

Abstract
One of the major hurdles to cure cancer lies in the low potency of currently available drugs, which could eventually be solved by using more potent therapeutic macromolecules, such as proteins or genes. However, although these macromolecules possess greater potency inside the cancer cells, the barely permeable cell membrane remains a formidable barrier to exert their efficacy. A widely used strategy is to use cell penetrating peptides (CPPs) to improve their intracellular uptake. Since the discovery of the first CPP, numerous CPPs have been derived from natural or synthesized products. Both in vitro and in vivo studies have demonstrated that those CPPs are highly efficient in transducing cargoes into almost all cell types. Therefore, to date, CPPs have been widely used for intracellular delivery of various cargoes, including peptides, proteins, genes, and even nanoparticles. In addition, recently, based on the successes of CPPs in cellular studies, their applications in vivo have been actively pursued. This review will focus on the advanced applications of CPP-based in vivo delivery of therapeutics (e.g., small molecule drugs, proteins, and genes). In addition, we will highlight certain updated applications of CPPs for intracellular delivery of nanoparticulate drug carriers, as well as several ‘smart’ strategies for tumor targeted delivery of CPP-cargoes.
Keywords: Cell penetrating peptides, Cancer, Macromolecule, Nanoparticle, In vivo
INTRODUCTION
In the past decades, numerous biomaterials have been newly developed and drawn enormous interest for their potential use in a wide variety of fields, encompassing medicine, biology, chemistry and tissue engineering.1, 2 To date, one of the specific fields that those biomaterials have made a significant impact is the cancer therapy. The intervention of biocompatible peptides (e.g., cell penetrating peptides (CPPs)), proteins, biopolymers and various nanocomposites in delivery of anti-cancer drugs has revolutionized the paradigm of drug delivery. 3–6
The first record of cancer can be traced to an Egyptian papyrus dating back to about 3000 B.C.7 Although cancer has been known for much of our recorded history, an effective cure for this life-threatening disease is yet to be found. Efforts during the past century have led to enormous advances in our understanding of cancer pathogenesis, detection techniques, surgical methods and also the development of anticancer drugs. Despite these advances, the primary treatment of cancer has yet been limited to chemotherapy intended for relief of pain or extension of life span, especially for metastatic cases. A major hurdle for treatment of cancer by current anticancer drugs lies in the poor efficacy and inadvertent systemic toxicity caused by the narrow therapeutic window of these drugs. To this regard, drug delivery systems (DDS) have been pursued to address the toxicity issue, and significant progress has been made in this area, resulting in the approval of several DDS formulated drugs for clinical applications. The second thrust of effort, meanwhile, has been concentrated on the search for new drug entities with greater efficacy. To this issue, macromolecules (e.g., protein, peptides or genes) have gained enormous interest due to their unmatched potency and specificity compared to current anticancer drugs. The delivery of these macromolecular drugs, however, remains a formidable challenge. Many of these macromolecular drugs act upon specific processes found within the cell which require internalization of the molecules and their delivery to appropriate cellular compartments. Yet, the greater size and lower lipophilicity of macromolecular drugs, compared to small molecule counterparts, hinders their efficient cellular internalization.
An interesting finding made by Frankel and Pabo in 1988 was that transcription transactivating protein (TAT) derived from HIV virus has the ability to penetrate cellular membranes.8 This discovery was a prelude to the identification and characterization of various cell penetrating peptides (CPPs) that possess the ability to enter cells. More interestingly, it was later found that the coupling of CPPs to different cargoes (e.g., small molecules, peptides or proteins, genes or nanoparticles) enabled efficient internalization of these cargoes into cells.9–13 The discovery of CPPs and their ability to ferry cargoes into cells has produced a revolutionary impact on the development of macromolecule-and nanoparticle-based treatments.
In this review, we will briefly discuss the nature of various CPP-mediated drug delivery, as well as the suggested mechanisms for their cellular uptake. We will then look at the application of the CPPs for cancer treatment, focusing specifically on advanced in vivo studies, followed by discussing a few ’smart’ strategies for targeted delivery of CPP-cargoes.
CELL PENETRATING PEPTIDES
CPPs, also known as protein transduction domains (PTDs), are a group of peptides sharing several common characteristics as follows.14–16 First, they are peptides consisting of less than 35 amino acid residues. Second, they can efficiently internalize into cells and also carry coupled cargoes into cells with them. Furthermore, they do not pose any reported cytotoxicity. The peptides which fall in this category originate from a wide variety of sources (e.g., humans, mice, viruses or synthesis). Based on their structural characteristics, CPPs can be simply divided into 2 classes: arginine-rich CPPs and amphipathic CPPs.17
Arginine rich CPPs
Commonly explored arginine-rich CPPs include TAT peptide (TATp), oligoarginine, penetratin, low molecular weight protamine (LMWP) and pVEC.10, 18–21 It is widely believed that the basic arginine and lysine residues of these peptides play a crucial role in the cell internalization process.
TATp is an 11-mer peptide (YGRKKRRQRRR) derived from the transcription transactivating protein (TAT) encoded by human immunodeficiency virus type 1 (HIV-1). A study of TATp (47–57) in 1998 revealed this to be the minimum TAT protein length that retained the cell entry capability.18 Since then, the TATp has become one of the most extensively studied CPPs.13, 15, 22, 23
Since the initial discovery of TATp, various researches have elucidated the critical role of cationic amino acids (i.e., arginine, lysine, and histidine) in the cell entry of TATp. Further studies have shown that oligoarginines composed of 8–10 arginine residues (Arg) can also efficiently internalize into cells.19 It was found that among the cationic amino acids, arginine possessed the greatest potential for cell penetration, possibly attributed to its more basic nature (pKa 12.5) and the formation of strong hydrogen bonds between the guanidine groups and anionic glycosaminoglycans on the cell membrane.24 Interestingly, it has also been demonstrated that nona-arginine (RRRRRRRRR) is the most efficient oligoarginine for the cell entry.19, 25 Further mechanistic studies showed that D-isoform and even the branched forms of oligoarginine retained the cell penetrating ability, which suggested the possible involvement of receptor-independent cell entry pathways.19
LMWP is a 14-mer peptide (VSRRRRRRGGRRRR) derived by thermolysin-mediated digestion of protamine.20 Compared with TATp, LMWP retains similar CPP motif in structure and has shown equivalent cell penetrating activity. LMWP, however, possesses several important and attractive characteristics compared to other CPPs, such as: (1) efficient mass production by simple enzymatic digestion of natural protamine; (2) proven low toxicity and immunogenicity in animal model studies; and (3) ensured safety as it originates from protamine, a widely used clinical heparin antidote.20 LMWP has been successfully utilized to translocate various cargos into different types of cells.20, 26–29
While TAT protein was the first identified cell permeable protein, the first CPP discovered was penetratin. Penetratin is a 16-mer peptide (RQIKIWFQNRRMKWKK) present in the third helix of Antennapedia homeodomain.4 Homeo-proteins are a class of transcription factors that bind DNA through a 60 amino acid length homeo-domain composed of three α-helices and a β-turn. Studies have shown that arginine and lysine residues are important for the cellular translocalization of penetratin.10 Also, tryptophan residues (Trp48 and 56) are reported to play an important role in the initiation of the translocation.10, 30, 31 Since the discovery of penetratin, a number of studies have been designed to use this CPP for delivery of various cargoes, including small molecules, proteins, oligonucleotides, peptide nucleic acid (PNA), and nanoparticles.32–34
pVEC, an 18-mer peptide (LLIILRRRIRKQAHAHSK) corresponding to residues (615-632) of the murine vascular endothelial cadherin (VE-cadherin), has been shown to translocate into various cell lines, even including the plant cells.21 A non-endocytic pathway for the cell entry has been proposed, as the rate of translocation is not affected by temperature or endocytosis inhibitors.21 Interestingly, pVEC was found to also possess antimicrobial activity at micro-molar concentrations.35
Amphipathic CPPs
Amphipathic CPPs are structurally distinguished from arginine-rich CPPs in that they have fewer, or completely lack of arginine residues and in that they are amphipathic in nature. Amphipathicity and lysine residues are considered to play a crucial role in the cellular translocation of this class of CPPs. They can be further divided into 2 groups: 1) peptides with primary amphipathicity (e.g., transportan, MPG, and pep-1) – having distinct hydrophobic and hydrophilic domains separately located in the peptide sequence;36–38 and 2) peptides with secondary amphipathicity (e.g. MAP) – having an α-helix structure with polar and non-polar residues located on the opposite sides.39
Transportan, a 27-mer chimeric peptide (GWTLNSAGYLLGKINLKALAALAKKIL) composed of 13 N-terminal residues of galanin neuropeptide with substitution of proline to lysine at 13th amino acid and 14 residues of mastoparan peptide from wasp venom, is able to translocate green fluorescence protein, antibody, or gold particles into cells.36 Interestingly, a truncated form of transportan named TP10 (AGYLLGKINLKALAALAKKIL) was found to also possess equivalent cell penetrating ability to the parent transportan.40 EL-Andaloussi et al. showed that a hybrid of TP10 and peptide nucleic acid (PNA), conjugated with a double stranded oligodeoxynucleotide against a transcription factor, Myc, was efficiently taken up by mouse neuroblastoma cells (N2a) and human breast cancer cells (MCF-7).41
Both the 27-mer MPG peptide (GALFLGFLGAAGSTMGAWSQPKKKRKV) and the 21-mer pep-1 peptide (KETWWETWWTEWSQPKKKRKV) are chimeric peptides with primary amphipathicity constructed of an N-terminal hydrophobic region and a C-terminal hydrophilic domain containing the nuclear localization signal (PKKKRKV) of the SV40 large T antigen. The hydrophobic region of MPG originates from HIV-gp41 coat protein and that of pep-1 consists of a tryptophan rich domain.37 It was suggested that the hydrophobic residues may contribute to the peptide’s interaction with, and destabilization of the cellular membrane. A truncated MPG (MPG-8: β-AFLGWLGAWGTMGWSPKKKRK-Cysteamide) was also reported to have the CPP activity.37, 38
Model amphipathic peptide (MAP) is an 18-mer peptide (KLALKLALKALKAALKLA) with secondary amphipathicity. Through mechanistic in vitro studies, MAP has been shown to internalize into cells by a non-endocytic pathway,42 and has successfully been used to deliver PNA into cells.43 Interestingly, it was found that MAP’s cell-internalization ability was retained even after depriving of its amphipathicity.42
CELL ENTRY MECHANISM OF CPPs
While the mechanism of CPP-mediated cell entry has been the subject of a large number of studies, there is still no consensus on the mechanism of this translocation process. A pioneering mechanism appeared to be a passive, energy-independent process that was not sensitive to endocytosis inhibitors.19, 21, 44 Recent studies re-evaluating the cellular entry mechanisms of CPPs or CPP-cargoes, however, have indicated the involvement of endocytosis.45 It has been suggested that properties of CPPs (e.g., amphipathicity or charge delocoalization) and the attached cargoes can affect the cell entry pathway.46, 47 Cell lines and incubation conditions have also been shown to affect the cell entry of CPPs.48 This review will briefly discuss two major intracellular uptake mechanisms of CPPs: the energy-independent (non-endocytosis) pathways and the endocytic pathways. These CPP internalization pathways are illustrated in Fig. 1.
FIGURE 1.

Schematic illustration of cell entry mechanism of CPPs. Reproduced with permission from Ref. 23.
Energy independent pathways
Different energy independent cell entry pathways for CPPs/CPP-cargoes have been suggested, such as the (1) inverted micelle model,49–51 (2) pore formation model,52 (3) carpet model,53, 54 and (4) membrane thinning model.55 The first step in all of these models requires the positively charged CPPs to interact with the negatively charged components of the cell membrane, such as heparan sulfate (HS) and the phospholipid bilayer. Binding of the CPPs on the lipid bilayer then leads to temporary destabilization of the plasma membrane. Subsequent steps in the internalization process vary depending upon the type and concentration of CPPs, cargoes, cell lines and incubation conditions.
The inverted micelle model was proposed based on NMR studies of the interaction of penetratin with phospholipid membranes. According to the hypothesized mechanism, the positively charged CPP interacts with the negative heads of the membrane phospholipids, followed by a reorganization of the lipid bilayer and membrane shuttling aided by interactions of the hydrophobic residues of the CPP with the membrane. Subsequently, the CPP moves into the bilayer while being entrapped in an inverted micelle, resulting in the move of this micelle to the opposite side of the bilayer and release of its contents directly into the cytosol. Because this model assumes the involvement of the hydrophobic residues of CPPs in the formation of the inverted micelle, it cannot be used to describe the translocation of CPPs that have no hydrophobic segments (e.g., TATp and oligoarginines). Moreover, Lewin et al.,56 based on their studies with CPP-conjugated iron oxide nanoparticles (~45 nm), raised serious doubts on the formation of such “inverted micelles”. Additionally, the inverted micelle structures have not been visualized microscopically.
The pore formation model, also called the barrel-stave model, describes one way in which antibacterial peptides weaken the bacterial membrane, which is followed by depolarization of the membrane. Pore formation is the result of bundles formed by the amphipathic α-helical structure of CPPs. In this arrangement, outwardly facing hydrophobic residues interact with the lipid membrane, while hydrophilic groups form a central lumen to create the pore.
The carpet model was described as a model for translocation of some antimicrobial peptides, but was also applied to describe CPP toxicity at higher concentrations.53, 54 In the carpet model, positively charged CPPs, monomeric or oligomeric, cover the negatively charged membrane in a carpet-like manner, which results in a change in the CPP secondary structure as the basic residues turn towards the membrane surface and the hydrophobic residues interact with the hydrophobic membrane core. When a concentration threshold is reached, the CPP permeates the membrane by locally disrupting the bilayer structure. Although this model has been suggested for CPP translocation, its dependence on hydrophobic residues seems to generate some controversy since most CPPs are primarily basic and hydrophilic.
An alternative to the carpet model is the so-called “membrane-thinning” effect, which was first suggested for a peptide toxin, magainin.55 In this model, following carpet formation by CPPs, perturbation caused by charge interaction in the outer leaflet results in a lateral rearrangement of negatively charged lipids, followed by a thinning of the membrane. Interaction, or aggregation, of CPPs on the membrane surface produce a reduction of the local surface tension and allows CPPs to intercalate within the membrane. The membrane then reseals after internalization of CPPs onto intracellular targets.
Although all these mechanisms explain certain aspects of membrane translocation by CPPs, none of them has provided a satisfactory or complete description of the internalization pathway applicable to all types of CPPs. Moreover, none of these models adequately predicts the cellular uptake of large cargoes (up to 100-fold their own size) mediated by these CPPs.
Endocytosis
Endocytosis, including phagocytosis and pinocytosis,57, 58 is a highly regulated process of internalization of solutes and fluids in the extracellular matrix. Phagocytosis is a complex process used to uptake large particles and pinocytosis is mainly used to bring in smaller particles. This pinocytosis can be further classified into four different pathways: 1) clathrin-mediated, 2) caveolae/lipid raft-mediated, 3) clathrin and caveolae-independent endocytosis and 4) macropinocytosis.59–61
According to most of the initial mechanistic studies, the cellular uptake of CPPs was considered to follow a non-endocytic pathway. Later studies, however, showed that experimental artifacts could affect the study results.59 For instance, the fixation of cells with methanol/formaldehyde could allow the redistribution of the CPPs bound on the cell surface, but not internalized. With further studies, there are now some agreements on the involvement of various types of endocytosis during the cell entry of CPPs or CPP-cargoes.40, 62
The endocytosis pathway for a particular CPP seems to strongly depend on its properties as well as that of the conjugated cargoes (type, size, charge or/and hydrophobicity). For example, TATp has been shown to enter cells via lipid raft-mediated endocytosis when conjugated to a protein, but through a clathrin-dependent endocytosis when the cargo was a fluorophore. When conjugated to large cargoes (MW >30 kDa), the CPP-cargoes entered cells via macropinocytosis. Macropinocytosis is an actin-dependent form of endocytosis that can occur in all cell lines at different rates.60, 61, 63, 64 It is distinguished from other types of endocytic pathways by the membrane ruffling event and the size of the vesicles formed.
APPLICATION OF CPPS AS DELIVERY VEHICLES FOR CANCER THERAPY
CPP-based drug delivery has been explored to treat various diseases, including neuronal disease, asthma, ischemia, diabetes, and cancer.22, 65–67 There are currently more than 300 published studies related to CPP-based drug delivery.15 The most principal application among all studies has been the cancer. As an efficient carrier, the CPP has been shown to successfully deliver cytotoxic drugs (e.g., small anticancer drugs, toxins, tumor suppressor proteins, siRNA, etc) into tumor cells to induce apoptosis.11, 20, 23, 68 Despite the success reported through in vitro studies, only a few studies have shown treatment efficacy in animal models. Here, we cover a few of the in vivo works conducted using CPPs to treat cancers. Additionally, recent progress in CPP-coupled intracellular delivery of nanoparticles is briefly mentioned.
CPP-coupled delivery of small molecule drugs
While small molecule anticancer drugs generally diffuse into tumor cells with great efficiency due to their small size and lipophilicity, there is a frequent occurrence of multidrug resistance (MDR) after repeated exposure of tumor cells to the same drugs.69 To overcome this problem, studies have explored the coupling of these drugs with CPPs. For an example, Dubikovskaya et al. conjugated octaarginine (R8) to taxol, an anticancer drug, via a disulfide bond, which displayed the effect of preferentially releasing taxol inside the cells.70 Both R8-taxol and taxol alone were tested against ovarian cancer models with OVCA-429 (taxol-sensitive) or OVCA-429T (taxol-resistant) cells implanted in the peritoneal cavity of mice. According to their results, R8-taxol and taxol had similar effects against taxol-sensitive tumors. However, when tested against a taxol-resistant tumor model, R8-taxol treated mice had significantly higher survival rates compared with mice treated with taxol alone. Interestingly, this study showed several more merits of coupling small molecule drugs with CPP, aside from the potential to overcome the MDR. One was the increased aqueous solubility of the drug, enabling use of the drug without adding surfactants (e.g., cremophore EL). The investigators also observed a high drug concentration near the injection site due to the increased adherence of CPP-drug to the surrounding tissues. Although this property might not be favorable for systemic administration of the CPP-drug, it can improve therapeutic efficacy of the drug in case of direct intra-tumor injection.
For enhanced cytotoxicity and targeted delivery of the anticancer drug, Lee et al. synthesized a chitosan/doxorubicin/TAT hybrid by chemically conjugating doxorubicin and TAT to the polymeric chitosan backbone. Compared with free doxorubicin or chitosan/doxorubicin without TAT, the chitosan/doxorubicin/TAT hybrid displayed more efficient cell internalization and, thereby, yielded significantly higher anti-tumor effects. This enhanced cell internalization of the chitosan/doxorubicin/TAT hybrid could further alter the biodistribution profiles of the doxorubicin, resulting in augmented tumor localization and eventually leading to significant inhibition of tumor growth in CT26 xenograft bearing mice.71 In addition, Myrberg et al. designed a chimeric peptide by coupling a breast tumor homing peptide (so-called ‘PEGA peptide’; 9-mer cyclic peptide: cCPGPEGAGC) with a CPP (pVEC) and then chemically conjugated this peptide to an anticancer drug, chlorambucil.72 In vitro studies showed higher cellular uptake and cytotoxicity level of PEGA-pVEC-chlorambucil conjugate against breast cancer cells, than chlorambucil alone. Further in vivo studies proved that fluorescence dye-labeled PEGA-pVEC could target the vessels of breast tumors with lower accumulation in non-tumor tissues, compared with pVEC alone. This study exemplifies a rational design for targeted delivery of CPP-coupled drugs. However, caution must be taken when applying targeting ligands with a CPP, as previous studies showed that CPP-mediated uptake was so efficient that it could abolish targeting specificity of the conjugated antibodies.73 In addition, Shokolenko et al. reported that the order of the targeting ligands and CPPs could affect the targeting efficiency of chimeric peptides.74
Not only CPP modification could enhance cellular internalization, but remarkably it could also augment the delivery of cargoes across the blood brain barrier (BBB) without compromising its integrity. Rousselle et al. showed that CPP (SynB1 or D-penetratin) modification enabled doxorubicin to bypass the P-glycoprotein-mediated efflux from the BBB, thereby increasing the transport of doxorubicin into brain parenchyma by approximately 20-fold. Interestingly, CPP modification also altered the tissue distribution profile of doxorubicin. Notably, compared with free doxorubicin, significantly lower (10-fold lower) concentration of CPP-modified doxorubicin was observed in the heart, which could potentially reduce the cardiotoxicity concerns.75
CPP-coupled delivery of therapeutic peptides or proteins
Apoptosis is induced by various cellular stresses and this process is regulated by certain tumor suppressor proteins, such as p53 or p16. In many human cancers, the tumor suppressor proteins are altered or dysfunctional due to mutations that occur during tumorigenesis.76, 77 Therefore, attempts have been made to treat cancer by restoring functions of these proteins by means of delivering the full length proteins or peptides that correspond to the crucial residues of these proteins.
Snyder et al. constructed a retro-inverso analogue of TATp-p53 C-terminus chimeric peptide.78 Tested against a terminal peritoneal carcinomatosis mouse model by intraperitoneal (i.p.) injection, the treatment group (mean survival time: >70 days) showed 6-fold increase of lifespan over mutant or vehicle treated mice (mean survival time: 10 days). Also, in a terminal peritoneal lymphoma model, treatment group showed 50% longer in survival time than those of the control groups (mean survival time for vehicle and mutant peptide treated groups: 35 and 33 days, respectively); with half of the mice surviving for more than 200 days. Another successful anti-tumor application was realized by using a chimeric peptide consisting of p53 peptide from its mdm-2-binding domain (residues 17–26; named PNC-28) and penetratin.79 Programmed release of PNC-28 from an osmotic pump implanted under the skin or in the peritoneal cavity effectively blocked the subcutaneously (s.c.) or i.p. implanted pancreatic tumor growth in vivo.
In an attempt to restore p16 activity, Hosotani et al. prepared ‘Trojan p16’ peptide which was a disulfide bridged conjugate consisting of 20 amino acid residues of the p16 protein and penetratin, and then tested the efficacy against a pancreatic tumor animal model.80 In both s.c. or i.p. implanted tumors, the Trojan p16-treated groups exhibited a significant inhibition of tumor growth (treatment group: 79 ± 17 mg vs. control vehicle only: 139 ± 15 mg vs. p16 only: 151 ± 15 mg vs. Trojan peptide only: 149 ± 12 mg) and an improved survival rate (mean survival time benefit of 6 days for treatment group) over the controls.
Fulda et al. attempted a different approach to induce apoptosis of tumor cells.81 These investigators prepared a chimeric peptide consisting of N-terminal 7 amino acid residues of second mitochondria-derived activator of caspase (SMAC) and TATp. SMACs are mitochondrial proteins which play important roles in mitochondrial regulation of apoptosis.82 Induced by apoptotic signals, SMACs are released from the mitochondria and deactivate the inhibitor of apoptosis proteins, allowing the apoptosis to proceed. In vitro studies showed that SMAC-TATp by itself was not effective enough to kill tumor cells, but could sensitize these cells to apoptotic stimuli, such as TNF-related apoptosis inducing ligand (TRAIL), CD95 or doxorubicin. In an intracranial U87 human glioma xenograft mouse model, delivery of a combination of SMAC-TATp and TRAIL significantly inhibited tumor growth, and complete eradication of the tumor was observed when 0.6 or 2 μg of TRAIL was administered with the SMAC-TATp.
A rather direct approach being explored to treat cancer is to deliver highly toxic enzyme drugs into tumor cells. Due to their repetitive mode of reaction, enzyme drugs offered an unmatched therapeutic potency over conventional, small cytotoxic drugs, rendering them an attractive candidate for cancer drug therapy. Gelonin, a plant origin toxin which belongs to ribosome inactivating proteins (RIPs), is highly potent in inhibiting protein translation with IC50 concentrations being at pico-molar levels.83 In spite of this exceptional potency, however, the anti-cancer activity of gelonin is rather negligible because it cannot efficiently internalize tumor cells. To overcome this membrane barrier, Park et al. prepared a chemical conjugate of gelonin with either TATp or LMWP.20 In cell culture studies, both the TATp- and LMWP-gelonin conjugates displayed strong anti-tumor activity when being tested against a mouse model harboring s.c. murine CT26 colorectal cancer cells. A complete inhibition of tumor growth was observed when the animals were injected with a total dose of 100 μg of either CPP-gelonin conjugates.
Apart from gelonin, caspase-3, asparaginase and a proapoptotic peptide (KLA; peptide sequence: KLAKLAKKLAKLAK) have also been conjugated with CPPs and studied in vivo. In the respective rat ascite model, acute lymphoblastic leukemia mouse model and lung carcinoma mouse s.c. model, CPP-caspase-3, CPP-asparaginase and CPP-KLA conjugates all yielded significantly improved survival rates over those of the control counterparts.29, 84, 85
CPP-coupled delivery of genes
Delivery of genes, antisense oligodeoxynucleotides (ODNs), or small interference RNA (siRNAs) could be a powerful method for cancer therapy.86–88 Indeed, RNA interference (RNAi) has emerged as a particularly attractive therapeutic strategy, due to its great specificity against the target gene and unmatched tumor-inhibiting efficacy. However, there remain many obstacles for the delivery of naked siRNA in vivo. A major hurdle of RNAi treatment lies in the poor cellular permeability of siRNA drugs, due to their strong charges and relatively large sizes. Various approaches have been attempted to overcome this obstacle, such as utilizing cationic liposomes, cationic polymers, or CPPs. Among these carriers, CPPs have drawn great interest primarily because of their demonstrated low toxicity under both in vitro and in vivo conditions.
The two major methods used to attach siRNA to CPP are via covalent conjugation or non-covalent complex formation.15 Although covalent conjugation may be the most ideal approach, it is technically difficult to conjugate and purify siRNA-CPP because of their strong charge-charge interactions – often leading to the formation of tightly bound complexes and aggregates. Cellular uptake of these ionic siRNA-CPP aggregates is much poorer comparing to that of the 1:1 siRNA-CPP covalent conjugates, because the exceeding negative charges of siRNA could easily neutralize the cationic amino acids of the CPP, rendering both siRNA and CPP to partially lose their biological functions. Despite such disadvantages, non-covalent, ionic complexation of siRNA with CPP has still been preferably employed in most of the published studies, primarily due to its simplicity. While most of the studies regarding siRNA-CPP have focused on proving the feasibility under in vitro conditions, the number of reports demonstrating success in animal models is continuously growing.
Vascular endothelial growth factor (VEGF) plays a major role in angiogenesis which is essential for the growth of tumors. Blocking the VEGF receptor (e.g., with an antibody targeted against the VEGF receptor – Bevacizumab)89 or silencing the VEGF gene are effective ways to treat cancers.90 Kim et al. used cholesterol-R9 conjugate as a carrier and prepared a complex with siRNA against VEGF (siVEGF).91 The complex was able to silence the VEGF in cellular studies and when tested in an s.c. CT26 murine colon cancer xenograft model, the cholesterol-R9/siVEGF complex yielded significant inhibition in the growth and vascularization of the tumor compared with the complex formulated with scrambled siRNA. Another successful application of siVEGF/CPP complex was performed by Choi et al., who prepared the complex and tested the anti-cancer activity in an s.c. SK-HEP1-LUC xenograft mice model.27 The siVEGF/LMWP complex significantly reduced the tumor size, which was already relatively large (700–750 mm3) when the treatment was started.
Cyclin B, a mitotic cyclin involved in mitosis by forming a complex with cyclin-dependent kinase, has also been of interest for RNAi treatment.92 Essential for cell division, cyclin B is often deregulated in tumors. Crombez et al. have prepared a siRNA against cyclin B1 in complex with a truncated version of MPG (named ‘MPG-8).93 The siRNA/MPG-8 complex was able to down-regulate cyclin B1 levels and induce arrest of the cell cycle, leading to inhibition of the proliferation of various human cancer cell lines. Furthermore, in an s.c. xenograft tumor model, significant inhibition of tumor growth was observed after intra-tumor or intravenous (i.v.) injection of the siRNA/MPG-8 complex.
Another attractive target for RNAi-based treatment is the epidermal growth factor receptor (EGFR) which is frequently altered in various tumors, including high-grade gliomas.94 Han et al. tested a complex of plasmid based EGFR siRNA (psiEGFR) and TATp-polyamidoamine dendrimer conjugated bacterial magnetic carrier against U251 glioma xenograft model via intra-tumor injection.95 The treatment group showed significant inhibition of tumor growth, which was equivalent to positive control group (lipofectamine 2000/psiEGFR). Michiue et al. devised a fusion peptide composing of a TATp and a double stranded RNA-binding domain (referred to as PTD-DRBD) that strongly binds to any double stranded siRNA. This versatile peptide was utilized as a carrier for siRNAs of EGFR and Akt2. In an intracerebral glioblastoma mouse model, these PTD-DRBD/siRNA complexes induced significant tumoricidal activity and led to increase in longevity.96 Kim et al. prepared a complex of siRNA against Human Epidermal Growth Factor Receptor 2 (HER2) and oligoarginine (R15) and tested the effect in a SK-COV3 ovarian xenograft tumor model via intra-tumor injection.95 Compared with the scrambled siRNA/R15 complex or naked siRNA, the siHER2/R15 complex displayed significant reduction of tumor growth.
CPP-coupled delivery of nanoparticles
Although the translocation mechanisms of CPPs/CPP-cargoes remain highly elusive, these peptides have been widely and successfully exploited for enhanced intracellular delivery of a variety of nanoparticles, such as magnetic nanoparticles,56, 97–101 lipid-based formulations,33, 102–107 micelles,108 gold nanoparticles,109, 110 quantum dots,111 and others.112–116 A number of such applications of CPP-mediated intracellular delivery of nanoparticles have been summarized in Table 1.
Table 1.
Summary of CPP modified nanoparticles and their cellular applications.
| Nanoparticles | CPPs | Cell lines and/or Effects | Refs |
|---|---|---|---|
| Liposomes | TATp, Penetratin, Octa-arginine | Calu-3 12- to 17-fold enhanced cell uptake of liposomes |
102 |
| TATp | COS-7 | 103 | |
| Poly-arginine | NCI-H446, A549, SK-MES-1 Enhanced siRNA intracellular delivery and tumor cell growth inhibition |
104 | |
| TAT, Penetratin | SK-BR-3, MCF-7, HTB 9, ADR, A431,C26 12-fold increase of Dox intracellular uptake |
33 | |
| TATp | LLC, BT20, H9C2 Enhanced cell uptake of liposomes |
107 | |
| CLIO (MION) | TATp | Mouse lymphocytes, human natural killer, HeLa, human hematopoietic CD34+, mouse neural progenitor C17.2, human lymphocytes CD4+, T-cells, B-cells, macrophages, stem cells MRI imaging and magnetic separation of homed cells |
56, 97–99 |
| pH sensitive PEG-polylactic acid micelles | TATp | MCF-7 Enhanced cytotoxicity at acidic environment (pH < 7) |
108 |
| Gold nanoparticles | TATp Other NLS peptides |
NIH3T3, HepG2, HeLa, hTERT-BJ1 Enhancedcytosolic and nuclear uptake of particles |
109,110 |
| Quantum dot-loaded micelles | TATp | MS1, lineage-negative bone marrow cells Dynamic imaging and tracking of labeled cells |
111 |
| PEI-PEG-TAT/DNA nanoplexes | TATp | SH-SY5Y Enhanced gene expression |
112 |
| TATp | A549 Enhanced gene transfection of lung cells |
113 | |
| Boron carbide nanoparticles | TATp | EL4, B16F10 Boron neutron capture therapy |
114 |
| Dendrimers | TATp, Penetratin | MDR3T3, MES-SA/Dx5 Enhanced ODN intracellular delivery |
115 |
| Solid lipid nanoparticles | DimericTATp | 16HBE14o- Enhanced gene transfection |
116 |
In 1999, Josephson et al. first reported the conjugation of CPPs to nanoparticles.97 In this study, TATp was conjugated to CLIOs (cross-linked iron oxide particles). The TATp-CLIO conjugate yielded efficient labeling of cells, offering the potential to function as a tool for MRI or in magnetic separation of homed cells in vivo. In all cell lines tested, the intracellular delivery of TATp-CLIO, with average size of 41 nm and TATp moieties of 6.7/particle, was about 100-fold higher than that of the control group (non-TATp conjugated CLIOs). Later on, the effect of TATp density on the particle surface and the sensitivity of MRI detection were investigated in mouse lymphocytes using different TATp/CLIO ratios.100 A non-linear increase profile in cellular uptake of TATp–CLIO was observed with increasing TATp/CLIO ratio. Compared with the control, a maximum of about 100-fold increase in cell uptake was achieved using 15 TATps/CLIO particles. Because of the advancement in intracellular uptake by the TATp moieties, cells could be monitored at 100-fold lower concentrations using a higher ratio of the TATp/CLIO conjugates.
Another successful study regarding to cell labeling by CPP modified nanoparticles was reported by Stroh et al. These investigators prepared TATp-modified micelles constructed with polyethylene glycol-phosphatidyl ethanolamine (PEG-PE) containing entrapped quantum dots.111 By incubation of these TATp-modified micelles with primary bone marrow lineage-negative cells, the cells could be successfully labeled ex vivo. When those quantum dot-labeled cells were injected through the carotid artery of MCaIV tumor bearing mice, their recruitment by the tumor vasculature could be clearly tracked via imaging of the quantum dots.
Modification of liposomes with CPPs such as TATp or penetratin was also proven successful in enhancing intracellular delivery over a variety of cell lines (rat cardiac myocyte H9C2 cells, murine Lewis lung carcinoma cells and human breast tumour BT20 cells).117 Direct interaction between CPPs and cell surface appeared to be essential for the cell entry, as shielding of CPPs severely inhibited the uptake. Results showed that approximately five CPPs per particle were sufficient to enhance the intracellular delivery of liposomes. Nevertheless, the efficiency in cell uptake of liposomes was proportional to the density of the conjugated CPPs. Interestingly, the kinetics of the uptake was dependent upon the type of the CPP and cell lines. Compared with the TATp–liposomes which exhibited relatively slow cell internalization, the penetratin-liposomes internalized into cells very rapidly and reached the maximum uptake within 1 hr.
An attractive application of CPP-modified nanoparticles lies in their use as a transfection agent. Combining the high binding capacity of the nanoparticles and great translocation ability of CPPs, highly efficient gene transfection is often observed. Torchilin et al. prepared a non-covalent complex between TATp-liposomes and DNA.105 Compared with the commonly used Lipotectin®, the TATp-liposome/DNA complex showed a significantly higher transfection and lower cytotoxicity in vitro, when incubated with mouse fibroblast NIH 3T3 cells and cardiac myocytes H9C2 cells. These complexes also displayed an efficient transfection by means of intra-tumor injection. A multifunctional envelope-type nanodevice (MEND), consisting of a condensed DNA core and a surrounding lipid envelope, was also synthesized for tumor gene delivery.118 The MEND containing octa-arginine on the envelope exhibited a 1000-fold higher transfection activity than the control DNA/(poly-L-lysine/lipid) complex.
Encouraged by the successes in CPPs for intracellular delivery of various nanoparticles in vitro, there has been rising interest in the use of CPP-conjugated nanoparticles for improved delivery of anti-cancer drugs in vivo. By combined effects of tumor targeting via enhanced permeability and retention (EPR) effect and CPP-mediated cell penetration, these nanoparticles can provide effective means for safer delivery of potentially toxic anti-cancer drugs with greater therapeutic efficacy. Balzeau et al. synthesized a lipid nanocapsule functionalized with a glioblastoma-specific CPP (NFL-TBS.40-63). When paclitaxel was loaded to this CPP-modified lipid nanocapsule and administered to GL261 glioma brain tumor bearing mice,119 it showed selective and efficient penetration into glioblastoma cells and yielded the best therapeutic effect among all the tested groups in inhibiting tumor growth (75%). Jiang et al. devised a dual-functional liposome consisting of pH-responsive CPP (R6H4) and exterior hyaluronic acid coating (readily degradable by hyaluronidase rich in certain tumor milieu).120 The negatively charged hyaluronic acid coating of the liposome was intended to mask the positive charge of CPPs during targeting and selectively expose the CPPs to tumor cells. Paclitaxel-loaded dual-functional liposome yielded significant tumor size reduction in Heps xenograft tumor mouse model, which excelled the therapeutic effect by clinically used Taxol®. In addition, a CPP (F3 peptide)-functionalized cisplatin-hydrogel nanoparticle (named “F3-Cis-Np”) was developed and evaluated for ovarian cancer treatment in both solid and i.p. tumor models.121 The results showed that F3-Cis-Np sufficiently and selectively bound to the tumor vessels and induced significant tumor regression via anti-angiogenesis.
SMART STRATEGIES FOR TUMOR-LOCALIZED DELIVERY OF CPP-COUPLED CARGOES
Even though CPPs themselves were not reported to produce toxicity below the level of hundreds of micro-molar concentration, nonspecific cellular internalization of CPP-coupled cargoes might result in unexpected toxicity caused by the cargoes. In an in vivo study, Schwarze et al. reported that TATp-β-galactosidase fusion protein could be found in various organs all over the body, including the brain.122 The general lack of the target specificity of CPPs demands that special considerations must be taken when delivering drugs with low tissue selectivity. Although the mechanism for CPP mediated cellular internalization is yet unclear, previous studies have shown that the initiation of the cell entry requires the interaction of CPPs with the cell surface glycosaminoglycans that are negatively charged. Based on this property, a key motif for achieving selective delivery of the CPP-drugs would be to utilize a “smart” strategy that could block the CPP activities during circulation until it reaches the target site, and then reverse the block at the target to expose the CPPs so that it could exert the cell penetrating activity locally.
To date, only a few DDS using the above strategy for targeted delivery of CPP-coupled cargoes have been established and tested in animal models. The blocking of CPPs was usually achieved by either directly forming complexes between cationic CPPs and anionic counterparts or by physically shielding CPPs with PEG chains. Different methods for reversing the block have been designed, including utilizing the tumor microenvironment or applying an external triggering agent.
ACPP (Activatable CPP) prodrug strategy
ACPP is a construct developed by Dr. Tsien and coworkers for selective delivery of CPPs.123–125 The scheme of the construct is depicted in Fig. 2. The construct is composed of a polycationic CPP (r9; nine D-form arginine residues) fused with an polyanionic counterpart (e8; eight D-form glutamate residues) via a peptide linker cleavable only by certain specific proteases (MMP-2 or 9) present in the tumors. While the linker is intact, r9 and e8 form an intra-molecular hairpin structure that neutralizes the CPP activity of r9. However, once the linker is cleaved in the presence of MMP-2 or 9, the r9 is dissociated from e8 due to a weakened interaction. Enhanced cellular uptake of ACPP was obvious in 2D and 3D cell culture models and even in human specimens, after cleavage of the linker by MMP-2 which was added externally or secreted from HT1080. In an in vivo study, investigators found that r9 could induce severe systemic toxicity with doses above 5 μmol/kg following i.v. injection, which eventually led to death of mice due to respiratory failure. However, injection of ACPP produced a much milder toxicity even with 4-fold of the tolerable dose.
FIGURE 2.

Schematic illustration of ACPPs. CPP is blocked by a polyanionic peptide covalently attached via a MMP2/9-cleavable linker. Cleavage of the linker induces exposure of CPP and enables cell internalization. Reproduced with permission from Ref. 123.
CPP-modified ATTEMPTS (Antibody targeted triggered electrically modified prodrug type strategy)
The ATTEMPTS is a drug delivery strategy utilizing active targeting and prodrug feature for selective delivery of macromolecules to the disease site.126 The scheme of the DDS is illustrated in Fig. 3. The underlying principle of this DDS is by forming a plasma-stable tight complex between the targeting component, the antibody-heparin conjugate, and the drug component, a CPP-coupled drug. The complex is formed by electrostatic interaction between the anionic heparin and the cationic CPP. With the complex formation, the charge of the CPP is reversibly masked by heparin, leading to an improved antibody targeting as well as plasma stability of CPP against endogenous proteases. The drug delivery strategy works in two steps. In the first step, the DDS is administered and allowed to accumulate in the tumor site via antibody-mediated targeting. In the second step, protamine is systemically injected as a triggering agent to reverse heparin inhibition on the CPP, when the tumor to non-tumor concentration ratio of the DDS is optimal. Protamine is a small cationic protein that has been used clinically as the antidote for heparin. Due to the stronger binding between heparin and protamine compared to that between heparin and the CPP, protamine triggers the release of the CPP-drug from the DDS, and the CPP-drug is now free to penetrate through the tumor cell membrane. The feasibility of the DDS was demonstrated in an in vitro study by Kwon et al.126 Based on the confocal microscopy and FACS results, cell uptake of LMWP-asparaginase conjugate was shown to be effectively blocked by heparin, and this blockage was successfully reversed by the addition of protamine. Further studies are currently ongoing in Yang’s lab by utilizing an anti-CEA antibody for targeting colorectal cancer and TATp modified-gelonin.
FIGURE 3.

Schematic diagram of the CPP-modified ATTEMPTS delivery system. Reproduced with permission from Ref. 126.
Reversal shielding by pH-sensitive PEG
A modified Doxil®-based nanocarrier (doxorubicin-loaded PEGylated liposome) was reported recently.127 This strategy was by incorporating a 2C5 monoclonal antibody with long-protective PEG chains as well as short PEG chains linked with TATp; both were linked via the pH-responsive hydrazone bond. Compared with the unmodified, commercial Doxil®, this multifunctional liposomal formulation significantly augmented the cytotoxicity and intracellular uptake of the drug, when being examined against four different cancer cell lines under the pre-exposure to low pH conditions. Other studies of multifunctional TATp-coupled liposomes and micelles by utilizing similar strategies were also reported elsewhere.128 At pH 7.5–8.0, these liposomes or micelles displayed very limited internalization by either NIH-3T3 or U-87 cells. However, in contrast, effective internalization into both NIH-3T3 and U-87 cells was achieved after brief incubation (15–30 min) at a low pH (pH 5.0–6.0) under which the nanocarriers shed their PEG shielding by hydrolysis of the hydrazone bonds.
TATp-modified and GFP-loaded liposomes were also prepared and studied in vivo by means of intra-tumor injection.129 The administration of liposomes without pH-degradable PEG coating resulted in very limited transfection of tumor cells, simply because of the TATp moieties were being shielded by the PEG chains. As a comparison, the administration of liposomes with pH-sensitive PEG showed a much higher transfection of tumor cells. Removal of the PEG shield by the acidic tumor environment led to the exposure of TATp on the liposome surface and, as a consequence, yielded much enhanced cellular uptake of the model gene, pEGFP-N1. The scheme of this approach is illustrated in Fig. 4.
FIGURE 4.
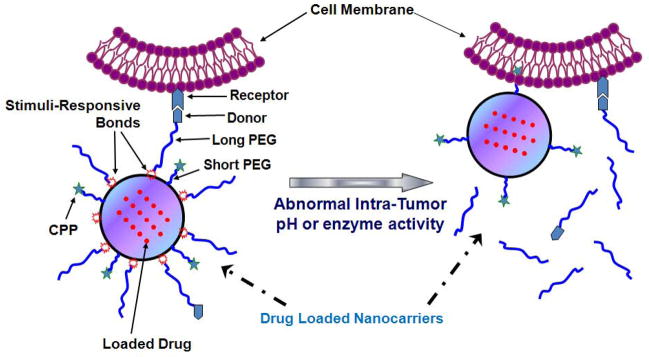
Schematic illustration of tumor-environment-responsive exposure of CPPs on ‘smart’ nanocarriers. Reproduced with permission from Ref. 13.
Tumor specific protease cleavable PEG shielding
In the tumor microenvironment, the levels of certain enzymes (so-called tumor specific proteases) such as MMP-2 or 9 are often elevated in the extracellular space of the tumors. This up-regulated enzyme activity provides a unique strategy to expose CPP molecules conjugated on the surface of nanoparticles selectively to tumor cells, by masking the nanoparticles with PEG chains that are cleavable by these tumor specific proteases. Harris et al. prepared a magnetic nanoparticle conjugated with PEG chains via MMP-2 cleavable peptide linkers and polyarginine peptides (NH2-RRRRGRRRRK(FITC)GC).101 When the veiled polyarginines under the PEG chains were exposed by cleavage of the PEG chains via MMP-2, 40-fold higher cell accumulation of nanoparticles were observed in vitro. The in vivo feasibility of this system was further confirmed by MRI and fluorescence imaging. The scheme of this approach is also depicted in Fig. 4.
CONCLUSION
CPPs have been exploited in a wide variety of theranostic applications, including the intracellular delivery of small anticancer drugs, macromolecules, and nanoparticle systems. Although the mechanism of CPP translocation remains unclear, new CPPs including modification of existing CPPs are being designed and tested, which will no doubt extend the therapeutic applications of CPPs. The potential of CPPs to deliver highly potent and highly specific macromolecular drugs is of particular interest. The ability of CPPs to enter cells of almost any type, however, still possesses significant toxicity concerns that must be appropriately addressed. To overcome such toxicity issues, different types of “smart” strategies have been designed to activate the CPPs only within diseased tissues. Although it may be too early to predict clinical applications of CPP-cargoes, the current achievements in the CPP-mediated delivery of drugs in vivo are inspiring, and the future of CPP-based effective and safe chemotherapy indeed looks very promising.
Acknowledgments
This work was supported in part by NIH R01 Grant CA114612 & NS066945. In addition, this work was partially sponsored by National Key Basic Research Program of China 973 Grant 2013CB932502, as well as Tianjin Municipal Science and Technology Commission Grant 12JCZDJC34000. Moreover, this research was also supported in part by Grant R31-2008-000-10103-01 from the World Class University (WCU) project of the MEST and NRF of South Korea.
References
- 1.Roach P, Eglin D, Rohde K, Perry CC. Modern biomaterials: a review - bulk properties and implications of surface modifications. J Mater Sci Mater Med. 2007;18:1263–77. doi: 10.1007/s10856-006-0064-3. [DOI] [PubMed] [Google Scholar]
- 2.Ratner BD, Bryant SJ. Biomaterials: where we have been and where we are going. Annu Rev Biomed Eng. 2004;6:41–75. doi: 10.1146/annurev.bioeng.6.040803.140027. [DOI] [PubMed] [Google Scholar]
- 3.Pridgen EM, Langer R, Farokhzad OC. Biodegradable, polymeric nanoparticle delivery systems for cancer therapy. Nanomedicine (Lond) 2007;2:669–80. doi: 10.2217/17435889.2.5.669. [DOI] [PubMed] [Google Scholar]
- 4.Hutmacher DW. Biomaterials offer cancer research the third dimension. Nat Mater. 2010;9:90–3. doi: 10.1038/nmat2619. [DOI] [PubMed] [Google Scholar]
- 5.Chopra S, Mahdi S, Kaur J, Iqbal Z, Talegaonkar S, Ahmad FJ. Advances and potential applications of chitosan derivatives as mucoadhesive biomaterials in modern drug delivery. J Pharm Pharmacol. 2006;58:1021–32. doi: 10.1211/jpp.58.8.0002. [DOI] [PubMed] [Google Scholar]
- 6.Snyder EL, Dowdy SF. Cell penetrating peptides in drug delivery. Pharm Res. 2004;21:389–93. doi: 10.1023/B:PHAM.0000019289.61978.f5. [DOI] [PubMed] [Google Scholar]
- 7.Hajdu SI. A note from history: landmarks in history of cancer, part 1. Cancer. 2011;117:1097–102. doi: 10.1002/cncr.25553. [DOI] [PubMed] [Google Scholar]
- 8.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–93. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 9.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci U S A. 1994;91:664–8. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–50. [PubMed] [Google Scholar]
- 11.Meade BR, Dowdy SF. Exogenous siRNA delivery using peptide transduction domains/cell penetrating peptides. Adv Drug Deliv Rev. 2007;59:134–40. doi: 10.1016/j.addr.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Mae M, Langel U. Cell-penetrating peptides as vectors for peptide, protein and oligonucleotide delivery. Curr Opin Pharmacol. 2006;6:509–14. doi: 10.1016/j.coph.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev. 2008;60:548–58. doi: 10.1016/j.addr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 14.Schwarze SR, Dowdy SF. In vivo protein transduction: intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol Sci. 2000;21:45–8. doi: 10.1016/s0165-6147(99)01429-7. [DOI] [PubMed] [Google Scholar]
- 15.Heitz F, Morris MC, Divita G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol. 2009;157:195–206. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindgren M, Hallbrink M, Prochiantz A, Langel U. Cell-penetrating peptides. Trends Pharmacol Sci. 2000;21:99–103. doi: 10.1016/s0165-6147(00)01447-4. [DOI] [PubMed] [Google Scholar]
- 17.Patel LN, Zaro JL, Shen WC. Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm Res. 2007;24:1977–92. doi: 10.1007/s11095-007-9303-7. [DOI] [PubMed] [Google Scholar]
- 18.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–7. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 19.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A. 2000;97:13003–8. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park YJ, Chang LC, Liang JF, Moon C, Chung CP, Yang VC. Nontoxic membrane translocation peptide from protamine, low molecular weight protamine (LMWP), for enhanced intracellular protein delivery: in vitro and in vivo study. FASEB J. 2005;19:1555–7. doi: 10.1096/fj.04-2322fje. [DOI] [PubMed] [Google Scholar]
- 21.Elmquist A, Lindgren M, Bartfai T, Langel U. VE-cadherin-derived cell-penetrating peptide, pVEC, with carrier functions. Exp Cell Res. 2001;269:237–44. doi: 10.1006/excr.2001.5316. [DOI] [PubMed] [Google Scholar]
- 22.Ezzat K, El Andaloussi S, Abdo R, Langel U. Peptide-based matrices as drug delivery vehicles. Curr Pharm Des. 2010;16:1167–78. doi: 10.2174/138161210790963832. [DOI] [PubMed] [Google Scholar]
- 23.Jarver P, Mager I, Langel U. In vivo biodistribution and efficacy of peptide mediated delivery. Trends Pharmacol Sci. 2010;31:528–35. doi: 10.1016/j.tips.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 24.Calnan BJ, Tidor B, Biancalana S, Hudson D, Frankel AD. Arginine-mediated RNA recognition: the arginine fork. Science. 1991;252:1167–71. doi: 10.1126/science.252.5009.1167. [DOI] [PubMed] [Google Scholar]
- 25.Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–40. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 26.Park YJ, Liang JF, Ko KS, Kim SW, Yang VC. Low molecular weight protamine as an efficient and nontoxic gene carrier: in vitro study. J Gene Med. 2003;5:700–11. doi: 10.1002/jgm.402. [DOI] [PubMed] [Google Scholar]
- 27.Choi YS, Lee JY, Suh JS, Kwon YM, Lee SJ, Chung JK, Lee DS, Yang VC, Chung CP, Park YJ. The systemic delivery of siRNAs by a cell penetrating peptide, low molecular weight protamine. Biomaterials. 2010;31:1429–43. doi: 10.1016/j.biomaterials.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 28.Moon C, Kwon YM, Lee WK, Park YJ, Yang VC. In vitro assessment of a novel polyrotaxane-based drug delivery system integrated with a cell-penetrating peptide. J Control Release. 2007;124:43–50. doi: 10.1016/j.jconrel.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwon YM, Chung HS, Moon C, Yockman J, Park YJ, Gitlin SD, David AE, Yang VC. L-Asparaginase encapsulated intact erythrocytes for treatment of acute lymphoblastic leukemia (ALL) J Control Release. 2009;139:182–9. doi: 10.1016/j.jconrel.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Roux I, Joliot AH, Bloch-Gallego E, Prochiantz A, Volovitch M. Neurotrophic activity of the Antennapedia homeodomain depends on its specific DNA-binding properties. Proc Natl Acad Sci U S A. 1993;90:9120–4. doi: 10.1073/pnas.90.19.9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christiaens B, Grooten J, Reusens M, Joliot A, Goethals M, Vandekerckhove J, Prochiantz A, Rosseneu M. Membrane interaction and cellular internalization of penetratin peptides. Eur J Biochem. 2004;271:1187–97. doi: 10.1111/j.1432-1033.2004.04022.x. [DOI] [PubMed] [Google Scholar]
- 32.Joliot A, Prochiantz A. Transduction peptides: from technology to physiology. Nat Cell Biol. 2004;6:189–96. doi: 10.1038/ncb0304-189. [DOI] [PubMed] [Google Scholar]
- 33.Tseng YL, Liu JJ, Hong RL. Translocation of liposomes into cancer cells by cell-penetrating peptides penetratin and tat: a kinetic and efficacy study. Mol Pharmacol. 2002;62:864–72. doi: 10.1124/mol.62.4.864. [DOI] [PubMed] [Google Scholar]
- 34.Dupont E, Prochiantz A, Joliot A. Penetratin story: an overview. Methods Mol Biol. 2011;683:21–9. doi: 10.1007/978-1-60761-919-2_2. [DOI] [PubMed] [Google Scholar]
- 35.Nan YH, Park IS, Hahm KS, Shin SY. Antimicrobial activity, bactericidal mechanism and LPS-neutralizing activity of the cell-penetrating peptide pVEC and its analogs. J Pept Sci. 2011;17:812–7. doi: 10.1002/psc.1408. [DOI] [PubMed] [Google Scholar]
- 36.Pooga M, Kut C, Kihlmark M, Hallbrink M, Fernaeus S, Raid R, Land T, Hallberg E, Bartfai T, Langel U. Cellular translocation of proteins by transportan. FASEB J. 2001;15:1451–3. doi: 10.1096/fj.00-0780fje. [DOI] [PubMed] [Google Scholar]
- 37.Morris MC, Vidal P, Chaloin L, Heitz F, Divita G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997;25:2730–6. doi: 10.1093/nar/25.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris MC, Depollier J, Mery J, Heitz F, Divita G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol. 2001;19:1173–6. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]
- 39.Oehlke J, Scheller A, Wiesner B, Krause E, Beyermann M, Klauschenz E, Melzig M, Bienert M. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim Biophys Acta. 1998;1414:127–39. doi: 10.1016/s0005-2736(98)00161-8. [DOI] [PubMed] [Google Scholar]
- 40.Fisher L, Soomets U, Cortes Toro V, Chilton L, Jiang Y, Langel U, Iverfeldt K. Cellular delivery of a double-stranded oligonucleotide NFkappaB decoy by hybridization to complementary PNA linked to a cell-penetrating peptide. Gene Ther. 2004;11:1264–72. doi: 10.1038/sj.gt.3302291. [DOI] [PubMed] [Google Scholar]
- 41.El-Andaloussi S, Johansson H, Magnusdottir A, Jarver P, Lundberg P, Langel U. TP10, a delivery vector for decoy oligonucleotides targeting the Myc protein. J Control Release. 2005;110:189–201. doi: 10.1016/j.jconrel.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 42.Scheller A, Wiesner B, Melzig M, Bienert M, Oehlke J. Evidence for an amphipathicity independent cellular uptake of amphipathic cell-penetrating peptides. Eur J Biochem. 2000;267:6043–50. doi: 10.1046/j.1432-1327.2000.01681.x. [DOI] [PubMed] [Google Scholar]
- 43.Oehlke J, Wallukat G, Wolf Y, Ehrlich A, Wiesner B, Berger H, Bienert M. Enhancement of intracellular concentration and biological activity of PNA after conjugation with a cell-penetrating synthetic model peptide. Eur J Biochem. 2004;271:3043–9. doi: 10.1111/j.1432-1033.2004.04236.x. [DOI] [PubMed] [Google Scholar]
- 44.Brugidou J, Legrand C, Mery J, Rabie A. The retro-inverso form of a homeobox-derived short peptide is rapidly internalised by cultured neurones: a new basis for an efficient intracellular delivery system. Biochem Biophys Res Commun. 1995;214:685–93. doi: 10.1006/bbrc.1995.2340. [DOI] [PubMed] [Google Scholar]
- 45.Koppelhus U, Awasthi SK, Zachar V, Holst HU, Ebbesen P, Nielsen PE. Cell-dependent differential cellular uptake of PNA, peptides, and PNA-peptide conjugates. Antisense Nucleic Acid Drug Dev. 2002;12:51–63. doi: 10.1089/108729002760070795. [DOI] [PubMed] [Google Scholar]
- 46.Maiolo JR, Ferrer M, Ottinger EA. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim Biophys Acta. 2005;1712:161–72. doi: 10.1016/j.bbamem.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 47.Tunnemann G, Martin RM, Haupt S, Patsch C, Edenhofer F, Cardoso MC. Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J. 2006;20:1775–84. doi: 10.1096/fj.05-5523com. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X, Wan L, Pooyan S, Su Y, Gardner CR, Leibowitz MJ, Stein S, Sinko PJ. Quantitative assessment of the cell penetrating properties of RI-Tat-9: evidence for a cell type-specific barrier at the plasma membrane of epithelial cells. Mol Pharm. 2004;1:145–55. doi: 10.1021/mp034014y. [DOI] [PubMed] [Google Scholar]
- 49.Prochiantz A. Getting hydrophilic compounds into cells: lessons from homeopeptides. Curr Opin Neurobiol. 1996;6:629–34. doi: 10.1016/s0959-4388(96)80095-x. [DOI] [PubMed] [Google Scholar]
- 50.Prochiantz A. Homeodomain-derived peptides. In and out of the cells. Ann N Y Acad Sci. 1999;886:172–9. doi: 10.1111/j.1749-6632.1999.tb09410.x. [DOI] [PubMed] [Google Scholar]
- 51.Derossi D, Calvet S, Trembleau A, Brunissen A, Chassaing G, Prochiantz A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J Biol Chem. 1996;271:18188–93. doi: 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- 52.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–95. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 53.Pouny Y, Rapaport D, Mor A, Nicolas P, Shai Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry. 1992;31:12416–23. doi: 10.1021/bi00164a017. [DOI] [PubMed] [Google Scholar]
- 54.Matsuzaki K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim Biophys Acta. 1999;1462:1–10. doi: 10.1016/s0005-2736(99)00197-2. [DOI] [PubMed] [Google Scholar]
- 55.Ludtke S, He K, Huang H. Membrane thinning caused by magainin 2. Biochemistry. 1995;34:16764–9. doi: 10.1021/bi00051a026. [DOI] [PubMed] [Google Scholar]
- 56.Lewin M, Carlesso N, Tung CH, Tang XW, Cory D, Scadden DT, Weissleder R. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol. 2000;18:410–4. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- 57.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 58.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 59.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278:585–90. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 60.Khalil IA, Kogure K, Futaki S, Harashima H. High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J Biol Chem. 2006;281:3544–51. doi: 10.1074/jbc.M503202200. [DOI] [PubMed] [Google Scholar]
- 61.Kaplan IM, Wadia JS, Dowdy SF. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Control Release. 2005;102:247–53. doi: 10.1016/j.jconrel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 62.Drin G, Cottin S, Blanc E, Rees AR, Temsamani J. Studies on the internalization mechanism of cationic cell-penetrating peptides. J Biol Chem. 2003;278:31192–201. doi: 10.1074/jbc.M303938200. [DOI] [PubMed] [Google Scholar]
- 63.Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–5. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 64.Fretz M, Jin J, Conibere R, Penning NA, Al-Taei S, Storm G, Futaki S, Takeuchi T, Nakase I, Jones AT. Effects of Na+/H+ exchanger inhibitors on subcellular localisation of endocytic organelles and intracellular dynamics of protein transduction domains HIV-TAT peptide and octaarginine. J Control Release. 2006;116:247–54. doi: 10.1016/j.jconrel.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 65.Dietz GP, Bahr M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci. 2004;27:85–131. doi: 10.1016/j.mcn.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 66.Snyder EL, Dowdy SF. Recent advances in the use of protein transduction domains for the delivery of peptides, proteins and nucleic acids in vivo. Expert Opin Drug Deliv. 2005;2:43–51. doi: 10.1517/17425247.2.1.43. [DOI] [PubMed] [Google Scholar]
- 67.Gros E, Deshayes S, Morris MC, Aldrian-Herrada G, Depollier J, Heitz F, Divita G. A non-covalent peptide-based strategy for protein and peptide nucleic acid transduction. Biochim Biophys Acta. 2006;1758:384–93. doi: 10.1016/j.bbamem.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 68.Rousselle C, Smirnova M, Clair P, Lefauconnier JM, Chavanieu A, Calas B, Scherrmann JM, Temsamani J. Enhanced delivery of doxorubicin into the brain via a peptide-vector-mediated strategy: saturation kinetics and specificity. J Pharmacol Exp Ther. 2001;296:124–31. [PubMed] [Google Scholar]
- 69.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–62. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 70.Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc Natl Acad Sci U S A. 2008;105:12128–33. doi: 10.1073/pnas.0805374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee JY, Choi YS, Suh JS, Kwon YM, Yang VC, Lee SJ, Chung CP, Park YJ. Cell-penetrating chitosan/doxorubicin/TAT conjugates for efficient cancer therapy. Int J Cancer. 2011;128:2470–80. doi: 10.1002/ijc.25578. [DOI] [PubMed] [Google Scholar]
- 72.Myrberg H, Zhang L, Mae M, Langel U. Design of a tumor-homing cell-penetrating peptide. Bioconjug Chem. 2008;19:70–5. doi: 10.1021/bc0701139. [DOI] [PubMed] [Google Scholar]
- 73.Jain M, Chauhan SC, Singh AP, Venkatraman G, Colcher D, Batra SK. Penetratin improves tumor retention of single-chain antibodies: a novel step toward optimization of radioimmunotherapy of solid tumors. Cancer Res. 2005;65:7840–6. doi: 10.1158/0008-5472.CAN-05-0662. [DOI] [PubMed] [Google Scholar]
- 74.Shokolenko IN, Alexeyev MF, LeDoux SP, Wilson GL. TAT-mediated protein transduction and targeted delivery of fusion proteins into mitochondria of breast cancer cells. DNA Repair (Amst) 2005;4:511–8. doi: 10.1016/j.dnarep.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 75.Rousselle C, Clair P, Lefauconnier JM, Kaczorek M, Scherrmann JM, Temsamani J. New advances in the transport of doxorubicin through the blood-brain barrier by a peptide vector-mediated strategy. Mol Pharmacol. 2000;57:679–86. doi: 10.1124/mol.57.4.679. [DOI] [PubMed] [Google Scholar]
- 76.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 77.Liggett WH, Jr, Sidransky D. Role of the p16 tumor suppressor gene in cancer. J Clin Oncol. 1998;16:1197–206. doi: 10.1200/JCO.1998.16.3.1197. [DOI] [PubMed] [Google Scholar]
- 78.Snyder EL, Meade BR, Saenz CC, Dowdy SF. Treatment of terminal peritoneal carcinomatosis by a transducible p53-activating peptide. PLoS Biol. 2004;2:E36. doi: 10.1371/journal.pbio.0020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Michl J, Scharf B, Schmidt A, Huynh C, Hannan R, von Gizycki H, Friedman FK, Brandt-Rauf P, Fine RL, Pincus MR. PNC-28, a p53-derived peptide that is cytotoxic to cancer cells, blocks pancreatic cancer cell growth in vivo. Int J Cancer. 2006;119:1577–85. doi: 10.1002/ijc.22029. [DOI] [PubMed] [Google Scholar]
- 80.Hosotani R, Miyamoto Y, Fujimoto K, Doi R, Otaka A, Fujii N, Imamura M. Trojan p16 peptide suppresses pancreatic cancer growth and prolongs survival in mice. Clin Cancer Res. 2002;8:1271–6. [PubMed] [Google Scholar]
- 81.Fulda S, Wick W, Weller M, Debatin KM. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med. 2002;8:808–15. doi: 10.1038/nm735. [DOI] [PubMed] [Google Scholar]
- 82.Vucic D, Deshayes K, Ackerly H, Pisabarro MT, Kadkhodayan S, Fairbrother WJ, Dixit VM. SMAC negatively regulates the anti-apoptotic activity of melanoma inhibitor of apoptosis (ML-IAP) J Biol Chem. 2002;277:12275–9. doi: 10.1074/jbc.M112045200. [DOI] [PubMed] [Google Scholar]
- 83.Stirpe F, Olsnes S, Pihl A. Gelonin, a new inhibitor of protein synthesis, nontoxic to intact cells. Isolation, characterization, and preparation of cytotoxic complexes with concanavalin A. J Biol Chem. 1980;255:6947–53. [PubMed] [Google Scholar]
- 84.Inoue M, Mukai M, Hamanaka Y, Tatsuta M, Hiraoka M, Kizaka-Kondoh S. Targeting hypoxic cancer cells with a protein prodrug is effective in experimental malignant ascites. Int J Oncol. 2004;25:713–20. [PubMed] [Google Scholar]
- 85.Kim HY, Kim S, Youn H, Chung JK, Shin DH, Lee K. The cell penetrating ability of the proapoptotic peptide, KLAKLAKKLAKLAK fused to the N-terminal protein transduction domain of translationally controlled tumor protein, MIIYRDLISH. Biomaterials. 2011;32:5262–8. doi: 10.1016/j.biomaterials.2011.03.074. [DOI] [PubMed] [Google Scholar]
- 86.Kushner DM, Silverman RH. Antisense cancer therapy: the state of the science. Curr Oncol Rep. 2000;2:23–30. doi: 10.1007/s11912-000-0007-y. [DOI] [PubMed] [Google Scholar]
- 87.Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 1999;286:950–2. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- 88.Lu PY, Xie FY, Woodle MC. siRNA-mediated antitumorigenesis for drug target validation and therapeutics. Curr Opin Mol Ther. 2003;5:225–34. [PubMed] [Google Scholar]
- 89.Los M, Roodhart JM, Voest EE. Target practice: lessons from phase III trials with bevacizumab and vatalanib in the treatment of advanced colorectal cancer. Oncologist. 2007;12:443–50. doi: 10.1634/theoncologist.12-4-443. [DOI] [PubMed] [Google Scholar]
- 90.Mei J, Gao Y, Zhang L, Cai X, Qian Z, Huang H, Huang W. VEGF-siRNA silencing induces apoptosis, inhibits proliferation and suppresses vasculogenic mimicry in osteosarcoma in vitro. Exp Oncol. 2008;30:29–34. [PubMed] [Google Scholar]
- 91.Kim WJ, Christensen LV, Jo S, Yockman JW, Jeong JH, Kim YH, Kim SW. Cholesteryl oligoarginine delivering vascular endothelial growth factor siRNA effectively inhibits tumor growth in colon adenocarcinoma. Mol Ther. 2006;14:343–50. doi: 10.1016/j.ymthe.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 92.Agarwal R, Gonzalez-Angulo AM, Myhre S, Carey M, Lee JS, Overgaard J, Alsner J, Stemke-Hale K, Lluch A, Neve RM, Kuo WL, Sorlie T, Sahin A, Valero V, Keyomarsi K, Gray JW, Borresen-Dale AL, Mills GB, Hennessy BT. Integrative analysis of cyclin protein levels identifies cyclin b1 as a classifier and predictor of outcomes in breast cancer. Clin Cancer Res. 2009;15:3654–62. doi: 10.1158/1078-0432.CCR-08-3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Crombez L, Morris MC, Dufort S, Aldrian-Herrada G, Nguyen Q, Mc Master G, Coll JL, Heitz F, Divita G. Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth. Nucleic Acids Res. 2009;37:4559–69. doi: 10.1093/nar/gkp451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kuan CT, Wikstrand CJ, Bigner DD. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr Relat Cancer. 2001;8:83–96. doi: 10.1677/erc.0.0080083. [DOI] [PubMed] [Google Scholar]
- 95.Han L, Zhang A, Wang H, Pu P, Jiang X, Kang C, Chang J. Tat-BMPs-PAMAM conjugates enhance therapeutic effect of small interference RNA on U251 glioma cells in vitro and in vivo. Hum Gene Ther. 2010;21:417–26. doi: 10.1089/hum.2009.087. [DOI] [PubMed] [Google Scholar]
- 96.Michiue H, Eguchi A, Scadeng M, Dowdy SF. Induction of in vivo synthetic lethal RNAi responses to treat glioblastoma. Cancer Biol Ther. 2009;8:2306–13. doi: 10.4161/cbt.8.23.10271. [DOI] [PubMed] [Google Scholar]
- 97.Josephson L, Tung CH, Moore A, Weissleder R. High-efficiency intracellular magnetic labeling with novel superparamagnetic-Tat peptide conjugates. Bioconjug Chem. 1999;10:186–91. doi: 10.1021/bc980125h. [DOI] [PubMed] [Google Scholar]
- 98.Dodd CH, Hsu HC, Chu WJ, Yang P, Zhang HG, Mountz JD, Jr, Zinn K, Forder J, Josephson L, Weissleder R, Mountz JM, Mountz JD. Normal T-cell response and in vivo magnetic resonance imaging of T cells loaded with HIV transactivator-peptide-derived superparamagnetic nanoparticles. J Immunol Methods. 2001;256:89–105. doi: 10.1016/s0022-1759(01)00433-1. [DOI] [PubMed] [Google Scholar]
- 99.Kaufman CL, Williams M, Ryle LM, Smith TL, Tanner M, Ho C. Superparamagnetic iron oxide particles transactivator protein-fluorescein isothiocyanate particle labeling for in vivo magnetic resonance imaging detection of cell migration: uptake and durability. Transplantation. 2003;76:1043–6. doi: 10.1097/01.TP.0000090164.42732.47. [DOI] [PubMed] [Google Scholar]
- 100.Zhao M, Kircher MF, Josephson L, Weissleder R. Differential conjugation of tat peptide to superparamagnetic nanoparticles and its effect on cellular uptake. Bioconjug Chem. 2002;13:840–4. doi: 10.1021/bc0255236. [DOI] [PubMed] [Google Scholar]
- 101.Harris TJ, von Maltzahn G, Lord ME, Park JH, Agrawal A, Min DH, Sailor MJ, Bhatia SN. Protease-triggered unveiling of bioactive nanoparticles. Small. 2008;4:1307–12. doi: 10.1002/smll.200701319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cryan SA, Devocelle M, Moran PJ, Hickey AJ, Kelly JG. Increased intracellular targeting to airway cells using octaarginine-coated liposomes: in vitro assessment of their suitability for inhalation. Mol Pharm. 2006;3:104–12. doi: 10.1021/mp050070i. [DOI] [PubMed] [Google Scholar]
- 103.Tseng YL, YL, Yagi N, Yano Y, Hatanaka K, Yokoyama Y, Okuno H. Synthesis and evaluation of a novel lipid-peptide conjugate for functionalized liposome. Bioorg Med Chem Lett. 2007;17:2590–3. doi: 10.1016/j.bmcl.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 104.Zhang C, Tang N, Liu X, Liang W, Xu W, Torchilin VP. siRNA-containing liposomes modified with polyarginine effectively silence the targeted gene. J Control Release. 2006;112:229–39. doi: 10.1016/j.jconrel.2006.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Torchilin VP, Levchenko TS, Rammohan R, Volodina N, Papahadjopoulos-Sternberg B, D’Souza GG. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome-DNA complexes. Proc Natl Acad Sci U S A. 2003;100:1972–7. doi: 10.1073/pnas.0435906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Marty C, Meylan C, Schott H, Ballmer-Hofer K, Schwendener RA. Enhanced heparan sulfate proteoglycan-mediated uptake of cell-penetrating peptide-modified liposomes. Cell Mol Life Sci. 2004;61:1785–94. doi: 10.1007/s00018-004-4166-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Torchilin VP, Rammohan R, Weissig V, Levchenko TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci U S A. 2001;98:8786–91. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sethuraman VA, Lee MC, Bae YH. A biodegradable pH-sensitive micelle system for targeting acidic solid tumors. Pharm Res. 2008;25:657–66. doi: 10.1007/s11095-007-9480-4. [DOI] [PubMed] [Google Scholar]
- 109.Tkachenko AG, Xie H, Liu Y, Coleman D, Ryan J, Glomm WR, Shipton MK, Franzen S, Feldheim DL. Cellular trajectories of peptide-modified gold particle complexes: comparison of nuclear localization signals and peptide transduction domains. Bioconjug Chem. 2004;15:482–90. doi: 10.1021/bc034189q. [DOI] [PubMed] [Google Scholar]
- 110.de la Fuente JM, Berry CC. Tat peptide as an efficient molecule to translocate gold nanoparticles into the cell nucleus. Bioconjug Chem. 2005;16:1176–80. doi: 10.1021/bc050033+. [DOI] [PubMed] [Google Scholar]
- 111.Stroh M, Zimmer JP, Duda DG, Levchenko TS, Cohen KS, Brown EB, Scadden DT, Torchilin VP, Bawendi MG, Fukumura D, Jain RK. Quantum dots spectrally distinguish multiple species within the tumor milieu in vivo. Nat Med. 2005;11:678–82. doi: 10.1038/nm1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Suk JS, Suh J, Choy K, Lai SK, Fu J, Hanes J. Gene delivery to differentiated neurotypic cells with RGD and HIV Tat peptide functionalized polymeric nanoparticles. Biomaterials. 2006;27:5143–50. doi: 10.1016/j.biomaterials.2006.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kleemann E, Neu M, Jekel N, Fink L, Schmehl T, Gessler T, Seeger W, Kissel T. Nano-carriers for DNA delivery to the lung based upon a TAT-derived peptide covalently coupled to PEG-PEI. J Control Release. 2005;109:299–316. doi: 10.1016/j.jconrel.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 114.Mortensen MW, Bjorkdahl O, Sorensen PG, Hansen T, Jensen MR, Gundersen HJ, Bjornholm T. Functionalization and cellular uptake of boron carbide nanoparticles. The first step toward T cell-guided boron neutron capture therapy. Bioconjug Chem. 2006;17:284–90. doi: 10.1021/bc050206v. [DOI] [PubMed] [Google Scholar]
- 115.Juliano RL. Intracellular delivery of oligonucleotide conjugates and dendrimer complexes. Ann N Y Acad Sci. 2006;1082:18–26. doi: 10.1196/annals.1348.011. [DOI] [PubMed] [Google Scholar]
- 116.Rudolph C, Schillinger U, Ortiz A, Tabatt K, Plank C, Muller RH, Rosenecker J. Application of novel solid lipid nanoparticle (SLN)-gene vector formulations based on a dimeric HIV-1 TAT-peptide in vitro and in vivo. Pharm Res. 2004;21:1662–9. doi: 10.1023/b:pham.0000041463.56768.ec. [DOI] [PubMed] [Google Scholar]
- 117.Torchilin VP. Cell penetrating peptide-modified pharmaceutical nanocarriers for intracellular drug and gene delivery. Biopolymers. 2008;90:604–10. doi: 10.1002/bip.20989. [DOI] [PubMed] [Google Scholar]
- 118.Nakamura Y, Kogure K, Futaki S, Harashima H. Octaarginine-modified multifunctional envelope-type nano device for siRNA. J Control Release. 2007;119:360–7. doi: 10.1016/j.jconrel.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 119.Balzeau J, Pinier M, Berges R, Saulnier P, Benoit JP, Eyer J. The effect of functionalizing lipid nanocapsules with NFL-TBS. 40-63 peptide on their uptake by glioblastoma cells. Biomaterials. 2013;34:3381–9. doi: 10.1016/j.biomaterials.2013.01.068. [DOI] [PubMed] [Google Scholar]
- 120.Jiang T, Zhang Z, Zhang Y, Lv H, Zhou J, Li C, Hou L, Zhang Q. Dual-functional liposomes based on pH-responsive cell-penetrating peptide and hyaluronic acid for tumor-targeted anticancer drug delivery. Biomaterials. 2012;33:9246–58. doi: 10.1016/j.biomaterials.2012.09.027. [DOI] [PubMed] [Google Scholar]
- 121.Winer I, Wang S, Lee YE, Fan W, Gong Y, Burgos-Ojeda D, Spahlinger G, Kopelman R, Buckanovich RJ. F3-targeted cisplatin-hydrogel nanoparticles as an effective therapeutic that targets both murine and human ovarian tumor endothelial cells in vivo. Cancer Res. 2010;70:8674–83. doi: 10.1158/0008-5472.CAN-10-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–72. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 123.Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci U S A. 2004;101:17867–72. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Olson ES, Aguilera TA, Jiang T, Ellies LG, Nguyen QT, Wong EH, Gross LA, Tsien RY. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr Biol (Camb) 2009;1:382–93. doi: 10.1039/b904890a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, Tsien RY. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc Natl Acad Sci U S A. 2010;107:4311–6. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kwon YM, Li YT, Liang JF, Park YJ, Chang LC, Yang VC. PTD-modified ATTEMPTS system for enhanced asparaginase therapy: a proof-of-concept investigation. J Control Release. 2008;130:252–8. doi: 10.1016/j.jconrel.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160:264–73. doi: 10.1016/j.jconrel.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sawant RM, Hurley JP, Salmaso S, Kale A, Tolcheva E, Levchenko TS, Torchilin VP. “SMART” drug delivery systems: double-targeted pH-responsive pharmaceutical nanocarriers. Bioconjug Chem. 2006;17:943–9. doi: 10.1021/bc060080h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kale AA, Torchilin VP. “Smart” drug carriers: PEGylated TATp-modified pH-sensitive liposomes. J Liposome Res. 2007;17:197–203. doi: 10.1080/08982100701525035. [DOI] [PMC free article] [PubMed] [Google Scholar]