

Abstract
Insulin receptor substrates (Irs1, 2, 3 and Irs4) mediate the actions of insulin/IGF1 signaling. They have similar structure, but distinctly regulate development, growth, and metabolic homeostasis. Irs2 contributes to central metabolic sensing, partially by acting in leptin receptor (LepRb)-expressing neurons. Although Irs4 is largely restricted to the hypothalamus, its contribution to metabolic regulation is unclear because Irs4-null mice barely distinguishable from controls. We postulated that Irs2 and Irs4 synergize and complement each other in the brain. To examine this possibility, we investigated the metabolism of whole body Irs4−/y mice that lacked Irs2 in the CNS (bIrs2−/−·Irs4−/y) or only in LepRb-neurons (Lepr∆Irs2·Irs4−/y). bIrs2−/−·Irs4−/y mice developed severe obesity and decreased energy expenditure, along with hyperglycemia and insulin resistance. Unexpectedly, the body weight and fed blood glucose levels of Lepr∆Irs2·Irs4−/y mice were not different from Lepr∆Irs2 mice, suggesting that the functions of Irs2 and Irs4 converge upon neurons that are distinct from those expressing LepRb.
Abbreviations: CNS, central nervous system; Irs2, insulin receptor substrate 2; Irs4, insulin receptor substrate 4; LepRb, leptin receptor; ERK, extracellular signal-regulated kinase; Socs3, suppressor of cytokine signaling-3; Stat3, signal transducer and activator of transcription 3; POMC, proopiomelanocortin; PI3K, phosphatidylinositol 3-kinase; ARC, arcuate nucleus of the hypothalamus
Keywords: Insulin receptor substrate 2, Insulin receptor substrate 4, Leptin, Obesity, Nutrient homeostasis, Energy balance
1. Introduction
The hypothalamus integrates signals from peripheral tissues and central nodes to regulate feeding, energy balance, nutrient flux, and counterregulatory responses that maintain nutrient homeostasis [1]. Insulin is secreted from the pancreatic beta-cells during meals to promote peripheral nutrient homeostasis [2]; however, it also modulates energy and glucose homeostasis by acting on hypothalamic and dopaminergic neurons [3]. By comparison, leptin produced by adipocytes is a dominant signal that informs the brain about peripheral energy stores [4]. Leptin binds to the long form of the leptin receptor (LepRb) found largely—but not entirely—on hypothalamic neurons [5]. LepRb neurons sense and integrate signals relevant to nutrient homeostasis to control satiety, energy balance and metabolism [4]. LepRb generates multiple downstream signals by activating JAK2 and recruiting SHP2, STAT3 and STAT5, along with SH2B, Irs1/2 and Irs4 [6,7].
The insulin receptor substrates (Irs1, 2, 3 and 4) are principle targets for the insulin and IGF1 receptor tyrosine kinases, which play a central role in somatic growth and metabolic regulation [8]. IRS-proteins are also phosphorylated by the receptors for some cytokines (IL4, IL9, IL13), growth hormone and leptin in various cells and tissues [7,9–14]. The IRS-proteins share a common structure, including an NH2-terminal pleckstrin homology (PH) domain followed by a phosphotyrosine binding (PTB) domain, and a tail containing many Tyr and Ser/Thr phosphorylation sites. The PH and PTB domains mediate recruitment to appropriate activated receptors in the plasma membrane [15,16]. Although the COOH-terminal amino acid sequences of the various IRS-proteins diverge significantly, multiple short tyrosine phosphorylation motifs can bind and activate similar SH2-domain containing proteins, including the type 1 phosphatidylinositol 3-kinase [17].
The various IRS-proteins have distinct physiologic functions. These differences in biological specificity among the various Irs-proteins may be conferred by sequence divergence or by differences in patterns of expression. Irs1 and Irs2 are widely expressed in mammalian tissues, whereas Irs3 is largely restricted to nonhuman adipose tissue (where it promotes adipogenesis [18,19]). Although some reports describe a role for Irs4 in regenerating liver and hepatocellular carcinoma [20–22], Irs4 is expressed mainly in the hypothalamus in healthy animals [23–25]. Potential overlap in the expression of Irs1 and Irs2 with Irs4 appears to occur mainly in neurons of the ventral hypothalamus [24]. Based upon genetic deletion experiments, Irs1 promotes embryonic and postnatal body growth, and mediates insulin sensitivity in classical insulin target tissues [26]. By comparison, Irs2 mediates several important functions that are not shared with Irs1—including pancreatic beta-cell growth and survival, CNS/hypothalamic nutrient sensing, endothelial cell function, and sensitivity to neurodegenerative disease [27–29]. Irs1−/− mice develop insulin resistance that is compensated by elevated circulating insulin and β-cell/islet growth, whereas Irs2−/− mice develop diabetes owing to the combined effects of insulin resistance and the progressive loss of pancreatic β-cells [30]. Together Irs1 and Irs2 apparently mediate some essential functions, since mice null for both genes die before weaning [8].
The deletion of Irs2 specifically in the CNS can extend life span while producing early-onset obesity, insulin resistance and glucose intolerance [31,32]. The metabolic phenotypes resulting from Irs2 ablation are at least in part attributable to Irs2 function in a relatively small subset of LepRb-expressing neurons in the brain [29]. In contrast, while Irs4 is highly conserved and displays a very specific and restricted pattern of expression that overlaps with LepRb cells in the ventral hypothalamus, the deletion of Irs4 alone has very mild or no effects upon energy balance or glucose metabolism in mice [33]. To reveal a potential physiological function for Irs4 in the CNS, we intercrossed mice without Irs2 in the brain (bIrs2−/−–mice) with whole-body Irs4−/y mice to generate compound male bIrs2−/−·Irs4−/y mice. We examined energy balance and metabolism in these mice, and also investigated whether there is a functional role for Irs4 in LepRb neurons.
2. Material and methods
2.1. Animals
Generation of Irs4-deficient mice. The Irs4 gene was obtained by screening a genomic DNA library derived from mouse 129/Sv embryonic stem (ES) cells. A DNA fragment was ligated to the pPNT vector 5′ to the neo cassette. The neo gene was flanked by EcoRv-Avr II fragment and Clal-Spel fragment derived from the Irs-4 regions 5′ and 3′ to the deleted coding region. The Irs4 targeting vector was introduced into R1 ES cells by electroporation. The transfectants were selected with neomycin (G418) and ganciclovir. Irs4+/− heterozygous ES cells were injected into C57BL/6 blastocysts. The chimeric male mice were bred with female wild-type C57BL/6 mice. Because the Irs-4 gene is on the X chromosome, this breeding yielded female mice heterozygous for Irs-4 disruption and male wild-type mice. The detailed breeding strategy is described in Section 3. Lepr∆Irs2, bIrs2−/− and Irs2L/L were described previously [29,31]. Mice were bred in our colony at Boston Childrens Hospital or at the Harvard School of Public Health. All animals were handled in accordance with all procedures approved by the appropriate Institutional Animal Care and Use Committee (IACUC). Animals were fed breeder chow diet containing 9 kcal %fat (Research diets, Inc).
2.2. Metabolic analysis
Lean and fat body mass were assessed by Dual-Energy X-ray Absorptiometry (DEXA, GE Lunar Corp.) as previously described [34]. Blood glucose levels were measured on random-fed or overnight-fasted animals in mouse-tail blood using Glucometer Elite (Bayer). Intraperitoneal glucose tolerance test was performed on mice fasted for 16 h overnight. Animals were then injected intraperitoneally with d-glucose (2 g/kg) and blood glucose levels were measured [35]. For insulin tolerance tests, mice were fasted for a 4-h period in the light cycle before ip injections of insulin (Humulin R, 0.8 U/kg) diluted in sterile saline. Blood glucose concentrations were measured at indicated time points. Blood insulin and leptin levels were determined on serum from tail vein bleeds using a Rat Insulin ELISA kit and Mouse Leptin ELISA kit (Crystal Chem. Inc.). For food intake measurements mice were housed individually and food intake was measured for 2 consecutive days.
2.3. Histology and morphometric analysis
Histological analysis was performed on various tissues isolated from the animals as previously described [36]. Morphometric analysis of gonadal white adipose tissue from 400 cells from 4 different animals per genotype was performed with NIH ImageJ software (http://rsb.info.nih.gov/ij/). The determination of islet area was done by measuring and counting islets from non-overlapping pictures that covered the entire pancreas section area. Measurements were corrected to the total area of the pancreas section in square microns, which was calculated using SPOT software.
2.4. Energy expenditure
As previously described [37], physical activity and energy expenditure were performed over a 72 h period with a Comprehensive Laboratory Animal Monitoring System (CLAMS, Oxymax Windows 3.0.3; Columbus Instruments, OH, USA). Mice were housed individually at room temperature (22 °C) under an alternating 12 h light/12 h dark cycle. Heat production was measured and analyzed by generalized linear regression to determine the energy expenditure.
2.5. RNA extraction and qPCR
Total RNA was extracted from brown adipose tissue or from hypothalamus using Trizol (Gibco BRL) and 1 µg samples were converted to cDNA using the iscript cDNA kit (Bio-Rad Laboratories Inc.). Sample cDNAs were analyzed in triplicate via quantitative RT-PCR for Pomc and Agrp in hypothalamus with customized primers as previously described [37]. Actin gene expression was used to normalize RNA content and the relative gene product amounts were reported as mean±SEM of several animals.
2.6. Statistical analysis
Unless otherwise stated mean values±SEM were used to make comparisons between 2 groups; significance was determined by a Student's t-test. A p-value less than 0.05 was considered statistically significant. Generalized linear regression (SPSS, v 19) was used to identify significant differences in body weight and energy expenditure.
3. Results
3.1. Generation of bIrs2−/−·Irs4−/y-mice
The Irs4 targeting vector was generated by standard methods and used for homologous recombination in 129/Sv embryonic stem cells (Figure S1). The Novartis Gene Atlas and Allen Brain Atlas confirm that Irs4 expression is largely restricted to the ventral hypothalamus, so the conventional whole body Irs4 knock-out is in practice restricted to the CNS (Figure S2) [38]. Because the Irs4 gene is on the X chromosome, there are no heterozygous knockout males. Heterozygous females and wild-type males were bred to obtain Irs4−/y males and Irs4+/− females as well as wild-type males and females. The Irs4−/y males and heterozygous females from this breeding were mated to obtain female heterozygous and Irs4−/− mice, as well as male wild-type and Irs4−/y mice. To focus our study on the interaction between Irs2 and Irs4 in the CNS, we utilized previously published nestin-cre transgenic mice intercrossed to loxP-flanked alleles to delete Irs2L/L specifically in neurons (bIrs2−/−) [31]. Mice with a combined deficiency of Irs2 and Irs4 were generated by intercrossing female bIrs2+/−·Irs4+/− mice with male bIrs2−/+·Irs4−/y mice to produce male bIrs2−/−·Irs4−/y and female bIrs2−/−·Irs4−/y mice, as well as control genotypes. There was no detectable embryonic lethality or early death associated with the Irs4 null phenotype, and the male Irs4−/y and female Irs4−/− mice were fertile and displayed normal size and a healthy appearance.
3.2. Growth and energy balance in bIrs2−/−·Irs4−/y mice
The body weight of male control (Irs2L/L), Irs4−/y, bIrs2−/−, or bIrs2−/−·Irs4−/y mice were monitored between 6 and 13 weeks of age to determine whether Irs4 interacts with neuronal Irs2 for body weight regulation. Compared to control mice (Irs2L/L), the whole body Irs4−/y mice grew to a normal size between 6 and 13 weeks of age (Figure 1A). As shown previously [31], bIrs2−/− mice were heavier and longer with significantly increased body fat compared with Irs2L/L mice (Figure 1A and D). Through 12 weeks of age, the lack of Irs4 had no effect on body weight compared against the control mice; however, male bIrs2−/−·Irs4−/y mice were 60% heavier than Irs2L/L and 20% heavier than bIrs2−/− animals (Figure 1A and B). Dual X-ray absorptometry (DEXA) confirmed that the obesity phenotype of bIrs2−/−·Irs4−/y mice was due to significantly increased adipose mass compared to obese bIrs2−/− mice (Figure 1D). Hematoxylin and eosin staining of 6-month-old mice confirmed that the adipocytes of bIrs2−/−·Irs4−/y mice were 20% larger (p<0.05) than bIrs2−/− mice (Figure 2A and B). Moreover, 9 month old bIrs2−/−·Irs4−/y-mice displayed hepatic steatosis similar to that of ob/ob mice; however, steatosis was never observed in Irs4−/y, bIrs2−/− or control mice (Figure 2C).
Figure 1.
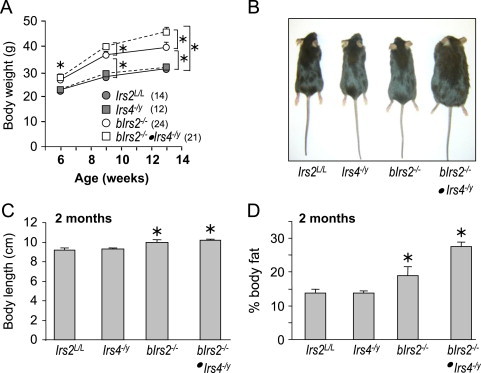
bIrs2−/−·Irs4−/y-mice are obese. (A) Average body weights of male bIrs2−/−·Irs4−/y-mice (open squares), Irs4−/y-mice (closed squares), bIrs2−/− mice (open circles) and control Irs2L/L mice (closed circles) on regular chow diet was determined in each age group and compared by generalized linear regression (SPSS, v19). The number of mice in each group is indicated in parentheses (mean±SD; *, Bonferroni p<0.001). (B) Representative image of 3-months-old male mice. (C) Body length and (D) percent body fat was determined by DEXA using 12-week-old mice (mean±SEM; n=8–10/genotype, *p<0.05 vs. controls).
Figure 2.
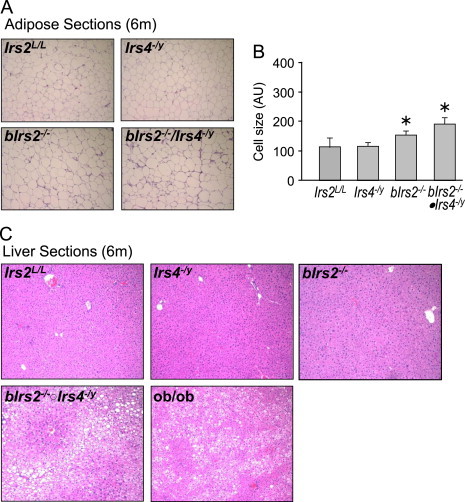
Representative H&E staining of (A) white adipose tissue (WAT) and (C) liver sections of 6-month-old bIrs2−/−·Irs4−/y-mice, Irs4−/y-mice, bIrs2−/− or Irs2L/L mice. Liver sections of 9-month-old ob/ob mice are also shown. (B) Morphometric analysis of adipocyte cell size in epididymal adipose tissue (n=5 animals per genotype; *p<0.05 vs. control).
Consistent with the graded adiposity of the bIrs2−/− and bIrs2−/−·Irs4−/y-mice, leptin concentrations were elevated in adult bIrs2−/−-mice and significantly greater in the bIrs2−/−·Irs4-/y mice; however, the leptin concentration was normal in the Irs4-/y-mice (Figure 3A). Food intake by chow-fed controls- and Irs4−/y-mice was indistinguishable, whereas bIrs2−/−-mice consumed more food (Figure 3B). The bIrs2−/−·Irs4−/y-mice consumed approximately 10% more food than the bIrs2−/−-mice during the 24-h test interval (Figure 3B). Thus, the lack of Irs4 augmented hyperphagia only when Irs2 was also absent from the CNS.
Figure 3.
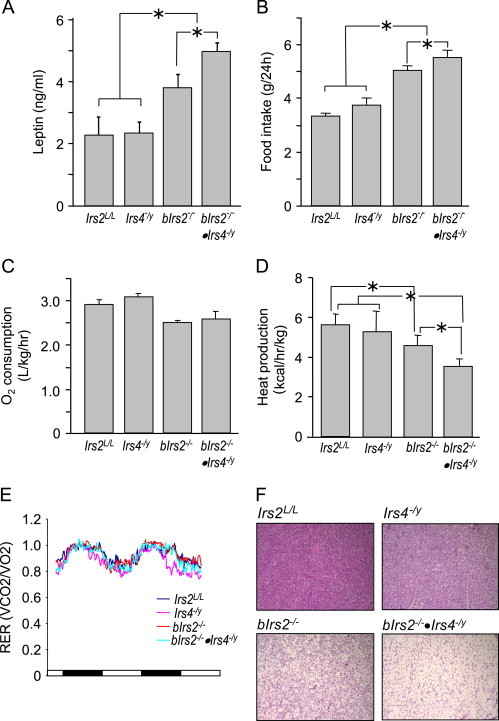
Energy expenditure in bIrs2−/−·Irs4−/y-mice. (A) Serum leptin levels of 9-month-old male bIrs2−/−·Irs4−/y-mice, Irs4−/y-mice, bIrs2−/− mice and Irs2L/L mice (n=8–10, mean±SEM; *, p<0.05 for indicated comparison). (B) Food intake over 24 h in 3-month-old male bIrs2−/−·Irs4−/y-mice, Irs4−/y mice, bIrs2−/−-mice or Irs2L/L-mice fed regular chow diet (mean±SEM; * p<0.05 for indicated comparison). Three month-old male mice of the indicated genotype were monitored for 72 h in the CLAMS (n=10/genotype) to assess (C) oxygen consumption (O2, l/kg/h), and (D) heat production (kcal/h/kg). Dark phase is presented (mean±SD; * p<0.001 for indicated comparison). Heat production was analyzed by generalized linear regression (SPSS, v19), controlling for the effect of body weight (Bonferroni, p<0.001) and (E) RER (respiratory exchange ratio) during the light and dark phases. (F) Representative H&E staining of brown adipose tissue (BAT) of 3-month-old male bIrs2−/−·Irs4−/y-mice, Irs4−/y-mice, bIrs2−/−-mice and Irs2L/L-mice.
Food intake and energy expenditure must be coordinately regulated to maintain energy balance and stable body weight. Irs2 was previously shown to alter energy homeostasis as young bIrs2−/− mice were less active and consumed less oxygen than age-matched control mice [31]. At 3 months of age, individually housed animals were monitored for 72 h in the Comprehensive Lab Animal Monitoring System (CLAMS). During the dark cycle, O2 consumption and heat generation were reduced similarly in bIrs2−/− and bIrs2−/−·Irs4−/y-mice compared to the normal parameters of Irs4−/y and control mice (Figure 3C and D). Respiratory exchange ratio (RER) was not different between the groups (Figure 3E). Nevertheless, compared against bIrs2−/− mice the hematoxylin and eosin stained brown adipose tissue (BAT) of adult 6-month-old bIrs2−/−·Irs4-/y-mice contained larger lipid-filled vacuoles that resembled white adipocytes (Figure 3F). Since Irs2 signaling was previously shown to play a role in BAT thermoregulation, these findings reveal a link between central Irs4 and BAT function that was exposed when Irs2 was deleted in the CNS [29].
3.3. Hypothalamic neuropeptide expression
To test whether the hyperphagia and obesity of bIrs2−/−·Irs4−/y-mice were related to changes in hypothalamic neuropeptide expression, we measured expression of Npy, Agrp, and Pomc mRNA in the ARC by qPCR [3]. The expression of mRNA encoding orexigenic neuropeptides (Npy and Agrp) was increased in bIrs2−/− and bIrs2−/−·Irs4−/y-mice, consistent with the observed hyperphagia of these animals (Figure 4A and B). By comparison, there were no significant changes in the expression of Pomc mRNA among the various genotypes (Figure 4C). Overall, the ratio between orexigenic (Agrp) and anorexigenic (Pomc) mRNAs was similarly increased in bIrs2-/- and bIrs2-/-·Irs4−/y mice compared to Irs4−/y and control mice (Figure 4D). Thus the mRNA expression of these hypothalamic neuropeptides was regulated by brain Irs2, but not influenced by Irs4.
Figure 4.
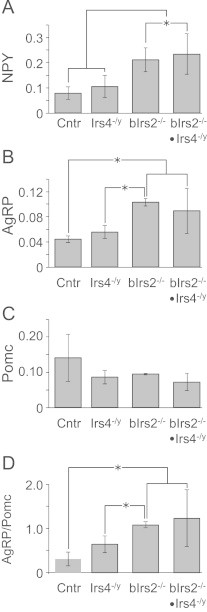
Levels of mRNA (relative to actin) by RT-PCR of Npy (A), Agrp (B) and Pomc (C) from hypothalamus of 3-month-old chow-fed male mice of the indicated genotypes. (D) Ratio between Agrp and Pomc mRNAs (from B, C) for the indicated genotypes. Data are presented as mean±SEM; * p<0.05 for indicated comparisons. (n=4–5).
3.4. Glucose homeostasis in bIrs2−/−·Irs4−/y-mice
Next, we investigated whether Irs4 synergizes with neuronal Irs2 to control systemic glucose homeostasis. Fasting glucose concentrations were normal in 3 month-old Irs4−/y mice, but slightly increased in bIrs2−/− mice and more dramatically increased in bIrs2−/−·Irs4−/y-mice (Figure 5A). Similarly, ad libitum-fed blood glucose values trended upward in the Irs4−/y and bIrs2−/− mice, but only reached significance in the bIrs2−/−·Irs4−/y-mice (Figure 5B). Whereas glucose tolerance was normal in Irs4−/y-mice and only slightly impaired in bIrs2−/−-mice, bIrs2−/−·Irs4−/y mice displayed dramatic glucose intolerance (Figure 5C). Furthermore, the bIrs2−/−·Irs4−/y-mice were resistant to the hypoglycemic effects of exogenous insulin, whereas the responses of bIrs2−/− and Irs4−/y mice were not significantly different from controls (Figure 5D). The HOMA2-IR confirmed that insulin resistance persists in 9 month old bIrs2−/−·Irs4-/y and bIrs2−/−-mice, whereas Irs4−/y mice displayed normal insulin sensitivity (Figure 5E). Consistent with these results, Irs4−/y-mice displayed normal insulin concentrations, whereas the bIrs2−/−-mice displayed slightly elevated circulating insulin at 9 months of age, whereas insulin concentrations in bIrs2−/−·Irs4−/y-mice were elevated approximately 5-fold (Figure 5F). The bIrs2−/− mice at 3.5 months displayed 3-fold greater β-cell mass, which was consistent with the demand for compensatory insulin secretion (Figure 6A). By comparison, the beta cell mass of 3.5 month old bIrs2−/− mice and 7.5 month old Irs4−/y mice was indistinguishable from the controls.
Figure 5.
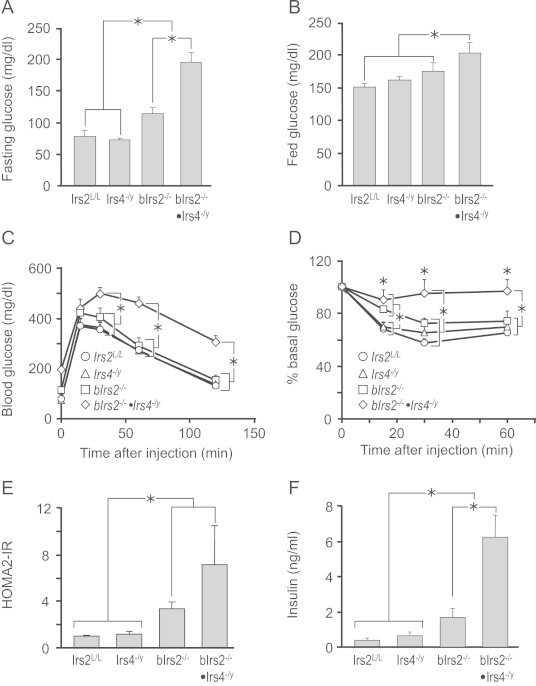
Glucose homeostasis in bIrs2−/−·Irs4−/y mice. (A) Fasted and (B) fed blood glucose for 3 month-old male mice. (C) Glucose tolerance test of 3 month-old male mice (n=10/genotype). (D) Insulin tolerance test of 4 month-old male mice (n=9–10/genotype). (E) HOMO2-IR index of insulin resistance (IR) and (F) fasting serum insulin levels (n=10/genotype) for 9 month-old male mice of the indicated genotypes. Data are expressed as mean±SEM. * p<0.05 for indicated comparisons.
Figure 6.
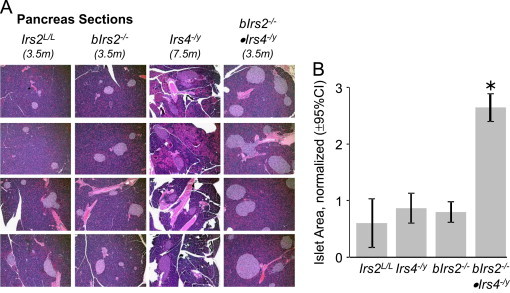
(A) Representative H&E staining of pancreatic sections of bIrs2−/−·Irs4−/y mice, Irs4−/y mice, bIrs2−/− mice and Irs2L/L mice at indicated ages. (B) Quantification of total pancreatic area occupied by β cells in bIrs2−/−·Irs4−/y mice, Irs4−/y mice, bIrs2−/− mice and Irs2L/L mice. Data are presented as mean±SEM; * p<0.05. (n=4–5) compared to other genotypes.
3.5. Irs4 effects on obesity are independent of leptin receptor expressing neurons
Leptin, a hormone produced by adipocytes, acts upon leptin receptor (LepRb) expressing neurons to modulate energy homeostasis [4]. Our recent observations show that deletion of Irs2 in LepRb neurons (Lepr∆Irs2-mice) increases body weight through the combined effects of hyperphagia and reduced energy consumption [29]. Since others have suggested that Irs4 mediates PI 3-kinase signaling in response to leptin stimulation [7], it is possible that the synergy between Irs4 and Irs2 occurs in LepRb neurons and that the deletion of Irs4 would increase the obesity observed in Lepr∆Irs2-mice. However, at 3 months of age the increased body weight of the Lepr∆Irs2-mice was not further increased in the Lepr∆Irs2·Irs4−/y mice (Figure 7A). Furthermore, while ad libitum-fed blood glucose levels were significantly elevated in the bIrs2−/−·Irs4−/y mice relative to all other groups (See Figure 5B, above), blood glucose levels remained unchanged among Lepr∆Irs2, Lepr∆Irs2·Irs4−/y and controls (Figure 7B). Thus, the synergy between brain Irs2 and Irs4 for body weight control occurs in neurons that are distinct from LepRb cells.
Figure 7.
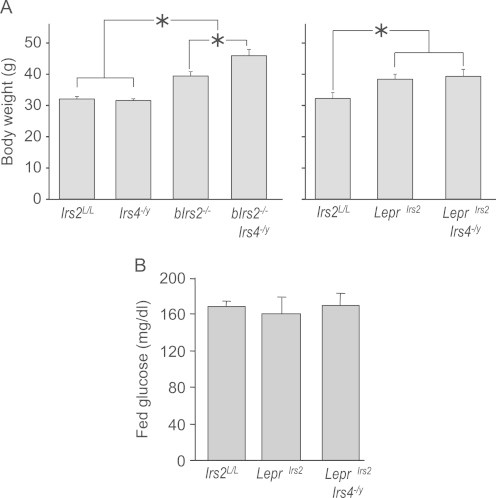
Lepr∆Irs2-mice and Lepr∆Irs2·Irs4−/y-mice display similar body weight and normal blood glucose levels. (A) Average body weights of 3 month-old male bIrs2−/−·Irs4−/y-mice, Irs4−/y mice, bIrs2−/− mice, Irs2L/L mice and Lepr∆Irs2-mice and Lepr∆Irs2·Irs4−/y-mice. (B) Fed blood glucose levels of 3 month-old male Irs2L/L mice, Lepr∆Irs2-mice and Lepr∆Irs2·Irs4−/y-mice. Mean±SD; n=6–8/genotype, *p<0.001 for indicated comparisons.
4. Discussion
Compared to Irs1 and Irs2, for which genetic ablation yields substantial phenotypes, the physiologic role of Irs4 has been difficult to establish because the deletion of Irs4 alone produces minimal physiologic perturbation [33]. Since Irs1 and Irs2 are widely expressed, while the expression of Irs4 is more narrowly distributed (largely in the hypothalamus, where Irs2 plays a crucial role in metabolic homeostasis), we reasoned that Irs2 and Irs4 might play synergistic and somewhat redundant roles. Indeed, insulin continues to stimulate the phosphorylation of AKT, a major IRS-protein-dependent insulin signaling pathway, in hypothalamic extracts from bIrs2−/− animals [31], suggesting the persistent and compensatory action of a second IRS-protein in the hypothalamus.
We thus examined the possibility of functional cooperation between Irs2 and Irs4 in the brain by combining whole body Irs4 deletion (which is functionally brain-restricted) with brain-specific Irs2 deletion thereby focusing on the tissue relevance for any synergistic action and avoiding the potentially confounding phenotypes associated with deletion of Irs2 in other tissues (such as the islet). Indeed, this analysis revealed important functional synergy between brain Irs2 and Irs4 in the regulation of energy balance and glucose homeostasis: To an extent far greater than either deletion alone, the brain-wide ablation of Irs2 and Irs4 in combination promoted obesity and glucose intolerance. The severe metabolic phenotype in the bIrs2-/-·Irs4-/y mice thus reveals an important physiological role for Irs4 that is obscured by the continued presence of Irs2 in the Irs4−/y-mice [33], much as the presence of Irs4 mitigates partially the phenotype produced by CNS-specific Irs2 deletion in bIrs2−/− animals. The concerted action of Irs2 and Irs4 contributes not only to overall adiposity and glucose intolerance, but plays an especially crucial role in the control of insulin action and hepatic lipid accumulation, as islet size, HOMA2-IR and hepatic steatosis are dramatically elevated in bIrs2−/−·Irs4−/y mice, but do not differ from controls in the single mutant (bIrs2−/− and Irs4−/y) mice.
Our current results, together with previous data, reveal separable functions for Irs2 and Irs4 expression in distinct neural subsets. The brain-wide ablation of Irs2 suffices to increase Agrp and Npy expression relative to Pomc, and promotes obesity and glucose intolerance. LepRb-restricted disruption of Irs2 expression similarly results in increased Agrp and Npy (and decreased Pomc) expression [29]. The Lepr∆Irs2-mice also display increased adiposity and decreased glucose tolerance relative to controls, although the magnitude of their metabolic phenotype is more modest than that observed in bIrs2−/− animals[29]. These findings suggest that while Irs2 signaling in LepRb cells modulates Agrp, Npy and Pomc expression and contributes to the control of glucose and energy homeostasis, Irs2 acts in other (non-LepRb) neurons to mediate additional aspects of metabolic control.
While Irs4 cooperates with Irs2 in the CNS to modulate energy balance and glucose homeostasis, Irs4 does not contribute to the control of Agrp, Npy, or Pomc expression; nor does the absence of Irs4 exacerbate the phenotype of Lepr∆Irs2-mice as it does the metabolic dysfunction of bIrs2−/− animals. Thus, our data are consistent with the idea that Irs4 synergizes with Irs2 in non-LepRb neurons to control energy balance and metabolism independently of LepRb neurons and the leptin-modulated ARC Pomc and AgRP/Npy neurons. In the future, it will be important to identify the relevant Irs2/Irs4-expressing non-LepRb cells to reveal their neurophysiologic function and independent roles in the control of metabolic homeostasis. Given the distribution of Irs4 and LepRb, it is tempting to speculate that the crucial Irs4-expressing neurons could lie in the paraventricular hypothalamic nucleus (PVH), which is crucial for energy balance and metabolism, but which contains little LepRb.
Importantly, while Irs4 may not synergize with Irs2 in LepRb neurons, it remains possible (especially given the distribution of Irs4 expression in the hypothalamus) that LepRb neurons contain Irs4. The lack of metabolic phenotype for Irs4−/y mice and lack of synergy between Irs2 and Irs4 in LepRb neurons suggests that if Irs4 plays a role in LepRb neurons, however, the function of Irs4 in LepRb cells must be redundant with a protein other than Irs2, such as Irs1. Thus, it may be useful to explore potentially redundant roles for Irs1 and Irs4 in the future, including in LepRb neurons.
Previous studies suggest that Irs4 might play a role in cellular leptin action [7]. Serum leptin concentrations are normal in Irs4−/y-mice, however. Furthermore, while leptin is elevated in bIrs2−/−·Irs4−/y relative to bIrs2−/− mice (and whole-body bIrs2+/−. Irs4−/y mice compared to controls [7]), the body weight of obese mice with double deletion of Irs4 and Irs2 in LepRb neurons (Lepr∆Irs2/Irs4−/y) was indistinguishable from the mice lacking only Irs2 in LepRb neurons (LeprΔIrs2). These results suggest that Irs4 effect on weight gain in the brain does not involve LepRb signaling. Indeed, despite the important role of Irs2 in LepRb neurons, and the hyperleptinemia of animals null for Irs2 in LepRb cells, leptin signals normally to decrease body weight in young Lepr∆Irs2-mice [29]. Thus, neither Irs2 nor Irs4 in LepRb neurons is required for leptin action, but rather presumably mediate crucial insulin signals in LepRb cells (Irs2) and non-LepRb cells (Irs2 and Irs4). Insulin action in the hypothalamus mediates important effects upon body weight and nutrient homeostasis through signaling cascades which regulate food intake, glucose and lipid metabolism, and energy expenditure [3]. Both Irs2 and Irs4 are expected to promote the PI3K→Akt ┤FoxO1 cascade in hypothalamic neurons, as elsewhere in the body; this pathway is known to play a crucial role in the hypothalamic control of metabolic regulation [39].
Overall, our results suggest that Irs2 and Irs4 synergize in non-LepRb neurons to mediate central insulin action, thereby controlling a variety of metabolically important phenotypes. While it contributes to the control of energy balance, Irs4 plays an especially crucial role in the control of whole-body insulin sensitivity and glucose homeostasis.
Conflict of interest
We wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Acknowledgments
This project was supported by NIH grants DK38712, DK55326, DK098655 and GM021700 (to MFW) and DK056731 and DK057768 (to MGM), and by the Ellison Foundation (to MFW).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at 10.1016/j.molmet.2013.10.004.
Appendix A. Supporting information
Supplementary data
References
- 1.Morris D.L., Cho K.W., Zhou Y., Rui L. SH2B1 enhances insulin sensitivity by both stimulating the insulin receptor and inhibiting tyrosine dephosphorylation of insulin receptor substrate proteins. Diabetes. 2009;58(9):2039–2047. doi: 10.2337/db08-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rhodes C.J., White M.F., Leahy J.L., Kahn S.E. Direct autocrine action of insulin on beta-cells: does it make physiological sense? Diabetes. 2013;62(7):2157–2163. doi: 10.2337/db13-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vogt M.C., Bruning J.C. CNS insulin signaling in the control of energy homeostasis and glucose metabolism – from embryo to old age. Trends in Endocrinology and Metabolism. 2013;24(2):76–84. doi: 10.1016/j.tem.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Myers M.G., Jr., Cowley M.A., Munzberg H. Mechanisms of leptin action and leptin resistance. Annual Review of Physiology. 2007;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- 5.Myers M.G., Jr., Munzberg H., Leinninger G.M., Leshan R.L. The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metabolism. 2009;9(2):117–123. doi: 10.1016/j.cmet.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris D.L., Rui L. Recent advances in understanding leptin signaling and leptin resistance. American Journal of Physiology – Endocrinology and Metabolism. 2009;297(6):E1247–E1259. doi: 10.1152/ajpendo.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wauman J., De Smet A.S., Catteeuw D., Belsham D., Tavernier J. Insulin receptor substrate 4 couples the leptin receptor to multiple signaling pathways. Molecular Endocrinology. 2008;22(4):965–977. doi: 10.1210/me.2007-0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White M.F. IRS proteins and the common path to diabetes. American Journal of Physiology – Endocrinology and Metabolism. 2002;283(3):E413–E422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- 9.Wang L.M., Keegan A.D., Li W., Lienhard G.E., Pacini S., Gutkind J.S. Common elements in interleukin 4 and insulin signaling pathways in factor dependent hematopoietic cells. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:4032–4036. doi: 10.1073/pnas.90.9.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welham M.J., Bone H., Levings M., Learmonth L., Wang L.M., Leslie K.B. Insulin receptor substrate-2 is the major 170-kDa protein phosphorylated on tyrosine in response to cytokines in murine lyphohemopoietic cells. Journal of Biological Chemistry. 1997;272(2):1377–1381. doi: 10.1074/jbc.272.2.1377. [DOI] [PubMed] [Google Scholar]
- 11.Johnston J.A., Wang L.M., Hanson E.P., Sun X.J., White M.F., Oakes S.A. Interleukins 2, 4, 7, and 15 stimulate tyrosine phosphorylation of insulin receptor substrates 1 and 2 in T cells. Potential role of JAK kinases. Journal of Biological Chemistry. 1995;270(48):28527–28530. doi: 10.1074/jbc.270.48.28527. [DOI] [PubMed] [Google Scholar]
- 12.Wang L.M., Michieli P., Lie W.R., Liu F., Lee C.C., Minty A. The insulin related substrate-1-related 4PS substrate but not the interleukin-2R gamma chain is involved in interleukin-13 mediated signal transduction. Blood. 1995;86(11):4218–4227. [PubMed] [Google Scholar]
- 13.Argetsinger L.S., Norstedt G., Billestrup N., White M.F., CarterSu C. Growth hormone, interferon-gamma, and leukemia inhibitory factor utilize insulin receptor substrate-2 in intracellular signaling. Journal of Biological Chemistry. 1996;271(46):29415–29421. doi: 10.1074/jbc.271.46.29415. [DOI] [PubMed] [Google Scholar]
- 14.Duan C., Li M., Rui L. SH2-B promotes insulin receptor substrate 1 (IRS1)- and IRS2-mediated activation of the phosphatidylinositol 3-kinase pathway in response to leptin. Journal of Biological Chemistry. 2004;279(42):43684–43691. doi: 10.1074/jbc.M408495200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yenush L., Makati K.J., SmithHall J., Ishibashi O., Myers M.G., White M.F. The pleckstrin homology domain is the principle link between insulin receptor and IRS-1. Journal of Biological Chemistry. 1996;271(39):24300–24306. doi: 10.1074/jbc.271.39.24300. [DOI] [PubMed] [Google Scholar]
- 16.Burks D.J., Wang J., Towery H., Ishibashi O., Lowe D., Riedel H. IRS pleckstrin homology domains bind to acidic motifs in proteins. Journal of Biological Chemistry. 1998;273(47):31061–31067. doi: 10.1074/jbc.273.47.31061. [DOI] [PubMed] [Google Scholar]
- 17.Fisher T.L., White M.F. Signaling pathways: the benefits of good communication. Current Biology. 2004;14(23):R1005–R1007. doi: 10.1016/j.cub.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 18.Laustsen P.G., Michael M.D., Crute B.E., Cohen S.E., Ueki K., Kulkarni R.N. Lipoatrophic diabetes in Irs1(−/−)/Irs3(−/−) double knockout mice. Genes and Development. 2002;16(24):3213–3222. doi: 10.1101/gad.1034802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bjornholm M., He A.R., Attersand A., Lake S., Liu S.C., Lienhard G.E. Absence of functional insulin receptor substrate-3 (IRS-3) gene in humans. Diabetologia. 2002;45(12):1697–1702. doi: 10.1007/s00125-002-0945-z. [DOI] [PubMed] [Google Scholar]
- 20.Escribano O., Fernandez-Moreno M.D., Zueco J.A., Menor C., Fueyo J., Ropero R.M. Insulin receptor substrate-4 signaling in quiescent rat hepatocytes and in regenerating rat liver. Hepatology. 2003;37(6):1461–1469. doi: 10.1053/jhep.2003.50245. [DOI] [PubMed] [Google Scholar]
- 21.Cuevas E.P., Escribano O., Chiloeches A., Ramirez R.S., Roman I.D., Fernandez-Moreno M.D. Role of insulin receptor substrate-4 in IGF-I-stimulated HEPG2 proliferation. Journal of Hepatology. 2007;46(6):1089–1098. doi: 10.1016/j.jhep.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 22.Cuevas E.P., Escribano O., Monserrat J., Martinez-Botas J., Sanchez M.G., Chiloeches A. RNAi-mediated silencing of insulin receptor substrate-4 enhances actinomycin D- and tumor necrosis factor-alpha-induced cell death in hepatocarcinoma cancer cell lines. Journal of Cellular Biochemistry. 2009;108(6):1292–1301. doi: 10.1002/jcb.22359. [DOI] [PubMed] [Google Scholar]
- 23.Fantin V.R., Lavan B.E., Wang Q., Jenkins N.A., Gilbert D.J., Copeland N.G. Cloning, tissue expression, and chromosomal location of the mouse insulin receptor substrate 4 gene. Endocrinology. 1999;140(3):1329–1337. doi: 10.1210/endo.140.3.6578. [DOI] [PubMed] [Google Scholar]
- 24.Numan S., Russell D.S. Discrete expression of insulin receptor substrate-4 mRNA in adult rat brain. Brain Research Molecular Brain Research. 1999;72(1):97–102. doi: 10.1016/s0169-328x(99)00160-6. [DOI] [PubMed] [Google Scholar]
- 25.Chiba T., Inoue D., Mizuno A., Komatsu T., Fujita S., Kubota H. Identification and characterization of an insulin receptor substrate 4-interacting protein in rat brain: implications for longevity. Neurobiology of Aging. 2009;30(3):474–482. doi: 10.1016/j.neurobiolaging.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 26.White M.F., Copps K.D., Ozcan U., Tseng Y.D. The molecular basis of insulin action. In: Jameson J.L., DeGroot L.J., editors. Endocrinology. 6th ed. Elsevier; Philadelphia: 2010. pp. 636–659. [Google Scholar]
- 27.White M.F. Regulating insulin signaling and beta-cell function through IRS proteins. Canadian Journal of Physiology and Pharmacology. 2006;84(7):725–737. doi: 10.1139/y06-008. [DOI] [PubMed] [Google Scholar]
- 28.Sadagurski M., Cheng Z., Rozzo A., Palazzolo I., Kelley G.R., Dong X. IRS2 increases mitochondrial dysfunction and oxidative stress in a mouse model of Huntington disease. Journal of Clinical Investigation. 2011;121(10):4070–4081. doi: 10.1172/JCI46305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sadagurski M., Leshan R.L., Patterson C., Rozzo A., Kuznetsova A., Skorupski J. IRS2 signaling in LepR-b neurons suppresses FoxO1 to control energy balance independently of leptin action. Cell Metabolism. 2012;15(5):703–712. doi: 10.1016/j.cmet.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White M.F. Insulin signaling in health and disease. Science. 2003;302(5651):1710–1711. doi: 10.1126/science.1092952. [DOI] [PubMed] [Google Scholar]
- 31.Taguchi A., Wartschow L.M., White M.F. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2007;317(5836):369–372. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- 32.Sadagurski M., White M.F. Integrating metabolism and longevity through insulin and IGF1 signaling. Endocrinology and Metabolism Clinics of North America. 2013;42(1):127–148. doi: 10.1016/j.ecl.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fantin V.R., Wang Q., Lienhard G.E., Keller S.R. Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction, and glucose homeostasis. American Journal of Physiology – Endocrinology and Metabolism. 2000;278(1):E127–E133. doi: 10.1152/ajpendo.2000.278.1.E127. [DOI] [PubMed] [Google Scholar]
- 34.Dong X.C., Copps K.D., Guo S., Li Y., Kollipara R., DePinho R.A. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metabolism. 2008;8(1):65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Withers D.J., Burks D.J., Towery H.H., Altamuro S.L., Flint C.L., White M.F. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nature Genetics. 1999;23(1):32–40. doi: 10.1038/12631. [DOI] [PubMed] [Google Scholar]
- 36.Dong X., Park S., Lin X., Copps K., Yi X., White M.F. Irs1 and Irs2 signaling is essential for hepatic glucose homeostasis and systemic growth. Journal of Clinical Investigation. 2006;116(1):101–114. doi: 10.1172/JCI25735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sadagurski M., Norquay L., Farhang J., D'Aquino K., Copps K., White M.F. Human IL6 enhances leptin action in mice. Diabetologia. 2010;53(3):525–535. doi: 10.1007/s00125-009-1580-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D. A gene atlas of the mouse and human protein-encoding transcriptomes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(16):6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niswender K.D., Morrison C.D., Clegg D.J., Olson R., Baskin D.G., Myers M.G., Jr. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes. 2003;52(2):227–231. doi: 10.2337/diabetes.52.2.227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data