

Abstract
Cell-directed changes in the ligand-binding affinity (‘activation’) of integrins regulate cell adhesion and migration, extracellular matrix assembly and mechanotransduction, thereby contributing to embryonic development and diseases such as atherothrombosis and cancer. Integrin activation comprises triggering events, intermediate signalling events and, finally, the interaction of integrins with cytoplasmic regulators, which changes an integrin’s affinity for its ligands. The first two events involve diverse interacting signalling pathways, whereas the final steps are immediately proximal to integrins, thus enabling integrin-focused therapeutic strategies. Recent progress provides insight into the structure of integrin transmembrane domains, and reveals how the final steps of integrin activation are mediated by integrin-binding proteins such as talins and kindlins.
Integrins play central roles in the biology of metazoa1 by controlling cell adhesion to the extracellular matrix (ECM) and cell migration, growth, differentiation and apoptosis. As a result, they contribute to the regulation of development, immunity, inflammation and haemo stasis, and to the development of diseases including autoimmunity, atherothrombosis and neoplasia1. Integrins are heterodimers of transmembrane α- and β-subunits1, which each have a large ectodomain, a single transmembrane domain and a generally short cytoplasmic tail (BOX 1). Integrin affinities for their cognate extracellular ligands, such as fibronectin, fibrinogen and collagen, are regulated by cellular signalling, resulting in integrin activation through ‘inside–out’ signalling1 (BOX 2). Consequently, integrin activation is important for a wide range of anchorage-dependent cellular events, such as platelet aggregation and leukocyte transmigration1. In addition to changes in adhesion, integrin activation can control the polarity of migrating cells and the assembly of the ECM, thereby regulating events such as tumour metastasis2. Blockade of integrin activation may therefore be useful in anti-adhesive therapies3. The broad biological and potentially therapeutic significance of integrin activation, and interest in this prototype of inside–out signalling, give rise to a fertile field of investigation. Here, we summarize recent progress and controversies in the study of integrin activation, focusing on the terminal events that lead to activation; that is, the ‘end game’. We do not consider the ability of integrins to signal into cells (‘outside–in’ signalling; BOX 2); instead, we emphasize recent advances that identify the cytoplasmic partners that trigger integrin activation, begin to explain how the association of these partners with integrins is regulated by signalling events, explain how these binding interactions activate integrins and identify transmembrane domain structural features that account for the ability of integrins to efficiently transmit signals across cell membranes.
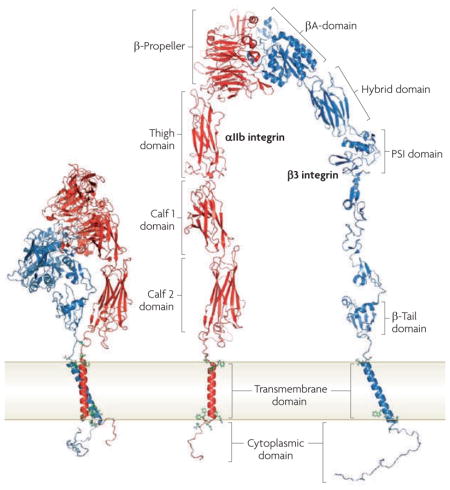
Box 1. Integrin domain structures.
Integrins are heterodimeric adhesive receptors consisting of an α- and a β-subunit. In mammals, there are 24 canonical integrins formed from combinations of 18 α-subunits and 8 β-subunits. The ‘bent conformation’ seen in crystal structures (see the figure; left) can be unfolded to facilitate visualization of the domains (see the figure; right). In most integrins the amino-terminal domain in the α- and β-integrin subunits (the β-propeller and the βA domain, respectively), assemble by non-covalent interactions to form a ‘head’ and provide a ligand binding site. In 8 α-integrin subunits (α1, α2, α10, α11, αL, αM, αX and αD), the αA domain, which is homologous to the βA domain of the β-integrin subunit, is inserted into the β-propeller domain and is the main ligand-binding site in these integrins. In integrins that lack an A domain, such as αIIbβ3 integrin, which is depicted here, the βA domain forms the main ligand-binding site. Note that the plexin, semaphorin and integrin (PSI) domain is at the N terminus of the β-integrin subunit, but is joined by disulphide bonds to more carboxy-terminal residues. The remaining C-terminal extracellular domains of each subunit comprise two long ‘legs’. The low affinity state of the integrin for its ligands is maintained by non-covalent interactions between the α- and β-integrin transmembrane and cytoplasmic domains. Figure is modified, with permission, from EMBO J REF. 44 © (2009) Macmillan Publishers Ltd. All rights reserved.
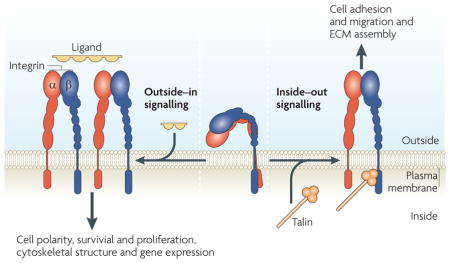
Box 2. Bidirectional integrin signalling.
There are two directions of integrin signalling, which have different biological consequences (see the figure). During ‘inside–out’ signalling, an intracellular activator, such as talin or kindlins, binds to the β-integrin tail, leading to conformational changes that result in increased affinity for extracellular ligands (integrin ‘activation’). The relationship between specific conformations and activation remains controversial. Inside–out signalling controls adhesion strength and enables sufficiently strong interactions between integrins and extracellular matrix (ECM) proteins to allow integrins to transmit the forces required for cell migration and ECM remodelling and assembly. Integrins also behave like traditional signalling receptors in transmitting information into cells by ‘outside–in’ signalling. Binding of integrins to their extracellular ligands changes the conformation of the integrin and, because many of the ligands are multivalent, contributes to integrin clustering. The combination of these two events leads to intracellular signals that control cell polarity, cytoskeletal structure, gene expression and cell survival and proliferation. Although we conceptually separate the two processes, they are often closely linked; for example, integrin activation can increase ligand binding, resulting in outside–in signalling. Conversely, ligand binding can generate signals that cause inside–out signalling.
Roles of conformation and clustering
Changes in the conformation of individual integrin heterodimers and clustering of heterodimers into oligomers can influence the binding of ligands4, the former through changes in single receptor affinity and the latter through increases in receptor valency that accompany integrin clustering4. Vigorous debate centres on the relative importance of each mechanism to integrin function4,5. Clarification of this debate has been hampered by inexact definitions of clustering and difficulties in quantifying integrin microclusters4. Conformational change and clustering are both likely to be important for integrin function, and their relative contributions might vary depending on the integrin, cell type and biological circumstances. For some integrins in circulating blood cells, such as αIIbβ3 integrin in platelets, changes in receptor conformation are the primary means of regulating receptor affinity and ligand binding in response to agonists.
Conformational changes
Much debate surrounds the nature of the extracellular domain conformational changes that underlie integrin activation and several excellent reviews have been devoted to this topic, which is not a main focus of this Review. In brief, integrin ectodomains can exist in bent ‘closed’ conformations, intermediate extended conformations with a closed head-piece, and extended ‘open’ conformations6,7. These may correspond to low affinity, activated, and activated and ligand occupied integrin conformers8, respectively, on cells9,10. The bent form can, in some circumstances, engage ligands such as fibronectin fragments11. This result is consistent with the idea that activation involves releasing a ‘deadbolt’ formed by an interface in the β-integrin subunit between the membrane-proximal β-tail domain and the α7 helix in the ligand-binding βA domain11,12 (BOX 1). Downwards displacement of this α7 helix leads to conformational activation of the βA domain13,14; thus, the deadbolt was proposed to prevent this displacement and block activation11,12. However, a structure of the αIIbβ3 integrin ectodomain lacked the deadbolt interface15. Furthermore, deletion of the loop connecting helices C and D (CD loop) in the β-tail domain, proposed to form the dead-bolt, failed to activate αIIbβ3 integrin or αVβ3 integrin16, casting further doubt on the deadbolt hypothesis. The conformations of integrins have been studied in isolated ectodomains, or fragments of these, potentially leading to artefacts owing to the release of constraints imposed by interactions of cytoplasmic or transmembrane domains17. Electron cryomicroscopy studies of intact integrins in detergent micelles provide insights18. However, the structure of the β3 integrin transmembrane domain can differ between detergent micelles and phospholipid bilayers19; the latter more accurately mimics biological membranes. Moreover, image selection bias is a potential problem in electron microscopy studies, and has been invoked11 as an explanation for discrepancies20. Electron cryomicroscopy tomography of lipid-embedded αIIbβ3 integrin revealed an average height of 11 nm, much less than the 19 nm height expected of a fully extended integrin6,21. Furthermore, addition of Mn2+, which activates integrins directly by interaction with cation coordination sites in the βA domain, did not change the height, indicating a lack of extension21 — a result in agreement with Förster resonance energy transfer (FRET) studies of αVβ3 integrin in living cells22. Steered molecular dynamic modelling15,23 and experimental studies24,25 suggest that force can contribute to integrin activation and extension. Furthermore, extension of unoccupied integrins may require either traction forces or collision with other membrane proteins15. It seems likely that a resolution of some of these hotly debated issues must take into account the relative strengths of negative stain electron microscopy versus electron cryomicroscopy26, and awaits structural studies of a lipid-embedded, full-length integrin activated in a physiologically relevant manner.
Clustering
Integrin clustering is defined as the interaction of heterodimers to form heterooligomers. It can be caused by inside–out signals that stimulate the recruitment of multivalent protein complexes to integrin cytoplasmic domains27,28,29, by binding of multivalent extracellular ligands to integrin ectodomains by the homodimerization of integrin transmembrane domains (α-to-α or β-to-β)30, or by the release of integrins from cytoskeletal constraints that leads to the free diffusion of integrins in the plane of the membrane31. Whatever the contribution to the binding of fibronectin or other adhesive ligands, integrin clustering is important for triggering outside–in signalling, integrin recycling32 and mechanotransduction by adhesion-based intracellular structures that contain integrins and associated molecules33. These intracellular structures include focal complexes and focal adhesions34 in adherent fibroblasts, immunological synapses and kinapses in activated T lymphocytes35, podosomes in adherent osteoclasts and macrophages, and invadopodia in cancer cells36. Technical issues currently limit the separation of integrin clustering from conformational change. Thus, cell adhesion assays typically reflect the combined effects of integrin conformation and valency regulation on adhesion strength25,33. Even the results of soluble ligand binding assays, the classical method to study integrin affinity modulation in non-adherent cells such as leukocytes and platelets, can be subject to ambiguity. For example, since most integrin ligands are multivalent, their binding may be influenced by the cellular regulation of integrin clustering. Furthermore, multimeric ligand binding itself may modify the nature of the bond between integrin and ligand through ligand-induced conformational changes8, microclustering37 and outside–in signalling38. Finally, because the application of force can prolong the bond lifetimes between integrins and their ligands25, this so-called ‘catch bond’ behaviour may erroneously be attributed to integrin clustering.
Advances in the detection of protein–protein interactions in living cells by FRET39, bioluminescence resonance energy transfer (BRET)37, image correlation spectroscopy40 and interferometric photoactivated localization microscopy41 promise to improve our understanding of integrin clustering at the nanoscale. As certain integrins are expressed at high density (for example, αIIbβ3 integrin molecules are < 200 Å apart in platelets38), spontaneous integrin microclusters may be favoured. FRET and BRET also show that MnCl2 activation of leukocyte αLβ2 integrin or platelet αIIbβ3 integrin fails to induce microclustering. Instead, microclustering requires the binding of multivalent ligands to these integrins and is enhanced by cytochalasins, presumably by releasing cytoskeletal constraints37,42.
Transmembrane domains: signalling conduits
Each α- or β-integrin subunit is a typical type 1 transmembrane protein with the amino terminus outside and a single transmembrane domain that connects to a carboxy-terminal cytoplasmic tail (BOX 1). The transmembrane domain is therefore an essential connection for the transmission of information across the membrane.
The topology of integrin transmembrane domains
Ulmer’s laboratory used NMR spectroscopy of the individual αIIb integrin and β3 integrin transmembrane domains and of the heterodimeric complex, to define their structure in phospholipid bicelles and to estimate the extent to which they are embedded in the membrane19,43,44 (FIG. 1a). Studies of the transmembrane domain of the αIIbβ3 integrin heterodimer subunits show that the β3 integrin transmembrane domain adopts a long helix19, whereas the αIIb integrin transmembrane domain folds into a shorter helix followed by a backbone reversal that packs Phe992–Phe993 against the transmembrane helix43(FIG. 1b,c). One important contribution of these studies was clarifying the membrane embedding of the α- and β-integrin transmembrane domains. Prediction methods placed45 the boundaries between transmembrane and cytoplasmic domains at conserved Lys or Arg residues that precede four to six apolar residues. Armulik and co-workers46 used enzymatic glycosylation mapping, a method that examines the efficiency of microsomal membrane glycosylation of Asn-X-(Thr/Ser) motifs (where X is any amino acid) placed at varying distances from the presumed transmembrane domain. They predicted that the conserved Lys residues and the C-terminal apolar residues in α1, α2, β1 and β2 integrin subunits are lipid-embedded. Protection from solvent water or paramagnetic relaxation of αIIb and β3 integrin transmembrane domains in bicelles confirmed the predictions of the glycosylation mapping studies19,43,44 (FIG. 1a). Consequently, for α-integrin subunits, the conserved Gly-Phe-Phe residues C-terminal to Lys-Arg are membrane embedded and terminate in a short transmembrane helix that is perpendicular to the plane of the membrane (FIG. 1b). The β3 integrin transmembrane domain is predicted to be tilted by ~ 25° relative to the plane of the membrane to enable side chains of corresponding hydrophobic residues in the β-subunit19,43,44 to maintain membrane embedding (FIG. 1b). This β3 integrin transmembrane helical tilt may also be favoured by the propensity of the positively charged side chain of a conserved membrane-embedded Lys-Arg to reside in proximity to the negatively charged phospholipid head-groups19. Mutational studies point to a crucial role for these membrane embedded, conserved apolar residues in both subunits in regulating integrin activation47–49, and the structural basis of the role of the transmembrane domain in activation has now become clear.
Figure 1. The structure of the αIIbβ3 integrin transmembrane complex enables inside–out signal transduction.

The models depicted are based on the average of an ensemble of 20 calculated simulated annealing NMR structures44 (Protein data bank identifier 2K9J). a | Sequences of the αIIb and β3 integrin transmembrane domains. The membrane- embedded segments, as assessed by NMR spectroscopy of integrins in phospholipid bicelles, are highlighted in blue. b | Ile966–Arg995 of αIIb integrin and Ile693–Asp723 of β3 integrin adopt well-structured conformations with a predicted crossing angle of 25°. c | Rotating the model in part b by 90° reveals the two discrete elements that mediate the principal interaction of the transmembrane domains. The β3 (left) or αIIb (right) integrin transmembrane domains are depicted as space-filling models, with the ribbon structure in the middle. Basic residues are blue and acidic residues are red. The association of α- and β-integrin transmembrane domains, through packing of Gly residues in the outer membrane leaflet, forms the outer membrane clasp. The novel assembly in the inner membrane leaflet extending into the membrane–cytosol interface forms the inner membrane clasp. d | The outer membrane clasp. Gly972 and Gly976 of αIIb integrin and Gly708 of β3 integrin are shown as atoms that form holes into which side chains from the apposing space filling model of β3 integrin pack (left). Gly708 of β3 integrin forms a hole into which αIIb integrin side chains pack (right). e | The αβ-integrin transmembrane interaction is stabilized by interhelical packing mediated by Phe992–Phe993 of αIIb integrin and then the electrostatic interaction of Arg995 of αIIb integrin with Asp723 of β3 integrin to form the inner membrane clasp. The left panel depicts a space-filling model of the β3 integrin transmembrane domain in which Asp723 of β3 integrin is shown in red and space-filling models of the Phe992, Phe993 and Arg995 side chains of αIIb integrin are shown. The right panel depicts a space-filling model of the αIIb integrin transmembrane domain in which Arg995 of αIIb integrin is shown in blue and space-filling models of the Trp715, Ile719 and Asp723 side chains of β3 integrin are shown.
αβ-Integrin cytoplasmic domain interactions and signalling
Interactions of integrin cytoplasmic domains with each other or cytoplasmic proteins lead to the long-range allosteric rearrangements of the integrins48,50 that underlie activation. Recent work provides new insights into how such rearrangements cross the membrane. The association of α- and β-integrin transmembrane and cytoplasmic domains regulates integrin signalling17,51–54. Mutational studies suggested that an electrostatic interaction between Asp723 of β3 integrin and Arg995 of αIIb integrin17 might constrain the C-termini of these integrins to inhibit activation. Subsequent studies indicated that mutations of the corresponding residues in β2 and β1 integrins could activate these integrins55,56; however, mutation of the Asp residue in β1 integrin produced no evident phenotype in mice57. Clasping the cytoplasmic or transmembrane domains together with artificial coiled coils inhibited activation15,58, as did linking the α- and β-integrin transmembrane domains with disulphides51. Nevertheless, efforts to identify interactions of isolated α- and β-integrin tails in aqueous solution by NMR spectroscopy59 were either unsuccessful, or reported differing structures of the αβ-integrin complex60,61.
αβ-Integrin transmembrane domains and activation
Mutational studies and molecular modelling suggest that interactions between the transmembrane domain of an α- and a β-integrin subunit are important in maintaining the low affinity inactive state, and that activation requires alteration of these transmembrane interactions17,51–54,62. Efforts to identify direct interactions between α- and β-integrin transmembrane domains have yielded differing results30,63,64. An affinity capture assay using mini integrins, which have only transmembrane and cytoplasmic domains (FIG. 2), was recently used in conjunction with NMR spectroscopy to reveal preferential heterodimeric association of αIIb integrin transmembrane cytoplasmic tails with those of β3 integrin by specific transmembrane interactions65. Furthermore, mutations in αIIb integrin (at Arg995) and β3 integrin (at Asp723) confirmed that an electrostatic interaction stabilizes the association between the αIIb and β3 integrin transmembrane tails. Finally, several transmembrane domain mutations that activate integrins reduce the αβ-integrin association. Thus, this affinity capture assay can be used to study interactions among transmembrane domains, and has documented the importance of αβ-integrin transmembrane interactions in integrin activation.
Figure 2. An affinity-capture method to study transmembrane domain interactions.
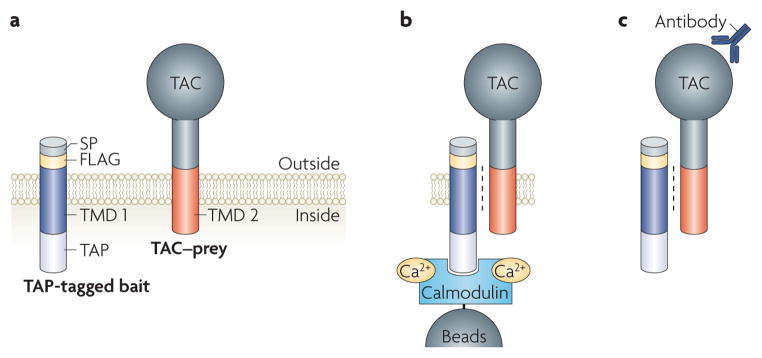
Use of an affinity-capture method reveals the preferential interaction of αIIb and β3 integrin transmembrane domains (TMDs). a | The transmembrane domain tail bait and prey constructs are depicted. The bait consists of the transmembrane and cytoplasmic domain fused to a FLAG tag (for detection), with a signal peptide at the amino terminus and a tandem affinity purification (TAP) tag at the carboxy terminus. The transmembrane and cytoplasmic domains of the prey are joined at the N terminus to the extracellular domain of an irrelevant type 1 membrane protein, such as the TAC subunit of the interleukin 2 receptor. b | Chinese hamster ovary cells are transiently transfected with baits and preys, cells are lysed and the bait is rapidly and efficiently captured through its TAP tag. Capture of the TAP tag with calmodulin beads is depicted. c | Bound preys are detected by western blotting using an anti-TAC antibody. SP, signal peptide.
αβ-Integrin transmembrane domain structure enables signalling
An NMR structure of the αβ-integrin transmembrane complex reveals that the transmembrane domains primarily associate through two structural elements, one in the inner membrane leaflet, which extends into the membrane–cytoplasmic interface, and the other at the outer leaflet of the lipid bilayer44 (FIG. 1c–e). In this way, perturbations at the cytoplasmic face, or separation of the legs of the ectodomain, can destabilize the αβ-integrin transmembrane dimer. Early models envisaged a coiled-coil-like arrangement for the αβ-integrin transmembrane complex62; however, the extended intersubunit interface of such a structure might be too stable to transmit perturbations at the cytoplasmic face to the ectodomain. The αIIbβ3 integrin transmembrane domain structure44 reveals that the dimer is stabilized by two structural assemblies, termed the inner membrane clasp (IMC) and outer membrane clasp (OMC) (FIG. 1c–e). The OMC is formed by the packing interactions of three Gly residues, Gly972 of αIIb integrin, Gly976 of αIIb integrin and Gly708 of β3 integrin, which cause the α- and β-integrin transmembrane helices to cross within their N-terminal halves at an angle of ~ 25° (FIG. 1d). Because of this crossing angle and their differing lengths, the αIIb and β3 integrin transmembrane helices would dissociate C-terminally to Lys712 of β3 integrin. However, this loss of contact is overcome by the placement of Phe992–Phe993 of αIIb integrin between the transmembrane domains, which brings the aromatic rings of these residues in proximity to the aromatic ring of Trp715 of β3 integrin, and by contacts between Ile719 of β3 integrin and Phe992–Phe993 of αIIb integrin. This structural motif brings Arg995 of αIIb integrin and Asp723 of β3 integrin into sufficient proximity to enable the electrostatic interactions that were predicted nearly 15 years ago17 to stabilize the IMC (FIG. 1e). Importantly, NMR structures calculated without using this electrostatic interaction as a distance restraint reveal an essentially identical IMC structure44. Furthermore, mutations that disrupt this electrostatic interaction lead to destabilization of the αβ-integrin transmembrane dimer, as shown by NMR spectroscopy in bicelles and affinity capture in mammalian cell membranes65. Rosetta modelling66 makes use of computer searches to identify short sequences of known structure for use in the prediction of protein structure from amino acid sequence. Thirty percent of the cluster 1 structures predicted by Rosetta modelling, combined with sparse restraints, indicate that Arg995 of αIIb integrin and Asp723 of β3 integrin are in proximity. Many of the Rosetta structures also indicate that the side chain of Lys716 of β3 integrin forms hydrogen bonds with αIIb integrin backbone carbonyl oxygens to stabilize the α- and β-integrin association. Substitution of this Lys with polar neutral or acidic residues or a bulky hydrophobic residue activated αIIbβ3 integrin, which was interpreted to provide support for this additional αβ-integrin interaction67. The IMC structure in phospholipid bicelles differs from that of the same region of the isolated cytoplasmic domains in aqueous solution60, suggesting that the distinct lipid tail-to-headgroup environment is important in driving IMC assembly. Indeed, the IMC structure formed by the αIIb and β3 integrin transmembrane cytoplasmic domains in a 50% acetonitrile/water solution closely resembles that of the isolated cytoplasmic domains in water68. Mutations that disrupt either the IMC or OMC destabilize the association of the αIIb and β3 integrin transmembrane domains44,65, providing experimental validation for the idea that both clasps are required to maintain the transmembrane complex. Thus, the binding of cytoplasmic proteins to the integrin intracellular domains can disrupt the IMC, in a manner that is described below for talin, destabilizing the transmembrane complex and resulting in rearrangements in the ectodomain that lead to integrin activation.
The structure of the integrin transmembrane domain described above was obtained with integrin transmembrane peptides in a model membrane, but how does it relate to the structure of an intact integrin in the plasma membrane? Rosetta modelling66 was combined with a few distance restraints provided by engineered disulphide bonds between introduced Cys mutations in the α- and β-integrin subunits to calculate seven clusters of (collections of similar) low energy models of the structure of the αIIbβ3 integrin transmembrane complex in mammalian cell membranes67. The centre structure of the most highly populated cluster is similar to the average structure calculated from NMR restraints obtained with αIIbβ3 integrin transmembrane peptides in bicelles44. The calculated NMR structures of the αIIb and β3 integrin transmembrane monomers were available before publication of the Rosetta model; however, the authors emphasized that those structures were not used to inform the Rosetta modelling67 or the selection of the representative structure. Similarly, models of the αIIbβ3 integrin transmembrane domain, derived by two different methods, both converged on the published NMR structure. These models exhibited close similarities with the averaged NMR structure; the root mean square deviation of α-carbons (or backbone carbons linked to both the amide and carbonyl groups in amino acids) was 1.1 and 1.6 Å from the averaged NMR structure69. Consequently, the modelling approaches used complementary methods to independently derive similar overall topographies to the NMR-derived structures of the integrin transmembrane domains in bicelles. Importantly, the sequences that form the IMC are highly conserved between integrins, suggesting that the mechanisms that regulate the IMC to induce integrin activation are likely to be shared. Indeed, the same cytoplasmic proteins (talins and kindlins) are involved in activating multiple classes of integrins (see below). Conversely, the OMC is less conserved in sequence, suggesting that the stability of the OMC might differ between integrins. These sequence variations may account for differences in transmembrane signalling among integrin classes.
How is integrin activation transmitted?
After the idea emerged that interactions between integrin cytoplasmic and transmembrane domains might maintain the low affinity state17, the idea followed that activation involves a rearrangement of these domains. Protein engineering studies established that enforcing the association of the integrin transmembrane or cytoplasmic domains with coiled coils or engineered disulphide bonds prevents integrin activation15,51,58,70. More importantly, elegant work showed that mutational activation of a recombinant integrin altered the formation of intersubunit disulphide bonds between Cys mutations in the outer portion of the transmembrane domain, suggesting that a complete separation of the transmembrane domains leads to integrin activation51. Oxidation-dependent disulphides formed in the activated integrin at 37°, but not at 0°. The authors emphasized that at 0° the mobility of these transmembrane domains would be greatly reduced, thus the lack of disulphide formation at this temperature indicates a loss of stable transmembrane domain association. In a subsequent paper67, the same group found that certain activating Cys substitutions did not prevent the formation of engineered disulphides at 0° in the transmembrane or cytoplasmic domains. Inclusion of these disulphides as restraints in the Rosetta calculations did not alter the predicted structures67. These results imply that the α- and β-integrin transmembrane domains continue to interact in integrins bearing these activating Cys substitutions. The authors have proposed that this seeming discrepancy may be because “The apparent lack of effect on cross-linking by these activating mutations may result from the use of 0°C and 37°C in cross-linking and activation assays, respectively” (REF. 67). Measures of the interactions of α- and β-integrin transmembrane domains showed that certain activating mutations can weaken but not completely disrupt their association44,65. Thus, whereas the idea that integrin activation requires complete disruption of transmembrane assembly is attractive, available evidence does not exclude other plausible rearrangements45.
Cytoplasmic activators of integrins
The idea that the integrin cytoplasmic domains are the trigger point for conformational changes that result in activation47,48 led to efforts to find cytoplasmic domain-binding proteins that might mediate this process. Many candidates have been identified71 and compelling evidence shows that talins and kindlins are major players.
Talins activate integrins
Studies in cultured cells showed that the binding of talin 1 to the cytoplasmic domain of the β-integrin subunit is a common step in β1 and β3 integrin activation in vitro72. Later studies extended this principle to β2 integrins in vitro73 and to mice, in which deletion of platelet talin 1 blocks activation of platelet β1 and β3 integrins74,75. Furthermore, insights from structural studies76 enabled the creation of mice in which β3 integrin–talin 1 binding was disrupted. These mice were defective in activating αIIbβ3 integrin3 and protected from pathological thrombosis, without experiencing the severe bleeding associated with complete loss of β3 integrin3. Thus, disrupting the β3 integrin–talin 1 interaction may offer an anti-thrombotic benefit by blocking integrin activation. In addition to activating integrins, talin 1 links integrins to filamentous actin (F-actin) and actin-binding proteins (reviewed in REF. 77), thereby linking the actin cytoskeleton to the ECM. More recent analysis of talin 1 structure has identified talin 1 and integrin mutants that have little effect on the binding of talin 1 to the β-integrin tail, or on talin 1 recruitment to integrin in cells, but do block the ability of talin 1 to induce activation78,79. These mutants offer the possibility of selectively disrupting the ability of talin 1 to activate integrins, without preventing integrin linkage to the cytoskeleton.
Talin 1 consists of a large C-terminal rod and an N-terminal head domain (THD) containing four subdomains: F0, F1, F2 and F3 (REFS 76,80,81). The F3 subdomain has a PTB domain, which contains a high affinity binding site for β-integrin tails and is sufficient to activate integrins81; other portions of the THD enhance activation82. A crystal structure of the THD F2 and F3 subdomains in complex with a 12 residue fragment from the mid-portion of the β3 integrin tail reveals that the F3–integrin interaction strongly resembles PTB domain interactions with peptide ligands76. Several other PTB domains bind to β3 integrin in a similar manner83, but talin 1 is uniquely designed to activate integrins because of an additional interaction between it and the membrane-proximal region of the β3 integrin cytoplasmic domain60,79,84,85. Mutations in the integrin or talin that block this interaction prevent integrin activation in cells. Thus, talin F3 interacts with β-integrin tails through a PTB-like interaction that is shared with many PTB domain-containing proteins and through a second interaction that is not shared with most of these PTB domains but is required for integrin activation.
How does the interaction of talin 1 with the membrane-proximal portion of the β-integrin tail lead to rearrangement of the integrin transmembranes to cause activation? Talin 1 binding destabilizes the interaction of the αIIb integrin transmembrane tail with the β3 integrin transmembrane tail60,65. Structure–function analysis of the talin F3 and β-integrin tail interaction79, together with the structure of the integrin transmembrane complex44, provide a compelling model to explain how talin 1 can alter the integrin transmembrane complex. First, binding of talin F3 stabilizes the helical structure of the membrane-proximal β3 integrin tail79 such that the β3 integrin transmembrane domain forms a continuous helix19. Second, the F3–β3 integrin interaction orients a group of Lys residues in F3 towards the negatively charged membrane phospholipid head groups. Mutation of some of these Lys residues disrupts activation79. An additional contribution may come from the asymmetric structure of the αIIbβ3 integrin transmembrane at the cytosolic face. In particular, the non-helical Phe992–Phe993 segment of αIIb integrin juxtaposes Arg995 of αIIb integrin and Asp723 of β3 integrin so that an electrostatic interaction can stabilize the transmembrane complex. Arg995 of αIIb integrin and Asp723 of β3 integrin are readily accessible to the THD, which could therefore prevent this electrostatic interaction (FIG. 1). Indeed, a recent structure of the F2–F3 region of a talin 1 paralogue, talin 2, in complex with the β1D integrin cytoplasmic domain revealed that talins can form a salt bridge with the conserved Asp residue of the β-integrin subunit (for example, Asp723 of β3 integrin), thus potentially disrupting its electrostatic interaction with the conserved Arg residue in the α-integrin subunit (for example Arg995 of αIIb integrin)86. This structure also identified additional basic residues in F2 that form a ‘membrane orientation patch’ that can interact with phospholipid head groups to enable talin to alter the tilt angle of the β-integrin transmembrane domain. The predicted capacity of talin to alter this tilt angle explains why talin binding is required for full activation72,79, even when the interaction of Arg995 of αIIb integrin with Asp723 of β3 integrin is prevented by mutation of Asp723 of β3 integrin. In sum, the THD is exquisitely engineered for activating integrins by binding to β-integrin tails through a PTB-like interaction and by engaging a membrane-proximal β-integrin tail site, which has three important consequences. First, it positions basic patches on talins for an extended electrostatic interaction with the phospholipid head groups of the membrane. Second, it favours the formation of a stable, continuous helix that spans the β-integrin transmembrane and the membrane-proximal portion of the tail, enabling talins to enforce an altered crossing angle on the β-integrin transmembrane domain. Third, talins may directly disrupt the conserved α-integrin Arg and β-integrin Asp interaction by forming a salt bridge with the β-integrin Asp. This unique combination of structural elements in talins, and complementary elements in integrins, explains why they are obligatory partners in the activation process.
Kindlins cooperate with talins
Talins are essential for integrin activation, but are they sufficient to activate integrins? Recent studies from model organisms and humans have established that another family of β-integrin-binding proteins, the kindlins, are important players in integrin activation. The ~ 76 kDa vertebrate kindlins include kindlin 1 (also known as FERMT1 and URP1), kindlin 2 (also known as FERMT2, MIG2 and URP3) and kindlin 3 (also known as FERMT3 and URP2). Each is structurally related to UNC-112, a Caenorhabditis elegans protein implicated in integrin-dependent muscle development87. Kindlins and UNC-112 contain a FERM domain near the C-terminus that is similar in sequence to the talin FERM domain, but is unique in that its F2 subdomain is interrupted by a pleckstrin homology domain88. The split FERM domain, and its F3 subdomain in particular, mediates the interaction of kindlins with β-integrin cytoplasmic tails. This interaction requires a region of the integrin tails (for example, Asn-X-X-Tyr in β1 and β3 integrins and Asn-X-X-Phe in β2 integrin; where X is any amino acid) that is distal to the talin-binding Asn-Pro-X-Tyr/Phe region, as well as a Ser or Thr in an 8–16 amino acid tract that separates these two regions89–91. Kindlin 2 and UNC-112 also interact, through conserved regions N-terminal to their FERM domain, with two additional proteins, migfilin and integrin-linked kinase, which are frequently found in adhesion complexes87,89,92.
Kindlin 1 deficiency in mice and humans causes Kindler syndrome — epithelial cell dysfunction leading to a skin blistering phenotype and gasterointestinal manifestations93–95. Morpholino knockdown of kindlin 2 in zebrafish embryos causes abnormalities of cardiac muscle development owing to defective cytoskeletal organization at sites of membrane attachment96, and kindlin 2 deficiency in mice causes peri-implantation lethality owing to detachment of the endoderm and epiblast from basement membranes89. Mice deficient in kindlin 3 die with diffuse haemorrhages and osteopetrosis shortly after birth97. Given the known integrin and cytoskeletal protein interaction partners of the kindlins, defects in bi directional integrin signalling probably underlie some of these severe phenotypes.
Studies of cells using knockdown or overexpression strategies indicate that kindlin 1, kindlin 2 and kindlin 3 are capable of regulating the activation of specific integrins, but only in concert with the interaction of talin 1 with the integrin cytoplasmic tail. For example, ligand binding to αIIbβ3 or α5β1 integrins in Chinese hamster ovary (CHO) cells is stimulated by overexpression of the THD. This activation of αIIbβ3 integrin, but not of α5β1 integrin, is increased by co-expression of kindlin 1 or kindlin 2 but not kindlin 3, and is decreased by small interfering RNA knockdown of endogenous kindlin 2. However, neither kindlin 1 nor kindlin 2 are stimulatory in the absence of THD89–91,93. In another study, loss of kindlin 1 from intestinal epithelial cells or a colon carcinoma cell line reduced talin-dependent β1 integrin activation and/or β1 integrin-mediated cell adhesion95. Thus, kindlins can co-activate integrins and talin 1, but their precise effects may vary with the kindlin, integrin and cell type involved.
Kindlin 3 in leukocyte and platelet integrin activation
Platelets that develop from mice with kindlin 3-deficient haematopoietic precursors exhibit defective activation of the αIIbβ3 integrin fibrinogen receptor and the α2β1 integrin collagen receptor, and impaired aggregation97. Kindlin 3-deficient platelets also show reduced adhesion to fibrinogen after direct activation of αIIbβ3 integrin by MnCl2, suggesting an additional defect in outside–in αIIbβ3 integrin signalling. Furthermore, the mice are resistant to mesenteric arteriolar thrombosis following vessel injury by FeCl3. In addition, kindlin 3 deficiency results in defective activation of neutrophil β2 integrins, as evidenced by reduced agonist-dependent binding of intercellular adhesion molecule 1 (ICAM1) and the inactive complement factor 3b fragment (iC3b) in vitro, and defective firm adhesion and arrest of neutrophils on activated endothelial cells in vivo98. These platelet and leukocyte integrin defects in kindlin 3-deficient mice phenocopy blood cell abnormalities in a rare human autosomal recessive disorder called leukocyte adhesion deficiency 1 (LAD1) variant (LAD 1v; also known as LAD3)99,100. This disorder is characterized by recurrent bleeding similar to that seen in individuals with Glanzmann thrombasthenia owing to a lack of αIIbβ3 integrin, and by a purulent bacterial infection and leukocytosis similar to that seen in individuals with LAD1 caused by a lack of β2 integrins. In contrast, LAD1v platelets and leukocytes express these integrins but exhibit an impairment of agonist-induced integrin activation.
Earlier studies had suggested that LAD1v is due to a splicing defect in the Ca2+- and DAG-regulated guanine nucleotide exchange factor 1 (CALDAG-GEFI; also known as RASGRP1) gene, resulting in reduced levels of its encoded protein RAP1 guanine nucleotide exchange factor (GEF) in haematopoietic cells101. This seemed reasonable because RAP1 is involved in integrin activation102–104, and CALDAG-GEFI-knockout mice105,106 exhibit defects, albeit partial, in platelet and leukocyte integrin activation. However, recent studies have now shown that kindlin 3 mutations are a cause of LADIv in several families, including some previously reported to be deficient in CALDAG-GEFI and others with normal levels of CALDAG-GEFI107–110. Affected individuals share a common haplotype involving a region on chromosome 11 that harbours mutations in kindlin 3 that result in a premature stop codon108–110. Primary haemato poietic cells or EBV-transformed lymphocytes from affected individuals exhibit reduced levels of kindlin 3 mRNA107 or absent kindlin 3 protein108,109. Various haematopoietic cells show defective agonist-induced binding of ligands to β1, β2 or β3 integrins99,107,108. Importantly, the integrin phenotype in kindlin 3-deficient cells is rescued by expression of recombinant kindlin 3 (REFS 107,108), and RNA interference-mediated knockdown of kindlin 3 in normal haematopoietic cells recapitulates the integrin phenotype108.
Whereas the evidence is clear that kindlins are key regulators of talin-dependent integrin activation by virtue of their association with β-integrin cytoplasmic domains, many questions remain (FIG. 3). Is the kindlin interaction with β-integrin cytoplasmic domains regulated and, if so, how? The kindlin-binding protein, migfilin, binds to filamin A. As filamin A can block talin 1 binding to β-integrin tails, what role, if any, does the shuttling of filamin A on and off integrins have in the ability of kindlins to co-activate integrins111,112 (for example, see FIG. 3c)? Is talin the direct integrin activator and kindlin the enabler, or is the reverse true (for example, see FIG. 3b)? Do all kindlins activate integrins in the same way? Are there yet unidentified integrin-binding proteins that are required for integrin activation in concert with talins and kindlins (for example, see FIG. 3a)? How do the kindlins participate in outside–in integrin signalling89?
Figure 3. Activators, such as talins and kindlins, bind to integrins to cause their activation.

a | In a direct model of integrin activation, both activators (A1 and A2) bind simultaneously to the integrin tail and, together, modify or disrupt the inner membrane clasp. Other proteins might be involved. In the other two general models, A1 is the primary activator and A2 is an ‘enabler’. b | In an indirect model, A2 regulates a signalling event (for example, synthesis of co-factors) that enables the activator (A1) to bind β-integrin and induce activation. c | In a displacement of an inhibitor model, A2–β-integrin binding displaces an inhibitor of A1, enabling A1 to bind and activate the integrin.
RIAM activates talin 1
Agonist stimulation (the triggering event) leads to integrin activation through many signalling intermediaries. If talin 1 binding is a common step in integrin activation, how do these signalling intermediaries regulate the talin–integrin interaction? Recent work has elucidated one such group of signalling intermediaries that are important in this activation — the Ras GTPases113. Ras proteins are small monomeric GTPases that cycle between the GTP-bound active form and the GDP-bound inactive form. GEFs promote Ras activity by exchanging bound GDP for GTP, whereas GTPase activating proteins (GAPs) enhance the hydrolysis of Ras-bound GTP to GDP114. The Ras subfamily members RAP1A and RAP1B stimulate integrin activation102–104. Knockout of RAP1B115 or its exchange factor CALDAG-GEFI116 in mice, results in the partial impairment of agonist-dependent fibrinogen binding to αIIbβ3 integrin and platelet aggregation. Several RAP1 effectors are implicated in integrin activation117–119. RAP1–GTP-interacting adaptor molecule (RIAM; also known as APBB1IP) is a RAP1 effector that is a member of the MIG10, RIAM and lamellipodin (MRL) family of adaptor proteins118. RIAM contains Ras association and pleckstrin homology domains and Pro-rich regions. In lymphoid cells, RIAM overexpression induces β1 and β2 integrin-mediated cell adhesion, and RIAM knockdown abolishes RAP1-dependent cell adhesion118. RIAM increases cellular F-actin content118, possibly through its interaction with ENA and VASP — related proteins that can promote actin polymerization to form F-actin. Whereas RIAM is enriched in haemato poietic cells, lamellipodin is a paralogue present in fibroblasts and other cells120.
Agonists do not efficiently activate recombinant αIIbβ3 integrin expressed in CHO cells; this observation led to a synthetic reconstruction of an integrin activation pathway in CHO cells. Its use in combination with forward and reverse genetics enabled the dissection of a pathway to integrin activation121. RAP1 activation induces the association of RAP1, RIAM and talin 1, which leads to αIIbβ3 integrin–talin 1 interactions. More recently, CHO cells transfected with the thrombin receptor proteinase- activated receptor 1 (PAR1; also known as F2R) enabled activation of αIIbβ3 integrin by a natural platelet agonist. Furthermore, bimolecular fluorescence complementation showed that RIAM overexpression stimulates, and RIAM knockdown blocks, talin 1 recruitment to αIIbβ3 integrin in living cells78. These studies facilitated the construction of a road map between receptor agonists and integrin activation (FIG. 4). Moreover, mapping studies identified short amphipathic helices in RIAM and lamellipodin that bind talin 1; joining these helical peptides to the membrane targeting sequences of RAP1 led to a minimal RAP1–RIAM module that was sufficient to recruit talin 1 to integrins and to activate the integrins122. Thus, RIAM functions as a scaffold that connects the membrane-targeting sequences in Ras GTPases to talin 1, thereby recruiting talin 1 to the plasma membrane and activating integrins. An intriguing alternative mechanism was identified in lymphocytes, in which WASP-family verprolin homologue 2 (WAVE2), an actin-nucleating protein, recruited vinculin to the immunological synapse, thereby recruiting talin 1 (REFS 123). Taken together, these studies raise the possibility of a general mechanism for integrin activation: talin-binding proteins that contain membrane-targeting motifs or that associate with proteins that possess such motifs can target talin 1 to integrins and induce activation.
Figure 4. A road map from thrombin receptors to αIIbβ3 integrin activation.
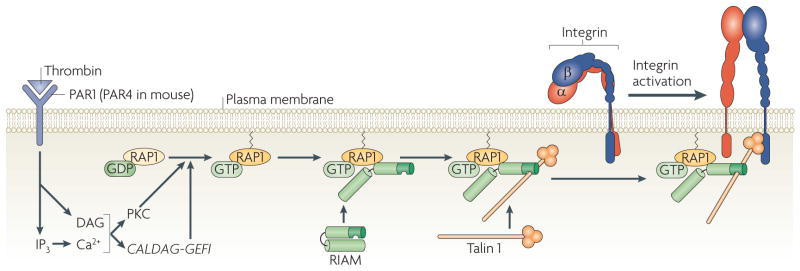
The schematic represents the minimal elements of one pathway of αIIbβ3 integrin activation by thrombin receptors, which were identified through the synthetic reconstruction of pathway components in Chinese hamster ovary cells and studies of gene-targeted platelets. Thrombin cleavage or ligand occupancy of the thrombin receptor proteinase-activated receptor 1 (PAR1; also known as F2R) in human platelets, or PAR4 receptors in mouse platelets, stimulates phospholipid hydrolysis, which results in the generation of inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 stimulates an increase in cytosolic free Ca2+, activating Ca2+- and DAG-regulated (CALDAG-GEFI; also known as RASGRP1), which in turn converts its encoded protein, RAP1, from a GDP-bound to an active GTP-bound form. Ca2+ and DAG also activate certain protein kinase C (PKC) isoforms, including PKCα, which among other actions may facilitate the activation of CALDAG-GEFI. Activation of RAP1 leads to recruitment of its effector, RAP1–GTP-interacting adaptor molecule (RIAM; also known as APBB1IP), and its binding partner, talin 1, to the plasma membrane. This enables talin binding to the β3 integrin tail and talin-induced activation of αIIbβ3 integrin. Kindlin 3 plays a crucial role in this process, but because its mechanistic role is uncertain, it is not depicted here.
α-Integrin subunit-specific activators
Talin 1 and kindlins bind to β-integrin subunits; however, early experiments pointed to an important role for the cytoplasmic domains of α-integrin subunits in regulating activation47,124. Whereas the protein sequences of the membrane proximal α-integrin subunits that form the IMC (for example, Gly-Phe-Phe-Lys-Arg) are well conserved, the sequences of the more distal α-integrin subunits are far more variable. Thus, because there are 18 α-integrin subunits, the complex literature on α-integrin subunit-binding proteins is too large to be thoroughly reviewed here. Nevertheless, a few outstanding examples will be mentioned. Naik, Parise and co-workers identified Ca2+- and integrin-binding protein 1 (CIB1) as an αIIb integrin tail-binding protein125 and subsequent work reported that it functions to oppose talin 1 binding, thereby serving as an inhibitor of activation126. Surprisingly, a lack of platelet CIB1 led to defective thrombosis and no increase in αIIbβ3 integrin activation127, possibly owing to compensation by CIB1 paralogues128. Similarly, Katagiri, Kinashi and co-workers identified regulator for cell adhesion and polarization enriched in lymphoid tissues (RAPL; also known as NORE1 and RASSF5) as a RAP1-binding protein that physically associates with αLβ2 integrin in an αL integrin-specific manner and regulates αLβ2 integrin-mediated adhesiveness129,130. More recent elegant in vivo studies131–133 show that RAPL regulates lymphocyte trafficking, in part through MST1 (also known as STK4), a STE20 kinase-like binding partner. In addition, these studies have also clarified the complementary roles of talin 1 and RAPL in the regulation of lymphocyte adhesion by RAP1 (REF. 131). It is clear that the diversity of α-integrin tail sequences and the consequent plethora of α-integrin-binding proteins will continue to be an exciting and fertile area for future investigation.
Endogenous suppressors of integrins
Negative regulators of integrin activation might be as important as positive regulators. In principle, negative regulation might occur at any step in the process of inside–out signalling. This is exemplified in platelets by the blockade of specific agonist pathways to αIIbβ3 integrin activation by aspirin, which inhibits cyclooxygenase, thus blocking synthesis of thromboxane, or by clopidogrel, which blocks P2Y12 ADP receptors134. Similarly, enforced activation of extracellular signal-regulated kinase 1 (ERK1; also known as MAPK3) and ERK2 (also known as MAPK1) by activated HRas suppresses integrin activation in many cell types135, an effect that may be pertinent to the changes in adhesion and ECM assembly of transformed cells136. ERK1 and ERK2 kinase activity is required for this suppression, and they exert their effects at the plasma membrane137. However, the relevant ERK1 and ERK2 substrate or substrates have not been identified.
Most pertinent to this review is the potential for the regulation of integrin activation at its final steps — through the interactions of talin 1 or kindlins with integrin tails. One example of this type of regulation may be the expression and localization to adhesion sites of phosphatidylinositol phosphate kinase type Iγ-90, a protein that competes with β-integrin tails for binding to talin 1 (REFS 138,139). Another example is the Tyr phosphorylation of β-integrin tails that is triggered by ligand binding to integrins, and mediated by Src family kinases140. Tyr in the membrane-proximal Asn-Pro-X-Tyr motif of β-integrin tails may exert multiple effects on cell adhesion through phosphorylation-dependent and phosphorylation-independent mechanisms141–144. Phosphorylation of this Tyr may serve as an integrin activation ‘off switch’ by interfering with required acidic and hydrophobic interactions between this region of the β-integrin tail and talin 1, thereby reducing the affinity of the interaction. Moreover, Tyr phosphorylation promotes the interaction of the β-integrin tail with competing PTB domain-containing proteins, such as docking protein 1 (DOK1), which, unlike talin 1, do not activate integrins83,145. Furthermore, integrin cytoplasmic domain-associated protein 1 (ICAP1; also known as ITGB1BP1) can bind to the β1A integrin tail and compete for talin 1 binding, thus blocking activation146. Another potential negative regulator of integrin activation is filamin A, the blockade of talin 1 binding to β-integrin tails by which may be regulated by kindlin through their mutual binding partner, migfilin111,147. To date, studies of negative regulation of integrin activation have been conducted largely with purified proteins or cell lines. Determining their bio logical significance will require further work in model organisms and humans.
Activation of integrins from the outside
Although the focus of this review is on the inside–out activation of integrins, integrins can be activated directly by extracellular factors, including ECM ligands, and ligand binding to integrins triggers outside–in signalling148. Non-physiological reducing agents such as dithiothreitol149 have been used experimentally for years to activate purified integrins and integrins in cells. For example, reducing agents activate αIIbβ3 integrin in platelets, a response attributed to disulphide exchange between selected Cys residues in the Cys-rich extracellular epidermal growth factor (EGF)-like domains of β3 integrin150. Disulphide exchange involving αIIbβ3 or αVβ3 integrins may occur during agonist-induced integrin activation and require thiol isomerases, such as protein disulphide isomerase or endoplasmic reticulum protein 5, or thiol isomerase activity intrinsic to β3 integrin149,151,152. However, the role of di sulphide exchange in integrin activation in cells, and how it relates to talin-dependent activation, will require more study.
Integrin affinity can also be modulated extrinsically by the binding of ligands. Even the binding of monovalent ligands, such as short Arg-Gly-Asp peptides, can induce conformational changes in integrin ectodomains153, as reported by ligand-induced binding site antibodies (anti-LIBS)154. FRET studies indicate that these conformational changes can be propagated across the plasma membrane, leading to alteration of the α- and β-integrin tails155. Consequently, inside–out and outside–in signalling responses are coupled by dynamic interactions of the integrins with proteins on both sides of the plasma membrane, and they are further modified by forces applied to integrins in adherent cells by virtue of integrin linkages with the ECM and the cytoskeleton.
Perspectives
Integrin activation has been studied for over two decades by a range of experimental techniques. Progress in this area has accelerated in recent years owing to studies using forward and reverse genetics, biochemistry and cell and structural biology. In addition, studies of integrin activation are a prime example of successful bidirectional information transfer between basic scientists and inquisitive clinicians making careful observations on patients with perplexing abnormalities of cell adhesion. Consequently, this field has come closer to a molecular understanding of the ‘end game’, the final cell signalling events that regulate activation at the level of integrin transmembrane and cytoplasmic domains. Predictably, new discoveries have led to new questions, such as the precise relationships between talins, kindlins and other regulatory proteins during integrin activation, and the structural basis of integrin activation in the context of intact integrin heterodimers in their native membrane environments. Thus, for integrinologists the end game is not the end of the game.
Acknowledgments
Researches quoted from our laboratories were supported by the National Institutes of Health. C.K. is the recipient of a postdoctoral fellowship from the American Institute for Cancer Research. We thank our colleagues for their understanding in cases where space limitations have forced us to cite or discuss their work less thoroughly than we would have liked to.
Glossary
- Valency
A term that refers to the number of chemical bonds between two atoms. Here it refers to the number of integrin-binding sites presented by a given adhesive ligand
- Microcluster
A loosely defined term that refers to a non-covalent oligomer of integrins on the cell surface that appears as a point source (that is, with a diameter < 100nm) in fluorescence microscopy
- Detergent micelle
A globular aggregate of amphipathic detergent in aqueous solution, with detergent hydrophilic ends facing outside and hydrophobic ends facing inside
- Förster resonance energy transfer
(FRET). A phenomenon in which one fluorophore (the donor) in its electronic excited state can transfer its energy to another fluorophore (the acceptor) in close proximity, so that excitation of the donor causes the acceptor to emit fluorescence. As the FRET only occurs when the distance between donor and acceptor is less than 10 nm, it is useful for monitoring interactions between two fluorophore-fused molecules
- Focal complex
A relatively small dot-like adhesion (~ 1 μm in width) mainly found in lamellipodia. It is a transient adhesion site during cell migration and can mature into a more stable focal adhesion
- Focal adhesion
A large (2–5 μm in width), elongated, oval-shaped protein complex found on the cell periphery, which connects the actin cytoskeleton (F-actin bundle) to the ECM and provides strong integrin-dependent adhesion
- Immunological synapse
A cell–cell junction between a T lymphocyte and an antigen presenting cell during T lymphocyte activation
- Podosome
A type of ECM contact that is different from focal complexes and focal adhesions. Podosomes have a core actin filament surrounded by a ring structure of integrin adhesive complexes. They are shorter than other ECM contacts in depth (~ 0.2–0.4 μm) and are typically found in monocytic lineages
- Invadopodium
A type of ECM contact that is different from focal complexes and focal adhesions but similar to podosomes. Invadopodia can extend up to ~ 8 μm, associate with ECM-degrading enzymes and are seen in transformed fibroblasts or malignant cells
- Bioluminescence resonance energy transfer
A FRET-like phenomenon in which bioluminescence generated by luciferase (the donor) can excite a nearby fluorophore (the acceptor)
- Image correlation spectroscopy
A method used to analyse molecular densities and rates of aggregation and diffusion of fluorescent molecules by autocorrelating the temporal (or spatial) fluctuation of intensities in confocal images
- Interferometric photoactivated localization microscopy
A recently developed fluorescent microscopy that provides 20 nm resolution in three dimensions, thus allowing single-molecule imaging
- Phospholipid bicelle
A planar disc-shaped particle made of a phospholipid mixture. The centre of the bicelle consists of two layers of phospholipids and the edge of the bicelle is covered by phospholipids with shorter lipid chains
- Microsomal membrane
A membrane vesicle that is generated by fragmentation of the endoplasmic reticulum
- Coiled coil
A protein structure generated by dimerization or multimerization of α-helices. These α-helices typically consist of repeats of two hydrophilic residues, followed by a hydrophobic residue that is buried into the binding interface in aqueous solution
- Pathological thrombosis
The formation of an occlusive mass of fibrin, platelets and leukocytes in a blood vessel, which results in diseases such as myocardial infarction and stroke
- PTB domain
(Phosphotyrosine binding domain). A protein domain that recognizes an Asn-Pro-X-Tyr motif that is found in most β-integrin tails
- FERM domain
(4.1 protein, ezrin, radixin and moesin homology domain). A common domain found in a number of proteins that mediate linkage of the cytoskeleton to the plasma membrane. The FERM domain often interacts with the cytoplasmic tail of transmembrane proteins
- Pleckstrin homology domain
A lipid-binding protein domain originally identified in Pleckstrin
- Mesenteric arteriolar thrombosis
Thrombosis that occurs in an arterial vessel of mesentery. Mesentery is the anatomical term indicating the layers of membrane that suspend the small intestine from the back wall of the abdomen
- Glanzmann thrombasthenia
A genetic bleeding disorder caused by a lack of αIIbβ3 integrin or by mutations that inhibit αIIbβ3 integrin function
- Amphipathic helix
An α-helix that contains hydrophilic amino acids on one side and hydrophobic amino acids on the other
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.Ginsberg MH, Partridge A, Shattil SJ. Integrin regulation. Curr Opin Cell Biol. 2005;17:509–516. doi: 10.1016/j.ceb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 3.Petrich BG, et al. The antithrombotic potential of selective blockade of talin-dependent integrin αIIbβ3 (platelet GPIIb-IIIa) activation. J Clin Invest. 2007;117:2250–2259. doi: 10.1172/JCI31024. Shows that the mutational inhibition of talin 1 binding to the β3 integrin tail prevents activation of αIIbβ3 integrin, resulting in protection from thrombosis without spontaneous pathological bleeding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carman CV, Springer TA. Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr Opin Cell Biol. 2003;15:547–556. doi: 10.1016/j.ceb.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Bazzoni G, Hemler ME. Are changes in integrin affinity and conformation overemphasized? Trends Biochem Sci. 1998;23:30–34. doi: 10.1016/s0968-0004(97)01141-9. [DOI] [PubMed] [Google Scholar]
- 6.Xiong JP, et al. Crystal structure of the extracellular segment of integrin αVβ3. Science. 2001;296:151–155. doi: 10.1126/science.1069040. The landmark high-resolution structure of the extracellular domain of αvβ3 integrin. [DOI] [PubMed] [Google Scholar]
- 7.Nishida N, et al. Activation of leukocyte β2 integrins by conversion from bent to extended conformations. Immunity. 2006;25:583–594. doi: 10.1016/j.immuni.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 8.Frelinger AL, III, et al. Occupancy of an adhesive glycoprotein receptor modulates expression of an antigenic site involved in cell adhesion. J Biol Chem. 1988;263:12397–12402. [PubMed] [Google Scholar]
- 9.Mould AP, Humphries MJ. Regulation of integrin function through conformational complexity: not simply a knee-jerk reaction? Curr Opin Cell Biol. 2004;16:544–551. doi: 10.1016/j.ceb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–611. doi: 10.1016/s0092-8674(02)00935-2. Proposes that integrin activation and molecular extension are linked. [DOI] [PubMed] [Google Scholar]
- 11.Adair BD, et al. Three-dimensional EM structure of the ectodomain of integrin αVβ3 in a complex with fibronectin. J Cell Biol. 2005;168:1109–1118. doi: 10.1083/jcb.200410068. An elegant electron microscopy study that unambiguously establishes that the bent integrin conformer can bind a macromolecular ligand. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong JP, Stehle T, Goodman SL, Arnaout MA. New insights into the structural basis of integrin activation. Blood. 2003;102:1155–1159. doi: 10.1182/blood-2003-01-0334. [DOI] [PubMed] [Google Scholar]
- 13.Emsley J, Knight CG, Farndale RW, Barnes MJ, Liddington RC. Structural basis of collagen recognition by integrin α2β1. Cell. 2000;101:47–56. doi: 10.1016/S0092-8674(00)80622-4. [DOI] [PubMed] [Google Scholar]
- 14.Luo BH, Takagi J, Springer TA. Locking the β3 integrin I-like domain into high and low affinity conformations with disulfides. J Biol Chem. 2004;279:10215–10221. doi: 10.1074/jbc.M312732200. [DOI] [PubMed] [Google Scholar]
- 15.Zhu J, et al. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol Cell. 2008;32:849–861. doi: 10.1016/j.molcel.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu J, Boylan B, Luo BH, Newman PJ, Springer TA. Tests of the extension and deadbolt models of integrin activation. J Biol Chem. 2007;282:11914–11920. doi: 10.1074/jbc.M700249200. A mutational analysis that shows that deletion of residues involved in the ‘deadbolt’ does not activate β3 integrins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes PE, et al. Breaking the integrin hinge: a defined structural constraint regulates integrin signaling. J Biol Chem. 1996;271:6571–6574. doi: 10.1074/jbc.271.12.6571. Proposes that αβ-integrin cytoplasmic domain interactions maintain the low affinity state. [DOI] [PubMed] [Google Scholar]
- 18.Adair BD, Yeager M. Three-dimensional model of the human platelet integrin αIIbβ3 based on electron cryomicroscopy and x-ray crystallography. Proc Natl Acad Sci USA. 2002;99:14059–14064. doi: 10.1073/pnas.212498199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lau TL, Partridge AW, Ginsberg MH, Ulmer TS. Structure of the integrin β3 transmembrane segment in phospholipid bicelles and detergent micelles. Biochemistry. 2008;47:4008–4016. doi: 10.1021/bi800107a. Provides a structure of a β-integrin transmembrane domain and defines its membrane insertion. [DOI] [PubMed] [Google Scholar]
- 20.Takagi J, Springer TA. Integrin activation and structural rearrangement. Immunol Rev. 2002;186:141–163. doi: 10.1034/j.1600-065x.2002.18613.x. [DOI] [PubMed] [Google Scholar]
- 21.Ye F, Liu J, Winkler H, Taylor KA. Integrin αIIbβ3 in a membrane environment remains the same height after Mn2+ activation when observed by cryoelectron tomography. J Mol Biol. 2008;378:976–986. doi: 10.1016/j.jmb.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiong JP, et al. Crystal structure of the complete integrin αVβ3 ectodomain plus an α/β transmembrane fragment. J Cell Biol. 2009;186:589–600. doi: 10.1083/jcb.200905085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puklin-Faucher E, Gao M, Schulten K, Vogel V. How the headpiece hinge angle is opened: New insights into the dynamics of integrin activation. J Cell Biol. 2006;175:349–360. doi: 10.1083/jcb.200602071. Uses steered molecular dynamics to predict that force could activate integrins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alon R, Ley K. Cells on the run: shear-regulated integrin activation in leukocyte rolling and arrest on endothelial cells. Curr Opin Cell Biol. 2008;20:525–532. doi: 10.1016/j.ceb.2008.04.003. A scholarly review that discusses the concepts of the force-induced integrin extension in the context of leukocyte function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedland JC, Lee MH, Boettiger D. Mechanically activated integrin switch controls α5β1 function. Science. 2009;323:642–644. doi: 10.1126/science.1168441. Provides direct proof that force can activate integrins. [DOI] [PubMed] [Google Scholar]
- 26.Ohi M, Li Y, Cheng Y, Walz T. Negative staining and image classification — powerful tools in modern electron microscopy. Biol Proced Online. 2004;6:23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Critchley DR, Gingras AR. Talin at a glance. J Cell Sci. 2008;121:1345–1347. doi: 10.1242/jcs.018085. [DOI] [PubMed] [Google Scholar]
- 28.Wu C. The PINCH-ILK-parvin complexes: assembly, functions and regulation. Biochim Biophys Acta. 2004;1692:55–62. doi: 10.1016/j.bbamcr.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Wu C. PINCH, N(i)ck and the ILK: network wiring at cell-matrix adhesions. Trends Cell Biol. 2005;15:460–466. doi: 10.1016/j.tcb.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 30.Li R, et al. Activation of integrin αIIbβ3 by modulation of transmembrane helix associations. Science. 2003;300:795–798. doi: 10.1126/science.1079441. [DOI] [PubMed] [Google Scholar]
- 31.Kucik DF. Rearrangement of integrins in avidity regulation by leukocytes. Immunol Res. 2002;26:199–206. doi: 10.1385/IR:26:1-3:199. [DOI] [PubMed] [Google Scholar]
- 32.Caswell PT, Norman JC. Integrin trafficking and the control of cell migration. Traffic. 2006;7:14–21. doi: 10.1111/j.1600-0854.2005.00362.x. [DOI] [PubMed] [Google Scholar]
- 33.Puklin-Faucher E, Sheetz MP. The mechanical integrin cycle. J Cell Sci. 2009;122:179–186. doi: 10.1242/jcs.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nature Rev Mol Cell Biol. 2009;10:21–33. doi: 10.1038/nrm2593. A comprehensive and thought-provoking review of the functions of focal adhesions. [DOI] [PubMed] [Google Scholar]
- 35.Dustin ML. The cellular context of T cell signaling. Immunity. 2009;30:482–492. doi: 10.1016/j.immuni.2009.03.010. A thorough review by a leader in the cell biological analysis of T cell integrin signalling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gimona M, Buccione R, Courtneidge SA, Linder S. Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol. 2008;20:235–241. doi: 10.1016/j.ceb.2008.01.005. A valuable analysis of the similarities and differences amongst these relatively less-studied adhesion structures. [DOI] [PubMed] [Google Scholar]
- 37.Buensuceso C, de Virgilio M, Shattil SJ. Detection of integrin αIIβb3 clustering in living cells. J Biol Chem. 2003;278:15217–15224. doi: 10.1074/jbc.M213234200. [DOI] [PubMed] [Google Scholar]
- 38.Coller BS, Shattil SJ. The GP IIb/IIIa (integrin αIIbβ3) odyssey: a technology driven saga of a receptor with twists, turns and even a bend. Blood. 2008;112:3011–3025. doi: 10.1182/blood-2008-06-077891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith EA, Bunch TA, Brower DL. General in vivo assay for the study of integrin cell membrane receptor microclustering. Anal Chem. 2007;79:3142–3147. doi: 10.1021/ac062008i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiseman PW, et al. Spatial mapping of integrin interactions and dynamics during cell migration by image correlation microscopy. J Cell Sci. 2004;117:5521–5534. doi: 10.1242/jcs.01416. [DOI] [PubMed] [Google Scholar]
- 41.Shtengel G, et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc Natl Acad Sci USA. 2009;106:3125–3130. doi: 10.1073/pnas.0813131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim M, Carman CV, Yang W, Salas A, Springer TA. The primacy of affinity over clustering in regulation of adhesiveness of the integrin αLβ2. J Cell Biol. 2004;167:1241–1253. doi: 10.1083/jcb.200404160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lau TL, Dua V, Ulmer TS. Structure of the integrin αIIb transmembrane segment. J Biol Chem. 2008;283:16162–16168. doi: 10.1074/jbc.M801748200. Reports the unusual structure of an α-integrin transmembrane domain and describes the membrane insertion of this domain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lau TL, Kim C, Ginsberg MH, Ulmer TS. The structure of the integrin αIIbβ3 transmembrane complex explains integrin transmembrane signalling. EMBO J. 2009;28:1351–1361. doi: 10.1038/emboj.2009.63. Provides the first structure of a heterodimeric transmembrane domain and shows how integrin transmembrane domains are designed for bidirectional signalling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williams MJ, Hughes PE, O’Toole TE, Ginsberg MH. The inner world of cell adhesion: integrin cytoplasmic domains. Trends Cell Biol. 1994;4:109–112. doi: 10.1016/0962-8924(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 46.Armulik A, Nilsson I, von Heijne G, Johansson S. Determination of the border between the transmembrane and cytoplasmic domains of human integrin subunits. J Biol Chem. 1999;274:37030–37034. doi: 10.1074/jbc.274.52.37030. Uses a creative transmembrane domain mapping strategy to identify the inner border of integrin transmembrane domains. [DOI] [PubMed] [Google Scholar]
- 47.O’Toole TE, et al. Modulation of the affinity of integrin αIIbβ3 (GPIIb-IIIa) by the cytoplasmic domain of αIIb. Science. 1991;254:845–847. doi: 10.1126/science.1948065. [DOI] [PubMed] [Google Scholar]
- 48.O’Toole TE, et al. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994;124:1047–1059. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hughes PE, O’Toole TE, Ylanne J, Shattil SJ, Ginsberg MH. The conserved membrane-proximal region of an integrin cytoplasmic domain specifies ligand binding affinity. J Biol Chem. 1995;270:12411–12417. doi: 10.1074/jbc.270.21.12411. [DOI] [PubMed] [Google Scholar]
- 50.Du X, et al. Long range propagation of conformational changes in integrin αIIbβ3. J Biol Chem. 1993;268:23087–23092. [PubMed] [Google Scholar]
- 51.Luo BH, Springer TA, Takagi J. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol. 2004;2:776. doi: 10.1371/journal.pbio.0020153. Uses disulphide cross-linking to establish the proximity of the α-integrin and β-integrin transmembrane domains and shows that stabilization of their interaction could prevent activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo BH, Carman CV, Takagi J, Springer TA. Disrupting integrin transmembrane domain heterodimerization increases ligand binding affinity, not valency or clustering. Proc Natl Acad Sci USA. 2005;102:3679–3684. doi: 10.1073/pnas.0409440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Partridge AW, Liu S, Kim S, Bowie JU, Ginsberg MH. Transmembrane domain helix packing stabilizes integrin αIIbβ3 in the low affinity state. J Biol Chem. 2005;280:7294–7300. doi: 10.1074/jbc.M412701200. [DOI] [PubMed] [Google Scholar]
- 54.Li W, et al. A push-pull mechanism for regulating integrin function. Proc Natl Acad Sci USA. 2005;102:1424–1429. doi: 10.1073/pnas.0409334102. References 52–54 show that point mutations in the integrin transmembrane domain could activate them, presumably by disrupting αβ-integrin transmembrane helix packing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu CF, Springer TA. The α subunit cytoplasmic domain regulates the assembly and adhesiveness of integrin lymphocyte function-associated antigen-1. J Immunol. 1997;159:268–278. [PubMed] [Google Scholar]
- 56.Imai Y, et al. Genetic perturbation of the putative cytoplasmic membrane-proximal salt bridge aberrantly activates α4 integrins. Blood. 2008;112:5007–5015. doi: 10.1182/blood-2008-03-144543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Czuchra A, Meyer H, Legate KR, Brakebusch C, Fassler R. Genetic analysis of β1 integrin “activation motifs” in mice. J Cell Biol. 2006;174:889–899. doi: 10.1083/jcb.200604060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu C, Takagi J, Springer TA. Association of the membrane proximal regions of the α and β subunit cytoplasmic domains constrains an integrin in the inactive state. J Biol Chem. 2001;276:14642–14648. doi: 10.1074/jbc.M100600200. [DOI] [PubMed] [Google Scholar]
- 59.Ulmer TS, Yaspan B, Ginsberg MH, Campbell ID. NMR analysis of structure and dynamics of the cytosolic tails of integrin αIIbβ3 in aqueous solution. Biochemistry. 2001;40:7498–7508. doi: 10.1021/bi010338l. [DOI] [PubMed] [Google Scholar]
- 60.Vinogradova O, et al. A structural mechanism of integrin αIIbβ3 “inside-out” activation as regulated by its cytoplasmic face. Cell. 2002;110:587–597. doi: 10.1016/s0092-8674(02)00906-6. Reports the association of the αIIb integrin and β3 integrin cytoplasmic domains, and that talin 1 could disrupt their interaction. [DOI] [PubMed] [Google Scholar]
- 61.Weljie AM, Hwang PM, Vogel HJ. Solution structures of the cytoplasmic tail complex from platelet integrin αIIb- and β3-subunits. Proc Natl Acad Sci USA. 2002;99:5878–5883. doi: 10.1073/pnas.092515799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gottschalk KE. A coiled-coil structure of the αIIbβ3 integrin transmembrane and cytoplasmic domains in its resting state. Structure. 2005;13:703–712. doi: 10.1016/j.str.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 63.Li R, et al. Oligomerization of the integrin αIIbβ3: Roles of the transmembrane and cytoplasmic domains. Proc Natl Acad Sci USA. 2001;98:12462–12467. doi: 10.1073/pnas.221463098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li R, et al. Dimerization of the transmembrane domain of integrin αIIb subunit in cell membranes. J Biol Chem. 2004;279:26666–26673. doi: 10.1074/jbc.M314168200. [DOI] [PubMed] [Google Scholar]
- 65.Kim C, Lau TL, Ulmer TS, Ginsberg MH. Interactions of platelet integrin αIIb and β3 transmembrane domains in mammalian cell membranes and their role in integrin activation. Blood. 2009;113:4747–4753. doi: 10.1182/blood-2008-10-186551. Describes a new method to study interactions among transmembrane domains and uses it to establish the primacy of αβ-integrin interactions among integrin transmembrane domains. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Das R, Baker D. Macromolecular modeling with Rosetta. Annu Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. Describes the principles of Rosetta modelling and its applications. [DOI] [PubMed] [Google Scholar]
- 67.Zhu J, et al. The structure of a receptor with two associating transmembrane domains on the cell surface: integrin αIIbβ3. Mol Cell. 2009;34:234–249. doi: 10.1016/j.molcel.2009.02.022. Combines Rosetta modelling with disulphide cross-linking to propose models of the αIIbβ3 integrin that include some clusters of transmembrane domain structures that resemble the structures reported in references 19, 43 and 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang J, et al. Structure of an integrin αIIbβ3 transmembrane-cytoplasmic heterocomplex provides insight into integrin activation. Proc Natl Acad Sci USA. 2009;106:17729–17734. doi: 10.1073/pnas.0909589106. A structure of the αIIbβ3 integrin transmembrane domain tails in a 50% acetonitrile/water solution, in which the IMC structure resembles that reported in reference 60 for the cytoplasmic tails in aqueous solution. Together with reference 44, this establishes the importance of the lipid–water interface in assembling the IMC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Metcalf DG, Kulp DW, Bennett JS, Degrado WF. Multiple approaches converge on the structure of the integrin αIIb/β3 transmembrane heterodimer. J Mol Biol. 2009;392:1087–1101. doi: 10.1016/j.jmb.2009.06.032. Structural modelling by two approaches provides αβ-integrin transmembrane domain models that converge on the NMR structure reported in reference 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu J, et al. Requirement of α and β subunit transmembrane helix separation for integrin outside-in signaling. Blood. 2007;110:2475–2483. doi: 10.1182/blood-2007-03-080077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu S, Calderwood DA, Ginsberg MH. Integrin cytoplasmic domain-binding proteins. J Cell Sci. 2000;113:3563–3571. doi: 10.1242/jcs.113.20.3563. [DOI] [PubMed] [Google Scholar]
- 72.Tadokoro S, et al. Talin binding to integrin tails: a final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. Shows that talin 1 binding to the β-integrin tail is required to activate β1 and β3 integrins in cells. [DOI] [PubMed] [Google Scholar]
- 73.Simonson WT, Franco SJ, Huttenlocher A. Talin1 regulates TCR-mediated LFA-1 function. J Immunol. 2006;177:7707–7714. doi: 10.4049/jimmunol.177.11.7707. [DOI] [PubMed] [Google Scholar]
- 74.Nieswandt B, et al. Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J Exp Med. 2007;204:3113–3118. doi: 10.1084/jem.20071827. Shows that deletion of talin 1 profoundly impairs integrin activation in platelets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petrich BG, et al. Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. J Exp Med. 2007;204:3103–3111. doi: 10.1084/jem.20071800. Shows that the platelet-specific deletion of talin 1 impairs integrin activation in platelets and leads to profound haemostatic defects. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Garcia-Alvarez B, et al. Structural determinants of integrin recognition by talin. Mol Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. Provides a high resolution structure of the F2–F3 domain of talin 1 in complex with a fragment of the β3 integrin tail, confirming that it is a PTB domain-like interaction, as predicted in reference 81. [DOI] [PubMed] [Google Scholar]
- 77.Critchley DR. Genetic, biochemical and structural approaches to talin function. Biochem Soc Trans. 2005;33:1308–1312. doi: 10.1042/BST0331308. [DOI] [PubMed] [Google Scholar]
- 78.Watanabe N, et al. Mechanisms and consequences of agonist-induced talin recruitment to platelet integrin αIIbβ3. J Cell Biol. 2008;181:1211–1222. doi: 10.1083/jcb.200803094. Reconstruction of the connections between thrombin stimulation and integrin activation, accompanied by the direct measurement of talin–integrin interactions in living cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wegener KL, et al. Structural basis of integrin activation by talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. The structure of the interface between talin 1 and the membrane-proximal region of the β-integrin cytoplasmic domain elucidates the mechanism by which talin 1 activates integrins. [DOI] [PubMed] [Google Scholar]
- 80.Rees DJG, Ades SE, Singer SJ, Hynes RO. Sequence and domain structure of talin. Nature. 1990;347:685–689. doi: 10.1038/347685a0. [DOI] [PubMed] [Google Scholar]
- 81.Calderwood DA, et al. The phosphotyrosine binding-like domain of talin activates integrins. J Biol Chem. 2002;277:21749–21758. doi: 10.1074/jbc.M111996200. [DOI] [PubMed] [Google Scholar]
- 82.Bouaouina M, Lad Y, Calderwood DA. The N-terminal domains of talin cooperate with the phosphotyrosine binding-like domain to activate β1 and β3 integrins. J Biol Chem. 2008;283:6118–6125. doi: 10.1074/jbc.M709527200. [DOI] [PubMed] [Google Scholar]
- 83.Calderwood DA, et al. Integrin β cytoplasmic domain interactions with phosphotyrosine-binding domains: a structural prototype for diversity in integrin signaling. Proc Natl Acad Sci USA. 2003;100:2272–2277. doi: 10.1073/pnas.262791999. Establishes that β-integrin tails bind to a wide variety of PTB domain-containing proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ulmer TS, Calderwood DA, Ginsberg MH, Campbell ID. Domain-specific interactions of talin with the membrane-proximal region of the integrin β3 subunit. Biochemistry. 2003;42:8307–8312. doi: 10.1021/bi034384s. [DOI] [PubMed] [Google Scholar]
- 85.Knezevic I, Leisner T, Lam S. Direct binding of the platelet integrin αIIb α3 (GPIIb-IIIa) to talin. J Biol Chem. 1996;271:16416–16421. doi: 10.1074/jbc.271.27.16416. [DOI] [PubMed] [Google Scholar]
- 86.Anthis NJ, et al. The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 2009;28:3623–3632. doi: 10.1038/emboj.2009.287. The first high resolution structure of talin F2–F3 in complex with a full-length integrin tail reveals that talin 1 forms a salt bridge with an Asp residue in the β-integrin tail and may therefore directly disrupt the salt bridge that stabilizes the IMC between α- and β-integrin subunit transmembrane domains. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mackinnon AC, Qadota H, Norman KR, Moerman DG, Williams BD. C-elegans PAT-4/ILK functions as an adaptor protein within integrin adhesion complexes. Curr Biol. 2002;12:787–797. doi: 10.1016/s0960-9822(02)00810-2. [DOI] [PubMed] [Google Scholar]
- 88.Moser M, Legate KR, Zent R, Fassler R. The tail of integrins, talin, and kindlins. Science. 2009;324:895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- 89.Montanez E, et al. Kindlin-2 controls bidirectional signaling of integrins. Genes. Dev. 2008;22:1325–1330. doi: 10.1101/gad.469408. Shows that the deletion of kindlin 2 profoundly impairs integrin function in fibroblasts and prevents recruitment of integrin-linked protein kinase (ILK) to focal adhesions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma YQ, Qin J, Wu C, Plow EF. Kindlin-2 (Mig-2): a co-activator of β3 integrins. J Cell Biol. 2008;181:439–446. doi: 10.1083/jcb.200710196. Shows that kindlin 2 could synergize with talin 1 to activate integrins, and that this function requires the interaction of kindlin 2 with the β-integrin tail. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harburger DS, Bouaouina M, Calderwood DA. Kindlin-1 and -2 directly bind the C-terminal region of β integrin cytoplasmic tails and exert integrin-specific activation effects. J Biol Chem. 2009;284:11485–11497. doi: 10.1074/jbc.M809233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tu Y, Wu S, Shi X, Chen K, Wu C. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell. 2003;113:37–47. doi: 10.1016/s0092-8674(03)00163-6. Shows that kindlin 2 is important for integrin function in cells and reports the discovery of migfilin. [DOI] [PubMed] [Google Scholar]
- 93.Kloeker S, et al. The Kindler syndrome protein is regulated by transforming growth factor-β and involved in integrin-mediated adhesion. J Biol Chem. 2004;279:6824–6833. doi: 10.1074/jbc.M307978200. Demonstrates that TGF-β induces kindlin 1 expression, and that kindlin 1 binds to β-integrin cytoplasmic domains. [DOI] [PubMed] [Google Scholar]
- 94.Lai-Cheong JE, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009;160:233–242. doi: 10.1111/j.1365-2133.2008.08976.x. [DOI] [PubMed] [Google Scholar]
- 95.Ussar S, et al. Loss of Kindlin-1 causes skin atrophy and lethal neonatal intestinal epithelial dysfunction. PLoS Genet. 2008;4:e1000289. doi: 10.1371/journal.pgen.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dowling JJ, et al. Kindlin-2 is an essential component of intercalated discs and is required for vertebrate cardiac structure and function. Circ Res. 2008;102:423–431. doi: 10.1161/CIRCRESAHA.107.161489. [DOI] [PubMed] [Google Scholar]
- 97.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nature Med. 2008;14:325–330. doi: 10.1038/nm1722. A breakthrough paper that shows that deletion of kindlin 3 profoundly impairs integrin activation in platelets. [DOI] [PubMed] [Google Scholar]
- 98.Moser M, et al. Kindlin-3 is required for β2 integrin-mediated leukocyte adhesion to endothelial cells. Nature Med. 2009;15:300–305. doi: 10.1038/nm.1921. Shows that kindlin 3 knockout impairs β2 integrin function in neutrophils. [DOI] [PubMed] [Google Scholar]
- 99.Kuijpers TW, et al. Leukocyte adhesion deficiency type 1 (LAD-1)/variant. A novel immunodeficiency syndrome characterized by dysfunctional β2 integrins. J Clin Invest. 1997;100:1725–1733. doi: 10.1172/JCI119697. Discovery of patients with a defect in the activation of β2 and β3 integrins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Manevich-Mendelson E, et al. Loss of Kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood. 2009;114:2344–2353. doi: 10.1182/blood-2009-04-218636. [DOI] [PubMed] [Google Scholar]
- 101.Alon R, Etzioni A. LAD-III, a novel group of leukocyte integrin activation deficiencies. Trends Immunol. 2003;24:561–566. doi: 10.1016/j.it.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 102.Boettner B, Van Aelst L. Control of cell adhesion dynamics by Rap1 signaling. Curr Opin Cell Biol. 2009;21:684–693. doi: 10.1016/j.ceb.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bos JL. Linking Rap to cell adhesion. Curr Opin Cell Biol. 2005;17:123–128. doi: 10.1016/j.ceb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 104.Kooistra MR, Dube N, Bos JL. Rap1: a key regulator in cell-cell junction formation. J Cell Sci. 2007;120:17–22. doi: 10.1242/jcs.03306. [DOI] [PubMed] [Google Scholar]
- 105.Crittenden JR, et al. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nature Med. 2004;10:982–986. doi: 10.1038/nm1098. [DOI] [PubMed] [Google Scholar]
- 106.Pasvolsky R, et al. A LAD-III syndrome is associated with defective expression of the Rap-1 activator CalDAG-GEFI in lymphocytes, neutrophils, and platelets. J Exp Med. 2007;204:1571–1582. doi: 10.1084/jem.20070058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Svensson L, et al. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nature Med. 2009;15:306–312. doi: 10.1038/nm.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Malinin NL, et al. A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nature Med. 2009;15:313–318. doi: 10.1038/nm.1917. Shows that activation of β1, β2 and β3 integrins is deficient in primary haematopoietic cells of kindlin 3-deficient humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kuijpers TW, et al. LAD-1/variant syndrome is caused by mutations in FERMT3. Blood. 2009;113:4740–4746. doi: 10.1182/blood-2008-10-182154. [DOI] [PubMed] [Google Scholar]
- 110.Mory A, et al. Kindlin-3: a new gene involved in the pathogenesis of LAD-III. Blood. 2008;112:2591. doi: 10.1182/blood-2008-06-163162. References 107–110 identify kindlin 3 mutations as the cause of defective integrin activation in cells of the hematopoietic lineage. [DOI] [PubMed] [Google Scholar]
- 111.Ithychanda S, et al. Migfilin: a molecular switch in regulation of integrin activation. J Biol Chem. 2009;284:4713–4722. doi: 10.1074/jbc.M807719200. Shows that migfilin competes with integrins for filamin binding, and proposes that kindlins act by recruiting migfilin to prevent filamin from competing with talin for integrin binding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lad Y, et al. Structural basis of the migfilin-filamin interaction and competition with integrin β tails. J Biol Chem. 2008;283:35154–35163. doi: 10.1074/jbc.M802592200. Shows that migfilin competes with integrins for filamin binding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kinbara K, Goldfinger LE, Hansen M, Chou FL, Ginsberg MH. Ras GTPases: integrins’ friends or foes? Nature Rev Mol Cell Biol. 2003;4:767–776. doi: 10.1038/nrm1229. [DOI] [PubMed] [Google Scholar]
- 114.Marshall CJ. Ras effectors. Curr Opin Cell Biol. 1996;8:197–204. doi: 10.1016/s0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- 115.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC., 2nd Rap1b is required for normal platelet function and hemostasis in mice. J Clin Invest. 2005;115:680–687. doi: 10.1172/JCI22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bergmeier W, et al. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J Clin Invest. 2007;117:1699–1707. doi: 10.1172/JCI30575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Katagiri K, Maeda A, Shimonaka M, Kinashi T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nature Immunol. 2003;4:741–748. doi: 10.1038/ni950. [DOI] [PubMed] [Google Scholar]
- 118.Lafuente EM, et al. RIAM, an Ena/VASP and Profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Dev Cell. 2004;7:585–595. doi: 10.1016/j.devcel.2004.07.021. A breakthrough paper that shows that RIAM is a Rap effector important in the activation of leukocyte integrins. [DOI] [PubMed] [Google Scholar]
- 119.Zhang Z, Rehmann H, Price LS, Riedl J, Bos JL. AF6 negatively regulates Rap1-induced cell adhesion. J Biol Chem. 2005;280:33200–33205. doi: 10.1074/jbc.M505057200. [DOI] [PubMed] [Google Scholar]
- 120.Krause M, et al. Lamellipodin, an Ena/VASP ligand, is implicated in the regulation of lamellipodial dynamics. Dev Cell. 2004;7:571–583. doi: 10.1016/j.devcel.2004.07.024. Discovery of the role of lamellipodin, a paralogue of RIAM, in lamellipodial dynamics. [DOI] [PubMed] [Google Scholar]
- 121.Han J, et al. Reconstructing and deconstructing agonist-induced activation of integrin αIIbβ3. Curr Biol. 2006;16:1796–1806. doi: 10.1016/j.cub.2006.08.035. Elucidates the molecular linkage between RAP1 and talins in integrin activation. [DOI] [PubMed] [Google Scholar]
- 122.Lee HS, Lim CJ, Puzon-McLaughlin W, Shattil SJ, Ginsberg MH. RIAM activates integrins by linking talin to Ras GTPase membrane-targeting sequences. J Biol Chem. 2009;284:5119–5127. doi: 10.1074/jbc.M807117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nolz JC, et al. WAVE2 regulates high-affinity integrin binding by recruiting vinculin and talin to the immunological synapse. Mol Cell Biol. 2007;27:5986–6000. doi: 10.1128/MCB.00136-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.O’Toole TE, et al. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994;124:1047–1059. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Naik UP, Patel PM, Parise LV. Identification of a novel calcium-binding protein that interacts with the integrin αIIb cytoplasmic domain. J Biol Chem. 1997;272:4651–4654. doi: 10.1074/jbc.272.8.4651. The discovery of CIB as an αIIb integrin tail-binding protein. [DOI] [PubMed] [Google Scholar]
- 126.Yuan W, et al. CIB1 is an endogenous inhibitor of agonist-induced integrin αIIbβ3 activation. J Cell Biol. 2006;172:169–175. doi: 10.1083/jcb.200505131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Naik MU, et al. CIB1 deficiency results in impaired thrombosis: the potential role of CIB1 in outside-in signaling through integrin αIIb β3. J Thromb Haemost. 2009;7:1906–1914. doi: 10.1111/j.1538-7836.2009.03581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Denofrio JC, Yuan W, Temple BR, Gentry HR, Parise LV. Characterization of calcium- and integrin-binding protein 1 (CIB1) knockout platelets: potential compensation by CIB family members. Thromb Haemost. 2008;100:847–856. [PMC free article] [PubMed] [Google Scholar]
- 129.Katagiri K, Maeda A, Shimonaka M, Kinashi T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nature Immunol. 2003;4:741–748. doi: 10.1038/ni950. Identification of RAPL as a regulator of αLβ2 integrin-mediated adhesion. [DOI] [PubMed] [Google Scholar]
- 130.Tohyama Y, et al. The critical cytoplasmic regions of the αL/β2 integrin in Rap1-induced adhesion and migration. Mol Biol Cell. 2003;14:2570–2582. doi: 10.1091/mbc.E02-09-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ebisuno Y, et al. Rap1 controls lymphocyte adhesion cascade and interstitial migration within lymph nodes in RAPL-dependent and -independent manners. Blood. 2009;115:804–814. doi: 10.1182/blood-2009-03-211979. Elucidation of the complementary roles of talins and RAPL in RAP1-regulated adhesion and migration of lymphocytes. [DOI] [PubMed] [Google Scholar]
- 132.Katagiri K, et al. Mst1 controls lymphocyte trafficking and interstitial motility within lymph nodes. EMBO J. 2009;28:1319–1331. doi: 10.1038/emboj.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Katagiri K, Imamura M, Kinashi T. Spatiotemporal regulation of the kinase Mst1 by binding protein RAPL is critical for lymphocyte polarity and adhesion. Nature Immunol. 2006;7:919–928. doi: 10.1038/ni1374. [DOI] [PubMed] [Google Scholar]
- 134.Coller BS. Platelet GPIIb/IIIa antagonists: the first anti-integrin receptor therapeutics. J Clin Invest. 1997;99:1467–1471. doi: 10.1172/JCI119307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hughes PE, et al. Suppression of integrin activation: a novel function of a Ras/Raf-initiated MAP kinase pathway. Cell. 1997;88:521–530. doi: 10.1016/s0092-8674(00)81892-9. [DOI] [PubMed] [Google Scholar]
- 136.Wierzbicka-Patynowski I, Schwarzbauer JE. The ins and outs of fibronectin matrix assembly. J Cell Sci. 2003;116:3269–3276. doi: 10.1242/jcs.00670. [DOI] [PubMed] [Google Scholar]
- 137.Chou FL, et al. PEA-15 binding to ERK1/2 MAP kinases is required for its modulation of integrin activation. J Biol Chem. 2003;278:52587–52597. doi: 10.1074/jbc.M309322200. [DOI] [PubMed] [Google Scholar]
- 138.Ling K, et al. Tyrosine phosphorylation of type Iγ phosphatidylinositol phosphate kinase by Src regulates an integrin-talin switch. J Cell Biol. 2003;163:1339–1349. doi: 10.1083/jcb.200310067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Calderwood DA, Tai V, Di Paolo G, De Camilli P, Ginsberg MH. Competition for Talin results in trans-dominant inhibition of integrin activation. J Biol Chem. 2004;279:28889–28895. doi: 10.1074/jbc.M402161200. [DOI] [PubMed] [Google Scholar]
- 140.Law DA, Nannizzi-Alalmo L, Phillips DR. Outside-in integrin signal transduction: αIIbβ3 (GPIIb-IIIa) tyrosine phosphorylation induced by platelet aggregation. J Biol Chem. 1996;271:10811–10815. doi: 10.1074/jbc.271.18.10811. [DOI] [PubMed] [Google Scholar]
- 141.Blystone SD, Williams MP, Slater SE, Brown EJ. Requirement of integrin β3 tyrosine 747 for β3 tyrosine phosphorylation and regulation of αvβ3 avidity. J Biol Chem. 1997;272:28757–28761. doi: 10.1074/jbc.272.45.28757. [DOI] [PubMed] [Google Scholar]
- 142.Chen H, et al. In vivo β1 integrin function requires phosphorylation-independent regulation by cytoplasmic tyrosines. Genes Dev. 2006;20:927–932. doi: 10.1101/gad.1408306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Law DA, et al. Integrin cytoplasmic tyrosine motif is required for outside-in αIIbβ3 signalling and platelet function. Nature. 1999;401:808–811. doi: 10.1038/44599. [DOI] [PubMed] [Google Scholar]
- 144.Sakai T, Jove R, Fassler R, Mosher DF. Role of the cytoplasmic tyrosines of β1A integrins in transformation by v-src. Proc Natl Acad Sci USA. 2001;98:3808–3813. doi: 10.1073/pnas.240456398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Oxley CL, et al. An integrin phosphorylation switch: the effect of β3 integrin tail phosphorylation on Dok1 and talin binding. J Biol Chem. 2008;283:5420–5426. doi: 10.1074/jbc.M709435200. [DOI] [PubMed] [Google Scholar]
- 146.Millon-Fremillon A, et al. Cell adaptive response to extracellular matrix density is controlled by ICAP-1-dependent β1-integrin affinity. J Cell Biol. 2008;180:427–441. doi: 10.1083/jcb.200707142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kiema T, et al. The molecular basis of filamin binding to integrins and competition with talin. Mol Cell. 2006;21:337–347. doi: 10.1016/j.molcel.2006.01.011. Reports the first structure of a β-integrin tail bound to filamin, explaining how talin and filamin could compete for binding to the integrin. [DOI] [PubMed] [Google Scholar]
- 148.Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Ann Rev Cell Dev Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- 149.Essex DW. Redox control of platelet function. Antioxid Redox Signal. 2008;11:191–225. doi: 10.1089/ars.2008.2322. [DOI] [PubMed] [Google Scholar]
- 150.Yan B, Smith JW. A redox site involved in integrin activation. J Biol Chem. 2000;275:39964–39972. doi: 10.1074/jbc.M007041200. [DOI] [PubMed] [Google Scholar]
- 151.O’Neill S, et al. The platelet integrin αIIbβ3 has an endogenous thiol isomerase activity. J Biol Chem. 2000;275:36984–36990. doi: 10.1074/jbc.M003279200. [DOI] [PubMed] [Google Scholar]
- 152.Jordan PA, et al. A role for the thiol isomerase protein ERP5 in platelet function. Blood. 2005;105:1500–1507. doi: 10.1182/blood-2004-02-0608. [DOI] [PubMed] [Google Scholar]
- 153.Du X, et al. Ligands “activate” integrin αIIbβ3 (platelet GPIIb-IIIa) Cell. 1991;65:409–416. doi: 10.1016/0092-8674(91)90458-b. [DOI] [PubMed] [Google Scholar]
- 154.Frelinger AL, III, Du X, Plow EF, Ginsberg MH. Monoclonal antibodies to ligand-occupied conformers of integrin αIIbβ3 (glycoprotein IIb-IIIa) alter receptor affinity, specificity, and function. J Biol Chem. 1991;266:17106–17111. [PubMed] [Google Scholar]
- 155.Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. Uses FRET to show that integrin activation is associated with a change in the cytoplasmic domains. [DOI] [PubMed] [Google Scholar]