

Abstract
The hormone ghrelin stimulates eating and helps maintain blood glucose upon caloric restriction. While previous studies have demonstrated that hypothalamic arcuate AgRP neurons are targets of ghrelin, the overall relevance of ghrelin signaling within intact AgRP neurons is unclear. Here, we tested the functional significance of ghrelin action on AgRP neurons using a new, tamoxifen-inducible AgRP-CreERT2 transgenic mouse model that allows spatiotemporally-controlled re-expression of physiological levels of ghrelin receptors (GHSRs) specifically in AgRP neurons of adult GHSR-null mice that otherwise lack GHSR expression. AgRP neuron-selective GHSR re-expression partially restored the orexigenic response to administered ghrelin and fully restored the lowered blood glucose levels observed upon caloric restriction. The normalizing glucoregulatory effect of AgRP neuron-selective GHSR expression was linked to glucagon rises and hepatic gluconeogenesis induction. Thus, our data indicate that GHSR-containing AgRP neurons are not solely responsible for ghrelin's orexigenic effects but are sufficient to mediate ghrelin's effects on glycemia.
Abbreviations: GHSR, growth hormone secretagogue receptor, ghrelin receptor; GOAT, ghrelin O-acyltransferase; CNS, central nervous system; Phox2b, paired-like homeobox 2b; AgRP, Agouti-related peptide; NPY, neuropeptide Y; GABA, gamma-aminobutyric acid; VGAT, vesicular GABA transporter; POMC, pro-opiomelanocortin; ARC, arcuate nucleus; BAC, bacterial artificial chromosome; VTA, ventral tegmental area; DG, dentate gyrus; NAc, nucleus accumbens; DVC, dorsal vagal complex; G6p, glucose-6 phosphatase; Pepck, phosphoenolpyruvate carboxykinase; Hnf4α, hepatocyte nuclear factor 4α; Foxo1, Forkhead box protein O1; Pcx, pyruvate carboxylase; GHRH, Growth-hormone-releasing hormone
Keywords: AgRP, Ghrelin, Ghrelin receptor, Food intake, Blood glucose homeostasis
1. Introduction
Ghrelin, the only known circulating hormone that stimulates appetite, is produced predominantly by a population of enteroendocrine “ghrelin” cells distributed sparsely throughout the gastric oxyntic mucosa [1,2]. Circulating ghrelin levels rise before meal initiation, following food restriction and after acute or chronic stress [3–5]. This physiological elevation, as well as pharmacologically-induced rises in ghrelin, promote both homeostatic and hedonic feeding behaviors [6–8]. Food intake in response to administered ghrelin has been demonstrated by both acute and chronic administration, using protocols in which ghrelin is given by one of several peripheral routes, intracerebroventricularly or by direct microinjection into the brain parenchyma [7–10]. The effects of ghrelin on both homeostatic and hedonic eating behaviors are well-established in rodents and humans [6,11–13] and occur via the growth hormone secretagogue receptor (GHSR), the only known ghrelin receptor [7,14].
In addition to its potent orexigenic effect, ghrelin signals and modulates glycemic state. For example, insulin-induced hypoglycemia elevates gastric ghrelin mRNA and exposure of cultured gastric ghrelin cells to low ambient glucose levels potentiates ghrelin release [15,16]. Furthermore, ghrelin administration to rodents increases blood glucose, raises glucagon and growth hormone levels, lowers insulin levels and attenuates insulin responses during glucose tolerance testing [17–20]. Conversely, genetic deletion and/or pharmacologic blockade of ghrelin, GHSR, and ghrelin O-acyltransferase (GOAT) lower blood glucose [10,17,20–23]. This effect seems most pronounced upon exposure to severely restricted food access, as evidenced by the progressive decline in fasting blood glucose to the point of near death in GOAT-knockout and ghrelin-knockout mice after 7 days daily access to only 40% of their usual daily calories [18,24].
These studies suggest an essential function for ghrelin in feeding and blood glucose regulation. However, the sites and mechanisms mediating these of ghrelin's actions are still poorly understood. The abundant expression of GHSRs in several distinct central nervous system (CNS) sites [25,26] and the induction of c-fos within several of these sites by ghrelin administration [7,27,28] suggests that the orexigenic and glucoregulatory effects of ghrelin may occur via its direct engagement of neurons within one or more CNS nuclei. Our group has begun to systematically map the CNS sites that directly mediate ghrelin action using a genetically-engineered “reactivatable” GHSR-null mouse model devoid of GHSR expression except in distinct cell populations where it occurs conditionally, at physiologic levels, upon exposure to Cre recombinase [10]. Using GHSR-null mice crossed to tyrosine hydroxylase-Cre mice, we have demonstrated that selective GHSR expression in catecholaminergic neurons is sufficient to partially mediate administered ghrelin-induced food intake and to completely restore both conditioned place preference for high fat diet (a reward-based eating behavior) and usual mood-related responses to chronic stress; however, it is not sufficient to normalize fasting blood glucose [29]. In contrast, we demonstrated that selective GHSR expression in hindbrain Phox2b-expressing neurons does not re-establish administered ghrelin-induced feeding but is sufficient to restore fasting blood glucose to wild-type levels [30]. These data suggest a distributed neuronal network that mediates different aspects of ghrelin action to varying degrees.
Among the many other CNS sites and neuronal subtypes through which ghrelin may act to affect eating and blood glucose, the hypothalamus, and more specifically AgRP/NPY/GABA-containing neurons which exist only in the arcuate nucleus (ARC) of the hypothalamus have been the most extensively studied. These neurons have long been recognized as having potent orexigenic effects when activated, for instance as achieved using designer receptors exclusively activated by designer drugs (DREADD) technology [31,32]. GHSRs are highly expressed in the ARC, predominantly on AgRP neurons [26,33]. Direct ghrelin microinjection into the ARC stimulates food intake, while chemical ablation of the ARC blunts the orexigenic actions of centrally delivered ghrelin [8,34,35]. Within AgRP neurons, ghrelin and/or GHSR agonists induce c-fos expression, augment transcription of the orexigenic neuropeptides AgRP and NPY, stimulate calcium influx, and directly depolarize resting membrane potential, all of which indicate that ghrelin activates these neurons [7,25,27,36–41]. Antibodies and antagonists of both AgRP and NPY abolish administered ghrelin-induced feeding [7,42]. AgRP neuron-specific deletion of VGAT, which disrupts presynaptic release of the inhibitory neurotransmitter GABA onto neighboring anorexigenic POMC neurons, results in a lean phenotype and also limits the usual orexigenic response to administered ghrelin [43]. More recent electrophysiology data suggests that the effect of ghrelin on AgRP neurons is indirect via activation of presynaptic glutamatergic inputs [44]. AgRP neurons also help mediate blood glucose homeostasis [45–49].
Despite these findings, the overall impact on feeding and blood glucose of AgRP neuronal GHSR expression in comparison to GHSR expression within other neuronal subtypes is still unclear. Previous attempts by our own group to direct ghrelin expression selectively to AgRP neurons at physiologic levels using the reactivatable GHSR-null mouse model crossed to transgenic AgRP-Cre mice [50] and “knock-in” AgRP-IRES-Cre mice [43,51] have failed due to presumed transient expression of AgRP – and thus, Cre recombinase – early in development, resulting in germline deletion of the loxP-flanked transcriptional blocking cassette present in the modified GHSR-null allele and a subsequent wild-type pattern of GHSR expression. To bypass these and other developmental issues, we now report a new AgRP-CreERT2 transgenic mouse line that allows for spatiotemporal gene manipulation specifically in ARC AgRP neurons following tamoxifen induction of Cre recombinase expression. Using crosses of the new AgRP-CreERT2 mice with the reactivatable GHSR-null mice, we have now successfully targeted GHSR expression to ARC AgRP neurons within adult mice, allowing us to investigate whether selective GHSR expression in AgRP neurons is sufficient to mediate the feeding and glucoregulatory actions of ghrelin.
2. Materials and methods
2.1. Animals
To generate AgRP-CreERT2 mice, standard recombineering techniques [52] were used to modify a bacterial artificial chromosome (BAC) containing regulatory elements of the mouse AgRP gene, which had previously been shown to drive reporter expression specifically in ARC AgRP neurons [50]. DNA sequences encoding a fusion protein of Cre recombinase with a mutated estrogen receptor (CreERT2, a gift from Dr. Pierre Chambon) were used to replace the coding sequence of the AgRP gene within the BAC. The BAC construct was then linearized and purified for pronuclear injections into fertilized one-cell stage embryos of C57B6/J mice, as performed by the UTSW Medical Center Transgenic Core Facility.
Transgenic founders were identified by PCR of tail genomic DNA with primers 5′-CCTGGCTACAGGAAGCAGTC-3′ and 5′-ATGTTTAGCTGGCCCAAATG-3′. For validation of transgene expression, AgRP-CreERT2 mice were bred to Rosa26-lox-STOP-lox-tdTomato reporter mice (B6J/N.Cg-Gt(ROSA)26Sortm14 (CAG-tdTomato); stock #007908; The Jackson Laboratory, Bar Harbor, ME). Six-week-old mice harboring both transgenes (R26Rtomato+, AgRP-CreERT2) were administered a daily dose of tamoxifen (150 mg/kg body weight, i.p.) or vehicle (corn oil) for 5 consecutive days. Ten days after the final injection of tamoxifen, expression of Cre activity was assessed by examining the pattern of tdTomato fluorescence, which only occurs upon Cre-mediated removal of a transcriptional stop cassette. Photomicrographs were produced with a Zeiss digital camera attached to a Zeiss Axioskop microcrope and a Dell desktop computer. An image editing software program, Adobe PhotoShop CS 6.0 (San Jose, CA), was used to adjust contrast and brightness.
Study animals including wild-type (2 wild-type GHSR alleles with 1 or no copies of AgRP-CreERT2), GHSR-null/AgRP-CreERT2 (2 GHSR-null alleles and 1 copy of AgRP-CreERT2), and GHSR-null (2 GHSR-null alleles without AgRP-Cre ERT2) littermates were generated using a previously-described breeding strategy [29]; all animals were on a pure C57BL6/J genetic background. Study animals were administered tamoxifen or corn oil, as above, between the ages of 5–7 weeks. Mice had free access to water and chow (2016 Teklad Global 16% Protein Rodent Diet, which provides 3.0 kcal/g [4.0 g% fat], Madison, WI) except as stated, and were housed in a temperature and humidity controlled environment under 12 h light/12 h dark. All experiments were approved by The University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee.
2.2. In situ hybridization histochemistry and immunohistochemistry
Mice (male and female) were deeply anesthetized and subsequently transcardially perfused with DEPC-treated 0.9% PBS and then 10% neutral-buffered formalin. Brains were extracted, post-fixed for 6 h at 4 °C and immersed in 30% sucrose (in DEPC-PBS) at 4 °C overnight. Brains were sectioned into 5 coronal series at a thickness of 25 μm using a sliding microtome, and stored at −20 °C until further processing. In situ hybridization histochemistry for GHSR mRNA was performed as reported previously using a 33P-labeled mouse GHSR riboprobe generated from a 916-bp fragment of cDNA that was amplified with GHSR specific primers (mGHSR1047, 5′-GTGGTGTTTG CTTTCAT CCTC-3′ and mGHSR1962, 5′-CATGCTCAAATTAAATGCATCC-3′), on mounted sections that had been air-dried and stored at −20 °C, as reported previously [29]. Immunohistochemistry was performed on free-floating sections using a rabbit anti-c-Fos antibody (1:30,000; Calbiochem/Oncogene, Temecula, CA; Catalog number PC38) and a biotin-SP donkey anti-rabbit IgG secondary antibody (Jackson ImmunoResearch, West Grove, PA, Catalog number 711-065-152), amplified by an ABC kit (1:500, Vectastain, Burlingame, CA, Catalog number PK-6100) and then followed by a metal enhanced DAB substrate kit (Thermo Scientific, Rockford, IL, Catalog number 34065), as previously described [53]. All histochemical analyses were performed on a minimum of 3 mice per group.
2.3. Brain punch and liver tissue collection and quantitative PCR analysis
Brains were removed from mice euthanized by live decapitation and sectioned into 1-mm coronal slices using a stainless steel mouse brain matrix on ice. Next, tissue punches from the ARC, ventral tegmental area (VTA), dentate gyrus (DG), and nucleus accumbens (NAc) (which were identified using a mouse brain atlas) were excised using a 15-g needle and stored at −80 °C until further processing. The left lobes of livers also were removed and stored at −80 °C. Total RNA was isolated using RNA STAT-60 (Tel-Test Inc., Friendswood, TX) and quantified using a Nanodrop Spectrophotometer (Thermo Scientific). Generation of cDNA was accomplished by treatment with RNase-free DNase (Promega, Madison, WI) and then reverse transcription with SuperScript II reagents (Invitrogen, Carlsbad, CA). Quantitative PCR was performed using SYBR Green (Bio-Rad Laboratories, Inc., Hercules, CA) and an Applied Biosystems 7900HT Fast Real-Time PCR System. Primer sets used (Supplementary Table 1) were previously validated [29,54]. The housekeeping genes 36B4 and 18S were used for brain tissue and liver, respectively, to normalize the expression of mRNA.
2.4. Determination of general metabolic parameters
Three weeks after the final tamoxifen injection, daily food intake and body weights were measured at 10:00 A.M. for 6 consecutive days. To determine food intake after a fast, regular chow was re-introduced to mice that had been fasted for 16 h, at 10:00 A.M. Then, food intake at 1 h, 2 h and 3 h was measured. Body composition analysis was performed using an EchoMRI-100™ (Echo Medical Systems, Houston, TX) on days 0 and 7 of the calorie restriction, at 4 P.M. Percent Body fat was calculated by dividing fat weight as determined with the EchoMRI-100™ by body weight as measured with a scale.
2.5. Ghrelin-induced food intake
Mice were acclimated to handling for 3 days prior to subcutaneous injection with either saline or acyl-ghrelin (2 mg/kg body weight) at 10:00 A.M. (4 h after the start of the daily light cycle). After injection, regular chow intake was determined at 2 h and 24 h later. Previously, this dose and route of administration had been shown to potently increase food intake after 30, 45, 90, 120 and 135 min and glucagon release after 30 min [5,29,30].
2.6. Blood glucose levels
Chow was removed from home cages at 6:00 P.M. (at the start of the daily dark cycle), and fasting blood glucose was measured 16 h later from tail blood using an AlphaTrak glucometer, at 10:00 A.M. “Fed” blood glucoses were measured from tail blood at 10:00 A.M. in ad lib-fed mice.
2.7. Chronic severe calorie restriction
Chronic severe calorie restriction was carried out as previously described [18]. Briefly, individually-housed 8-week-old male mice were monitored for daily food intake for 1 week, and an average daily consumption was calculated. Thereafter, the mice were provided 40% of their average daily consumption at 6 P.M. every day for 1 week. Before providing the food, blood glucoses were measured daily, as above.
2.8. Measurement of glucagon and insulin levels
Mice were euthanized and blood was collected from the inferior vena cava into EDTA-coated tubes containing p-hydroxymercuribenzoic acid (final concentration, 1 mM) and aprotinin (final concentration, 0.028 mg/ml). The separated plasma was stored at −80 °C until processing. Insulin was assayed by ELISA (Crystal Chem, Inc., Downers Grove, IL, Catalog number 90080) and glucagon was measured by RIA (Millipore, Billerica, MA, Catalog number GL-32K) using 10 µl and 100 µl plasma, respectively.
2.9. Data analysis
All data are shown as mean±SEM. The effects of genotype and treatment on ghrelin-induced acute food intake and blood glucose levels were determined using a two-way ANOVA followed by a secondary one-way ANOVA analysis when significant genotype×treatment interactions were detected. The effects of genotype on daily food intake, plasma insulin and glucagon, and mRNA levels were determined using a one-way ANOVA. Repeated measures two-way ANOVA was used to analyze the effect of genotype on blood glucose over the 7-day course of chronic severe calorie restriction. Data were transformed before analysis when unequal variance among groups was detected via Bartlett's test. A Tukey post-hoc test was used in all two-way and one-way ANOVA analyses. GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA) was used.
3. Results
3.1. Generation of GHSR-null mice with re-expression of endogenous GHSR selectively in AgRP neurons of adult mice
To investigate the sufficiency of AgRP neurons in mediating ghrelin's actions, we generated mice with GHSR expression only in AgRP neurons. This necessitated the design of a novel AgRP-CreERT2 transgenic mouse line that allowed for spatiotemporal targeting of Cre recombinase selectively to AgRP neurons upon tamoxifen delivery (Figure 1A). BAC engineering was employed to generate the AgRP-CreERT2 targeting construct such that the coding region for a Cre recombinase-mutant estrogen receptor fusion protein was inserted immediatedly downstream of the start codon of AgRP, thereby allowing both the transcription and translation of the tamoxifen-inducible Cre recombinase to be controlled by AgRP regulatory elements. We identified several founder lines with inducible Cre activity, as confirmed by injecting 6-week-old Cre reporter mice (R26Rtomato+, AgRP-CreERT2) with a single daily dose of tamoxifen or vehicle for 5 consecutive days. Expression of tdTomato, was detected in the ARC 10 days after the last tamoxifen treatment. Recombination was strictly dependent upon tamoxifen treatment as no Cre activity could be detected in the absence of tamoxifen (Figure 1B). Importantly, Cre activity was not detected in other regions of the CNS after tamoxifen injections (Supplementary Figure 1). The transgenic line that showed the highest levels of recombination after tamoxifen treatment (total number of tdTomato positive neurons in ARC) was used for subsequent metabolic analyses.
Figure 1.
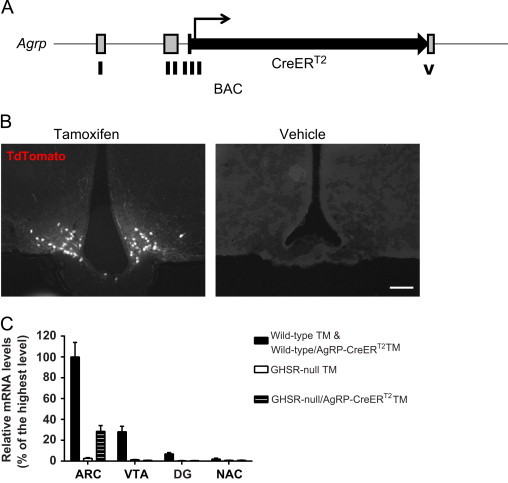
Generation of a mouse model with selective GHSR expression in AgRP neurons. (A) Schematic diagram of the derivation of AgRP-CreERT2 mice. The coding sequence of AgRP, found in exons III and IV, was replaced by the coding sequence of a CreERT2 fusion protein. (B) The desired spatiotemporal expression of cre recombinase in AgRP-CreERT2 mice was validated by a cross to Rosa26-lox-STOP-lox-tdTomato reporter mice; tdTomato expression (white neurons) was detected selectively in the ARC of tamoxifen (TM)-injected mice but not in vehicle-injected mice (n=3 per group; representative photomicrographs shown; scale bar 100 µm). (C) Ghsr mRNA is expressed in the ARC and VTA of wild-type and wild-type/AgRP-CreERT2 mice but not in GHSR-null animals as expected, as determined using qRT-PCR; tamoxifen-induced Cre-mediated re-expression of Ghsr mRNA is only observed in the ARC of GHSR-null/AgRP-CreERT2 mice, but not other regions (mean±SEM, n=4–6 per group).
Following its validation, the AgRP-CreERT2 transgenic line was crossed to a GHSR-null mouse model, in which the Ghsr locus is modified by the insertion of a loxP-flanked transcriptional blocking cassette [10], enabling tamoxifen-dependent reactivation of physiological levels of GHSR expression selectively in AgRP neurons. Study mice derived from this genetic cross included wild-type mice (which contain GHSRs in all usual GHSR expression sites, without the AgRP-CreERT2 transgene), wild-type/AgRP-CreERT2 mice (which contain GHSRs in all usual GHSR expression sites, with the AgRP-CreERT2 transgene), GHSR-null mice (which lack GHSRs), and GHSR-null/AgRP-CreERT2 mice (which express endogenous levels of GHSRs only upon tamoxifen exposure in AgRP-containing neurons programmed to express both GHSR and AgRP). Mice of these 4 genotypes were administered either tamoxifen or vehicle beginning at 5–7 weeks of age, for 5 days, and then either were tested by quantitative PCR or histochemistry for the predicted GHSR expression or were characterized metabolically.
The predicted pattern of tamoxifen-dependent, ARC-restricted GHSR expression within GHSR-null/AgRP-CreERT2 mice was confirmed by real-time quantitative reverse transcription PCR (qRT-PCR) on tissue punches from different brain regions and by in situ hybridization histochemistry on coronal brain sections. In particular, within tamoxifen-treated GHSR-null/AgRP-CreERT2 mice, GHSR expression was observed by qRT-PCR in the ARC but not in other tested brain sites including the VTA, DG and NAc (Figure 1C). Of note, GHSR expression levels within ARC tissue punches were not as high as those in the corresponding wild-type+wild-type/AgRP-CreERT2 ARC tissue punches. This is probably due to the observation that Cre activity does not appear to be present in all AgRP neurons and the fact that GHSR is expressed in both AgRP neurons and non-AgRP neurons in the ARC of wild-type animals. [29,33,40,55]. These qRT-PCR findings were further supported by in situ hybridization histochemistry studies using a previously validated antisense GHSR-specific riboprobe. Reactivation of GHSR expression was observed in GHSR-null/AgRP-CreERT2 mice in a limited fashion within the ARC, but not within the VTA or dorsal vagal complex (DVC), which all normally highly express GHSR (Supplementary Figure 2). Of note, detectable binding of the antisense GHSR riboprobe was observed in the Edinger-Westphal nucleus of both GHSR-null mice [as had been reported previously, [10,29]] and GHSR-null/AgRP-CreERT2 mice. GHSR expression was not observed in the ARC of vehicle-treated GHSR-null/AgRP-CreERT2 mice (data not shown).
3.2. Ghrelin-induced acute feeding is partially restored in adult GHSR-null mice with reactivated AgRP neuron-selective GHSR expression
To examine the physiological significance of AgRP neuron expression of GHSRs in feeding regulation, we first monitored intake of standard rodent chow, body weight and body composition in tamoxifen-treated male and female animals that were 8–10 weeks of age. No differences among the wild-type, wild-type/AgRP-CreERT2, GHSR-null and GHSR-null/AgRP-CreERT2 GHSR mice were observed in average daily food intake, % change in body weight over 6-day period, or % body fat (Supplementary Figure 3A–C). Also, rebound hyperphagia following a 16-h fast occurred to a similar degree in all 4 tamoxifen-treated groups (Supplementary Figure 3D). Next, we measured food intake at 2 h (acute) and 24 h following subcutaneous ghrelin or saline administration to male and female animals. As expected, ghrelin potently stimulated acute feeding in both wild-type and wild-type/AgRP-CreERT2 groups, but not in GHSR-null mice. Acute feeding in response to ghrelin was partially restored (about 65% of the wild-type level) in GHSR-null/AgRP-CreERT2 mice (Figure 2A and B). Restoration of ghrelin-induced acute food intake was not observed in vehicle-injected GHSR-null/AgRP-CreERT2 mice, in which the Cre recombinase is inactive (Figure 2C). The acute orexigenic effects of ghrelin became less apparent over time, as food intake measured 24 h after ghrelin or saline injection was comparable among the four different study groups (Supplementary Figure 3E). The disappearance of ghrelin's stimulatory effect on food intake 24 h post-injection has previously been described in rats [8].
Figure 2.
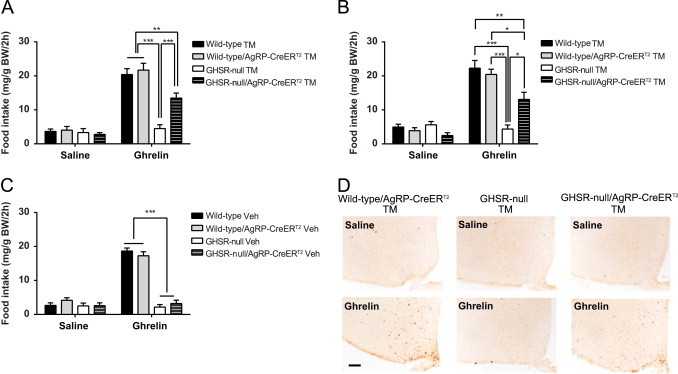
GHSR-expressing AgRP neurons partially mediate ghrelin's orexigenic action. (A) Ghrelin-induced 2 h-food intake is partially restored in male GHSR-null/AgRP-CreERT2 mice with tamoxifen administration (mean±SEM, n=7–12 per group). (B) Ghrelin-induced 2 h-food intake is also partially restored in female GHSR-null/AgRP-CreERT2 mice with tamoxifen administration (mean±SEM, n=7 per group). (C) Ghrelin-induced acute food intake is not restored in male GHSR-null/AgRP-CreERT2 mice with vehicle injection (mean±SEM, n=3–7 per group). (D) Induction of c-fos (dark orange cell bodies) in response to ghrelin in ARC of wild-type/AgRP-CreERT2 and GHSR-null/AgRP-CreERT2 mice but not in GHSR-null mice or in response to saline (n=3 per group, representative photomicrographs shown; scale bar 100 µm). *P<0.05; **P<0.01; ***P<0.001.
3.3. Arcuate c-fos induction in ghrelin-treated mice with AgRP neuron-selective GHSR expression
To further explore the mechanism underlying this ghrelin-induced acute feeding response, the expression of c-fos, a marker for neuronal activation, was examined 2 h after s.c. ghrelin or saline administration. Expression of c-fos in the ARC was robustly induced by administered ghrelin in tamoxifen-treated wild-type/AgRP-CreERT2 mice and GHSR-null/AgRP-CreERT2 mice but not in GHSR-null mice. No c-fos induction was observed in the ARC of saline-administered, tamoxifen-treated mice of the same genotypes (Figure 2D). Notably, unlike the more generalized c-fos induction within the mediobasal hypothalamus of wild-type/AgRP-CreERT2 mice, the hypothalamic c-fos induction in GHSR-null/AgRP-CreERT2 mice was restricted to the ARC (Figure 2D). Moreover, c-fos induction in the dorsal vagal complex was inconsistently observed in ghrelin-injected wild-type and wild-type/AgRP-CreER T2 mice (data not shown).
3.4. Fasting plasma glucose levels restored in mice with AgRP neuron-selective GHSR expression
Blood glucose decreases normally occur in wild-type mice following an overnight fast, and these falls become exaggerated in GHSR-null mice [17,29,30]. To determine the role of AgRP neuronal GHSR expression in the blood glucose response to fasting, we measured blood glucose in both tamoxifen-treated (male and female) and vehicle-treated (male) mice of the above-described four genotypes. No differences among the four genotypes were observed in ad-lib fed mice. In contrast, blood glucoses of overnight fasted GHSR-null mice were lower than those of overnight fasted wild-type mice. Notably, blood glucoses in both male and female mice with selective GHSR re-expression in AgRP neurons returned to wild-type levels (Figure 3A and B). This normalization of blood glucose was observed in GHSR-null/AgRP-CreERT2 mice only upon tamoxifen treatment and not in vehicle-treated animals (Figure 3C).
Figure 3.
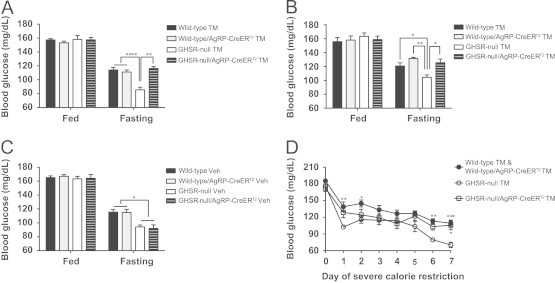
GHSR-expressing AgRP neurons mediate the glucoregulatory action of ghrelin. (A) Fasting blood glucose levels are restored to wild-type levels in male GHSR-null/AgRP-CreERT2 mice with tamoxifen administration (mean±SEM, n=7–12 per group). (B) Fasting blood glucose levels are also restored to wild-type levels in female GHSR-null/AgRP-CreERT2 mice with tamoxifen administration (mean±SEM, n=7 per group). (C) Fasting blood glucose levels are not restored in male GHSR-null/AgRP-CreERT2 mice with vehicle (Veh) injection (mean±SEM, n=3–7 per group). (D) Blood glucose levels during severe calorie restriction are restored to wild-type levels in male tamoxifen-treated GHSR-null/AgRP-CreERT2 mice (mean±SEM, n=5–12 per group). *P<0.05; **P<0.01; ***P<0.001.
3.5. Fasting glucagon levels restored in mice with AgRP neuron-selective GHSR expression
In order to investigate potential mechanisms by which AgRP neuronal GHSRs help maintain blood glucose homeostasis, levels of fasting plasma glucagon and insulin in our study groups were determined. The reduced fasting glucagon observed in GHSR-null mice [17] was normalized to wild-type levels upon induction of Cre recombinase expression selectively in AgRP neurons. No changes in fasting insulin levels were observed in GHSR-null mice as compared to wild-type control mice, just as had been reported previously [17], nor were differences in fasting insulin observed in tamoxifen-treated GHSR-null/AgRP-CreERT2 mice (Figure 4A and B). These data suggest that a glucagon response helps maintain blood glucose after an overnight fast, and that ghrelin-responsive AgRP neurons are sufficient to mediate this glucagon response.
Figure 4.
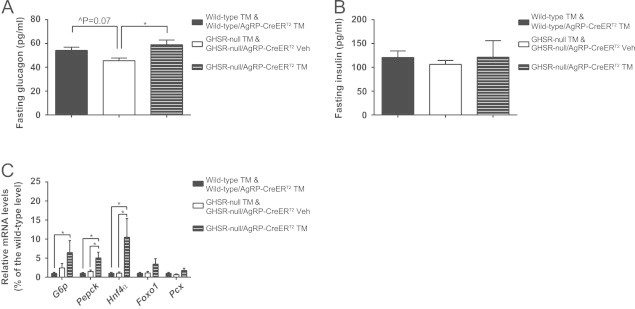
AgRP neuronal expression of GHSR restores fasting glucagon levels and increases fasting-induced liver gluconeogenesis. (A) Fasting glucagon levels are restored in male GHSR-null/AgRP-CreERT2 mice with tamoxifen administration (mean±SEM, n=8–14 per group). (B) Fasting insulin levels are similar for all genotypes (mean±SEM, n=8–14 per group). (C) Expression of hepatic gluconeogenesis genes after overnight fasting, as determined using qRT-PCR, is increased in male GHSR-null/AgRP-CreERT2 mice with tamoxifen injection (mean±SEM, n=4–8 per group). ∧P=0.07; *P<0.05.
3.6. Increased gluconeogenic gene expression in livers of mice with AgRP neuron-selective GHSR expression
To test whether hepatic gluconeogenesis contributes to the restoration of fasting glucose levels in tamoxifen-treated GHSR-null/AgRP-CreERT2 mice, mRNA levels of several gluconeogenic genes in livers from fasted mice were measured. These genes included glucose-6 phosphotase (G6p), phosphoenolpyruvate carboxykinase (Pepck), hepatocyte nuclear factor 4α (Hnf4α), Forkhead box protein O1 (Foxo1) and pyruvate carboxylase (Pcx). We found increased expression of G6p, Pepck and Hnf4α in tamoxifen-treated GHSR-null/AgRP-CreERT2 mice as compared to both GHSR-null and wild-type littermates (Figure 4C). These results suggest a compensatory mechanism in which selective AgRP neuronal GHSR re-expression increases hepatic gluconeogenesis to prevent the drop in blood glucose otherwise induced by overnight caloric restriction food in GHSR-null mice [17].
3.7. Restored glucose levels under chronic severe calorie restriction in mice with AgRP neuron-selective GHSR expression
The function of AgRP neuronal expression of GHSRs in maintaining glucose homeostasis was also tested under a more chronic and severe caloric restriction paradigm. When mice were provided access to only 40% of their usual calories daily for 7 days, GHSR-null mice experienced a more pronounced drop in blood glucose than wild-type mice and wild-type mice with AgRP-CreERT2 (Figure 3D), as previously demonstrated in two other models of ghrelin signaling deficiency (GOAT-knockout mice and ghrelin-knockout mice) [18,24]. This exacerbated fall in blood glucose upon chronic, severe caloric restriction was rescued by tamoxifen-induced GHSR re-expression in AgRP neurons (Figure 3D).
4. Discussion
AgRP neurons localized to the ARC have long been hypothesized to play a central role in the orexigenic effects of ghrelin. Some of the strongest supporting evidence for this hypothesis includes observations of ghrelin-stimulated AgRP neuron electrical activity in hypothalamic slices [25] and abrogated ghrelin-stimulated food intake in mice with ablated AgRP neurons [41,46,56,57]. However, until the current study, the overall impact of direct ghrelin engagement of intact AgRP neurons on food intake had not yet been adequately assessed in comparison with its direct action on other neuronal cell types. Nor, to our knowledge, had AgRP neurons been assessed at all in regards to the blood glucose-related actions of ghrelin. Here, we have demonstrated the sufficiency of AgRP neuronal expression of GHSRs in mediating a partial orexigenic response to administered ghrelin and in mediating ghrelin's protective glucoregulatory actions occurring in the settings of an overnight fast and a more chronic caloric restriction protocol.
There are several noteworthy points of discussion stemming from the current study. To the best of our knowledge, the tamoxifen-inducible AgRP-CreERT2 transgenic mouse line is the first to allow Cre activity to be triggered in AgRP neurons in a temporal manner. When crossed with the GHSR-null mouse, it becomes the first reported mouse model that directly tests the impact of AgRP neuron-selective GHSR expression on mouse feeding behaviors and glycemic states. Our novel animal model shows direct proof of the hypothesis established for years that ghrelin directly activates GHSR-expressing AgRP neurons to induce acute food intake in mice. In particular, in mice with AgRP neuron-selective GHSR expression, ghrelin-induced acute feeding was intermediate to that observed in wild-type mice and GHSR-null mice. Although we cannot rule out the possibility that the partial nature of this restoration of ghrelin-induced feeding is due to an incomplete rescue of GHSRs in AgRP neurons, Aponte et al. have previously described that optogenetic activation of only a small percentage of AgRP neurons (800 out of ~3800, or rather ~22% of the total number of AgRP neurons) can induce a maximal feeding response [46]. In our animal model, the GHSR mRNA level achieved was about 30% of the wild-type level. Since this 30% re-expression was limited to AgRP neurons and excluded other ARC neuronal cell populations known to express GHSR in wild-type mice (including catecholaminergic neurons, POMC neurons and GHRH neurons) [29,33,40,55], we presume that we were able to achieve GHSR expression in a number of AgRP neurons above the threshold required to maximally stimulate feeding [46]. As such, GHSR expression selectively in AgRP neurons appears insufficient for the entire acute orexigenic response to administered ghrelin.
The incomplete restoration of ghrelin-induced eating in tamoxifen-treated GHSR-null/AgRP-CreERT2 mice also might stem from the developmental stage of the mice at which GHSR expression was achieved. In particular, GHSR-null/AgRP-CreERT2 mice lacked GHSR expression until 5–7 weeks of age, as dictated by the timing of tamoxifen exposure. The timing of this selective re-expression could potentially impact the ability of the ARC AgRP neurons to form their usual neurocircuits and also could potentially impact the development of compensatory neurocircuits [45,58]. Indeed, such time-dependent effects on ARC neurocircuits have been shown previously. For instance, leptin's neurotrophic effects on mediobasal hypothalamic development are restricted to a critical neonatal period [58]. Also, unlike the lack of significant body weight or food intake phenotypes in mice with genetic AgRP expression achieved since inception or in neonates, rapid starvation ensues when AgRP expression is deleted in adult mice [45,47,59]. Similar time-sensitive neurotrophic actions have been predicted for ghrelin [60]. It is thus also of interest that the orexigenic actions of administered ghrelin were present at all in the tamoxifen-treated GHSR-null/AgRP-CreERT2 mice, given that GHSR expression was lacking until early adulthood. The new AgRP-CreERT2 mouse model should serve as a powerful tool for future studies investigating the impact of ghrelin and other signals on the development of AgRP neuron-containing neurocircuitry.
Despite only partial restoration of ghrelin-induced feeding in mice with GHSR expression selectively in AgRP neurons, there was a full normalization of the overnight fasting-induced and severe caloric restriction-induced exacerbated falls in blood glucose that otherwise occur in GHSR-deleted animals. Two interrelated glucoregulatory processes were identified as being likely downstream of AgRP neuronal engagement by ghrelin: glucagon release and hepatic gluconeogenesis. In particular, the lowered plasma glucagon levels associated with the relative hypoglycemia in overnight fasted GHSR-null mice [17] were normalized in mice with AgRP neuron-selective GHSR expression. Also, after the overnight fast, levels of several hepatic gluconeogenesis gene transcripts observed in the livers of mice with selective GHSR expression in AgRP neurons were elevated as compared to both wild-type and GHSR-null mice. These results suggest that a ghrelin-induced, AgRP neuron-mediated rise in glucagon helps to mediate the usual blood glucose response to overnight fasting, and furthermore that ghrelin-engaged AgRP neurons can compensate for presumed other missing glucoregulatory actions by inducing higher expression of hepatic gluconeogenesis genes. These other glucoregulatory processes downstream of ghrelin could include those normally induced by direct ghrelin action on Phox2b-containing hindbrain neurons, which also can fully mediate ghrelin's defense of blood glucose following an overnight fast [30]. A growth hormone response has previously been shown to partially mediate ghrelin's glucoregulatory actions during the severe caloric restriction protocol [18]. The capacity of ghrelin to modulate glucagon and insulin action by direct interaction with its receptors on pancreatic alpha and beta cells, respectively, also might be part of the usual glucoregulatory response to caloric restriction [17,19].
The new data presented here, when paired with previous data obtained using mice with tyrosine hydroxylase-Cre-mediated and Phox2b-Cre-mediated re-expression of GHSRs, supports the model of a distributed neuronal network mediating different aspects of ghrelin action. Whereas AgRP neurons and catecholaminergic neurons each mediate a partial orexigenic response to administered ghrelin, neither neuronal group is sufficient to mediate the entire ghrelin response on food intake, nor do Phox2b neurons directly mediate this response. In contrast, whereas AgRP neurons and Phox2b neurons each mediate a full ghrelin-induced glucoregulatory response to an overnight fast, catecholamiergic neurons do not. This distributed network of central sites at which ghrelin acts to have its varied effects is reminiscent of what has been shown using similar types of genetic mouse models to determine those neuronal subtypes sufficient and/or required for the varied actions of leptin, serotonin and melanocortin [52,61–63]. We speculate that the involvement of multiple regions of the CNS in ghrelin's orexigenic and glucoregulatory actions help form a fail-safe mechanism whereby if one site should become compromised, other sites are available to mediate these critical functions.
Conflict of interest
None declared.
Acknowledgments
This work was supported by the National Institutes of Health (R01DA024680 and R01MH085298 to J.M.Z., R01DK071320, P01DK088761, R01 DK088423 and R37DK053301 to J.K.E, T32DK007307-33 to A.U., T32DA007290 to A.K.W., K01DK098317 to E.D.B.), by the National Health and Medical Research Council (APP1030337 to Zane Andrews and to J.M.Z.), by the Davis Foundation in Eating Disorder Research to C.L. and by the American Diabetes Association 07-11-MN-16 to J.K.E. and T.L.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.molmet.2013.10.001.
Appendix A. Supplementary materials
Supplementary material
References
- 1.Kojima M., Hosoda H., Date Y., Nakazato M., Matsuo H., Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 2.Sakata I., Nakano Y., Osborne-Lawrence S., Rovinsky S.A., Lee C.E., Perello M. Characterization of a novel ghrelin cell reporter mouse. Regulatory peptides. 2009;155:91–98. doi: 10.1016/j.regpep.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cummings D.E., Purnell J.Q., Frayo R.S., Schmidova K., Wisse B.E., Weigle D.S. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50:1714–1719. doi: 10.2337/diabetes.50.8.1714. [DOI] [PubMed] [Google Scholar]
- 4.Lutter M., Krishnan V., Russo S.J., Jung S., McClung C.A., Nestler E.J. Orexin signaling mediates the antidepressant-like effect of calorie restriction. Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2008;28:3071–3075. doi: 10.1523/JNEUROSCI.5584-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lutter M., Sakata I., Osborne-Lawrence S., Rovinsky S.A., Anderson J.G., Jung S. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nature Neuroscience. 2008;11:752–753. doi: 10.1038/nn.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perello M., Zigman J.M. The role of ghrelin in reward-based eating. Biological Psychiatry. 2012;72:347–353. doi: 10.1016/j.biopsych.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakazato M., Murakami N., Date Y., Kojima M., Matsuo H., Kangawa K. A role for ghrelin in the central regulation of feeding. Nature. 2001;409:194–198. doi: 10.1038/35051587. [DOI] [PubMed] [Google Scholar]
- 8.Wren A.M., Small C.J., Abbott C.R., Dhillo W.S., Seal L.J., Cohen M.A. Ghrelin causes hyperphagia and obesity in rats. Diabetes. 2001;50:2540–2547. doi: 10.2337/diabetes.50.11.2540. [DOI] [PubMed] [Google Scholar]
- 9.Wang L., Saint-Pierre D.H., Tache Y. Peripheral ghrelin selectively increases Fos expression in neuropeptide Y-synthesizing neurons in mouse hypothalamic arcuate nucleus. Neuroscience Letters. 2002;325:47–51. doi: 10.1016/s0304-3940(02)00241-0. [DOI] [PubMed] [Google Scholar]
- 10.Zigman J.M., Nakano Y., Coppari R., Balthasar N., Marcus J.N., Lee C.E. Mice lacking ghrelin receptors resist the development of diet-induced obesity. Journal of Clinical Investigation. 2005;115:3564–3572. doi: 10.1172/JCI26002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cummings D.E. Ghrelin and the short- and long-term regulation of appetite and body weight. Physiology & Behavior. 2006;89:71–84. doi: 10.1016/j.physbeh.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 12.Karra E., O'Daly O.G., Choudhury A.I., Yousseif A., Millership S., Neary M.T. A link between FTO, ghrelin, and impaired brain food-cue responsivity. Journal of Clinical Investigation. 2013;123:3539–3551. doi: 10.1172/JCI44403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uchida A., Zigman J.M., Perello M. Ghrelin and eating behavior: evidence and insights from genetically-modified mouse models. Frontiers in Neuroscience. 2013;7:121. doi: 10.3389/fnins.2013.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howard A.D., Feighner S.D., Cully D.F., Arena J.P., Liberator P.A., Rosenblum C.I. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science. 1996;273:974–977. doi: 10.1126/science.273.5277.974. [DOI] [PubMed] [Google Scholar]
- 15.Toshinai K., Mondal M.S., Nakazato M., Date Y., Murakami N., Kojima M. Upregulation of Ghrelin expression in the stomach upon fasting, insulin-induced hypoglycemia, and leptin administration. Biochemical and Biophysical Research Communications. 2001;281:1220–1225. doi: 10.1006/bbrc.2001.4518. [DOI] [PubMed] [Google Scholar]
- 16.Sakata I., Park W.M., Walker A.K., Piper P.K., Chuang J.C., Osborne-Lawrence S. Glucose-mediated control of ghrelin release from primary cultures of gastric mucosal cells. American Journal of Physiology – Endocrinology and Metabolism. 2012;302:E1300–1310. doi: 10.1152/ajpendo.00041.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chuang J.C., Sakata I., Kohno D., Perello M., Osborne-Lawrence S., Repa J.J. Ghrelin directly stimulates glucagon secretion from pancreatic alpha-cells. Molecular Endocrinology. 2011;25:1600–1611. doi: 10.1210/me.2011-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao T.J., Liang G., Li R.L., Xie X., Sleeman M.W., Murphy A.J. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7467–7472. doi: 10.1073/pnas.1002271107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dezaki K., Hosoda H., Kakei M., Hashiguchi S., Watanabe M., Kangawa K. Endogenous ghrelin in pancreatic islets restricts insulin release by attenuating Ca2+ signaling in beta-cells: implication in the glycemic control in rodents. Diabetes. 2004;53:3142–3151. doi: 10.2337/diabetes.53.12.3142. [DOI] [PubMed] [Google Scholar]
- 20.Dezaki K., Kakei M., Yada T. Ghrelin uses Galphai2 and activates voltage-dependent K+ channels to attenuate glucose-induced Ca2+ signaling and insulin release in islet beta-cells: novel signal transduction of ghrelin. Diabetes. 2007;56:2319–2327. doi: 10.2337/db07-0345. [DOI] [PubMed] [Google Scholar]
- 21.Longo K.A., Charoenthongtrakul S., Giuliana D.J., Govek E.K., McDonagh T., Qi Y. Improved insulin sensitivity and metabolic flexibility in ghrelin receptor knockout mice. Regulatory Peptides. 2008;150:55–61. doi: 10.1016/j.regpep.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 22.Sun Y., Asnicar M., Saha P.K., Chan L., Smith R.G. Ablation of ghrelin improves the diabetic but not obese phenotype of ob/ob mice. Cell Metabolism. 2006;3:379–386. doi: 10.1016/j.cmet.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 23.Dezaki K., Sone H., Koizumi M., Nakata M., Kakei M., Nagai H. Blockade of pancreatic islet-derived ghrelin enhances insulin secretion to prevent high-fat diet-induced glucose intolerance. Diabetes. 2006;55:3486–3493. doi: 10.2337/db06-0878. [DOI] [PubMed] [Google Scholar]
- 24.Li R.L., Sherbet D.P., Elsbernd B.L., Goldstein J.L., Brown M.S., Zhao T.J. Profound hypoglycemia in starved, ghrelin-deficient mice is caused by decreased gluconeogenesis and reversed by lactate or fatty acids. Journal of Biological Chemistry. 2012;287:17942–17950. doi: 10.1074/jbc.M112.358051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cowley M.A., Smith R.G., Diano S., Tschop M., Pronchuk N., Grove K.L. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron. 2003;37:649–661. doi: 10.1016/s0896-6273(03)00063-1. [DOI] [PubMed] [Google Scholar]
- 26.Zigman J.M., Jones J.E., Lee C.E., Saper C.B., Elmquist J.K. Expression of ghrelin receptor mRNA in the rat and the mouse brain. Journal of Comparative Neurology. 2006;494:528–548. doi: 10.1002/cne.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence C.B., Snape A.C., Baudoin F.M., Luckman S.M. Acute central ghrelin and GH secretagogues induce feeding and activate brain appetite centers. Endocrinology. 2002;143:155–162. doi: 10.1210/endo.143.1.8561. [DOI] [PubMed] [Google Scholar]
- 28.Faulconbridge L.F., Grill H.J., Kaplan J.M., Daniels D. Caudal brainstem delivery of ghrelin induces fos expression in the nucleus of the solitary tract, but not in the arcuate or paraventricular nuclei of the hypothalamus. Brain Research. 2008;1218:151–157. doi: 10.1016/j.brainres.2008.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chuang J.C., Perello M., Sakata I., Osborne-Lawrence S., Savitt J.M., Lutter M. Ghrelin mediates stress-induced food-reward behavior in mice. Journal of Clinical Investigation. 2011;121:2684–2692. doi: 10.1172/JCI57660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott M.M., Perello M., Chuang J.C., Sakata I., Gautron L., Lee C.E. Hindbrain ghrelin receptor signaling is sufficient to maintain fasting glucose. PloS One. 2012;7:e44089. doi: 10.1371/journal.pone.0044089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krashes M.J., Koda S., Ye C., Rogan S.C., Adams A.C., Cusher D.S. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. Journal of Clinical Investigation. 2011;121:1424–1428. doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu T., Wang Q., Berglund E.D., Tong Q. Action of neurotransmitter: a key to unlock the AgRP neuron feeding circuit. Frontiers in Neuroscience. 2012;6:200. doi: 10.3389/fnins.2012.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willesen M.G., Kristensen P., Romer J. Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology. 1999;70:306–316. doi: 10.1159/000054491. [DOI] [PubMed] [Google Scholar]
- 34.Bagnasco M., Tulipano G., Melis M.R., Argiolas A., Cocchi D., Muller E.E. Endogenous ghrelin is an orexigenic peptide acting in the arcuate nucleus in response to fasting. Regulatory Peptides. 2003;111:161–167. doi: 10.1016/s0167-0115(02)00283-5. [DOI] [PubMed] [Google Scholar]
- 35.Tamura H., Kamegai J., Shimizu T., Ishii S., Sugihara H., Oikawa S. Ghrelin stimulates GH but not food intake in arcuate nucleus ablated rats. Endocrinology. 2002;143:3268–3275. doi: 10.1210/en.2002-220268. [DOI] [PubMed] [Google Scholar]
- 36.Kamegai J., Tamura H., Shimizu T., Ishii S., Sugihara H., Wakabayashi I. Central effect of ghrelin, an endogenous growth hormone secretagogue, on hypothalamic peptide gene expression. Endocrinology. 2000;141:4797–4800. doi: 10.1210/endo.141.12.7920. [DOI] [PubMed] [Google Scholar]
- 37.Hewson A.K., Dickson S.L. Systemic administration of ghrelin induces Fos and Egr-1 proteins in the hypothalamic arcuate nucleus of fasted and fed rats. Journal of Neuroendocrinology. 2000;12:1047–1049. doi: 10.1046/j.1365-2826.2000.00584.x. [DOI] [PubMed] [Google Scholar]
- 38.Kohno D., Gao H.Z., Muroya S., Kikuyama S., Yada T. Ghrelin directly interacts with neuropeptide-Y-containing neurons in the rat arcuate nucleus: Ca2+ signaling via protein kinase A and N-type channel-dependent mechanisms and cross-talk with leptin and orexin. Diabetes. 2003;52:948–956. doi: 10.2337/diabetes.52.4.948. [DOI] [PubMed] [Google Scholar]
- 39.Seoane L.M., Lopez M., Tovar S., Casanueva F.F., Senaris R., Dieguez C. Agouti-related peptide, neuropeptide Y, and somatostatin-producing neurons are targets for ghrelin actions in the rat hypothalamus. Endocrinology. 2003;144:544–551. doi: 10.1210/en.2002-220795. [DOI] [PubMed] [Google Scholar]
- 40.Dickson S.L., Luckman S.M. Induction of c-fos messenger ribonucleic acid in neuropeptide Y and growth hormone (GH)-releasing factor neurons in the rat arcuate nucleus following systemic injection of the GH secretagogue, GH-releasing peptide-6. Endocrinology. 1997;138:771–777. doi: 10.1210/endo.138.2.4907. [DOI] [PubMed] [Google Scholar]
- 41.Andrews Z.B., Liu Z.W., Walllingford N., Erion D.M., Borok E., Friedman J.M. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454:846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asakawa A., Inui A., Kaga T., Yuzuriha H., Nagata T., Ueno N. Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin. Gastroenterology. 2001;120:337–345. doi: 10.1053/gast.2001.22158. [DOI] [PubMed] [Google Scholar]
- 43.Tong Q., Ye C.P., Jones J.E., Elmquist J.K., Lowell B.B. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nature Neuroscience. 2008;11:998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Y., Atasoy D., Su H.H., Sternson S.M. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell. 2011;146:992–1003. doi: 10.1016/j.cell.2011.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luquet S., Perez F.A., Hnasko T.S., Palmiter R.D. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 46.Aponte Y., Atasoy D., Sternson S.M. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nature Neuroscience. 2011;14:351–355. doi: 10.1038/nn.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gropp E., Shanabrough M., Borok E., Xu A.W., Janoschek R., Buch T. Agouti-related peptide-expressing neurons are mandatory for feeding. Nature Neuroscience. 2005;8:1289–1291. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- 48.Konner A.C., Janoschek R., Plum L., Jordan S.D., Rother E., Ma X. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metabolism. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 49.Ren H., Orozco I.J., Su Y., Suyama S., Gutierrez-Juarez R., Horvath T.L. FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell. 2012;149:1314–1326. doi: 10.1016/j.cell.2012.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaelin C.B., Xu A.W., Lu X.Y., Barsh G.S. Transcriptional regulation of agouti-related protein (Agrp) in transgenic mice. Endocrinology. 2004;145:5798–5806. doi: 10.1210/en.2004-0956. [DOI] [PubMed] [Google Scholar]
- 51.Fukuda M., Jones J.E., Olson D., Hill J., Lee C.E., Gautron L. Monitoring FoxO1 localization in chemically identified neurons. Journal of Neuroscience: the Official Journal of the Society for Neuroscience. 2008;28:13640–13648. doi: 10.1523/JNEUROSCI.4023-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dhillon H., Zigman J.M., Ye C., Lee C.E., McGovern R.A., Tang V. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 53.Perello M., Sakata I., Birnbaum S., Chuang J.C., Osborne-Lawrence S., Rovinsky S.A. Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biological Psychiatry. 2010;67:880–886. doi: 10.1016/j.biopsych.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng Y., Wang Z.V., Tao C., Gao N., Holland W.L., Ferdous A. The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. Journal of Clinical Investigation. 2013;123:455–468. doi: 10.1172/JCI62819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tannenbaum G.S., Lapointe M., Beaudet A., Howard A.D. Expression of growth hormone secretagogue-receptors by growth hormone-releasing hormone neurons in the mediobasal hypothalamus. Endocrinology. 1998;139:4420–4423. doi: 10.1210/endo.139.10.6330. [DOI] [PubMed] [Google Scholar]
- 56.Luquet S., Phillips C.T., Palmiter R.D. NPY/AgRP neurons are not essential for feeding responses to glucoprivation. Peptides. 2007;28:214–225. doi: 10.1016/j.peptides.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 57.Bugarith K., Dinh T.T., Li A.J., Speth R.C., Ritter S. Basomedial hypothalamic injections of neuropeptide Y conjugated to saporin selectively disrupt hypothalamic controls of food intake. Endocrinology. 2005;146:1179–1191. doi: 10.1210/en.2004-1166. [DOI] [PubMed] [Google Scholar]
- 58.Bouret S.G., Draper S.J., Simerly R.B. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004;304:108–110. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- 59.Wu Q., Boyle M.P., Palmiter R.D. Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell. 2009;137:1225–1234. doi: 10.1016/j.cell.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steculorum S.M., Bouret S.G. Developmental effects of ghrelin. Peptides. 2011;32:2362–2366. doi: 10.1016/j.peptides.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Williams K.W., Elmquist J.K. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nature Neuroscience. 2012;15:1350–1355. doi: 10.1038/nn.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu Y., Elmquist J.K., Fukuda M. Central nervous control of energy and glucose balance: focus on the central melanocortin system. Annals of the New York Academy of Sciences. 2011;1243:1–14. doi: 10.1111/j.1749-6632.2011.06248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Williams K.W., Scott M.M., Elmquist J.K. Modulation of the central melanocortin system by leptin, insulin, and serotonin: co-ordinated actions in a dispersed neuronal network. European Journal of Pharmacology. 2011;660:2–12. doi: 10.1016/j.ejphar.2010.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material