of the autophagic pathway at least partially underlies the mechanism of NAS-induced tolerance to cerebral ischemia. For the first time, we found that the neuroprotective effects of NAS might be associated, in part, with autophagy inhibition under stress conditions.
Our findings of an antiapoptotic role of NAS against cerebral ischemia are consistent among environmentally stressed primary neurons in vitro, OHSC systems ex vivo, and in MCAO mice in vivo. In addition, our data also suggest that NAS reduces PCN and PHN cell death, with stronger effects in PCNs. We demonstrated that the mechanism underlying the protective effects of NAS consists of antiapoptotic action on the mitochondria to inhibit the dissipation of the ΔΨm, mPTP opening, mitochondrial morphology alterations in intact cells, as well as the subsequent release of apoptogenic factors cyto. c/Smac/AIF from mitochondria into cytosol, forestalling the caspase cascades that lead to cell death.
There are at least two major pathways for cyto. c release: mPT and the mitochondrial apoptosis-induced channel (MAC) formed in the outer mitochondrial membrane (Dejean et al., 2006a,b). The finding that NAS slows H2O2-induced dissipation of the ΔΨm and mPTP opening in intact cells suggests that NAS-mediated inhibition of the release of apoptogenic factors cyto. c/Smac/AIF in primary neurons may involve the mPT pathway. MAC serves a regulatory function, as it precedes morphological changes associated with apoptosis (Cotran et al., 1999). MAC is located in the outer mitochondrial membrane and is regulated by various proteins including the mammalian Bcl-2 family of antiapoptotic genes (Dejean et al., 2006a). Bax and/or Bak form the pore, whereas Bcl-2, Bcl-xL, or Mcl-1 inhibits its formation. The finding that NAS fails to affect Ca2+-induced mPT in isolated mitochondria (i.e., NAS does not mediate its protective effect by acting upon basic mitochondrial physiology in isolated brain mitochondria) is most consistent with a scenario in which the effects of NAS mediate outside the inner mitochondrial membrane/mitochondrial matrix, including potential targets, such as Bcl-2 family members (e.g., Bax) and the elements involved in release of proapoptotic proteins, and/or temporally before the mitochondrial events involved in mPT induction.
Considering that the required concentrations of neuroprotective antioxidants in clinical trials may be high, the potential toxicity of compounds must be taken into account. It has been reported that NAS, as a phenolic antioxidant agent, was nontoxic to cultivated cells at concentrations up to 200 μm (Moosmann and Behl, 1999). Our tests on isolated brain mitochondria indicated that 30 μm NAS (the highest concentration used) is not toxic to mitochondria, further indicating that NAS has a relatively wide window of effective doses in experimental models. In addition, compounds considered good candidates for neuroprotective antioxidants in vivo should readily cross the BBB. Parenterally administered NAS has been reported to show detectable effects on the CNS in animals (Winters et al., 1984; Cohen et al., 1996), thus NAS may cross the BBB with required specific transport (Moosmann and Behl, 1999). Our LC/MS data showing the high level of NAS in the MCAO mouse brain further indicates that NAS can cross the BBB. Additionally, intraperitoneal injection of NAS inhibits cerebral ischemia-induced injury in vivo not only with pretreatment, but also with post-treatment of MCAO mice, suggesting its clinical relevance. Neuroprotective effects, low toxicity, and access to the CNS by crossing the BBB may make NAS an attractive agent for further human evaluation.
Footnotes
This work was supported by grants from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (X.W.), the Bill and Melinda Gates Foundation (X.W.), the Muscular Dystrophy Association (X.W.), and the ALS Therapy Alliance (X.W.).
The authors declare no competing financial interests.
References
- Aguiar LM, Macedo DS, de Freitas RM, de Albuquerque Oliveira A, Vasconcelos SM, de Sousa FC, de Barros Viana GS. Protective effects of N-acetylserotonin against 6-hydroxydopamine-induced neurotoxicity. Life Sci. 2005;76:2193–2202. doi: 10.1016/j.lfs.2004.09.035. [DOI] [PubMed] [Google Scholar]
- Azad MB, Chen Y, Henson ES, Cizeau J, McMillan-Ward E, Israels SJ, Gibson SB. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy. 2008;4:195–204. doi: 10.4161/auto.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai JZ, Lipski J. Differential expression of TRPM2 and TRPV4 channels and their potential role in oxidative stress-induced cell death in organotypic hippocampal culture. Neurotoxicology. 2010;31:204–214. doi: 10.1016/j.neuro.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Baranov SV, Stavrovskaya IG, Brown AM, Tyryshkin AM, Kristal BS. Kinetic model for Ca2+-induced permeability transition in energized liver mitochondria discriminates between inhibitor mechanisms. J Biol Chem. 2008;283:665–676. doi: 10.1074/jbc.M703484200. [DOI] [PubMed] [Google Scholar]
- Büki A, Okonkwo DO, Wang KK, Povlishock JT. Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci. 2000;20:2825–2834. doi: 10.1523/JNEUROSCI.20-08-02825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LK, Johnson EM., Jr Cyclosporin A inhibits caspase-independent death of NGF-deprived sympathetic neurons: a potential role for mitochondrial permeability transition. J Cell Biol. 2002;157:771–781. doi: 10.1083/jcb.200112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Wood A, Bowlby MR. Brain slices as models for neurodegenerative disease and screening platforms to identify novel therapeutics. Curr Neuropharmacol. 2007;5:19–33. doi: 10.2174/157015907780077105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Z, Bonvento G, Lacombe P, Hamel E. Serotonin in the regulation of brain microcirculation. Prog Neurobiol. 1996;50:335–362. doi: 10.1016/S0301-0082(96)00033-0. [DOI] [PubMed] [Google Scholar]
- Cotran RS, Kumar V, Collins T, Robbins SL. Robbins pathologic basis of disease. Philadelphia: W.B. Saunders; 1999. [Google Scholar]
- Cui D, Wang L, Qi A, Zhou Q, Zhang X, Jiang W. Propofol prevents autophagic cell death following oxygen and glucose deprivation in PC12 cells and cerebral ischemia-reperfusion injury in rats. PLoS One. 2012;7:e35324. doi: 10.1371/journal.pone.0035324. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dejean LM, Martinez-Caballero S, Kinnally KW. Is MAC the knife that cuts cytochrome c from mitochondria during apoptosis? Cell Death Differ. 2006a;13:1387–1395. doi: 10.1038/sj.cdd.4401949. [DOI] [PubMed] [Google Scholar]
- Dejean LM, Martinez-Caballero S, Manon S, Kinnally KW. Regulation of the mitochondrial apoptosis-induced channel, MAC, by BCL-2 family proteins. Biochim Biophys Acta. 2006b;1762:191–201. doi: 10.1016/j.bbadis.2005.07.002. [DOI] [PubMed] [Google Scholar]
- De Simoni A, Yu LM. Preparation of organotypic hippocampal slice cultures: interface method. Nat Protoc. 2006;1:1439–1445. doi: 10.1038/nprot.2006.228. [DOI] [PubMed] [Google Scholar]
- Domañska-Janik K, Buzañska L, Dłuzniewska J, Kozłowska H, Sarnowska A, Zabłocka B. Neuroprotection by cyclosporin A following transient brain ischemia correlates with the inhibition of the early efflux of cytochrome C to cytoplasm. Brain Res Mol Brain Res. 2004;121:50–59. doi: 10.1016/j.molbrainres.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. N Engl J Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L, Su L, Zhang Y. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PloS one. 2012;7:e46092. doi: 10.1371/journal.pone.0046092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Ha JH, Noh HS, Shin IW, Hahm JR, Kim DR. Mitigation of H2O2-induced autophagic cell death by propofol in H9c2 cardiomyocytes. Cell Biol Toxicol. 2012;28:19–29. doi: 10.1007/s10565-011-9202-x. [DOI] [PubMed] [Google Scholar]
- Higgins GC, Devenish RJ, Beart PM, Nagley P. Autophagic activity in cortical neurons under acute oxidative stress directly contributes to cell death. Cell Mol Life Sci. 2011;68:3725–3740. doi: 10.1007/s00018-011-0667-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahani-Asl A, Cheung EC, Neuspiel M, MacLaurin JG, Fortin A, Park DS, McBride HM, Slack RS. Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J Biol Chem. 2007;282:23788–23798. doi: 10.1074/jbc.M703812200. [DOI] [PubMed] [Google Scholar]
- Jang SW, Liu X, Pradoldej S, Tosini G, Chang Q, Iuvone PM, Ye K. N-acetylserotonin activates TrkB receptor in a circadian rhythm. Proc Natl Acad Sci U S A. 2010;107:3876–3881. doi: 10.1073/pnas.0912531107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- Li P, Yang X, Wasser M, Cai Y, Chia W. Inscuteable and Staufen mediate asymmetric localization and segregation of prospero RNA during Drosophila neuroblast cell divisions. Cell. 1997;90:437–447. doi: 10.1016/S0092-8674(00)80504-8. [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Friberg H, Ferrand-Drake M, Wieloch T. Blockade of the mitochondrial permeability transition pore diminishes infarct size in the rat after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1999;19:736–741. doi: 10.1097/00004647-199907000-00002. [DOI] [PubMed] [Google Scholar]
- Moosmann B, Behl C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc Natl Acad Sci U S A. 1999;96:8867–8872. doi: 10.1073/pnas.96.16.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagley P, Higgins GC, Atkin JD, Beart PM. Multifaceted deaths orchestrated by mitochondria in neurones. Biochim Biophys Acta. 2010;1802:167–185. doi: 10.1016/j.bbadis.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Nopparat C, Porter JE, Ebadi M, Govitrapong P. The mechanism for the neuroprotective effect of melatonin against methamphetamine-induced autophagy. J Pineal Res. 2010;49:382–389. doi: 10.1111/j.1600-079X.2010.00805.x. [DOI] [PubMed] [Google Scholar]
- Oxenkrug G, Requintina P, Bachurin S. Antioxidant and antiaging activity of N-acetylserotonin and melatonin in the in vivo models. Ann N Y Acad Sci. 2001;939:190–199. doi: 10.1111/j.1749-6632.2001.tb03626.x. [DOI] [PubMed] [Google Scholar]
- Pan R. Autophagy in cerebral ischemia: therapist or killer. J Biomol Res Ther. 2013;2:1000e111. doi: 10.4172/2167-7956.1000e111. [DOI] [Google Scholar]
- Pandya RS, Mao L, Zhou H, Zhou S, Zeng J, Popp AJ, Wang X. Central nervous system agents for ischemic stroke: neuroprotection mechanisms. Cent Nerv Syst Agents Med Chem. 2011;11:81–97. doi: 10.2174/187152411796011321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya RS, Zhu H, Li W, Bowser R, Friedlander RM, Wang X. Therapeutic neuroprotective agents for amyotrophic lateral sclerosis. Cell Mol Life Sci. 2013;70:4729–4745. doi: 10.1007/s00018-013-1415-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Cozzi A, Peruginelli F, Leonardi P, Meli E, Pellicciari R, Moroni F. 1-Aminoindan-1,5-dicarboxylic acid and (S)-(+)-2-(3′-carboxybicyclo[1.1.1] pentyl)-glycine, two mGlu1 receptor-preferring antagonists, reduce neuronal death in in vitro and in vivo models of cerebral ischaemia. Eur J Neurosci. 1999;11:3637–3647. doi: 10.1046/j.1460-9568.1999.00786.x. [DOI] [PubMed] [Google Scholar]
- Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, Chiarugi A, Thomas SS, Kohane DS, Korsmeyer SJ, Moskowitz MA. BID mediates neuronal cell death after oxygen/ glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci U S A. 2001;98:15318–15323. doi: 10.1073/pnas.261323298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle AK, Iannotti F, Wilde GJ, Chad JE, Seeley PJ, Sundstrom LE. Neuroprotection by both NMDA and non-NMDA receptor antagonists in in vitro ischemia. Brain Res. 1997;755:36–46. doi: 10.1016/S0006-8993(97)00089-9. [DOI] [PubMed] [Google Scholar]
- Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–389. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy. 2010;6:482–494. doi: 10.4161/auto.6.4.11737. [DOI] [PubMed] [Google Scholar]
- Sheng R, Liu XQ, Zhang LS, Gao B, Han R, Wu YQ, Zhang XY, Qin ZH. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy. 2012;8:310–325. doi: 10.4161/auto.18673. [DOI] [PubMed] [Google Scholar]
- Shi R, Weng J, Zhao L, Li XM, Gao TM, Kong J. Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neurosci Ther. 2012;18:250–260. doi: 10.1111/j.1755-5949.2012.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjö BK. Pathophysiology and treatment of focal cerebral ischemia: Part II. Mechanisms of damage and treatment. J Neurosurg. 1992;77:337–354. doi: 10.3171/jns.1992.77.3.0337. [DOI] [PubMed] [Google Scholar]
- Soane L, Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell death in cerebral ischemia. In: Reed JC, editor. Apoptosis physiology and pathology. Cambridge: Cambridge UP; 2011. [Google Scholar]
- Stavrovskaya IG, Baranov SV, Guo X, Davies SS, Roberts LJ, 2nd, Kristal BS. Reactive gamma-ketoaldehydes formed via the isoprostane pathway disrupt mitochondrial respiration and calcium homeostasis. Free Radic Biol Med. 2010;49:567–579. doi: 10.1016/j.freeradbiomed.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-M. [DOI] [PubMed] [Google Scholar]
- Strasser U, Fischer G. Protection from neuronal damage induced by combined oxygen and glucose deprivation in organotypic hippocampal cultures by glutamate receptor antagonists. Brain Res. 1995;687:167–174. doi: 10.1016/0006-8993(95)00519-V. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Wang X. The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS Neurosci Ther. 2009;15:345–357. doi: 10.1111/j.1755-5949.2009.00105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bauer JH, Li Y, Shao Z, Zetoune FS, Cattaneo E, Vincenz C. Characterization of a p75(NTR) apoptotic signaling pathway using a novel cellular model. J Biol Chem. 2001;276:33812–33820. doi: 10.1074/jbc.M010548200. [DOI] [PubMed] [Google Scholar]
- Wang X, Bregegere F, Soroka Y, Kayat A, Redziniak G, Milner Y. Enhancement of Fas-mediated apoptosis in ageing human keratinocytes. Mech Ageing Dev. 2004;125:237–249. doi: 10.1016/j.mad.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhu S, Pei Z, Drozda M, Stavrovskaya IG, Del Signore SJ, Cormier K, Shimony EM, Wang H, Ferrante RJ, Kristal BS, Friedlander RM. Inhibitors of cytochrome c release with therapeutic potential for Huntington's disease. J Neurosci. 2008;28:9473–9485. doi: 10.1523/JNEUROSCI.1867-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Figueroa BE, Stavrovskaya IG, Zhang Y, Sirianni AC, Zhu S, Day AL, Kristal BS, Friedlander RM. Methazolamide and melatonin inhibit mitochondrial cytochrome C release and are neuroprotective in experimental models of ischemic injury. Stroke. 2009;40:1877–1885. doi: 10.1161/STROKEAHA.108.540765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Sirianni A, Pei Z, Cormier K, Smith K, Jiang J, Zhou S, Wang H, Zhao R, Yano H, Kim JE, Li W, Kristal BS, Ferrante RJ, Friedlander RM. The melatonin MT1 receptor axis modulates mutant Huntingtin-mediated toxicity. J Neurosci. 2011;31:14496–14507. doi: 10.1523/JNEUROSCI.3059-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- Winters WD, Petit JP, Lakin ML, Miller CH. Effects of amine pretreatment on ketamine catatonia in pinealectomized or hypophysectomized animals. J Pharmacol Exp Ther. 1984;230:69–74. [PubMed] [Google Scholar]
- Wölfler A, Abuja PM, Schauenstein K, Liebmann PM. N-acetylserotonin is a better extra- and intracellular antioxidant than melatonin. FEBS Lett. 1999;449:206–210. doi: 10.1016/S0014-5793(99)00435-4. [DOI] [PubMed] [Google Scholar]
- Young KW, Piñon LG, Bampton ET, Nicotera P. Different pathways lead to mitochondrial fragmentation during apoptotic and excitotoxic cell death in primary neurons. J Biochem Mol Toxicol. 2010;24:335–341. doi: 10.1002/jbt.20343. [DOI] [PubMed] [Google Scholar]
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- Yu S, Zheng J, Jiang Z, Shi C, Li J, Du X, Wang H, Jiang J, Wang X. Protective effect of N-acetylserotonin against acute hepatic ischemia-reperfusion injury in mice. Int J Mol Sci. 2013;14:17680–17693. doi: 10.3390/ijms140917680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WH, Wang X, Narayanan M, Zhang Y, Huo C, Reed JC, Friedlander RM. Fundamental role of the Rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death. Proc Natl Acad Sci U S A. 2003;100:16012–16017. doi: 10.1073/pnas.2534856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WH, Wang H, Wang X, Narayanan MV, Stavrovskaya IG, Kristal BS, Friedlander RM. Nortriptyline protects mitochondria and reduces cerebral ischemia/hypoxia injury. Stroke. 2008;39:455–462. doi: 10.1161/STROKEAHA.107.496810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang X, Baranov SV, Zhu S, Huang Z, Fellows-Mayle W, Jiang J, Day AL, Kristal BS, Friedlander RM. Dipyrone inhibits neuronal cell death and diminishes hypoxic/ischemic brain injury. Neurosurgery. 2011;69:942–956. doi: 10.1227/NEU.0b013e318222afb2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Cook A, Kim J, Baranov SV, Jiang J, Smith K, Cormier K, Bennett E, Browser RP, Day AL, Carlisle DL, Ferrante RJ, Wang X, Friedlander RM. Melatonin inhibits the caspase-1/cytochrome c/caspase-3 cell death pathway, inhibits MT1 receptor loss and delays disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2013;55:26–35. doi: 10.1016/j.nbd.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]