

Abstract
Learning-induced modulation of neuronal intrinsic excitability is a metaplasticity mechanism that can impact the acquisition of new memories. Although the amygdala is important for emotional learning and other behaviors, including fear and anxiety, whether learning alters intrinsic excitability within the amygdala has received very little attention. Fear conditioning was combined with intracellular recordings to investigate the effects of learning on the intrinsic excitability of lateral amygdala (LA) neurons. To assess time-dependent changes, brain slices were prepared either immediately or 24-h post-conditioning. Fear conditioning significantly enhanced excitability of LA neurons, as evidenced by both decreased afterhyperpolarization (AHP) and increased neuronal firing. These changes were time-dependent such that reduced AHPs were evident at both time points whereas increased neuronal firing was only observed at the later (24-h) time point. Moreover, these changes occurred within a subset (32%) of LA neurons. Previous work also demonstrated that learning-related changes in synaptic plasticity are also evident in less than one-third of amygdala neurons, suggesting that the neurons undergoing intrinsic plasticity may be critical for fear memory. These data may be clinically relevant as enhanced LA excitability following fear learning could influence future amygdala-dependent behaviors.
The amygdala is critical for the processing of emotional stimuli (LeDoux 2000). Plasticity within the amygdala is essential for storage of emotional memories (Johansen et al. 2011), and in the manifestation of emotional disorders (Rauch et al. 2000; Mahan and Ressler 2012). Although synaptic plasticity within the amygdala has been investigated extensively (McKernan and Shinnick-Gallagher 1997; Rogan et al. 1997; Rumpel et al. 2005), intrinsic plasticity has received relatively little attention (but see Rosenkranz et al. 2010; Motanis et al. 2012).
Intrinsic plasticity is a change in the intrinsic firing properties of a neuron that regulates two important aspects of learning and memory. First, modulation of intrinsic excitability can allow neuronal ensembles to enter a “learning mode” (Saar et al. 1998). Once neurons enter this learning mode, acquisition of skills or learning dependent on these ensembles is accelerated. For example, acquisition of an olfactory discrimination task enhances intrinsic excitability of hippocampal neurons in a transient manner, and acquisition of a hippocampus-dependent task, such as the Morris water maze, is enhanced during this period of increased excitability (Zelcer et al. 2006). Furthermore, pharmacological or genetic manipulations that increase or decrease intrinsic excitability can enhance or impair learning, respectively (Han et al. 2007; Zhou et al. 2009; Santini and Porter 2010; Santini et al. 2012). Thus, intrinsic excitability influences the strength of the new learning. Second, neurons with greater intrinsic excitability are more likely to be a part of the memory engram than neighboring neurons (Han et al. 2009, Zhou et al. 2009). Specifically, overexpression of CREB (cyclic AMP response element-binding protein) enhances intrinsic excitability of lateral amygdala (LA) neurons and biases them to be preferentially recruited into the memory trace. Hence, intrinsic plasticity can modulate the strength of learning and its allocation to a specific subset of neurons.
Intrinsic plasticity has been demonstrated following many learning and memory paradigms in a variety of brain structures. These include modulation of intrinsic excitability within hippocampus (Moyer et al. 1996; Oh et al. 2003; Zelcer et al. 2006), piriform cortex (Saar et al. 1998; Cohen-Matsliah et al. 2009) as well as the infralimbic cortex (Santini et al. 2008). Although fear learning modulates excitability of neurons in hippocampus and infralimbic cortex (Santini et al. 2008; Kaczorowski and Disterhoft 2009; McKay et al. 2009; Song et al. 2012), whether fear conditioning leads to modulation of intrinsic excitability of lateral amygdala neurons has yet to be investigated.
In the current study, we investigated whether learning causes modulation of intrinsic excitability of LA neurons using fear conditioning, an amygdala-dependent task. To assess any time-dependent effects of fear learning on intrinsic excitability, acute brain slices were collected either immediately or 24 h following fear conditioning, and intracellular recordings were obtained from LA neurons. Our data suggest that fear learning enhances intrinsic excitability in a subset of LA neurons in a time-dependent manner. This is the first demonstration of a learning-related modulation of intrinsic excitability of LA neurons.
Results
Behavioral training
To determine the effects of fear conditioning on intrinsic excitability of LA neurons, rats were trained on an auditory fear conditioning paradigm. Specifically, this particular long-delay fear conditioning paradigm was chosen as we have previously demonstrated that this training protocol leads to high levels of cued fear but very low levels of background context fear (Detert et al. 2008). This allowed us to study the effects of cued fear while minimizing the confounding effects of context fear learning. Additionally, a subset of rats was tested the following day to ensure fear memory recall was consistent with that previously observed (see Fig. 1A).
Figure 1.

Experimental design used to study learning-related changes in the amygdala. (A) Behavioral groups. Rats were divided into five groups: two control groups (naive [N = 12]) and CS-alone [N = 4]) and three experimental groups (Cond-1hr [N = 8], Cond-24hr-Probe [N = 9], and Cond-24hr [N = 3]; see Materials and Methods for details). (B) Electrophysiological recordings. Right panel is a schematic of a typical coronal brain slice showing the location of the lateral amygdala (LA). Left panel is a photomicrograph of a brain slice showing the location of a typical recording electrode (inset is a neurobiotin-filled LA pyramidal neuron; scale, 40 µm).
To assess memory, the percentage of time spent freezing during the baseline and the conditioned stimulus (CS) presentations for the training and testing sessions was measured. During behavioral training (day 1), rats from the two conditioned groups, Cond-1hr and Cond-24hr (probed and nonprobed rats) showed comparable acquisition of fear conditioning while the levels of freezing for CS-alone rats remained low (see Fig. 2A). A repeated-measures ANOVA revealed a significant effect of training trial (F(4.86,102.11) = 9.1, P < 0.001, Greenhouse–Geisser corrected), of group (F(2,21) = 5.6, P < 0.05), and a training trial by group interaction (F(9.72,102.11) = 2.5, P < 0.05, Greenhouse–Geisser corrected). Post hoc analysis indicated that CS-alone rats froze significantly less than the Cond-1hr and Cond-24hr group throughout the training session (P < 0.01 and P < 0.05, respectively). Average percent freezing for Cond-1hr and Cond-24hr were not significantly different (P = 0.355). A follow-up ANOVA and post hoc analysis confirmed that while baseline and CS trial 1 freezing were not significantly different between groups, CS-alone rats froze significantly less than Cond-1hr and Cond-24hr rats for trials 2–7 (all P-values <0.05). Thus, analysis of percent freezing during training session indicates both Cond-1hr and Cond-24hr groups acquired fear conditioning but the CS-alone group did not.
Figure 2.

Rats readily acquire long-delay fear conditioning. (A) Training. During the fear conditioning session on day 1, CS-alone rats (N = 4) froze significantly less than Cond-1hr (N = 8) and Cond-24hr (N = 12) rats. The Cond-1hr and Cond-24hr rats froze at comparable levels. The Cond-24hr group includes both probed and not probed rats. The high average freezing for 10th trial for CS-alone group (43.75%) is likely immobility due to time in the chamber (∼1 h) as the average freezing for the 45-sec period preceding the 10th CS presentation was also relatively high (47.19%). (B) Testing. During the probe test on day 2, Cond-24hr-Probe rats (N = 9) froze significantly more than CS-alone rats (N = 4). Baseline freezing was low for both CS-alone and Cond-24hr-Probe rats. (*) P < 0.05 relative to baseline freezing, (B) baseline freezing, (CS) freezing during CS presentation.
Behavioral testing
Analyses of percent time freezing during probe test on day 2 revealed a significant effect of time (baseline vs. CS probe, F(1,11) = 37.3, P < 0.001), group (F(1,11) = 35.9, P < 0.001) as well as a group by time interaction (F(1,11) = 29.0, P < 0.001) (see Fig. 2B). Follow-up analyses revealed that both CS-alone and Cond-24hr-Probe rats froze little during baseline (0 ± 0% and 11 ± 3.60%, respectively; t(8) = −3.2, P = 0.01, equal variances not assumed). While CS-alone rats expressed little freezing during CS presentation (0.59 ± 0.59%), Cond-24hr-Probe rats froze significantly more during the CS probe (72.84 ± 7.74%) as compared to baseline (t(3) = −1.0, P = 0.39 and t(8) = −9.3, P < 0.001, respectively). Furthermore, the time spent freezing during CS probe was significantly greater for the Cond-24hr-Probe rats than the CS-alone rats (t(8.09) = −9.3, P < 0.001, equal variances not assumed). These data indicate that conditioned but not CS-alone rats display robust learning-specific fear memory to the CS presentation.
Fear conditioning reduces post-burst afterhyperpolarization
The post-burst afterhyperpolarization (AHP) is a measure of intrinsic excitability and is mainly composed of multiple calcium-dependent potassium currents that make up the fast, medium, and slow AHP (Madison and Nicoll 1984; Storm 1990). Intracellular recordings from LA neurons revealed a significant reduction in the post-burst AHP following fear conditioning. The AHP amplitude was measured at several time points following the offset of current injection (0.01–4.0 sec) (see Fig. 3A,B). A repeated-measures ANOVA indicated a significant effect of time (F(2.24,147.85) = 163.4, P < 0.001, Greenhouse–Geisser corrected), group (F(2,66) = 3.8, P < 0.05) as well as a group by time interaction (F(4.48,147.85) = 2.4, P < 0.05, Greenhouse–Geisser corrected). To establish the time points at which the AHP amplitude was significantly different between groups, a one-way ANOVA was performed for the AHP amplitudes at various time points. The AHP amplitude was significantly different between groups from 0.5 sec to 4 sec after current offset (all values P < 0.05, except P = 0.053 at 3.5 sec). Post hoc comparisons revealed that the AHP amplitude was significantly reduced in LA neurons from both the Cond-1hr as well as the Cond-24hr groups relative to the Control rats (all values P < 0.05). Similar group effects on post-burst AHP area were also observed (F(2,66) = 4.1, P < 0.05) (see Fig. 3C and Table 1). Post hoc comparisons confirmed that area of the post-burst AHP is significantly reduced in LA neurons from the Cond-1hr and the Cond-24hr rats in comparison to Control rats (P < 0.05). The slow AHP (sAHP) in LA neurons corresponds to the period of several hundred milliseconds up to 6 sec following current offset (Faber and Sah 2002). These results indicate that fear conditioning leads to a reduction in the slow AHP within 1 h of fear conditioning in LA neurons, and that these changes persist for up to 24 h.
Figure 3.

Long-delay fear conditioning reduces the post-burst AHP in lateral amygdala pyramidal neurons. (A) Time course of the post-burst AHP amplitude. Neurons from Cond-1hr (n = 13) and Cond-24hr rats (n = 28) had a significantly smaller AHP relative to Control rats (n = 28) at all points between 0.5 and 4.0 sec following current offset. (B) Representative traces of the post-burst AHP. Voltage sweeps illustrate LA pyramidal neurons from fear conditioned rats have smaller AHPs compared to Control rats (scale: 1 mV, 100 msec). (C) Area of the post-burst AHP. Bar graphs illustrate that the AHP area is significantly smaller in neurons from Cond-1hr and Cond-24hr compared to Control rats. (*) P < 0.05 relative to LA neurons from Control rats.
Table 1.
Summary of learning-related changes on intrinsic excitability of LA neurons
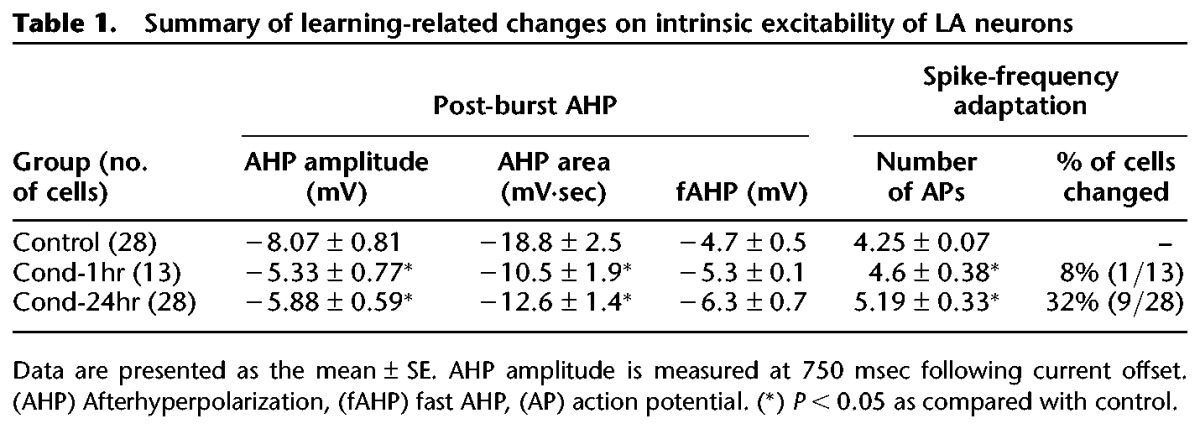
In addition to the sAHP, fast and medium AHP currents also contribute to neuronal excitability. In LA neurons, medium AHP is evident during the period of tens to few hundred milliseconds following action potential (AP) discharge (Faber and Sah 2002). Although we observed a strong trend toward reduced medium AHP in neurons from Cond-1hr and Cond-24hr groups relative to Control, these changes did not reach statistical significance (P < 0.1, 200–500 msec following current offset) (see Fig. 3A). We also measured the fast AHP amplitude following the first action potential during the AHP measurement and found no significant changes in fast AHP amplitude between groups (F(2,66) = 2.37, P = 0.1) (see Table 1). Thus, fear conditioning reduces sAHP but not fast AHP and may lead to a reduction in medium AHP as well.
Fear conditioning reduces spike-frequency adaptation
Spike-frequency adaptation (or accommodation), another measure of intrinsic excitability, is the reduction of neuronal firing despite the presence of a prolonged depolarizing current. Changes in the post-burst AHP are often accompanied by changes in spike-frequency adaptation (Madison and Nicoll 1984; Faber et al. 2001). To quantify spike-frequency adaptation, the current used for the post-burst AHP measurement was extended over a 1-sec duration and the number of action potentials (APs) elicited were counted. A one-way ANOVA revealed a significant effect of group for spike-frequency adaptation (F(2,66) = 3.8, P < 0.05) (see Fig. 4; Table 1). Post hoc tests indicate that LA neurons from Cond-24hr rats fired significantly more APs than those from Control rats (P < 0.01). Surprisingly, the number of APs elicited in LA neurons from Cond-1hr rats did not differ significantly when compared with those from Control or Cond-24hr rats. These data indicate that fear conditioning-induced reductions in spike-frequency adaptation of LA neurons are time-dependent and follow a different time course than sAHP changes.
Figure 4.
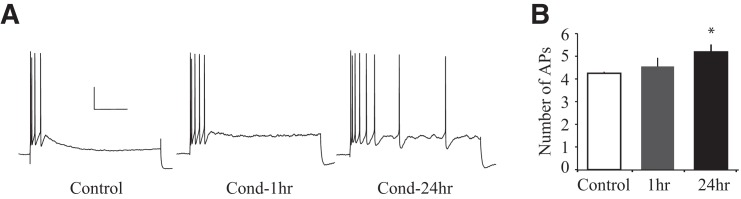
Long-delay fear conditioning reduces spike-frequency adaptation in a time-dependent manner. (A) Representative traces illustrating spike-frequency adaptation in response to a prolonged current injection. Note that LA pyramidal neurons from Control rats (n = 28) but not Cond-24hr rats (n = 28) display remarkable spike-frequency adaptation. Scale: 20 mV, 0.2 sec. (B) Average number of action potentials (APs) during prolonged current injection. LA pyramidal neurons from Cond-24hr rats fire significantly more APs than those from Control rats. Neurons from Cond-1hr rats (n = 13) are not significantly different from any other group. (*) P < 0.05 relative to LA neurons from Control rats.
Relationship between post-burst AHP and spike-frequency adaptation
Figure 4 illustrates that the time course over which learning-related changes in spike-frequency adaptation emerge is slower than that of the post-burst AHP (see Fig. 3), suggesting dissociation between the two measures of intrinsic excitability. To further explore the relationship between these two measures of intrinsic excitability, the AHP amplitude was plotted as a function of the number of APs elicited during a long current injection (see Fig. 5). For neurons from Control and Cond-1hr rats, no significant correlation was observed between the AHP amplitudes (at any time point after current offset) and the number of APs elicited (all P-values >0.05) (Fig. 5A,B). This lack of correlation is expected as the sAHP amplitude influences AP firing toward the later part of the neuronal response to a prolonged current injection (Faber and Sah 2005); LA neurons from Control and Cond-1hr rats displayed remarkable spike-frequency adaptation and rarely fired during the latter part of the current injection. In contrast, for neurons from the Cond-24hr rats the AHP amplitude was significantly correlated with the number of APs elicited (for AHP amplitude 0.08–3.75 sec after current offset; all P-values <0.05) (see Fig. 5C). Similar results were found for area of the post-burst AHP, with no significant correlation for neurons from Control (r = 0.232, P > 0.05) and Cond-1hr rats (r = 0.363, P > 0.05), but a significant correlation emerged at 24 h (r = 0.644, P < 0.001). These data indicate that although the post-burst AHP is not correlated with spike-frequency adaptation under control conditions and immediately post-conditioning, a significant correlation emerged 24 h post-conditioning.
Figure 5.

Fear conditioning modulates the relationship between the sAHP and spike-frequency adaptation. The post-burst AHP amplitude (measured at 750 msec following current offset) is not correlated with number of action potentials fired during a prolonged current injection for (A) Control (n = 28) and (B) Cond-1hr (n = 13) rats. (C) In contrast, AHP amplitude is significantly correlated with number of action potentials for Cond-24hr rats (n = 28). (*) P < 0.05.
Factors other than the sAHP can modulate spike-frequency adaptation. Indeed, for LA neurons, spike-frequency adaptation is controlled by both sIAHP (voltage-insensitive calcium-dependent potassium currents that contribute to the sAHP) and ID (voltage-dependent potassium currents) (Faber and Sah 2005). To determine whether a modulation of ID underlies the observed reduction in spike-frequency adaptation, we measured the initial spike frequencies and interspike intervals (ISIs), a characteristic dependent on ID in LA neurons (Faber and Sah 2005). A one-way ANOVA revealed no significant effect of fear conditioning on ISIs for the three pairs of APs (F(2,66) = 1.1, 1.2, 1.3 for 1st, 2nd and 3rd pair of APs, respectively; all P-values >0.05). Similar results were observed for the spike frequencies as well (data not presented). Thus, it is unlikely that a reduction in ID was responsible for the learning-related changes in spike-frequency adaptation.
Dissociation between the sAHP and spike-frequency adaptation has also been observed in other structures, such as the hippocampus (Moyer et al. 1996) and basolateral amygdala (BLA) (Motanis et al. 2012). For example, using trace eye-blink conditioning in rabbits, a significant reduction has been observed in the sAHP as well as spike-frequency adaptation of hippocampal neurons as early as 1 h following acquisition. While spike-frequency adaptation begins to revert to baseline levels by the third day after learning, the sAHP changes last until the seventh day (Moyer et al. 1996). Thus, the dissociation between the sAHP and spike-frequency adaptation changes observed in the current study is consistent with that from previous studies.
Fear conditioning enhances intrinsic excitability in a subset of LA neurons
We next examined whether the enhancement in the intrinsic excitability occurs in all LA neurons or whether such changes are present in only a subset of neurons. The cumulative frequency distribution of the number of APs elicited from LA neurons indicates that the curve for Cond-24hr rats is shifted to the right relative to that from Control or Cond-1hr rats (see Fig. 6A). Significantly, the rightward shift in Cond-24hr rats is only evident for a part of the curve indicating only a subset of LA neurons change as a result of conditioning. LA neurons from Control rats displayed remarkable spike-frequency adaptation with none of the recorded neurons firing more than five APs (e.g., see Figs. 4 and 5A). To quantify a learning-related change in spike-frequency adaptation, any neuron firing more APs than the most excitable Control neuron (five APs) was defined as having changed. We used the spike-frequency adaptation data and not the AHP measurement data for this analysis, as the variability in AHP data from Control rats was too large to make such an analysis informative (see Fig. 5A for the range of AHP measurements in Control rats). Using this criterion (AP firing), we find that 8% of neurons from Cond-1hr rats (one of 13) and 32% of neurons from Cond-24hr rats (nine of 28) displayed a learning-related change (see Fig. 6B). Hence, fear conditioning enhances intrinsic excitability, but only in a subset of LA neurons.
Figure 6.

Fear conditioning enhances intrinsic excitability in a subset of lateral amygdala neurons. (A) Cumulative frequency distribution of “number of action potentials (APs)” for LA neurons. Number of APs elicited in LA neurons from Cond-24hr rats (n = 28) is shifted to the right relative to neurons from Control (n = 28) or Cond-1hr (n = 13) rats. The rightward shift in Cond-24hr rats is only evident for a part of the curve indicating only a subset of LA neurons change as a result of conditioning. (B) Frequency histogram for number of APs. Approximately 32% of the LA neurons (9/28) have enhanced intrinsic excitability (i.e., those that fire more APs than the most excitable LA neuron from a Control rat) 24 h following fear conditioning. For both graphs, dashed line indicates the maximum number of APs fired by a LA neuron from a Control rat.
To confirm whether the reduction in spike-frequency adaptation was a result of fear conditioning and not sampling error, we performed a shuffling analysis on the data from the spike-frequency adaptation experiment. Briefly, data (number of APs fired) from all the recorded LA neurons were shuffled and then randomly assigned to the three groups (group sizes were the same as in the actual experiment). The number of cells that met the criterion for learning-related changes (more than five APs) was quantified for each group. For 1000 such iterations, none of the runs resulted in our observed learning-related changes (P < 0.001). Thus, it is extremely unlikely that such a distribution would result from sampling error alone and indicates that a subset of LA neurons underwent a time-dependent learning-specific enhancement of intrinsic excitability.
Next, we confirmed whether learning-related changes in the AHP were also restricted to a subset of neurons. We divided LA neurons from Cond-24hr rats into Cond-24hr “changed” or “unchanged” based on the above-mentioned criteria. As expected, spike-frequency adaptation was significantly reduced in Cond-24hr changed neurons relative to neurons from Control, Cond-1hr, and Cond-24hr unchanged groups (F(3,65) = 22.0, P < 0.001; post hoc P < 0.001) (see Fig. 7A). More importantly, the size of the AHP was also selectively reduced in Cond-24hr changed neurons relative to Cond-24hr unchanged groups. The AHP amplitude (from 0.09 sec to 4 sec after current offset) was significantly reduced in Cond-24hr changed (all P values <0.05) but not Cond-24hr unchanged group (all P values >0.08) relative to Controls (see Fig. 7B). Notice that the time period of 0.09–4 sec comprises both medium as well as slow AHP. Hence, both medium and slow AHP are reduced in Cond-24hr changed neurons relative to Controls. Similarly, AHP area was also reduced in Cond-24hr changed but not the Cond-24hr unchanged group relative to Controls (F(3,65) = 5.4, P < 0.01; post hoc P < 0.001 and P = 0.326, respectively) (see Fig. 7C). In contrast to medium and sAHP, and as previously reported (see Table 1), the fast AHP was unaltered between groups (F(3,65) = 2.25, P = 0.09). Therefore, spike-frequency adaptation as well as medium and slow AHP reduction was restricted to a subset (∼32%) of LA neurons at 24-h post-conditioning.
Figure 7.

Learning-related changes in lateral amygdala at 24 h after fear conditioning are exclusively observed in a subset of neurons. (A) Spike-frequency adaptation in LA neurons is reduced only in neurons that were defined as “changed” (n = 9). Notice that LA neurons classified as “unchanged” (n = 19) were virtually identical with those from control animals (n = 28). (B) The AHP amplitude (from 0.09–4.0 sec following current offset) was reduced in Cond-24hr changed but not Cond-24hr unchanged neurons relative to Control group. (C) Area of the post-burst AHP is also significantly smaller for Cond-24hr changed but not Cond-24hr unchanged neurons relative to neurons from the Control group. (D) The Cond-24hr changed neurons are LA pyramidal neurons. In addition to electrophysiological criteria, representative neurobiotin cell fills from Cond-24hr changed neurons confirm that these are LA projection neurons because (1) they either have a prominent apical dendrite and a characteristic pyramidal morphology (left panel) or (2) numerous spines on the dendrites (right panel). (Inset) A higher magnification image of a dendrite from the area in the white box in the right panel. Scale, left panel, 25 µm; right panel, 25 µm; inset, 10 µm. (*) P < 0.05.
The above data suggest that the Cond-24hr changed neurons represent a distinct subset of LA neurons within the Cond-24hr rats. In order to confirm that Cond-24hr neurons only differed in their firing properties and did not represent a distinct neuronal subtype (e.g., LA interneurons) we used electrophysiological and morphological criteria. Using electrophysiological parameters such as spike-frequency adaptation, input resistance, spike half width, and fast AHP, we were able to confirm that Cond-24 changed neurons (as well as other recorded neurons) were LA pyramidal neurons (Tables 1 and 2; Materials and Methods). Furthermore, we obtained complete neurobiotin cell fills from five of the Cond-24hr changed neurons (see Fig. 7D). These cells were confirmed as pyramidal neurons either due to the presence of large soma (∼300 µm2), prominent apical dendrite, or numerous dendritic spines (Faber et al. 2001; Sosulina et al. 2006, 2010). Last, the shuffling analysis described above supports the idea that such a distribution of spike-frequency adaptation as observed in neurons from Cond-24hr rats could not have resulted from biased sampling of a different neuronal type in Cond-24hr rats. Thus, the electrophysiological and morphological analyses confirmed that the all recorded neurons, including Cond-24hr changed neurons, represent LA pyramidal neurons.
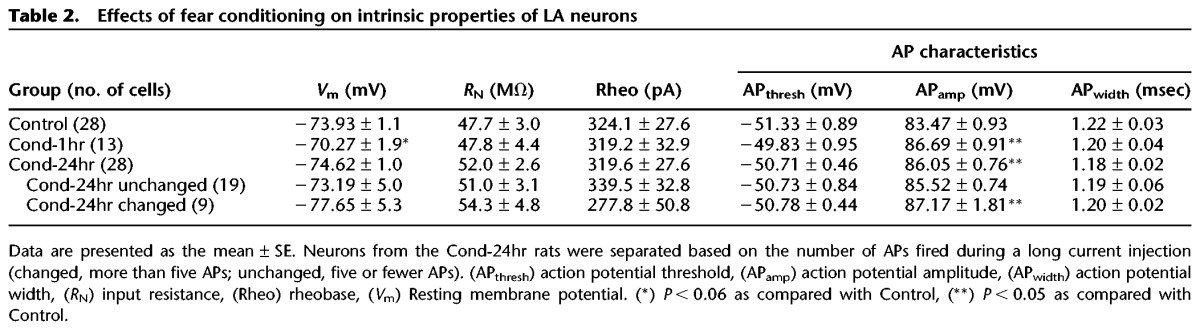
Table 2.
Effects of fear conditioning on intrinsic properties of LA neurons
Passive membrane and AP properties
A change in spike-frequency adaptation can result from a change in basic membrane properties. Passive membrane properties like input resistance (RN) were not significantly different between groups (F(3,65) = 0.538, P = 0.66) (see Table 2). A one-way ANOVA indicated a significant effect of group on resting membrane potential (RMP) (F(3,65) = 3.08, P < 0.05). However, post hoc tests indicated only a small and nonsignificant decrease in RMP of neurons from Cond-1hr group relative to those from Controls (P = 0.06). Additionally, the current injection amplitude used to perform the AHP and spike-frequency adaptation studies also did not differ between groups (F(3,65) = 0.85, P = 0.47). The threshold for AP initiation and rheobase were also unaltered as a result of fear conditioning (F(3,65) = 0.49, P = 0.69 and F(3,65) = 0.03, P = 0.758, respectively). Taken together, these data suggest that the changes in spike-frequency adaptation in Cond-24hr changed neurons were not a result of changes in basic membrane properties.
In addition to number of APs, properties of a single AP can also contribute to neuronal excitability. Analysis of AP properties from the first AP evoked during the AHP measurements revealed that AP amplitude increased following fear conditioning (F(3,65) = 2.66, P = 0.055). Post hoc tests revealed that AP amplitude was significantly higher in LA neurons from Cond-1hr as well as Cond-24hr changed neurons relative to those from Control rats (P < 0.05). In contrast, AP half-width remained unchanged following fear conditioning (F(3,65) = 0.34, P = 0.8). The increased AP amplitude could result from increased Na+ and Ca2+ conductances and lead to enhanced synaptic gain as well facilitation of synaptic plasticity (Varela et al. 2012).
Discussion
The current study combined auditory fear conditioning with electrophysiological recordings to investigate learning-related changes in the intrinsic excitability of LA neurons. We demonstrate for the first time that: (1) fear conditioning enhances intrinsic excitability of LA neurons, (2) the increased excitability of LA neurons is time-dependent, and (3) these changes are only observed in approximately one-third of LA neurons. These data indicate that learning an amygdala-dependent task can enhance local intrinsic excitability. Such intrinsic plasticity may serve as a metaplasticity mechanism (Sehgal et al. 2013) and enhance future amygdala-dependent learning.
Fear conditioning enhances intrinsic excitability within LA
The amygdala is important for attributing emotional salience to environmental stimuli. Within the amygdala, polymodal sensory inputs first converge in LA. The information is then processed and relayed onto various output structures, including the central nucleus, resulting in different aspects of the behavioral response (LeDoux 2000). Thus, LA acts as the gateway to the rest of the amygdala. It follows that plasticity within LA, especially a change in neuronal excitability, is uniquely capable of modulating behavioral responses to emotional stimuli.
Intrinsic plasticity has been demonstrated following many learning paradigms in numerous brain structures (e.g., Moyer et al. 1996; Thompson et al. 1996; Saar et al. 1998; Oh et al. 2003; Zelcer et al. 2006). Among these, trace or context fear conditioning enhances intrinsic excitability of hippocampal neurons (Kaczorowski and Disterhoft 2009; McKay et al. 2009; Song et al. 2012). Fear conditioning and extinction also modulate the intrinsic excitability of infralimbic medial prefrontal cortex (mPFC) neurons in a bidirectional manner (Santini et al. 2008). The present study is the first to demonstrate that intrinsic excitability is enhanced in LA neurons following fear conditioning.
Few studies have assessed intrinsic plasticity within the amygdala. For example, using in vivo intracellular recordings in an anesthetized rat preparation, Rosenkranz and Grace (2002) demonstrated that olfactory fear conditioning enhanced intrinsic excitability (increased input resistance that lasted for at least 10 min) of LA neurons. In contrast, a recent report indicates that while olfactory fear learning reduces excitability of basolateral amygdala (BLA) neurons, olfactory reward learning enhances BLA excitability (Motanis et al. 2012). It is unclear why fear learning would cause contrasting changes in LA vs. BLA; however, it is possible that these subnuclei respond differentially to the valence of emotional stimuli (Davis et al. 2010). Our data demonstrating enhanced LA excitability following auditory fear conditioning are consistent with olfactory fear conditioning studies in LA (Rosenkranz and Grace 2002).
The amygdala is critical for cued as well as context fear conditioning (Phillips and LeDoux 1992). To minimize the confounding effect of background context fear conditioning, we deliberately chose a long-delay fear conditioning paradigm that results in high cued freezing but low levels of context freezing (Detert et al. 2008). Hence, the effect of fear learning on LA intrinsic plasticity observed in the current study can be attributed to cued fear. Whether context fear conditioning also leads to LA intrinsic plasticity is an interesting question worthy of future studies.
Although the current study did not specifically control for the effect of unconditioned stimulus (US) presentations on LA intrinsic excitability, we do not think our observed changes could result from US presentations alone. Repeated footshock (US) presentations in the absence of learning are a rodent model for stress (Valenti et al. 2011) that can result in plasticity within LA neurons. Indeed, chronic but not acute stress leads to enhanced excitability of LA neurons (Rosenkranz et al. 2010). However, for several reasons it is unlikely that our results on LA neuronal excitability are due to the effect of stress. Most important among these are that a single session of fear conditioning is akin to acute stress which does not lead to changes in LA excitability. Additionally, the degree of intrinsic plasticity following chronic stress (reduced RMP, increased input resistance, reduced sAHP, and spike-frequency adaptation) was more extensive than that seen in the current study. It is possible that within amygdala, learning-related intrinsic plasticity facilitates future learning, whereas more extensive intrinsic plasticity following chronic stress may also contribute to emotional disorders like PTSD (Roozendaal et al. 2009).
Moreover, the popular controls for US presentation, namely pseudoconditioning (or explicitly unpaired CS and US presentations), lead to inhibitory conditioning to the CS, i.e. animals receiving unpaired CS and US presentations encode CS as a safety signal (Rescorla and Lolordo 1965) and display distinct forms of plasticity within amygdala (Amano et al. 2010). In addition, unpaired animals show higher context fear than those receiving paired CS–US presentations (Phillips and LeDoux 1994). These factors make pseudoconditioning a poor control for fear conditioning studies investigating learning-related neurophysiological changes in the amygdala.
Fear conditioning enhances intrinsic excitability in a time-dependent manner
Previous studies have revealed that learning-related modulation of intrinsic excitability is transient (e.g., Moyer et al. 1996; Thompson et al. 1996). To assess the time course of excitability changes in LA neurons, intrinsic excitability was measured either immediately or 24-h post-conditioning. While sAHP was reduced at 1-h and 24-h post-conditioning (Fig. 3), spike-frequency adaptation was reduced at 24 h only (Fig. 4). Furthermore, the sAHP was correlated with spike-frequency adaptation only at the 24-h time point (Fig. 5). Such a dissociation between the time course for AHP and spike-frequency changes following learning has been observed in BLA (Motanis et al. 2012) and hippocampus (Moyer et al. 1996). The changes in AHP usually outlast (Moyer et al. 1996; Motanis et al. 2012) and may also precede changes in spike-frequency adaptation (present study), indicating other factors may contribute to learning-related changes in spike-frequency adaptation.
Intrinsic excitability is enhanced in a subset of LA neurons
Approximately one-third of the recorded LA neurons display learning-related intrinsic plasticity following fear conditioning (Figs. 6 and 7). Interestingly, synaptic plasticity, as measured by the incorporation of GluR1-containing AMPA receptors, is also evident in a similar proportion of LA neurons following fear conditioning (Rumpel et al. 2005). Such synaptic plasticity is necessary for memory formation, suggesting that ∼30% of LA neurons participate in the fear memory trace. Indeed, 20%–30% of the LA neurons are activated during acquisition, as well as recall, of fear memory, and selective ablation of these neurons erases the fear memory (Han et al. 2007, 2009; Reijmers et al. 2007). Thus, the fear memory engram within LA comprises less than one-third of the neuronal population.
Implications of intrinsic plasticity for learning
Although the size of the neuronal population displaying intrinsic plasticity corresponds to the size of the engram within LA, it is unlikely that enhanced intrinsic excitability actually codes for the memory. In support, behavioral expression of a hippocampus-dependent trace eye-blink conditioning memory outlasts changes in the intrinsic excitability of hippocampal neurons, indicating that intrinsic excitability is not required for memory expression per se (Moyer et al. 1996). However, the duration of enhanced intrinsic excitability does correlate with the period of enhanced learning. For example, olfactory learning results in transient enhancement of hippocampal intrinsic excitability and facilitated acquisition of Morris water maze (Zelcer et al. 2006). Similar results have been observed in the piriform cortex with olfactory learning (Cohen-Matsliah et al. 2009). Thus, unlike synaptic plasticity that may serve as the engram, intrinsic plasticity is likely a mechanism that facilitates acquisition of new learning.
If intrinsic excitability regulates the strength of learning, then interventions that reduce the AHP or enhance intrinsic excitability should enhance learning. Indeed, within amygdala β-adrenergic receptor antagonists increase the sAHP (Faber and Sah 2002) and impair acquisition and reconsolidation of fear conditioning memory (Debiec and Ledoux 2004; Bush et al. 2010; Muravieva and Alberini 2010). Modulation of CREB expression within LA enhances both intrinsic excitability (Zhou et al. 2009) and fear learning (Han et al. 2007). Similarly, enhanced noradrenergic and cholinergic transmission decreases the sAHP, increases spike firing, and enhances mPFC-dependent learning (Mueller et al. 2008; Santini and Porter 2010; Santini et al. 2012). Finally, nimodipine, an L-type Ca2+ channel blocker, enhances hippocampal intrinsic excitability (Moyer et al. 1992) and improves the acquisition of trace eye-blink conditioning in aged rabbits (Deyo et al. 1989). Therefore, manipulations that enhance intrinsic excitability also enhance learning of mPFC, hippocampus, and amygdala-dependent tasks.
Intrinsic plasticity has been implicated as a metaplasticity mechanism (Abraham and Bear 1996; Abraham 2008). Metaplasticity refers to the plasticity of synaptic plasticity, whereby synaptic or cellular activity results in a change in the ability to induce subsequent synaptic plasticity (Abraham and Bear 1996). Enhanced intrinsic excitability following trace fear conditioning has been correlated with a learning-specific facilitation of subsequent LTP (Song et al. 2012). Supporting this, currents underlying the sAHP can act as adjustable gain control mechanisms by shunting excitatory glutamatergic transmission (Moyer et al. 1996; Sah and Bekkers 1996). Therefore, intrinsic plasticity in the form of sAHP reductions can enhance baseline synaptic transmission. Moreover, inhibition of the sAHP also facilitates the induction of LTP in the hippocampus (Sah and Bekkers 1996) and the mPFC (Zaitsev and Anwyl 2012). Thus, enhanced intrinsic excitability in a subset of neurons could lead to enhanced synaptic plasticity in these neurons.
If synaptic plasticity is the physical basis for the memory trace, then cells with higher neuronal excitability would be more likely to code new memories. Accordingly, up-regulation of CREB expression within a subset of LA neurons enhances their neuronal excitability, leads to greater fear conditioning-related LTP (Zhou et al. 2009), and increases their likelihood to be a part of the memory trace (Han et al. 2007, 2009; Zhou et al. 2009). Thus, by enhancing synaptic facilitation, enhanced excitability could bias the allocation of new memories to a subset of neurons.
Conclusions
We demonstrate that fear conditioning leads to a time-dependent enhancement in the intrinsic excitability of LA neurons. These changes occur in a subset of the LA neuronal population and could predispose these neurons for future memory formation. Such enhanced excitability of LA neurons could result in greater amygdalar output to structures such as PFC, central amygdala, and nucleus accumbens. This could result in greater emotional affect manifesting itself as increased stress and elucidates a mechanism by which fear memories could potentiate emotional learning, as well as disorders like PTSD, that result from pathological forms of such learning.
Materials and Methods
Subjects
Male Sprague Dawley rats (age = 3.31 ± 0.24 mo, n = 36) were individually housed in clear plastic cages. Rats were maintained in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited facility on a 14-h light/10-h dark cycle with ad libitum access to food and water. All procedures were conducted in accordance with the University of Wisconsin-Milwaukee Animal Care and Use Committee (ACUC) and NIH guidelines.
Apparatus
Fear conditioning chambers
Fear conditioning was conducted in an apparatus previously described (Song et al. 2012). Briefly, Plexiglas and stainless-steel chambers (30.5 × 25.4 × 30.5 cm, Coulbourn Instruments) with a standard grid floor consisting of 26 parallel steel rods (5 mm diameter and 6 mm spacing) and located in a sound-attenuating box were used. The floor of the chamber was connected to a precision adjustable shock generator (Coulbourn Instruments) for delivery of a scrambled footshock, the unconditioned stimulus (US). A ventilation fan produced a constant background noise of about 58 dB (measured by a sound level meter, Realistic, A scale, model #33-2050) inside the sound attenuating box. The chamber was illuminated by a miniature incandescent white lamp (28 V, type 1819) and was wiped with a 5% ammonium hydroxide solution prior to each training session to provide a distinct olfactory cue. During training, the room lights were left on (illumination 20.9 lux) for the entire session.
Testing chambers
An additional Plexiglas chamber served as a novel context for the auditory cue test. This chamber was located within a separate sound-attenuating box located in the same room. The test chamber was physically different from the training chamber in that it was a hexagonal chamber, the floor was black-painted Plexiglas, and it was illuminated with an infrared light. In addition, the tray below the test chamber floor contained clean bedding and the test chamber was wiped with 2% acetic acid prior to each test session to provide a different olfactory stimulus from that used during training. The room lights were turned off (illumination 0.2 lux) for the entire testing session.
Behavioral groups
Training
Rats were randomly divided into five groups (see Fig. 1A). The two Control groups were either experimentally naive (never handled, Naive, n = 12) or presented with the conditioned stimulus (CS) alone (CS alone, n = 4). The CS-alone group received 10 CS presentations on day 1 and a single CS presentation on day 2 and served as a control for CS presentation. On day 1, the three experimental groups received one 10-trial session of auditory long-delay fear conditioning using a 45-sec CS (80-dB white noise) followed by a 1-sec 1-mA footshock US, and a 5.2-min intertrial interval (ITI). Brain slices from the Cond-1hr rats (n = 8) and the Cond-24hr rats (n = 3) were obtained immediately or 24 h following fear conditioning, respectively. Rats in the Cond-24hr-Probe group (n = 9) were tested for fear memory recall using a single CS presentation 24 h following fear conditioning (see behavioral testing below), and brain slices were obtained immediately after testing.
Testing
On day 2 (24 h after training), Cond-24hr-Probe and CS-alone rats received a brief CS test session in a novel context. After a 2-min baseline, both groups of rats received one 45-sec CS presentation. Rats were removed 2 min after the CS presentation and brain slices were obtained immediately following behavioral testing.
No significant differences were found between the naive and CS-alone groups on any measure of intrinsic excitability (data not presented). Thus, the data from these two groups were combined into a Control group. Similarly, the Cond-24hr and Cond-24hr-Probe rats did not differ significantly during acquisition of fear conditioning as well as on any measure of intrinsic excitability (data not presented). Thus, data from Cond-24hr and Cond-24hr-Probe rats were combined and represented as the Cond-24hr group throughout the rest of the manuscript.
Analyses of behavioral data
The training and testing sessions were recorded using a remote CCTV video camera (model #WV-BP334, Panasonic Corp.) mounted to the top of each behavioral chamber. The video data were fed to a PC running FreezeFrame 2.04. Freezing was defined as the absence of all movement except that required for respiration (Blanchard and Blanchard 1969) and a 1-sec bout of immobility was scored as freezing using FreezeView 2.04 (Actimetrics Software, Coulbourn Instruments).
Slice preparation
Brain slices were prepared within 1 h of the conditioning or testing session by an individual blind to the training condition. Rats were deeply anesthetized with isoflurane and decapitated. The brain was quickly removed and placed in ice-cold oxygenated (95% O2/5% CO2) aCSF (composition in mM: 124 NaCl, 2.8 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, and 20 dextrose). The brain was then blocked and 400 µm-thick coronal brain slices were cut in aCSF at ∼0°C using a vibrating microtome (Model 3000, Vibratome, or VT1200, Leica). Only slices that were located between 1.88 and 3.30 mm posterior to bregma, thus containing lateral amygdala, were used (Paxinos and Watson 1998). Slices were then transferred to a holding chamber (Moyer and Brown 1998) containing oxygenated aCSF at room temperature (21°C–23°C). For electrophysiological measurements, slices were transferred as needed to an interface-type recording chamber (Warner Instrument), where they were perfused with oxygenated aCSF at 32°C and allowed to recover for 1 h prior to starting an experiment. A subset of recordings was performed in presence of 10 µM picrotoxin. Since there were no significant differences between recordings performed with or without picrotoxin, the data have been combined.
Electrophysiological recordings
All recordings were obtained using a MultiClamp 700B amplifier system (Molecular Devices). Experiments were controlled by PClamp 10 software running on a PC, and the data were acquired using the Digidata 1440A acquisition system. All recording electrodes were pulled from thin-walled capillary glass (A-M Systems) using a Sutter Instruments P97 puller. The voltage signals were filtered at 0.5–2 kHz and digitized at 20 kHz. Figure 1B shows a photograph of a coronal slice with typical intracellular recording site noted.
Intrinsic excitability measurements
Somatic intracellular recordings were obtained from LA neurons using sharp microelectrodes filled with 3 M potassium acetate and 20 mM KCl (40–100 MΩ), and a subset of recordings also included 2% neurobiotin. Only cells with a stable resting membrane potential (RMP) between −60 and −85 mV, overshooting action potentials, and an input resistance (RN) >30 MΩ were used. To minimize the influence of voltage-dependent changes on membrane conductances, all cells were studied at a membrane potential near −65 mV (≤0.6 nA constant current injection, if necessary). Measures of intrinsic excitability were recorded under current clamp using the following protocol:
Voltage–current (V-I) relations were obtained using 400-msec current steps (range, −1.0 nA to rheobase) and plotting the plateau voltage deflection against current amplitude. Neuronal input resistance (RN) was determined from the slope of the linear fit of that portion of the V-I plot where the voltage sweeps did not exhibit sags or active conductance.
The post-burst afterhyperpolarization (AHP; 3× at 20-sec intervals) was evoked using a 100-msec depolarizing current injection just sufficient to elicit a burst of four action potentials. The same current injection did not always lead to the exact same neuronal response, and sometimes the current pulse used resulted in more than or fewer than four action potentials, usually three or five. In such a case, AHP measurements were continued until three sweeps with four APs each were obtained from a neuron. Both the amplitude (at various time points) and integrated area of the AHP were measured. Fast AHP was measured by subtracting the action potential threshold from the peak of the afterhyperpolarization following the first evoked action potential (Faber and Sah 2002; Santini et al. 2008). The medium AHP was measured as the AHP amplitude from few tens to 500 msec following current offset (Faber and Sah 2002). The slow AHP was measured 500 msec and onward following the current offset. Action potential (AP) characteristics were analyzed from the first AP evoked during the AHP measurements. AP amplitude (APamp) was measured relative to the baseline and AP width (APwidth) was measured at one-half the APamp. Interspike intervals were measured for the four action potentials.
Spike-frequency adaptation (accommodation; 3× at 30-sec intervals) was studied using a 1-sec depolarizing current injection of the same stimulus intensity used to study the AHP. For each sweep, the number of action potentials elicited was counted. Similar to AHP measurements, data from sweeps that resulted in fewer than four APs were discarded.
Resting membrane potential (Vrest) was calculated as the difference in membrane potential before and after withdrawing the microelectrode from the neuron. For a given slice, only data from one cell were recorded. The slice was changed after five penetrating tracks were made or after the completion of the first recording. All data points represent average measurements obtained for one neuron.
Characterization of LA pyramidal neurons
In order to confirm that all the neuronal recordings were obtained from LA pyramidal neurons, we used electrophysiological criteria (spike-frequency adaptation, input resistance, spike half width, and fast AHP). The near complete spike-frequency adaptation in a majority of recorded neurons (59/69) is consistent with the properties of LA projection neurons (Faber et al. 2001; Sosulina et al. 2006, 2010) and allowed easy characterization of these neurons. Additional criteria were used to eliminate the possibility that the remaining neurons that did not display complete spike-frequency adaptation (10/69, especially Cond-24hr changed neurons [n = 9]) could represent interneurons. This is especially important because LA class V interneurons have been reported to display some spike-frequency adaptation as well as the slow and medium AHP—properties distinct from other interneuron types but similar to pyramidal neurons (Sosulina et al. 2010). However, class V interneurons can be distinguished from projection neurons in LA by their high input resistance (about twice that of LA projection neurons) and their smaller fast AHPs (Sosulina et al. 2006, 2010). The mean and the range of input resistance were virtually identical between the Cond-24hr changed and other neurons (see Table 2). Moreover, there was a nonsignificant increase in fast AHP amplitude in Cond-24hr changed neurons relative to others (see Table 1) which further eliminates the possibility that the Cond-24hr neurons could be class V interneurons. Thus, electrophysiological properties confirmed that all recorded neurons were LA pyramidal neurons.
Neurobiotin staining
A subset of neurons (n = 20) was filled with neurobiotin to confirm the position and identity of pyramidal cells in LA. For these recordings, sharp electrodes were filled with 2% (wt/vol) neurobiotin (SP-1120, Vector Laboratories) dissolved in 3 M potassium acetate and 20 mM KCl (40–100 MΩ). After obtaining intrinsic excitability measures (as previously mentioned), neurobiotin was injected iontophoretically by using depolarizing current pulses (300 msec, 500–1500 pA) delivered at 1.67 Hz for 10 to 20 min. Slices were then fixed in 10% neutral-buffered formalin at 4°C for 1 to 3 d before further processing. To visualize LA neurons, slices were incubated in 3% H2O2/10% methanol for 45 min, washed with PBS for 10 min (3×), followed by 0.25% Triton X-100/2% BSA for 60 min. The slices were then incubated with 1:500 streptavidin Alexa Fluor 488 (Invitrogen) for 120 min in the dark, and washed with PBS for 10 min (3×). They were mounted onto slides, coverslipped with Ultra Cruz Mounting Medium (Santa Cruz Biotechnology), and sealed with nail polish. The neurons were viewed and photographed using a fluorescence microscope (BX51WI, Olympus) at 20× or Leica TCS SP2 confocal system. All neurons visualized were confirmed as LA pyramidal neurons. Neurons were classified as pyramidal when either a prominent apical dendrite (n = 5) or large soma and dendritic spines (n = 15) were detected. Representative neurobiotin-filled LA pyramidal neurons are shown in Figures 1B and 7D.
Statistical analyses
The overall treatment effects were examined using either a repeated-measures ANOVA, one-way ANOVA, or t-tests using SPSS 13.0 (SPSS). A repeated-measures ANOVA was used to compare freezing levels across training trials and the AHP across time for each group of rats. A Fisher's PLSD test was used for post hoc comparisons following significant main effects (α = 0.05), unless otherwise noted. All data are expressed as mean ± SEM. Statistical analyses were performed on cells.
Acknowledgments
This work was supported by a Research Growth Initiative from the University of Wisconsin–Milwaukee (J.R.M.), NIA grant R03-AG042814 (J.R.M.) and Quincy Bioscience (J.R.M.). We thank Chenghui Song, Alex M. Girgis, Tyler Bula, Joseph C. Aitken, Erin L. Adams, and Hiroyuki Miyawaki for technical assistance.
References
- Abraham WC 2008. Metaplasticity: Tuning synapses and networks for plasticity. Nat Rev Neurosci 9: 387. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Bear MF 1996. Metaplasticity: The plasticity of synaptic plasticity. Trends Neurosci 19: 126–130 [DOI] [PubMed] [Google Scholar]
- Amano T, Unal CT, Pare D 2010. Synaptic correlates of fear extinction in the amygdala. Nat Neurosci 13: 489–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard RJ, Blanchard DC 1969. Crouching as an index of fear. J Comp Physiol Psychol 67: 370–375 [DOI] [PubMed] [Google Scholar]
- Bush DE, Caparosa EM, Gekker A, Ledoux J 2010. β-Adrenergic receptors in the lateral nucleus of the amygdala contribute to the acquisition but not the consolidation of auditory fear conditioning. Front Behav Neurosci 4: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Matsliah SI, Rosenblum K, Barkai E 2009. Olfactory-learning abilities are correlated with the rate by which intrinsic neuronal excitability is modulated in the piriform cortex. Eur J Neurosci 30: 1339–1348 [DOI] [PubMed] [Google Scholar]
- Davis FC, Johnstone T, Mazzulla EC, Oler JA, Whalen PJ 2010. Regional response differences across the human amygdaloid complex during social conditioning. Cereb Cortex 20: 612–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiec J, Ledoux JE 2004. Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience 129: 267–272 [DOI] [PubMed] [Google Scholar]
- Detert JA, Kampa ND, Moyer JR Jr 2008. Differential effects of training intertrial interval on acquisition of trace and long-delay fear conditioning in rats. Behav Neurosci 122: 1318–1327 [DOI] [PubMed] [Google Scholar]
- Deyo RA, Straube KT, Disterhoft JF 1989. Nimodipine facilitates associative learning in aging rabbits. Science 243: 809–811 [DOI] [PubMed] [Google Scholar]
- Faber ES, Sah P 2002. Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci 22: 1618–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Sah P 2005. Independent roles of calcium and voltage-dependent potassium currents in controlling spike frequency adaptation in lateral amygdala pyramidal neurons. Eur J Neurosci 22: 1627–1635 [DOI] [PubMed] [Google Scholar]
- Faber ES, Callister RJ, Sah P 2001. Morphological and electrophysiological properties of principal neurons in the rat lateral amygdala in vitro. J Neurophysiol 85: 714–723 [DOI] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, Neve RL, Guzowski JF, Silva AJ, Josselyn SA 2007. Neuronal competition and selection during memory formation. Science 316: 457–460 [DOI] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Hsiang HL, Buch T, Waisman A, Bontempi B, Neve RL, Frankland PW, Josselyn SA 2009. Selective erasure of a fear memory. Science 323: 1492–1496 [DOI] [PubMed] [Google Scholar]
- Johansen JP, Cain CK, Ostroff LE, LeDoux JE 2011. Molecular mechanisms of fear learning and memory. Cell 147: 509–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski CC, Disterhoft JF 2009. Memory deficits are associated with impaired ability to modulate neuronal excitability in middle-aged mice. Learn Mem 16: 362–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE 2000. Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184 [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA 1984. Control of the repetitive discharge of rat CA 1 pyramidal neurones in vitro. J Physiol 354: 319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahan AL, Ressler KJ 2012. Fear conditioning, synaptic plasticity and the amygdala: Implications for posttraumatic stress disorder. Trends Neurosci 35: 24–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay BM, Matthews EA, Oliveira FA, Disterhoft JF 2009. Intrinsic neuronal excitability is reversibly altered by a single experience in fear conditioning. J Neurophysiol 102: 2763–2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P 1997. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390: 607–611 [DOI] [PubMed] [Google Scholar]
- Motanis H, Maroun M, Barkai E 2012. Learning-induced bidirectional plasticity of intrinsic neuronal excitability reflects the valence of the outcome. Cereb Cortex. 10.1093/cercor/bhs394 [DOI] [PubMed] [Google Scholar]
- Moyer JR Jr., Brown TH 1998. Methods for whole-cell recording from visually preselected neurons of perirhinal cortex in brain slices from young and aging rats. J Neurosci Methods 86: 35–54 [DOI] [PubMed] [Google Scholar]
- Moyer JR Jr., Thompson LT, Black JP, Disterhoft JF 1992. Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J Neurophysiol 68: 2100–2109 [DOI] [PubMed] [Google Scholar]
- Moyer JR Jr., Thompson LT, Disterhoft JF 1996. Trace eyeblink conditioning increases CA1 excitability in a transient and learning-specific manner. J Neurosci 16: 5536–5546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller D, Porter JT, Quirk GJ 2008. Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J Neurosci 28: 369–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muravieva EV, Alberini CM 2010. Limited efficacy of propranolol on the reconsolidation of fear memories. Learn Mem 17: 306–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Kuo AG, Wu WW, Sametsky EA, Disterhoft JF 2003. Watermaze learning enhances excitability of CA1 pyramidal neurons. J Neurophysiol 90: 2171–2179 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C 1998. The rat brain in stereotaxic coordinates. Academic Press, San Diego: [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE 1992. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106: 274–285 [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE 1994. Lesions of the dorsal hippocampal formation interfere with background but not foreground contextual fear conditioning. Learn Mem 1: 34–44 [PubMed] [Google Scholar]
- Rauch SL, Whalen PJ, Shin LM, McInerney SC, Macklin ML, Lasko NB, Orr SP, Pitman RK 2000. Exaggerated amygdala response to masked facial stimuli in posttraumatic stress disorder: A functional MRI study. Biol Psychiatry 47: 769–776 [DOI] [PubMed] [Google Scholar]
- Reijmers LG, Perkins BL, Matsuo N, Mayford M 2007. Localization of a stable neural correlate of associative memory. Science 317: 1230–1233 [DOI] [PubMed] [Google Scholar]
- Rescorla RA, Lolordo VM 1965. Inhibition of avoidance behavior. J Comp Physiol Psychol 59: 406–412 [DOI] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE 1997. Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390: 604–607 [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McEwen BS, Chattarji S 2009. Stress, memory and the amygdala. Nat Rev Neurosci 10: 423–433 [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA 2002. Dopamine-mediated modulation of odour-evoked amygdala potentials during pavlovian conditioning. Nature 417: 282–287 [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Venheim ER, Padival M 2010. Chronic stress causes amygdala hyperexcitability in rodents. Biol Psychiatry 67: 1128–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R 2005. Postsynaptic receptor trafficking underlying a form of associative learning. Science 308: 83–88 [DOI] [PubMed] [Google Scholar]
- Saar D, Grossman Y, Barkai E 1998. Reduced after-hyperpolarization in rat piriform cortex pyramidal neurons is associated with increased learning capability during operant conditioning. Eur J Neurosci 10: 1518–1523 [DOI] [PubMed] [Google Scholar]
- Sah P, Bekkers JM 1996. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: Implications for the integration of long-term potentiation. J Neurosci 16: 4537–4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Porter JT 2010. M-type potassium channels modulate the intrinsic excitability of infralimbic neurons and regulate fear expression and extinction. J Neurosci 30: 12379–12386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Quirk GJ, Porter JT 2008. Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J Neurosci 28: 4028–4036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Sepulveda-Orengo M, Porter JT 2012. Muscarinic receptors modulate the intrinsic excitability of infralimbic neurons and consolidation of fear extinction. Neuropsychopharmacology 37: 2047–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal M, Song C, Ehlers VL, Moyer JR Jr 2013. Learning to learn—intrinsic plasticity as a metaplasticity mechanism for memory formation. Neurobiol Learn Mem 105: 186–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Detert JA, Sehgal M, Moyer JR Jr 2012. Trace fear conditioning enhances synaptic and intrinsic plasticity in rat hippocampus. J Neurophysiol 107: 3397–3408 [DOI] [PubMed] [Google Scholar]
- Sosulina L, Meis S, Seifert G, Steinhauser C, Pape HC 2006. Classification of projection neurons and interneurons in the rat lateral amygdala based upon cluster analysis. Mol Cell Neurosci 33: 57–67 [DOI] [PubMed] [Google Scholar]
- Sosulina L, Graebenitz S, Pape HC 2010. GABAergic interneurons in the mouse lateral amygdala: A classification study. J Neurophysiol 104: 617–626 [DOI] [PubMed] [Google Scholar]
- Storm JF 1990. Potassium currents in hippocampal pyramidal cells. Prog Brain Res 83: 161–187 [DOI] [PubMed] [Google Scholar]
- Thompson LT, Moyer JR Jr., Disterhoft JF 1996. Transient changes in excitability of rabbit CA3 neurons with a time course appropriate to support memory consolidation. J Neurophysiol 76: 1836–1849 [DOI] [PubMed] [Google Scholar]
- Valenti O, Lodge DJ, Grace AA 2011. Aversive stimuli alter ventral tegmental area dopamine neuron activity via a common action in the ventral hippocampus. J Neurosci 31: 4280–4289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela JA, Wang J, Christianson JP, Maier SF, Cooper DC 2012. Control over stress, but not stress per se increases prefrontal cortical pyramidal neuron excitability. J Neurosci 32: 12848–12853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitsev AV, Anwyl R 2012. Inhibition of the slow afterhyperpolarization restores the classical spike timing-dependent plasticity rule obeyed in layer 2/3 pyramidal cells of the prefrontal cortex. J Neurophysiol 107: 205–215 [DOI] [PubMed] [Google Scholar]
- Zelcer I, Cohen H, Richter-Levin G, Lebiosn T, Grossberger T, Barkai E 2006. A cellular correlate of learning-induced metaplasticity in the hippocampus. Cereb Cortex 16: 460–468 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, Neve R, Poirazi P, Silva AJ 2009. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci 12: 1438–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]