

Abstract
Antidepressant drugs and psychotherapy combined are more effective in treating mood disorders than either treatment alone, but the neurobiological basis of this interaction is unknown. To investigate how antidepressants influence the response of mood-related systems to behavioral experience, we used a fear-conditioning and extinction paradigm in mice. Combining extinction training with chronic fluoxetine, but neither treatment alone, induced an enduring loss of conditioned fear memory in adult animals. Fluoxetine treatment increased synaptic plasticity, converted the fear memory circuitry to a more immature state, and acted through local brain-derived neurotrophic factor. Fluoxetine-induced plasticity may allow fear erasure by extinction-guided remodeling of the memory circuitry. Thus, the pharmacological effects of antidepressants need to be combined with psychological rehabilitation to reorganize networks rendered more plastic by the drug treatment.
Antidepressant drugs have been used for over 5 decades to treat depression and anxiety disorders, and their actions have been connected to effects on the monoamines serotonin and norepinephrine (1). Clinical experience has shown that a combination of antidepressant treatment with psychotherapy is more effective than either treatment alone (2), but the neurobiological basis of this combined effect is not known. Recently, antidepressants have been shown to enhance neuronal plasticity in hippocampus and cortex (3-5), but it remains unclear whether these effects are linked to their anti-depressant actions.
Currently, anxiety and fear disorders, including phobias and posttraumatic stress disorder (PTSD), are usually treated by either exposure therapies (6) or pharmacological treatments, most often using serotonin selective reuptake inhibitor (SSRI) antidepressants (7). Exposure therapy extinguishes or suppresses fear responses by repeatedly exposing the subject to the fear-inducing stimulus. This has been successfully modeled in both human and animals by using Pavlovian fear-conditioning paradigm, where a neutral conditioned stimulus (CS, a tone) begins to elicit fear after association with a noxious unconditioned stimulus (US), but that fear response is extinguished after repeated exposure to the CS without the US (8-10). However, although such extinction training in humans suffering from anxiety disorders and adult rodents initially reduces fear responses, this is typically followed by spontaneous recovery over time and fear renewal upon later reexposure to the CS (11, 12).
Extinction training during a critical period in juvenile mice leads to permanent fear erasure (13, 14). Because the antidepressant fluoxetine (Flx) reactivates a critical periodlike plasticity in the visual cortex (5), we used the fear-conditioning and extinction paradigm to investigate whether Flx might reactivate juvenile-like plasticity in the fear-conditioning network and therefore, when combined with extinction training, induce long-term fear erasure in adult mice.
First, Flx was given for 3 weeks before fear-conditioning and extinction training (Fig. 1A). Flx did not influence fear acquisition or locomotion (Fig. 1B and figs. S1 and S2), although it did cause faster extinction (fig. S1). To explore whether this fear reduction was permanent, we used two techniques that increase fear after extinction: spontaneous recovery of fear and renewal induced through CS presentations in extinction and conditioning contexts, respectively. One week after the end of the extinction training, control mice showed significant spontaneous recovery and fear renewal (Fig. 1B). However, both recovery and renewal were attenuated in mice treated with Flx (Fig. 1B). Importantly, Flx-treated mice not exposed to extinction training maintained elevated levels of freezing, a conditioned fear response (Fig. 1B), and control experiments indicated that potential Flx-induced locomotor and freezing differences did not account for these results (fig. S2).
Fig. 1.
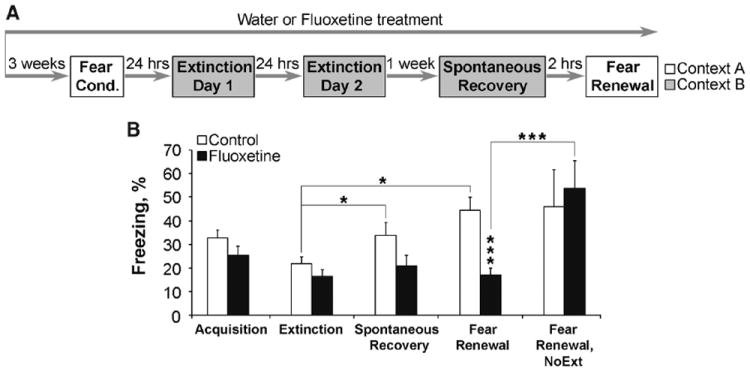
Chronic Flx treatment before fear conditioning leads to fear erasure when combined with extinction training. (A) Flx treatment started 3 weeks before and continued throughout the experiment. (B) Control and Flx groups (n = 31 to 34 mice per group) exhibited similar levels of fear acquisition (extinction day 1, first block of 2 CS) and extinction (extinction day 2, last block of 2 CS). One week later (n = 21 per group) only control group showed spontaneous recovery and fear renewal. In the fear renewal test, the Flx-extinction group froze less than either control-extinction or Flx–no-extinction group (NoExt, n = 5). *P < 0.05, **P < 0.01, ***P < 0.001. Error bars indicate mean ± SEM.
Next, we investigated whether Flx treatment could also lead to fear erasure when given in a more clinically relevant manner, that is, after fear conditioning. After successful acquisition, the mice were given either Flx or water for 2 weeks, then both groups were exposed to extinction training; 7 days later, the mice were tested for spontaneous recovery and fear renewal (Fig. 2A). Although both Flx-treated and control mice acquired extinction (Fig. 2B), the Flx-treated mice again showed faster extinction (fig. S3). Whereas the control mice showed clear fear renewal and a tendency to spontaneous recovery, the Flx-treated mice showed no signs of renewal or spontaneous recovery (Fig. 2B). Again, mice not exposed to extinction training showed enhanced freezing, regardless of their treatment group (Fig. 2B). Lastly, we examined the effect of Flx on fear reinstatement, where, after successful extinction in the fear-conditioning context (fig. S4), the mice were exposed to a foot shock five times without a CS and tested for freezing after a tone 24 hours later (Fig. 2A). Control mice showed a robust fear reinstatement, but freezing in mice receiving Flx was significantly reduced (Fig. 2C) (15).
Fig. 2.
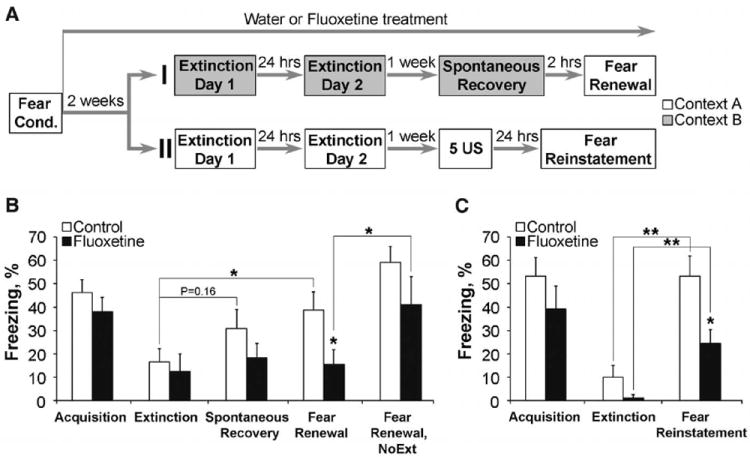
Extinction training is effective in combination with chronic Flx treatment after fear conditioning. (A) After fear conditioning and 2 weeks of Flx treatment, mice were subjected to fear renewal (protocol I) or fear reinstatement (protocol II). 5 US indicates five unsignaled foot shocks. (B) Fear renewal: Control and Flx groups (n = 10 per group) exhibited similar levels of fear acquisition and extinction. One week later, only control group showed elevated spontaneous recovery and significant fear renewal. The Flxextinction group froze less than either control-extinction or Flx–no-extinction groups (NoExt, n = 7). (C) Fear reinstatement: Although both groups exhibited increased levels of freezing when compared with the extinguished levels, the Flx group showed lower levels of fear reinstatement (P < 0.05). N = 8 per group. *P < 0.05, **P < 0.01. Error bars indicate mean ± SEM.
Flx reactivates developmental-like plasticity in the visual cortex and dentate gyrus (5, 16). We thus asked whether Flx similarly affected fear networks. Perineuronal nets (PNNs) develop as pups acquire the ability to learn and extinguish fear (13, 14, 17), and PNN disruption in the adult basolateral amygdala (BLA) before, but not after, fear conditioning leads to fear erasure after extinction (14). Although Flx treatment differs from PNN disruption in influencing fear renewal when given either before or after acquisition (14), we assessed whether disruption of PNNs might contribute to the drug’s ability to facilitate extinction. Control and Flx-treated mice had similar numbers of PNN-positive neurons in the BLA, hippocampal CA1 area, and infralimbic cortex (IL) (Fig. 3, A and B, and table S1), suggesting that Flx treatment does not influence fear renewal by disrupting PNNs. However, Flx treatment reduced the percentage of PNN neurons expressing parvalbumin in the BLA and CA1 (Fig. 3, A and B), whereas no differences were found in PNN-positive interneurons containing calbindin or calretinin (fig. S5 and table S1). Because PNNs are particularly enriched around parvalbumin-positive fast-spiking neurons (18), these data suggest that Flx treatment selectively shifted the parvalbumin- and PNN-containing neurons toward an immature state (18). Expression of polysialynated neuronal cell-adhesion molecule (PSA-NCAM), which is expressed in immature cortical cells and reduced with maturation of the postnatal brain (19), was increased by Flx treatment in the BLA (Fig. 3C). Conversely, Flx treatment reduced the expression of K+-Cl− cotransporter KCC2 (Fig. 3D), which increases during postnatal development (20). These data suggest that Flx reactivates juvenile-like plasticity in the fear-conditioning network, allowing the erasure of conditioned fear when extinction training is given.
Fig. 3.
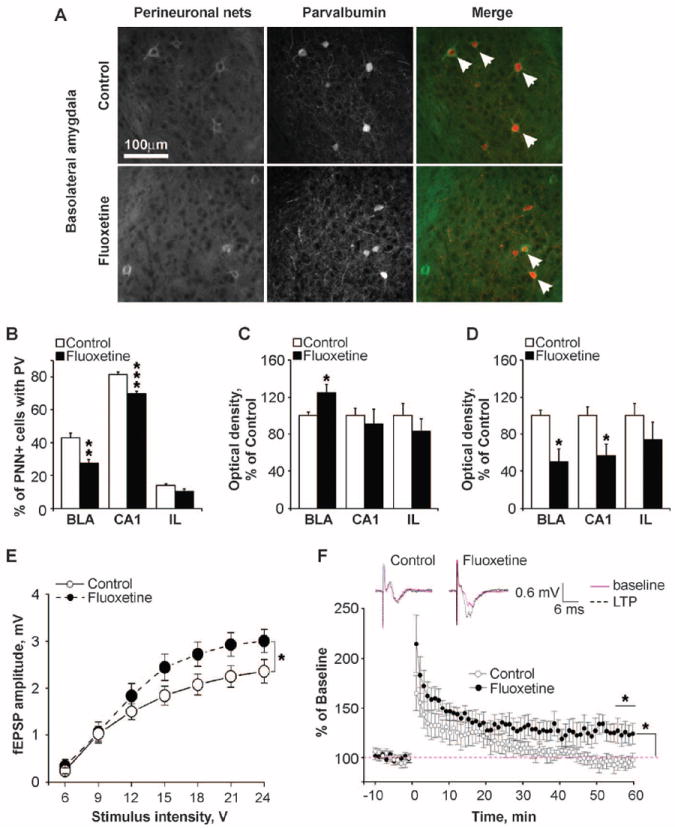
Flx treatment enhances plasticity in the fear memory circuitry. (A) Representative PNNs and parvalbumin (PV) immunostaining in the BLA. Arrows, double-positive neurons. (B to D) Chronic Flx decreased the percentage of PNN-containing neurons with parvalbumin (B), increased PSA-NCAM expression (C), and decreased KCC2 levels (D) in the fear circuit. N = 6 per group. (E) Input-output function for fEPSPs in LA evoked by EC stimulation. Flx treatment increased the average amplitude of fEPSPs above control levels. N = 12 to 15 per group. (F) Effect of high-frequency stimulation (two 1-s stimuli at 100 Hz) of EC afferents on LA fEPSPs. In control animals, EC tetanization caused short-term synaptic potentiation, but there was no potentiation at 1 hour; conversely, in fluoxetine-treated animals tetanization resulted in LTP at 1 hour. N = 8 per group. Inset shows example traces before and 1 hour after tetanization. *P < 0.05, **P < 0.01 versus respective control group. Error bars indicate mean ± SEM.
To further test the hypothesis that chronic Flx increases plasticity in the neural circuitry underlying fear conditioning and extinction, we prepared brain slices from Flx-treated and control mice and measured basal synaptic transmission and synaptic plasticity in the lateral amygdala (LA), a brain area critical for fear learning and extinction (8, 9). Field excitatory postsynaptic potentials (fEPSPs) were recorded in response to stimulation of the external capsule (EC) (fig. S6, A and B) (21). At baseline, Flx treatment resulted in an overall increase in the amplitude of evoked fEPSPs (Fig. 3E), as previously reported in the dentate gyrus (16, 22). Paired pulse ratios were unchanged, indicative of normal release probability (fig. S6C).
To investigate synaptic plasticity, we measured fEPSP potentiation in horizontal brain slices, where, with γ-aminobutyric acid–mediated (GABAergic) transmission left intact, EC tetanization typically induces little or no long-term potentiation (LTP) (21). In vehicle-treated mice 1 hour after tetanization, fEPSPs were unchanged from control levels (96.3 ± 5.1%, mean ± SEM). However, in Flx-treated mice fEPSPs were increased to 122.8 ± 8.8% of baseline (Fig. 3F). Increased cortical input and synaptic plasticity in LA may facilitate associational learning in specific extinction circuits or suppress fear expression by recruiting inhibitory interneurons that project to the amygdalar central nucleus (10, 23).
Lastly, we sought to determine the pathways that Flx activates to alter circuit plasticity. Chronic Flx treatment increases brain-derived neurotrophic factor (BDNF) expression in many brain areas (3) and BDNF infusion into prefrontal cortex accelerates fear extinction in rats (24), whereas both humans and mice carrying the met allele of the BDNF Val66Met polymorphism show impaired extinction (25). Flx treatment after fear conditioning significantly increased BDNF mRNA level in the BLA, and the activity-dependent BDNF transcript 1 expression was increased in the BLA and hippocampus (Fig. 4A).
Fig. 4.
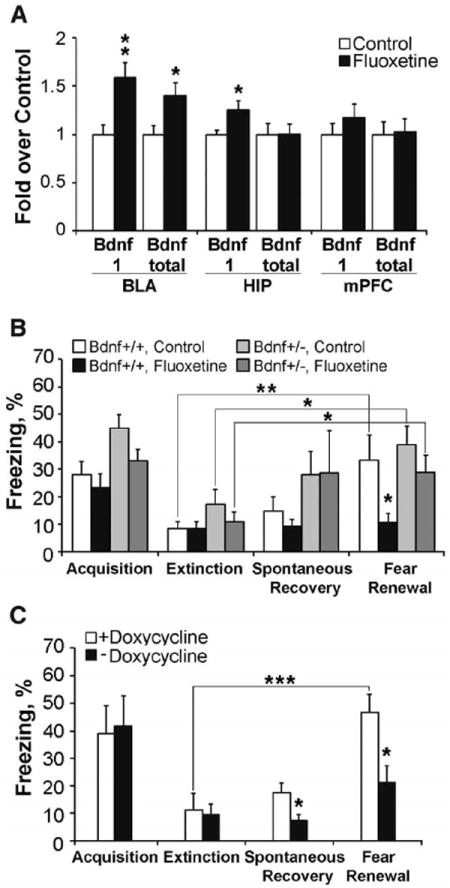
BDNF regulates fear memory erasure. (A) Chronic Flx after fear conditioning increased the BDNF exon-1 and total BDNF mRNA levels in the BLA and BDNF exon-1 level in the hippocampus (HIP) but not in the prefrontal cortex (mPFC). N = 6 to 8 per group. (B) Flx prevented fear renewal in BDNF+/+ mice (see protocol I, Fig. 2A) but failed to erase fearful memory in BDNF+/− mice. N = 10 or 11 per group. (C) BDNF-virus experiment: After successful fear acquisition and extinction, doxycycline treatment was terminated to induce BDNF overexpression in amygdala, which resulted in a significant reduction in freezing level in the fear renewal test. N = 6 or 7 per group. *P < 0.05, **P < 0.01, *** P < 0.001 versus control groups. Error bars indicate means ± SEM.
Because mice heterozygous for the BDNF null allele (BDNF+/−) are insensitive to Flx treatment in behavioral models of depression and anxiety (3, 26), we tested whether BDNF+/− mice (C57Bl/6J background) responded differentially to Flx in the fear-conditioning paradigm. Flx again prevented fear renewal in the wild-type mice, but in BDNF+/− littermates trained to fully extinguish the fear response, the Flx effect was absent as indicated by elevated levels of freezing 1 week after extinction (Fig. 4B and fig. S7). To test whether BDNF was acting predominately in the amygdala, we used doxycycline-regulatable lentiviral infection to overexpress BDNF locally in the BLA from the end of extinction onward (figs. S8 and S9). BDNF-overexpressing mice did not show fear renewal, whereas control mice did (Fig. 4C).
This study demonstrates that long-term loss of fearful memories can be induced through a combination of SSRI pharmacotherapy and extinction training, which neither treatment alone achieved. Fear conditioning and extinction were used as a model for exposure therapy for PTSD. Although combining pharmacological and psychological treatments has been suggested for PTSD, few clinical studies have been conducted (27). Other methods of increasing extinction have also been identified and include treatment with d-cycloserine (10, 12, 28) and yohimbine (29) and a reminder CS given before the extinction (30, 31), all of which lead to a long-term erasure of conditioned fear in both rodents and humans. However, the present results provide evidence for a unique mechanism of fear reduction, suggesting that Flx treatment reactivates juvenile-like plasticity in the amygdala (13, 14), as has previously been demonstrated in the visual cortex (5) and dentate gyrus (16). This reactivated plasticity then may allow behavioral experience, such as extinction training, to reshape maladapted networks to better adjust to the environment (4, 5). These data, therefore, provide a putative neurobiological basis for the enhanced effect of combining drug and psychological treatments and support the hypothesis that the chemical effect produced by administering antidepressants alone will not give full clinical benefit. Instead, drug treatments need to be combined with psychotherapy or other kinds of social rehabilitation to optimize their mood-elevating effects (4).
Supplementary Material
Acknowledgments
We thank O. Nikkilä, T. Rantamäki, V. Võikar, and M. Kislin for technical help; Jean-Luc Dreyer for the gift of BDNF lentivirus; F. Battaglia for help with electrophysiology; C. Rivera for the KCC2 antibody; and J. Sallinen, Orion Pharma, for the gift of fluoxetine. E.C. is a shareholder of Hermo Pharma, Incorporated. This work was supported by Sigrid Jusélius Foundation and Academy of Finland Center of Excellence program (E.C.) and NIH grants DC003906 and NIH-DC009910 (R.M.S.).
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/334/6063/1731/DC1
Materials and Methods
Figs. S1 to S9
Table S1
References (32-39)
References and Notes
- 1.Belmaker RH, Agam G. N Engl J Med. 2008;358:55. doi: 10.1056/NEJMra073096. [DOI] [PubMed] [Google Scholar]
- 2.Pampallona S, Bollini P, Tibaldi G, Kupelnick B, Munizza C. Arch Gen Psychiatry. 2004;61:714. doi: 10.1001/archpsyc.61.7.714. [DOI] [PubMed] [Google Scholar]
- 3.Castrén E, Rantamäki T. Dev Neurobiol. 2010;70:289. doi: 10.1002/dneu.20758. [DOI] [PubMed] [Google Scholar]
- 4.Castrén E. Nat Rev Neurosci. 2005;6:241. doi: 10.1038/nrn1629. [DOI] [PubMed] [Google Scholar]
- 5.Maya Vetencourt JF, et al. Science. 2008;320:385. doi: 10.1126/science.1150516. [DOI] [PubMed] [Google Scholar]
- 6.Bisson J, Andrew M. Cochrane Database Syst Rev. 2007;3 doi: 10.1002/14651858.CD003388.pub3. CD003388. [DOI] [PubMed] [Google Scholar]
- 7.Stein DJ, Ipser J, McAnda N. CNS Spectr. 2009;14(suppl. 1):25. [PubMed] [Google Scholar]
- 8.Milad MR, Rauch SL, Pitman RK, Quirk GJ. Biol Psychol. 2006;73:61. doi: 10.1016/j.biopsycho.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 9.LeDoux JE. Annu Rev Neurosci. 2000;23:155. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 10.Mahan AL, Ressler KJ. Trends Neurosci. doi: 10.1016/j.tins.2011.06.007. published online 26 July, 2011. [DOI] [Google Scholar]
- 11.Yehuda R, LeDoux J. Neuron. 2007;56:19. doi: 10.1016/j.neuron.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Quirk GJ, Mueller D. Neuropsychopharmacology. 2008;33:56. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JH, Richardson R. Biol Psychiatry. 2010;67:297. doi: 10.1016/j.biopsych.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Gogolla N, Caroni P, Lüthi A, Herry C. Science. 2009;325:1258. doi: 10.1126/science.1174146. [DOI] [PubMed] [Google Scholar]
- 15.Deschaux O, Spennato G, Moreau JL, Garcia R. Psychopharmacology. 2011;215:231. doi: 10.1007/s00213-010-2134-y. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi K, et al. Proc Natl Acad Sci U S A. 2010;107:8434. doi: 10.1073/pnas.0912690107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullivan RM, Landers M, Yeaman B, Wilson DA. Nature. 2000;407:38. doi: 10.1038/35024156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balmer TS, Carels VM, Frisch JL, Nick TA. J Neurosci. 2009;29:12878. doi: 10.1523/JNEUROSCI.2974-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gascon E, Vutskits L, Kiss JZ. Brain Res Brain Res Rev. 2007;56:101. doi: 10.1016/j.brainresrev.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 20.Rivera C, et al. Nature. 1999;397:251. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 21.Drephal C, Schubert M, Albrecht D. Neurobiol Learn Mem. 2006;85:272. doi: 10.1016/j.nlm.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Wang JW, David DJ, Monckton JE, Battaglia F, Hen R. J Neurosci. 2008;28:1374. doi: 10.1523/JNEUROSCI.3632-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herry C, et al. Eur J Neurosci. 2010;31:599. doi: 10.1111/j.1460-9568.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- 24.Peters J, DieppaPerea LM, Melendez LM, Quirk GJ. Science. 2010;328:1288. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soliman F, et al. Science. 2010;327:863. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saarelainen T, et al. J Neurosci. 2003;23:349. doi: 10.1523/JNEUROSCI.23-01-00349.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hetrick SE, Purcell R, Garner B, Parslow R. Cochrane Database Syst Rev. 2010;7 doi: 10.1002/14651858.CD007316.pub2. CD007316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker DL, Ressler KJ, Lu KT, Davis M. J Neurosci. 2002;22:2343. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmes A, Quirk GJ. Trends Pharmacol Sci. 2010;31:2. doi: 10.1016/j.tips.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monfils MH, Cowansage KK, Klann E, LeDoux JE. Science. 2009;324:951. doi: 10.1126/science.1167975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schiller D, et al. Nature. 2010;463:49. doi: 10.1038/nature08637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.