

Abstract
Capuramycin and its congeners have been considered important lead molecules for the development of a new drug for multidrug-resistant (MDR) Mycobacterium tuberculosis infections. Extensive structure-activity relationship studies of capuramycin to improve the efficacy have been limited due to difficulty in selective chemical modifications of the desired position(s) of the natural product with biologically interesting functional groups. We have developed efficient syntheses of capuramycin and its analogs using new protecting groups, which are derived from the chiral (chloro-4-methoxyphenyl) (chlorophenyl) methanols, for the uridine ureido nitrogen and primary alcohol. The chiral non-racemic (2,6-dichloro-4-methoxyphenyl) (2,4-dichlorophenyl) methanol derivative is a useful reagent to resolve rac-3-amino-1,3-dihydro-5-phenyl-2H-1,4-benzodiazepin-2-one, whose (S)-configuration isomer plays a significant role in improving the mycobactericidal activity of capuramycin.
Introduction
The emergence of multidrug-resistant (MDR) strains of Mycobacterium tuberculosis (Mtb) seriously threatens tuberculosis (TB) control and prevention efforts.[1] Moreover, people who are HIV-AIDS patients are susceptible to TB infection.[2] There are significant problems associated with treatment of AIDS and Mtb co-infected patients. Rifampicin and isoniazid [a key component of the DOTS (Directly Observed Treatment, Short-course) therapy] induce the cytochrome P450 3A4 enzyme in liver which shows significant interactions with protease inhibitors for HIV infections.[3] In addition, rifampicin strongly interacts with non-nucleoside reverse transcriptase inhibitors. Thus, clinicians avoid starting Highly Active Antiretroviral Therapy (HAART), which consists of three or more highly potent reverse transcriptase inhibitors and protease inhibitors, until the TB infection has been cleared.[4] Thus, there are significant needs and interests in developing new TB drugs. However, over the last 40 years, only bedaquiline (Sirturo™), an ATP synthase inhibitor, was approved for the treatment of MDR-Mtb infections as a monotherapeutic agent in 2012.[5] The ultimate goal of the development of the treatment of MDR-Mtb strains is to find novel antibacterial agents which 1) interfere with unexploited bacterial molecular targets, 2) can shorten a TB drug regimen (one-month to three-month regimen), 3) can apply to combination TB chemotherapy, and 4) do not interfere with ability of HAART to treat HIV patients who are co-infected with Mtb.
Since peptidoglycan (PG) is an essential bacterial cell wall polymer, the machinery for PG biosynthesis provides a unique and selective target for antibiotic action. However, only a few enzymes in PG biosynthesis such as the penicillin binding proteins (PBPs) have been extensively studied.[6] Thus, the enzymes associated with the early PG biosynthesis enzymes [MurA, B, C, D, E, and F, MraY (phospho-MurNAc-pentapeptide translocase or translocase I), and MurG] are considered to be a source of unexploited drug targets.[7] Our interest in unexploited molecular targets related to PG biosynthesis is MraY,[8] which catalyzes the transformation of UDP-N-acylmuramyl-L-alanyl-γ-D-glutamyl-meso-diaminopimelyl-D-alanyl-D-alanine (Park’s nucleotide) to prenylpyrophosphoryl-N-acylmuramyl-L-Ala-γ-D-glu-meso-DAP-D-Ala-D-Ala (lipid I).[9] MraY is inhibited by nucleoside-based complex natural products such as muraymycins,[10] liposidomycin,[11], caprazamycin,[12] pacidamycin,[13] capuramycin,[14], and other related natural products.[15] Capuramycin (1) and its analogs exhibited significant mycobacterial growth inhibitory activities in vitro and in vivo (Figure 1) and very low toxicity in mice.[16] Moreover, capuramycin killed Mtb much faster than other first-line TB drugs (>90 % of the bacilli were killed within 48 h), and thus could dramatically reduce the time frame for effective anti-TB chemotherapy. Therefore, capuramycin and its congeners have been considered important lead molecules for the development of a new drug for MDR-Mtb infections.
Figure 1.
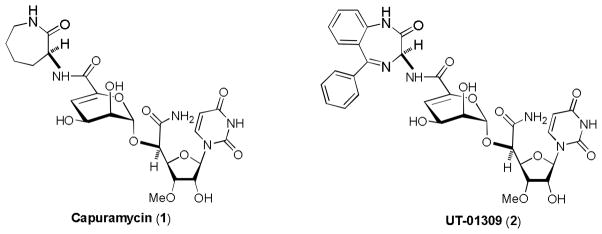
Structures of Capuramycin (1) and UT-01309 (2).
Since discovery of capuramycin as a specific spectrum antimycobacterial agent, extensive SAR studies of capuramycins have been limited because of difficulty in modifying the complex natural product at the desired position(s) with a wide range of functional groups. Accordingly, it is essential to establish a concise and convergent synthesis of capuramycin that is amenable to synthesis of analogs for SAR studies. The first total synthesis of capuramycin was reported in 1994 by Knapp et al. Their synthesis requires 22 linear steps from diisopropylidene-D-glucofuranose, and relatively lengthy synthesis of the manno-pyranuronate glycosyl donor.[17] We have developed a concise synthesis of capuramycin in which the intact molecule can be synthesized in 13 steps from the known intermediate 3 (Scheme 1).[18] Although each step in our previously reported capuramycin synthesis is a high-yielding conversion when applied to small to medium scale, several steps are not ideal for the synthesis of a large amount of capuramycin and its analogs for in vivo studies using rodents.
Scheme 1.

Previously reported syntheses of capuramycin.
α-Mannosylation of 5 with the thioglycoside 6 requires the diluted conditions (0.05 M) and long reaction time (12–16 h). Selective deacetylation at the 6″-position of 7a have to be stopped at around 30–70% conversion in order to avoid the over-reactions and the recovered starting material is recycled to perform the same reaction multiple times. Hydrogenolytic cleavage of the benzyloxymethyl (BOM) group of the uridine ureido nitrogen via heterogeneous conditions often yields the over-reduced product of which the C5–C6 double bond of the uracil moiety was saturated.[19] Recently, we identified a new capuramycin analog UT-01309 (2) possessing (S)-3-amino-1,4-benzodiazepine-2-one [(S)-13], which showed an improved antimycobacterial activity (2.5 μg/mL vs 12.0 μg/mL for 1 against M. tuberculosis).[20] Significantly, UT-01309 is active against drug-resistant M. tuberculosis and did not exhibit cytotoxicity against Vero monkey kidney cells and HepG2 human hepatoblastoma cells even at 250 μg/mL concentrations (vide infra). Thus, we are very interested in in vivo evaluation of 2 in comparison with capuramycin (1) and other related molecules. Herein, we report improved synthesis of capuramycin (1) and its analog UT-01309 (2) via 1) novel protecting groups for the uridine ureido nitrogen and primary alcohol, and 2) the chiral carbonate reagent for the resolution of rac-3-amino-1,4-benzodiazepine-2-one (13).
Results and Discussions
Our synthetic strategy to improve the syntheses of capuramycin (1) and capuramycin analog, UT-01309 (2) is illustrated in Scheme 2. We have developed new protecting groups, (2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methyl [monomethoxytetrachlorodiphenylmethyl (MTPM)] and (2,6-dichloro-4-methoxyphenyl)(2,4,6-O-diphenylmethyl trichloroacetimidateophenyl) methoxymethyl [monomethoxydiphenylmethoxylmethyl (MDPM)] for primary alcohols and ureido nitrogens, respectively.[21] These protecting groups showed a significant relative stability against a wide variety of conditions utilized for the syntheses of natural and unnatural products. However, the MTPM and MDPM protecting groups can conveniently be deprotected by using 30% TFA in CH2Cl2. The use of these protecting groups for the uridine ureido nitrogen (3-position) and the primary alcohol (6″-position) will significantly improve the synthesis of capuramycin analogs (Scheme 2). (S)-3-Amino-1,4-benzodiazepin-2-one [(S)-13] is an important functional group to improve antimycobacterial activity of capuramycin. For our SAR studies of capuramycin analogs, it is desirable to have a versatile resolution protocol of racemic amino acids which are not available commercially. We expected that the optically pure carbonate (S)-14, derived from the unsymmetrical (2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methanol, can resolve a diastereomixture of the carbamates via convenient chromatography and can readily be deprotected under mild conditions.[22]
Scheme 2.

Improved synthetic strategy for capuramycin analogs.
Synthesis of (2S)-uridyl-hydroxyacetonitrile 10 and mannosyl donor imidate 11
MDPM and MTPM groups have significant advantages over the other ordinal protecting groups for the syntheses of capuramycin analogs in that these new protecting groups 1) are stable to a wide variety of acids, 2) are not susceptible to hydrogenation conditions, and 3) can be deprotected efficiently by solvolytic cleavage with 30% TFA at room temperature within 2 h without addition of a cation scavenger.[21] We synthesized over 10 gram of MDPMCl (16) and MTPM-imidate (22) according to the established procedures.21 The uridine ureido nitrogen was protected with MDPMCl (16) in the presence of DBU to afford the MDPM-protected uridine 9 in 95% yield (Scheme 3). Selective alkylation of 9 at the secondary alcohol (3′-position) was achieved via a SnCl2-mediated methylation condition to yield the desired mono-methyl derivative in 60% yield.[23] Selective chloroacetylation of the primary alcohol of the diol was performed with ClCH2CO2H, EDCI, NaHCO3, and glyceroacetonide-Oxyma (17) in 5% H2O-CH3CN to give rise to 18 in 98% yield.[24] The regiochemistry of 18 was unequivocally determined by extensive 1H-NMR decoupling studies and the 2D NOESY experiments.[25] Albeit the ordinal esterification conditions (e.g. ClCH2CO2H, DCC, DMAP in CH2Cl2 or ClCH2COCl, pyridine in CH2Cl2) provided a mixture of 18 and the over-reaction product, we did not observe the formation of secondary alcohol ester under the conditions applied to the synthesis of 18. Acetylation of the secondary alcohol of 18 followed by the removal of the chloroacetyl group with thiourea in MeOH afforded 19 in 95% overall yield.[26] The primary alcohol of 19 was oxidized under Pfitzner-Moffatt conditions (DCC, Cl2CHCO2H, DMSO-CH2Cl2) to provide the corresponding aldehyde 20, which was utilized without purification.[27] We have extensively studied cyanohydrin formation reactions of the uridyl-aldehyde derivatives using TMSCN.[28] In all cases, Lewis acid-promoted trimethylsilylcyanations of the uridyl-aldehydes furnished a mixture of the TMS-protected cyanohydrins in favor of undesired (R)-configuration products (e.g. 21) in low yields. Lewis base-catalyzed trimethylsilylcyanations (e.g. Ph3PO, NMO, BABCO, cinchona alkaloids) did not provide the products due to the fact that the uridyl-aldehydes were not stable against Lewis and Brønsted bases. In previous studies we observed that the Ti-mediated conditions gave the desired (S)-configuration cyanohydrin as the major product with satisfactory yield.[18] Similarly, TMSCN addition of 20 with 10 mol% of Ti(OiPr)4 in CH2Cl2-H2O (1%) provided a mixture of 10 and 21 in 90 % yield with the 10/21 ratio of 1.5–2.0:1 after desilylation.[29] In our recent studies, we found that trimethylsilylcyanation reaction of 20 with 1,3-bis(2-(dimethylamino)ethyl)thiourea also gave a mixture of 10 and 21 with compatible selectivity and yield to the reaction with Ti(OiPr)4 in CH2Cl2-H2O (1%). Moreover, we observed that hydrocyanation of 20 with BzCN in DMSO-H2O afforded a 2 : 1 mixture of 10 and 21 in 95% yield from 19.[30] Because water-catalyzed hydrocyanation with BzCN is operationally simple and high-yield conversion, we decided to scale-up the conversion of 19 to 10 with this condition and the undesired stereochemistry of 21 was inverted via a modified Mitsunobu reaction [DIAD, TPP, ClCH2CO2H, pyridine (1:1:1:1)]. The chloroacetyl group of the ester was selectively deprotected with thiourea in MeOH. Thus, we could achieve the synthesis of the mannosyl acceptor 10 in 7 steps from uridine (15) with 34% overall yield without the process of the inversion (21→10) or in 9 steps with 45% overall yield including the Mitsunobu reaction followed by deprotection.
Scheme 3.

Syntheses of the glycosyl donor 10 and acceptor 11.
The mannosyl donor 11 was synthesized in 4 steps from α-benzyl glycoside 22 (Scheme 3). The primary acetate of 22 was selectively deprotected with [tBu2SnCl(OH)]2[31] and the generated alcohol was protected with MTPM-imidate 23 in the presence of TMSOTf to afford 24 in 93% overall yield.[21] Hydrogenolytic cleavage of the anomeric benzyl ether followed by the imidate-formation reaction provided 11 in 95% overall yield.[32]
Mannosylation of the cyanohydrin 10 with 11
In our previous capuramycin synthesis, we have screened an effective promoter to catalyze the thioglycoside 6 for the mannosylation of 5 (Scheme 1). We found that α-selective mannosylation of 5 with 6 was achieved via the combination of NIS and AgBF4 in CH2Cl2 (at 0.05 μM concentrations).[18],[33] Interestingly, the NIS/AgBF4 promoted mannosylation of 5 provided the orthoester 25 within 15 min, which underwent the rearrangement within 16 h to afford 7a exclusively in 90% yield. The orthoester 25 could be distinguished from 7a in 1H-NMR spectra of the crude reaction mixture; 25 showed a characteristic chemical shift of 1.78 ppm (CH3).[34] All triflate ion associated-glycosylations with 6 (e.g. NIS/TfOH or NBS/TfOH) yielded a mixture of α- and β-mannosides.[35] Under the NIS/AgBF4 promoted conditions, mannosylation of the MDPM-protected 10 with the thioglycoside 26 did not provide the desired product 12. The acceptor 10 was stable under the NIS/AgBF4 conditions, however, the thioglycoside 26 was completely consumed to form the complex mixtures. Albeit mannosylation of 10 with α-mannopyranose 2,3,4,6-tetraacetate 1-(2,2,2-trichloroethanimidate) (27) did not provide the desired product 7b, TMSOTf- and BF3•OEt2-catalyzed mannosylation of 10 with the imidate 11 afforded the desired product 12 in 45% and 75% yield, respectively. It is worth noting that the mannosylation with 11 could be achieved at high concentrations in short reaction times compared to the mannosylation of 5 with 6 under the conditions of NIS/AgBF4. We confirmed that mannosylations of 10 with 11 were reproducible at any concentration between 0.1–0.5 M and applied to a gram scale synthesis of 12.
Resolution of racemic 3-amino-1,4-benzodiazepine-2-one
We identified that in vitro antimycobacterial activity of capuramycin (1) was improved by the replacement of the (S)-3-aminoazepan-2-one moiety of 1 with (S)-3-amino-1,4-benzodiazepine-2-one [(S)-13] (vide supra). In order to synthesize enough quantities of UT-01309 (2) for in vivo studies, it was desirable to establish an efficient resolution method of racemic 3-amino-1,4-benzodiazepine-2-one [(±)-13]. Due to the fact that 3-amino-1,4-benzodiazepine-2-ones are important building blocks[36] for the development of several therapeutic areas (e.g. respiratory syncytial virus), resolution methods of racemic of 3-amino-1,4-benzodiazepine-2-one [(±)-13] have been reported by several groups.[37] However, most reported protocols provide separations of the diastereomers formed by amide-forming reactions with optically active amino acids, and only a few reports have demonstrated the resolution of (±)-13 with readily cleavable chiral agents. Sherrill et al. reported the resolution of 13 with the p-nitrophenyl carbonate of (R)-α-methyl benzyl alcohol. In their procedure, the carbamate auxiliary was cleaved with HBr (gas) in CH2Cl2 and the generated by-product, (1-bromoethyl)benzene, needed to be removed by recrystallization.[38]
We envisioned the resolution of (±)-13 with the chiral carbonate (S)-14, which was originally developed as a new chiral derivatizing agent for determination of absolute configuration of amino acids (Scheme 5).[21c] Carbamate formation of (±)-13 with (S)-14 was achieved by using iPr2NEt in a mixture of acetone and H2O (3/1). Gratifyingly, the generated diastereomers could be purified by silica gel chromatography to afford 28 and 29 in 98% yield (approximately 49% each). As shown in Figure 2, we have reported that the absolute configurations of a wide range of amino acids can be determined by only analyzing the carbamate nitrogen protons of (S)-14 and (R)-14 derivatives in 1H-NMR spectra. In all cases, the nitrogen protons of carbamates derived from L-amino acids and (S)-14 were shifted downfield relative to those obtained with L-amino acid-(R)-14 derivatives.[22] In 1H-NMR, the chemical shifts of 29 should appear identical to those of the antipode of 29 (ent-29) (Figure 2). Thus, the Δδ(S-R) value of the Nα protons of 28 and ent-29 should determine the absolute stereochemistry of 28. The Δδ(Nα28-Nα29) value was +0.03 and thus the absolute stereochemistry of 28 was assigned to be L-configuration (R for 3-amino-1,4-benzodiazepine-2-one) as shown in Scheme 5. The diastereomeric excesses (des) of purified 28 and 29 were determined by HPLC to be >99.0%. Removal of the carbamate auxiliaries of 28 and 29 was achieved by 20% TFA in CH2Cl2 to afford (S)-13 and (R)-13 in >95% yields. The chiral auxiliary was recovered as racemic trifluoroacetate 30 in quantitative yield. The absolute configurations of (S)-13 and (R)-13 were unequivocally confirmed by the comparison of optical rotations of those with the reported values for (S)-13 and (R)-13.[37c]
Scheme 5.
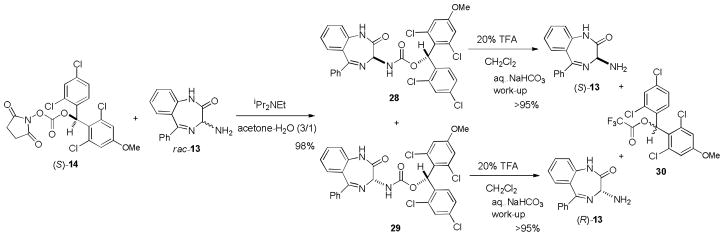
Resolution of rac-3-amino-1,4-benzodiazepine-2-one.
Figure 2.
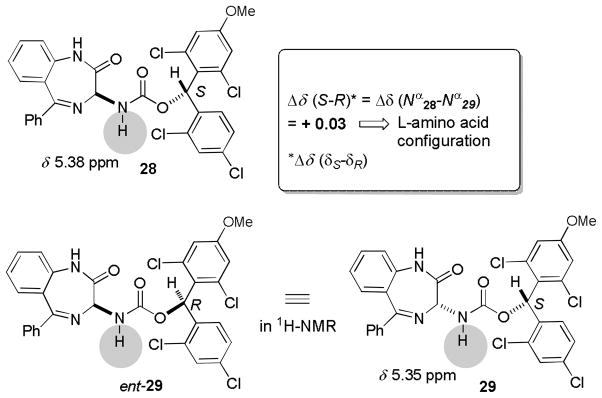
Absolute configurations of 28 and 29.
Syntheses of capuramycin and UT-01309
We have reported the synthesis of capuramycin (1) from 7 in 7 steps (Scheme 1).[18] The use of MDPM and MPTM protecting groups for the uridine ureido nitrogen and primary alcohol could significantly improve the synthesis of 8. As summarized in Scheme 6, capuramycin (1) and UT-01309 (2) were synthesized in 6 steps from 12. The improved scheme required only three times of purifications by chromatography in the total number of synthetic steps (Scheme 6). The cyano group of 12 was hydrated using InCl3-aldoxime in toluene, furnishing the corresponding primary amide.[39] Without further purification, the primary amide was subjected to simultaneous deprotections of the MDPM and MPTM groups with 30% TFA in CH2Cl2 to afford 8 in greater than 95% overall yield for two steps. We could achieve the synthesis of over 1 gram of 8 via the new protecting group strategy summarized in Schemes 3, 4, 5 and 6. The conversions of 8 to capuramycin (1) and UT-01309 (2) were carried out via the previously reported procedures except for the amide-forming reactions.[18] Oxidation-elimination reactions of 8 using SO3•pyridine in a biphasic solvent system (DMSO/Et3N = 3/1) provided the α,β-unsaturated aldehyde 31.[40] The aldehyde 31 was oxidized to the corresponding carboxylic acid 32 by Pinnick oxidation (NaClO2, 2-methyl-2-butene).[41] The resulting crude carboxylic acid was coupled with (S)-aminocaprolactam (33) using an amide forming reaction in water media [glyceroacetonide-Oxyma (17), EDCI, NaHCO3 in H2O] to yield 33 in 80–85% overall yield from 8.[42] In our previous synthesis of 1, HOAt (1-hydroxy-7-azabezotriazole) was used as a peptide-coupling additive to couple the segments 32 and 33. HOAt and some other by-products generated in the coupling conditions (EDCI, HOAt, NMM (N-methylmorpholine) in DMF) were difficult to separate from the desired product by a standard column chromatography. On the contrary, in the glyceroacetonide-Oxyma (17) / EDCI-mediated coupling reaction, simple basic and acidic aqueous work-up procedures could remove all reagents utilized in the reactions to afford the coupling product 34 in high yield with excellent purity. Saponification of 34 by using LiOH in THF-H2O provided capuramycin (1) in greater than 95% yield. Similarly, UT-01309 (2) was synthesized using (S)-13 instead of 33 in the synthesis of 1 (Scheme 6). The purity of synthetic products, 1 and 2 were determined to be >99% by reverse-phase HPLC analyses.
Scheme 6.

Synthesis of capuramycin and UT-01309.
Scheme 4.

Mannosylation of the cyanohydrins.
In vitro biological evaluation of UT-01309
UT-01309 (2) was identified by cell-based assays of a small optimized library of capuramycin analogs. In vitro biological activities of 2 synthesized here were evaluated against Mtb MraY (IC50) and a series of bacteria including Mycobacterium spp. The IC50 value of 2 against Mtb MraY was 5.5 nM (1: IC50 18 nM against Mtb MraY). UT-01309 did not exhibit growth inhibitory activity against a series of Gram-positive and -negative bacteria including S. aureus, E. faecalis, E. coli, K. pneumonia, and P. aeruginosa even at 400 μg/mL concentrations. UT-01309 showed bactericidal activities specific to Mycobacterium spp. UT-01309 killed M. tuberculosis (H37Rv) completely at 2.5 μg/mL concentrations whereas capuramycin required 12.0 μg/mL. UT-01309 showed the MIC value of 6.5 μg/mL against M. smegmatis. Significantly, UT-01309 is active against drug-resistant M. tuberculosis (e.g. M. tuberculosis H37Rv INHr and M. tuberculosis H37Rv RFPr), and did not exhibit cytotoxicity against Vero monkey kidney cells and HepG2 human hepatoblastoma cells even at 250 μg/mL concentrations.
Conclusion
In summary, we present an improved synthesis of capuramycin (1) and its analog UT-01309 (2), a promising investigational drug lead for MDR-Mycobacterium tuberculosis infections. MDPM and MPTM protecting groups for the uridine ureido nitrogen and primary alcohol could improve the overall efficiency of syntheses of 1 and 2. The synthetic scheme reported here enables us to synthesize gram-quantities of the key intermediate 8 for the syntheses of a series of capuramycin analogs; 8 could be synthesized in 9 steps from uridine (15) in 32% overall yield. In addition, we have demonstrated an efficient resolution of racemic 3-amino-1,4-benzodiazepine-2-one [(±)-13] with the chiral carbonate (S)-14 to yield (S)-13, an important building block to improve in vitro biological activity of capuramycin. We will evaluate UT-01309 (2) in vivo using an infected mouse model and study toxicity and PK/PD profile of 2. These data will be reported elsewhere.
Experimental Section
General
All reagents and solvents were of commercial grade and were used as received without further purification unless otherwise noted. Tetrahydrofuran (THF) and diethyl ether (Et2O) were distilled from sodium benzophenone ketyl under an argon atmosphere prior to use. Methylene Chloride (CH2Cl2), acetonitrile (CH3CN), benzene, toluene and triethylamine (Et3N) were distilled from calcium hydride under an Argon atmosphere. Flash chromatography was performed with Whatman silica gel (Purasil 60 Å, 230–400 Mesh). Analytical thin-layer chromatography was performed with 0.25 mm coated commercial silica gel plates (EMD, Silica Gel 60F254) visualizing at 254 nm, or developed with ceric ammonium molybdate or anisaldehyde solutions by heating on a hot plate. 1H-NMR spectral data were obtained using 400, and 500 MHz instruments. 13C-NMR spectral data were obtained using 100 and125 MHz instruments. For all NMR spectra, δ values are given in ppm and J values in Hz.
(2,6-Dichloro-4-methoxyphenyl)(2,4,6-trichlorophenyl)-methoxy methyl chloride (16)
(2,6-Dichloro-4-methoxyphenyl)-(2,4,6-trichlorophenyl)-methoxymethyl methyl sulfide was synthesized according to the procedure previously reported.7a To a stirred solution of (2,6-dichloro-4-methoxyphenyl)-(2,4,6-trichlorophenyl)-methoxymethyl methyl sulfide (11.18 g, 25.0 mmol) in CH2Cl2 (63.0 mL) was added sulfuryl chloride (2.0 mL, 25.0 mmol) at rt. The reaction mixture was stirred for 1 h and all volatiles were evaporated to provide the crude product as oil which was pure enough for the next reaction (10.45 g, 96%). 1H NMR (400 MHz, CDCl3): δ= 7.33 (s, 2H), 6.88 (s, 2H), 6.77 (s, 1H), 5.57 (q, J = 6.4 Hz, 2H), 3.80 (s, 3H); 13C NMR (100 MHz, CDCl3): δ= 159.6, 136.8, 136.6, 134.4, 131.7, 129.7, 124.3, 115.6, 80.1, 55.8; IR: υ̃ = 3473, 1445, 1309 cm−1; elemental analysis calcd (%) for C15H10Cl6O2: C, 41.42; H, 2.32; Cl, 48.91. Found: C, 41.81; H, 2.41; Cl, 48.97.
3-[(2,6-Dichloro-4-methoxy-phenyl)-(2,4,6-trichloro-phenyl)-methoxymethyl]-1-(3,4-dihydroxy-5-hydroxymethyl-tetrahydro-furan-2-yl)-1H-pyrimidine-2,4-dione(9)
To a stirred solution of uridine (10.98 g, 45.0 mmol) in DMF (120 mL) at 0 °C, DBU (9.0 mL, 60.0 mmol) and 16 (13.08 g, 30.0 mmol) were added. After 1 h at 0 °C, the reaction was quenched by addition of MeOH (24 mL). All volatiles were evaporated in vacuo and the crude product was purified by silica gel chromatography with CHCl3/MeOH (95:5) to afford 9 as an oil (17.62 g, 95%). Rf = 0.3 (10% MeOH/CHCl3); 1H NMR (400 MHz, CDCl3): δ= 7.67 (d, J = 6.8 Hz, 1H), 7.30 (d, J = 3.6 Hz, 2H), 6.83 (d, J = 4.8 Hz, 2H), 6.57 (s, 1H), 5.77 (d, J = 8.4 Hz, 1H), 5.59 (m, 3H), 4.32 (m, 2H), 4.24 (s, 1H), 3.97 (d, J = 12.0 Hz, 1H), 3.90 (s, 1H), 3.83 (m, 1H), 3.78 (s, 3H), 3.05 (s, 1H), 2.20 (s, 1H); 13C NMR (100 MHz, CDCl3): δ= 162.9, 159.4, 151.9, 140.1, 136.7, 134.1, 132.5, 129.5, 125.2, 115.5, 101.6, 93.2, 85.7, 77.9, 74.8, 70.4, 69.1, 61.7, 55.7, 36.6, 31.5; IR: υ̃ = 3435, 1719, 1665, 1440, 1081 cm−1; HRMS (ESI+) m/z calcd for C24H22Cl5N2O8: 640.9819; found: 640.9825.
Synthesis of 18
To a stirred solution of 9 (12.8 g, 20.0 mmol) in DMF (300 mL) was added SnCl2 (1.91 g, 10.0 mmol). The reaction mixture was heated to 50 °C followed by addition of CH2N2 (150 mL, 60.0 mmol, 0.4 M in Et2O). After 1h, all volatiles were evaporated in vacuo. The selectivity ratio and yield of the mono-methyl ethers were determined by 1H-NMR analyses of the crude mixture to be 3:2 ratio in favor of the desired product. The crude product was dissolved in 5% H2O/MeCN (1.0 mL). Glyceroacetonide-Oxyma 17 (6.7 g, 30.0 mmol), EDCI (5.7 g, 30.0 mmol), chloroacetatic acid (3.72 g, 40.0 mmol), and NaHCO3 (10.1 g, 120.0 mmol) were added to the reaction mixture. After 3 h, the reaction was quenched with aq. NaHCO3. The aqueous layer was extracted with EtOAc (2x). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo to yield the desired ester 18 (8.6 g, 59% over two steps) as a colorless liquid. Rf = 0.3 (30% hexanes/AcOEt); 1H NMR (500 MHz, CDCl3): δ= 7.29 (d, J = 7.5 Hz, 1H), 7.20 (s, 2H), 6.76, (s, 2H), 6.50 (s, 1H), 5.71 (d, J = 7.5 Hz, 1H), 5.50-5.46 (m, 3H), 4.45 (m, 1H), 4.34 (m, 2H), 4.15 (m, 1H), 4.04 (m, 2H), 3.96 (m, 1H), 3.71 (s, 3H), 3.42 (s, 3H); 13C NMR (100 MHz, CDCl3): δ= 166.9, 162.7, 159.3, 151.2, 140.3, 136.7, 134.0, 132.6, 129.5, 125.3, 115.5, 102.1, 79.7, 78.9, 77.8, 72.7, 69.2, 65.0, 58.9, 55.7, 40.6; IR: υ̃ = 3442, 1711, 1660, 1445, 1309, 1070 cm−1; HRMS (ESI+) m/z calcd for C27H24C6N2NaO9: 754.9481; found 754.9484.
Synthesis of 19
The ester 18 above was dissolved in pyridine/Ac2O (2:1, 200 mL) and stirred at rt. Upon completion, all volatiles were evaporated in vacuo to afford the desired acetate. The crude material was dissolved in MeOH (200 mL) and thiourea (3.8 g, 50.0 mmol) was added. The reaction mixture was stirred at 50 °C for 4 h and cooled to rt. All volatiles were evaporated in vacuo. Purification by silica gel column chromatography with hexanes/AcOEt (1:1) yielded the desired product 19 as an oil (7.8 g, 95% over two steps). Rf = 0.4 (30% hexanes/AcOEt); 1H NMR (500 MHz, CDCl3): δ= 7.48 (d, J = 7.5 Hz, 1H), 7.30 (s, 2H), 6.83 (s, 2H), 6.57 (s, 1H), 5.77 (d, J = 7.5 Hz, 1H), 5.66 (s, 1H), 5.57 (bs, 2H), 5.44 (bs, 1H), 4.18 (bs, 1H), 4.11 (bs, 1H), 4.00 (d, J = 11.5 Hz, 1H), 3.80 (s, 1H), 3.77 (s, 3H), 3.41 (s, 3H), 2.25 (bs, 1H), 2.16 (s, 3H); 13C NMR (100 MHz, CDCl3): δ= 170.5, 162.6, 159.3, 151.4, 140.3, 136.7, 134.0, 132.6, 129.5, 125.3, 115.5, 102.3, 91.4, 83.0, 80.9, 77.8, 70.1, 69.2, 61.3, 59.0, 55.7, 20.8; IR: υ̃ = 3445, 1719, 1665, 1440, 1302, 1081 cm−1; HRMS (ESI+) m/z calcd for C27H25Cl5N2NaO9: 720.9871; found 720.9875.
Synthesis of 10
To a stirred solution of 19 (5.75 g, 8.0 mmol) in CH2Cl2/DMSO (1:1, 80 mL) at 0 °C was added DCC (4.0 g, 20.0 mmol) and dichloroacetic acid (1.02 g, 8.0 mmol). After 1 h at 0 °C, the reaction mixture was diluted with CH2Cl2 (60 mL) and washed with NaHCO3 (aq.). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo to give the crude aldehyde which was used directly in the next step after passing through a SiO2 pad. To a stirred solution of the crude aldehyde in DMSO/H2O (4/1, 80 mL) was added BzCN (1.58 g, 12.0 mmol). After being stirred for 12 h at rt, NaHCO3 (aq.) was added followed by AcOEt. The aqueous layer was extracted with EtOAc (2x). The combined organic extracts were dried over Na2SO4 and concentrated. The resulting crude material was purified by silica gel chromatography with AcOEt/hexanes (2:3) to give 10 (3.7 g, 63%) and 21 (1.88 g, 32%). Data for 10: Rf = 0.4 (60% AcOEt/hexanes); 1H NMR (500 MHz, CDCl3): δ= 7.31 (d, J = 7.5 Hz, 2H), 6.84 (d, J = 10.0 Hz, 2H), 6.56 (s, 1H), 5.83 (dd, J = 5.5 Hz, 1H), 5.58 (m, 1H), 5.40 (bs, 1H), 5.34 (bs, 1H), 5.23 (bs, 1H), 4.67 (d, J = 11.0 Hz, 1H), 4.55 (bs, 1H), 4.35 (s, 1H), 3.77 (s, 3H), 3.40 (s, 3H), 2.18 (s, 3H); 13C NMR (100 MHz, CDCl3): δ= 170.2, 162.2, 159.4, 151.7, 142.2, 136.7, 134.2, 132.3, 129.6, 124.9, 117.2, 115.6, 103.1, 95.1, 84.2, 78.5, 70.4, 69.3, 61.7, 59.3, 55.8, 43.1, 21.9; IR: υ̃ = 3378, 1755, 1724, 1676, 1463, 1238 cm−1; HRMS (ESI+) m/z calcd for C28H24Cl5N3NaO9: 745.9823; found 745.9826. Data for 21: Rf = 0.45 (60% AcOEt/hexanes); 1H NMR (500 MHz, CDCl3): δ= 7.31 (d, J = 5.5 Hz, 2H), 6.83 (s, 2H), 6.55 (m, 1H), 5.81 (dd, J = 4.5 Hz, 1H), 5.69 (m, 0.5H), 5.57 (m, 2.5H), 5.49 (m, 0.5H), 5.42 (m, 0.5H), 4.77 (m, 1H), 4.71 (bs, 0.5H), 4.49 (bs, 0.5H), 4.36-4.29 (m, 2H), 3.77 (s, 3H), 3.45 (s, 1.5H), 3.43 (s, 1.5 H), 2.19 (s, 1.5H); 2.16 (s, 1.5 H); 13C NMR (100 MHz, CDCl3): δ= 169.9, 162.7, 159.4, 151.4, 140.0, 136.7, 134.1, 132.3, 129.5, 125.1, 117.3, 115.5, 102.7, 83.6, 81.1, 77.9, 73.5, 61.4, 59.4, 55.7, 42.4, 23.4; IR: υ̃ = 3378, 1755, 1724, 1676, 1463, 1238 cm−1; HRMS (ESI+) m/z calcd for C28H24Cl5N3NaO9: 745.9823; found 745.9826.
Synthesis of 10 via a Mitsunobu reaction
To a stirred solution of 21 (72.0 mg, 0.10 mmol), ClCH2COOH (10.0 mg, 0.10 mmol), Ph3P (26.0 mg, 0.10 mmol), and pyridine (8.0 μL, 0.10 mmol) in toluene (1 mL) was added DIAD (22.0 mg, 0.10 mmol). After 4 h at rt, all volatiles were removed in vacuo and the crude ester was purified by silica gel chromatography. To a stirred solution of the ester in MeOH (2 mL), thiourea (38.0 mg, 0.50 mmol) was added and the reaction mixture was heated to 50 °C. After 4 h at 50 °C, the reaction was cooled down to rt and MeOH was evaporated in vacuo. The residue was purified by silica gel chromatography with hexanes/AcOEt (1:1) to give 10 (68.0 mg, 90%) as a colorless oil. This reaction was performed for 21 (1.5 g, 2.01 mmol).
Synthesis of 26
To a stirred solution of 6 (9.0 g, 20.0 mmol) in MeOH (200 mL), was added the [tBu2SnCl(OH)]2 (0.58 g, 1.0 mmol). Upon completion, the reaction mixture was concentrated in vacuo and filtered through a silica gel plug and concentrated to yield the free alcohol in quantitative yield. To the free alcohol in CH2Cl2 (400 mL) at 0 °C was added the imidate 237b (11.9 g, 24.0 mmol) and TMSOTf (1.0 mL, 12.0 mmol) was added. After 2 h at 0 °C, the reaction mixture was quenched with aq. sat. NaHCO3. The aqueous layer was extracted with CH2Cl2 (2x) and the combined organic extracts was washed with brine, dried over Na2SO4 and evaporated. Purification of the crude material by silica gel chromatography afforded 26 (13.7 g, 92% over two steps) as a colorless liquid. Rf = 0.5 (30% AcOEt/hexanes); 1H NMR (500 MHz, CDCl3): δ= 7.83 (dd, J = 8.5 Hz, 1H), 7.37 (m, 2H), 7.18 (dd, J = 7.5 Hz, 1H), 7.00 (d, J = 8.0 Hz, 2H), 6.83 (d, J = 5.5 Hz, 2H), 6.21 (s, 0.5H), 6.15 (s, 0.5H), 5.47 (s, 1H), 5.39-5.23 (m, 3H), 4.63-4.55 (m, 1H), 3.78 (d, J = 6.5 Hz, 3H), 3.64 (m, 1H), 3.57 (d, J = 9.5 Hz, 1H), 2.32 (s, 1.5H), 2.13 (s, 1.5H), 2.07 (s, 1.5H), 2.01 (s, 1.5H), 2.00 (s, 1.5H), 1.97 (s, 3H); 13C NMR (100 MHz, CDCl3): δ= 169.9, 159.5, 137.5, 137.2, 136.4, 133.4, 132.7, 132.4, 131.7, 131.6, 129.9, 129.8, 129.0, 126.1, 125.5, 125.0, 115.2, 86.0, 76.1, 71.2, 70.4, 69.7, 69.4, 68.2, 67.1, 55.7, 25.7; IR: υ̃ = 3050, 1742, 1613, 1481 cm−1; HRMS (ESI+) m/z calcd for C33H32Cl4NaO9S: 769.0389; found 769.0387.
Synthesis of 24
To a stirred solution of 22 (13.2 g, 30.0 mmol) in MeOH (300 mL) was added the [tBu2SnCl(OH)]2 catalyst (0.87 g, 1.5 mmol). Upon completion, the reaction mixture was concentrated in vacuo and filtered through a silica gel plug and concentrated to yield the free alcohol in 100% yield. To a stirred solution of the primary alcohol (12.0 g, 30.0 mmol) and the imidate 237b (16.3 g, 33.0 mmol) in CH2Cl2 (300 mL) at 0 °C was added TMSOTf (1.0 mL, 6.0 mmol) dropwise. After being stirred for 2 h, the reaction mixture was quenched with aq. sat. NaHCO3. The aqueous layer was extracted with CH2Cl2 (2x) and the combined organic extract was washed with brine and dried over Na2SO4. The evaporation of all volatiles in vacuo gave the crude product which was purified by silica gel chromatography to afford 24 (21.9 g, 98%) as a colorless liquid. Rf = 0.5 (30% AcOEt/hexanes); 1H NMR (500 MHz, CDCl3): δ= 7.91 (m, 1H), 7.35-7.18 (m, 7H), 6.85 (s, 2H), 6.23 (d, J = 6.5 Hz, 1H), 5.40-5.31 (m, 2H), 5.27 (s, 1H), 5.22 (m, 1H), 4.85 (d, J = 7.5 Hz, 1H), 4.71 (m, 1H), 4.52 (d, J = 11.5 Hz, 1H), 4.06 (m, 1H), 3.80 (s, 3H), 3.69-3.56 (m, 2H), 2.13 (s, 1.5H), 2.10 (s, 1.5H), 1.99 (s, 1.5H), 1.98 (s, 1.5H), 1.96 (s, 1.5H), 1.95 (s, 1.5H); 13C NMR (100 MHz, CDCl3): δ= 170.1, 170.0, 169.9, 169.7, 159.5, 137.4, 137.2, 136.4, 133.5, 132.5, 131.6, 129.1, 128.5, 128.2, 126.1, 125.6, 125.1, 115.2, 96.4, 96.0, 76.9, 70.4, 69.7, 69.3, 69.0, 68.4, 67.6, 66.9, 60.4, 55.7, 20.8; IR: υ̃ = 3055, 1744, 1615, 1484 cm−1; HRMS (ESI+) m/z calcd for C33H32Cl4NaO10: 753.0618; found 753.0615.
Synthesis of 11
To a stirred solution of 24 (10.8 g, 15.0 mmol) in MeOH (600 mL) was added Pd/C (4.5 g, 10 wt %) under N2. H2 gas was introduced via double-folded balloon and the reaction mixture was stirred for 4h under H2. Upon completion, the solution was filtered through Celite and eluted with AcOEt. The organic solvent was evaporated to form the crude product which was used directly without further purification. The crude product was dissolved in dry CH2Cl2 followed by the addition of CCl3CN (15.0 mL) and DBU (0.45 mL). Upon completion, all volatiles were evaporated in vacuo. Purification by silica gel chromatography afforded the desired product 11 as colorless oil (11.2 g, 95%). Rf = 0.7 (30% AcOEt/hexanes); 1H NMR (500 MHz, CDCl3): δ= 8.71 (s, 1H), 7.87 (dd, J = 1H)7.28-7.23 (m, 2H), 6.83 (s, 2H), 6.26 (d, J = 8.5 Hz, 1H), 6.19 (s, 1H), 5.48-5.40 (m, 3H), 4.23 (m, 1H), 3.78 (s, 3H), 3.74 (m, 1H), 3.64 (m, 1H), 2.18 (s, 1.5H), 2.16 (s, 1.5H), 2.01 (s, 3H), 1.97 (s, 1.5H), 1.94 (s, 1.5H). 13C NMR (100 MHz, CDCl3): δ= 169.9, 169.8, 169.4, 159.7, 159.5, 137.3, 136.4, 133.4, 132.3, 131.6, 131.4, 129.0,128.8, 126.1, 126.1, 125.8, 125.4, 125.0, 115.2, 94.6, 94.3, 90.6, 76.5, 75.8, 72.8, 71.8, 69.0, 68.2, 68.0, 67.1, 66.0, 55.8, 55.6, 20.7; IR: υ̃ = 3050, 1755, 1680, 1622, 1480 cm−1; HRMS (ESI+) m/z calcd for C28H26Cl7N NaO10: 805.9245; found 805.9249.
Synthesis of 12
To a stirred solution of 10 (2.90 g, 4.0 mmol) and 11 (6.26 g, 8.0 mmol) in dry CH2Cl2 (50 mL) was added MS 3 Å (10.0 g). The reaction was stirred for 30 min. at rt. The reaction mixture was cooled to −5 °C, followed by dropwise addition of BF3•OEt2 (1.48 mL, 12.0 mmol). After being stirred for 3 h at −5 °C, the reaction was quenched with NaHCO3 (aq.). The reaction mixture was passed through SiO2 pad and eluted with CH2Cl2. The Organic layer was separated and dried over Na2SO4 and concentrated in vacuo. Purification by silica gel chromatography afforded 12 (4.04 g, 75%) as an oil. Rf = 0.45 (60% AcOEt/hexanes); 1H NMR (500 MHz, CDCl3): δ= 7.98 (dd, J = 21 Hz, 1H), 7.30 (s, 4H), 7.25 (s, 1H), 6.84 (s, 4H), 6.56 (s, 1H), 6.27 (s, 0.5H), 6.17 (s, 0.5H), 5.94 (d, J = 7.5 Hz, 1H), 5.88 (m, 1H), 5.58 (m, 3H), 5.39 (bs, 1H), 5.25 (d, J = 10.5 Hz, 1H), 5.22 (s, 1H), 5.00 (d, J = 20.5 Hz, 1H), 4.60 (m, 1H), 4.38 (s, 1H), 4.19 (m, 1H), 3.82 (m, 1H), 3.79 (s, 3H), 3.78 (s, 3H), 3.66 (m, 1H), 3.45 (s, 3H), 2.20 (s, 3H), 2.18 (s, 1.5 H), 2.16 (s, 1.5H), 2.11 (s, 1.5H), 2.06 (s, 1.5H), 2.02 (s, 3H), 1.97 (s, 1.5H), 1.96 (s, 1.5H). 13C NMR (100 MHz, CDCl3): δ= 169.6, 162.3, 159.6, 149.8, 137.1, 136.9, 135.8, 134.4, 133.9, 133.0, 131.7, 131.1, 129.7, 129.3, 126.3, 125.1, 124.3, 115.4, 114.7, 103.8, 95.9, 89.7, 89.3, 80.9, 80.173.1, 72.1, 71.8, 68.9, 68.1, 65.7, 63.5, 59.2, 55.8, 29.7, 20.6; IR: υ̃ = 3338, 2921, 2250, 1737, 1669, 1465, 1221 cm−1; HRMS (ESI+) m/z calcd for C54H48Cl9N3NaO18: 1367.9968; found 1367.9975.
Synthesis of 8
To a stirred solution of 12 (2.42 g, 1.8 mmol) in toluene (180 mL) was added InCl3 (0.4 g, 1.8 mmol) and acetaldoxime (0.67 mL, 10.8 mmol). The reaction mixture was heated at 70 °C for 4 h. Upon completion, the reaction was cooled to rt and all volatiles were evaporated. The crude material was passed through a short SiO2 pad. The amide was dissolved in TFA/CH2Cl2 (1:2, 75 mL) and stirring was continued for 1 h at rt. The reaction mixture was concentrated in vacuo. The crude product was purified by silica gel chromatography to afford the product 8 as an amorphous solid (1.1 g, 96% over two steps). Rf = 0.3 (95% CHCl3/MeOH); [α]D20 = +75 (c = 0.4 in MeOH); 1H NMR (500 MHz, CD3OD): δ= 7.83 (d, J = 8.0 Hz, 1H), 5.98 (d, J = 1.5 Hz, 1H), 5.91 (d, J = 8.5 Hz, 1H), 5.52 (s, 1H), 5.40 (t, J = 5.0 Hz, 1H), 5.28 (m, 2H), 5.01 (s, 1H), 4.49 (d, J = 3.0 Hz, 1H), 4.40 (m, 1H), 4.20 (t, J = 4.0 Hz, 1H), 3.91, (bs, 1H), 3.64-3.58 (m, 3H), 3.40 (s, 3H), 2.13 (s, 6H), 2.04 (s, 3H), 2.00 (s, 3H); 13C NMR (100 MHz, CD3OD): δ= 172.8, 172.0, 171.7, 171.6, 166.3, 152.2, 142.1, 103.9, 98.2, 89.4, 83.8, 79.4, 76.7, 75.2, 73.9, 71.1, 70.5, 67.2, 62.0, 59.4, 20.8, 20.6; IR: υ̃ = 3413, 1710, 1680, 1223, 1066 cm−1; HRMS (ESI+) m/z calcd for C25H33N3NaO16: 654.1753; found 654.1746.
(2-Oxo-5-phenyl-2, 3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)-carbamic acid (2,6-dichloro-4-methoxy-phenyl)-(2,4-dichloro-phenyl)-methyl ester (28, 29)
Racemic 3-amino-1,3-dihydro-5-phenyl-2H-1,4-benzodiazepin-2-one [(±)-13] was synthesized according the reported procedure.14 To a stirred solution of (±)-13 (25.0 mg, 0.10 mmol) in acetone/H2O (3:1, 3 mL) at rt was added (S)-(2,6-dichloro-4-methoxyphenyl)-(2,4-dichlorophenyl-methyl-N-succinimidyl carbonate (S)-14 (58.0 mg, 0.20 mmol) and iPr2NEt (70.0 μL, 0.40 mmol). Upon completion after 4h, the reaction mixture was concentrated in vacuo to remove acetone. The crude material was partitioned between AcOEt (5 mL) and HCl (1 N, 5 mL). The water phase was extracted with AcOEt (2x). The combined organic extracts was dried over Na2SO4, and concentrated in vacuo. Purification by silica gel chromatography (hexanes/acetone = 1:3) afforded the desired diastereomers 28 and 29 as an amorphous solid (31.0 mg each, 98% total yield). This reaction was performed with 1 gram of rac-13 to provide 28 (1.24 g). Data for 28: Rf = 0.34 (95% CHCl3/MeOH); [α]D20 = +77 (c = 0.2 in MeOH); 1H NMR (400 MHz, CD3OD): δ= 7.55-7.49 (m, 5H), 7.45 (m, 2H), 7.36 (m, 4H), 7.25-7.14 (m, 3H), 6.91 (s, 2H), 6.71 (m, 1H), 5.38 (d, J = 8.8 Hz, 1H), 3.82 (s, 3H); 13C NMR (100 MHz, CDCl3): δ= 168.6, 168.2, 160.0, 154.9, 138.6, 137.4, 134.5, 133.6, 132.4, 131.6, 130.9, 130.1, 129.9, 128.5, 127.9, 126.7, 125.1, 124.5, 121.6, 115.5, 71.9, 69.5, 56.0; IR: υ̃ = 3441, 1936, 1711, 1413, 1354, 1222, 1150 cm−1; HRMS (ESI+) m/z calcd for C30H21Cl4N3NaO4: 652.0154; found 652.0150; HPLC: retention time, 8.5 min (des >99%). Data for 29: Rf = 0.30 (95% CHCl3:MeOH); [α]D20 = +181 (c = 0.2 in MeOH); 1H NMR (400 MHz, CD3OD): δ= 7.48 (d, J = 16.8 Hz, 1H), 7.46 (m, 4H), 7.40 (m, 1H), 7.38 (m, 5H), 7.20 (m, 2H), 7.12 (m, 1H), 6.86 (s, 2H), 6.82 (m, 1H), 5.35 (d, J = 8.0 Hz, 1H), 3.78 (s, 3H); 13C NMR (100 MHz, CDCl3): δ= 170.6, 168.1, 159.7, 154.7, 138.4, 137.2, 134.4, 133.4, 132.2, 131.5, 130.7, 129.9, 129.7, 128.3, 127.7, 126.4, 124.8, 124.3, 121.3, 115.3, 71.7, 69.0, 55.7; IR: υ̃ = 3441, 1936, 1711, 1413, 1354, 1222, 1150 cm−1; HRMS (ESI+) m/z calcd for C30H21Cl4N3NaO4: 652.0154; found 652.0151; HPLC: retention time, 8.0 min (des >99%).
(S)-3-Amino-1,3-dihydro-5-phenyl-2H-1,4-benzodiazepin-2-one [(S)-13]
The carbamate 28 (30.0 mg, 0.05 mmol) was dissolved in TFA/ CH2Cl2 (1:4, 2 mL) under N2. After 1 h at rt, the reaction mixture was concentrated in vacuo. The residue was partitioned between NaHCO3 (aq.) and CHCl3/MeOH (10:1). The aqueous layer was back extracted with CHCl3/MeOH (10:1). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. Purification of the crude material by silica gel chromatography afforded the desired product (S)-13 (12.0 mg, 95%) as an amorphous solid and the byproduct ester 29 (24.0 mg, 100%) as an oil. Data for (S)-13: [α]D20 = −220 (c = 0.2 in CH2Cl2); 1H NMR (400 MHz, DMSO-d6): δ= 10.74 (bs, 1H), 7.64 (m, 1H), 7.50 (m, 5H), 7.33 (m, 2H), 7.25 (m, 1H), 4.29 (s, 1H), 2.60 (bs, 2H); 13C NMR (100 MHz, DMSO-d6): δ= 170.5, 164.7, 138.8, 138.6, 131.6, 130.1, 129.3, 128.2, 126.6, 122.8, 121.2, 70.4; IR: υ̃ = 3389, 2935, 1688, 1519, 1251, 1081 cm−1; HRMS (ESI+) m/z calcd for C15H13N3NaO: 274.0956; found 274.0958. Data for 30: Rf = 0.6 (95% hexanes/AcOEt); 1H NMR (500 MHz, CDCl3): δ= 7.70 (s, 1H), 7.46 (s, 1H), 7.25 (s, 2H), 6.94 (s, 2H), 3.83 (s, 3H); 13C NMR (100 MHz, CDCl3): δ= 172.0, 160.5, 136.8, 135.8, 134.7, 130.9, 126.9, 122.1, 115.6, 74.3, 55.9; IR: υ̃ = 1721, 1438, 1410, 1325 cm−1; HRMS (ESI+) m/z calcd for C16H9Cl4F3NaO3: 470.9126; found 470.9124.
Synthesis of 35
To a vigorously stirred solution of the alcohol 8 (0.20 g, 0.32 mmol) in dry DMSO (10 mL) and dry Et3N (5 mL) was added a solution of SO3•Py (0.252 g, 1.60 mmol) in dry DMSO (5 mL) at 20 °C under N2. After 1 h at rt, the reaction mixture was quenched with water (0.1 mL). The DMSO and all volatiles were removed by evaporation in vacuo to give the crude aldehyde 31 which was used without purification in the next step. To a vigorously stirred solution of crude aldehyde 31 in tBuOH (0.8 mL) and 2-methyl-2-butene (0.60 mL) at rt was added a solution of NaH2PO4 (11.0 mg, 0.10 mmol) and NaClO2 (9.0 mg 0.10 mmol) in H2O (0.8 mL). After 1 h at rt, the reaction mixture was extracted with AcOEt, then CHCl3/MeOH (10:1). The combined organic extracts was dried over Na2SO4 and concentrated in vacuo to give the crude acid 32. To a stirred solution of the crude acid 32 (55.0 mg, 96.0 3mol) and (S)-13 (48.0 mg, 192.0 3mol) in DMF/H2O (2:1, 3 mL) was added EDCI (90.0 mg, 0.48 mmol), glyceroacetonide-Oxyma 17 (0.114 g, 0.48 mmol) and NaHCO3 (0.102 g, 1.20 mmol) sequentially. After 4 h at rt, all volatiles were evaporated and the resulting slurry was partitioned between AcOEt and NaHCO3 (aq.), the aqueous layer was extracted with AcOEt (3x). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo to give the crude product which was purified by silica gel chromatography to afford 35 (66.7 mg, 85% from 8) as an amorphous solid. [α]D20 = +99 (c = 0.2 in MeOH); 1H NMR (500 MHz, CD3OD): δ= 7.91 (s, 1H), 7.86 (m, 1H), 7.61 (t, J = 7.5 Hz, 1H), 7.53 (m, 2H), 7.43 (m, 2H), 7.31 (m, 2H), 7.22 (m, 1H), 5.98 (s, 2H), 5.96 (s, 1H), 5.50 (s, 1H), 5.41 (s, 1H), 5.09 (m, 1H), 4.97 (m, 1H), 4.74 (m, 2H), 4.37 (s, 1H), 4.18 (m, 1H), 3.89 (m, 2H), 3.76 (m, 2H), 3.44 (s, 1H), 3.41 (s, 3H), 2.14 (s, 3H), 2.11 (s, 3H), 2.05 (s, 3H); 13C NMR (100 MHz, CDCl3): δ= 172.3, 171.6, 169.3, 166.1, 152.1, 141.5, 140.1, 133.4, 132.1, 131.8, 130.9, 129.4, 128.7, 124.7, 122.5, 104.0, 98.1, 88.7, 82.3, 82.9, 79.5, 76.7, 75.9, 73.5, 71.8, 70.9, 70.4, 65.1, 62.2, 59.5, 20.6; IR: υ̃ = 3389, 2935, 1688, 1519, 1251, 1081cm−1; HRMS (ESI+) m/z calcd for C38H38N6NaO15: 841.2293; found 841.2296.
Synthesis of UT-01309 (2)
To a stirred solution of 34 (13.0 mg, 16.0 μmol) in THF/H2O (10:1, 0.4 mL) at 0 °C was added LiOH (0.08 mL, 1 M in H2O). After being stirred for 1 h at 0 °C, the reaction mixture was quenched with THF/AcOH (10:1, 0.08 mL). All volatiles were evaporated in vacuo. Purification by silica gel PTLC (MeOH/CHCl3, 1:2) afforded 2 (10.60 mg, 95%) as an amorphous solid. Rf = 0.4 (70% CHCl3/MeOH); [α]D20 = +85 (c = 0.1 in MeOH); 1H NMR (500 MHz, CD3OD): δ= 7.88 (d, J = 8.5 Hz, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.41 (m, 3H), 7.33 (m, 2H), 7.26-7.17 (m, 3H), 6.00 (d, J = 4.0 Hz, 1H), 5.81 (d, J = 3.0, 1H), 5.64 (d, J = 8.5 Hz, 1H), 5.33 (s, 1H), 5.19 (d, J = 6.0 Hz, 1H), 4.67 (s, 1H), 4.43 (d, J = 6.5 Hz, 1H), 4.35 (d, J = 5.5 Hz, 1H), 4.3 (m, 1H), 3.89 (t, J = 5.5 Hz, 1H), 3.74 (t, J = 4.0 Hz, 1H), 3.40 (s, 3H); 13C NMR (100 MHz, CD3OD): δ= 179.1, 173.8, 166.3, 164.9, 152.3, 142.0, 140.0, 133.6, 132.2, 131.9, 131.0, 129.4, 128.6, 124.8, 122.7, 102.7, 100.7, 91.1, 83.6, 80.0, 76.3, 75.8, 74.1, 72.7, 71.5, 70.6, 68.3, 62.8, 58.4; IR: υ̃ = 3411, 2933, 1696, 1515, 1279 cm−1; HRMS (ESI+) m/z calcd for C32H32N6NaO12: 715.1976; found 715.1972.
Synthesis of 34
To a stirred solution of the acid 32 (55.0 mg, 96.0 3mol) and 32 (31.0 mg, 192.0 3mol) in H2O (1.0 mL) was added EDCI (90.0 mg, 0.48 mmol), glyceroacetonide-Oxyma 17 (0.114 g, 0.48 mmol) and NaHCO3 (0.102 g, 1.20 mmol) sequentially. After being stirred for 4 h at rt, all volatiles were evaporated and the resulting slurry was partitioned between EtOAc and aq. NaHCO3, the aqueous layer was extracted with AcOEt (3x). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo to give the crude product. For data collections, a portion was purified by silica gel chromatography to afford 34 as an amorphous solid (57.0 mg, 85% from 8). [α]D20 = +103 (c = 0.3 in CHCl3); 1H NMR (500 MHz, CDCl3): δ= 9.72 (broad, 1H), 7.96 (d, J = 6.8 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.39, (broad, 1H), 7.20 (broad, 1H), 6.23 (broad, 1H), 6.05 (d, J = 3.2 Hz, 1H), 5.80 (s, 1H), 5.69 (t, J = 3.6 Hz, 1H), 5.49 (s, 1H), 5.32-5.26 (m, 2H), 4.61 (dd, J = 7.2, 10.8 Hz, 1H), 4.55 (s, 1H), 4.39 (d, J = 5.6 Hz,1H), 4.01 (s, 1H), 3.30 (s, 2H), 3.25 (s, 3H), 2.11 (s, 6H), 2.06 (s, 3H), 1.86 (m, 2H), 1.60 (m, 3H), 1.40 (m, 1H); 13C NMR (100 MHz, CDCl3): δ= 176.1, 170.3, 170.2, 170.12, 170.08, 169.03, 169.00, 163.5, 159.1, 150.8, 144.5, 140.6, 104.1, 103.8, 98.0, 82.0, 73.3, 65.2, 63.1, 59.1, 52.0, 42.4, 31.6, 28.8, 20.95, 20.88, 20.83; IR: υ̃ = 3379, 2930, 1691, 1509, 1250, 1070 cm−1; HRMS (ESI+) m/z calcd for C29H8N5NaO15: 718.2178; found 718.2186.
Synthesis of Capuramycin (1)
To a stirred solution of 34 (11.0 mg, 16.0 μmol) in THF/H2O (10:1, 0.4 mL) at 0 °C was added LiOH (0.08 mL, 1 M in H2O). After being stirred for 1 h at 0 °C, the reaction mixture was quenched with THF/AcOH (10:1, 0.08 mL). All volatiles were evaporated in vacuo. Purification by silica gel PTLC (MeOH/CHCl3, 1:2) afforded the desired product 1 (8.60 mg, 95%) as an amorphous solid. Rf = 0.4 (70% CHCl3/MeOH); [α]D20 = +98 (c = 0.1 in H2O); 1H NMR (400 MHz, CD3OD): δ= 7.71 (d, J = 8.0 Hz, 1H), 5.97 (s, 1H), 5.82 (d, J = 8.0 Hz, 1H), 5.73 (s, 1H), 5.35 (s, 1H), 4.59 (d, J = 11.2 Hz, 2H), 4.47 (s, 1H), 4.44 (d, J = 4.8 Hz, 1H), 4.34 (s, 1H), 4.15 (s, 1H), 3.71 (t, J = 4.8 Hz, 1H), 3.26 (s, 3H), 1.94-1.57 (m, 6H), 1.32 (m, 2H). 13C NMR (100 MHz, D2O): δ= 176.3, 173.0, 166.1, 161.4, 151.2, 141.5, 141.0, 109.4, 101.8, 99.3, 90.1, 81.6, 78.1, 75.5, 71.9, 64.7, 61.7, 57.8, 52.2, 41.4, 30.3, 27.3; IR: υ̃ = 3411, 2933, 1696, 1515, 1279 cm−1; HRMS (ESI+) m/z calcd for C23H31N5NaO12: 592.1867; found 592.1864.
Acknowledgments
The National Institutes of Health is greatly acknowledged for financial support of this work (AI084411). We also thank University of Tennessee for generous financial support. NMR data were obtained on instruments supported by the NIH Shared Instrumentation Grant.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.
References
- 1.a) Grenet K, Guillemot D, Jarlier V, Moreau B, Dubourdieu S, Ruimy R, Armand-Lefevre L, Brau P, Andremont A. Emerg Infect Dis. 2004;10:1150–1153. doi: 10.3201/eid1006.031015. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cohen J. Science. 2004;306:1872. doi: 10.1126/science.306.5703.1872. [DOI] [PubMed] [Google Scholar]
- 2.Godfrey-Faussett P. AIDS. 2003;17:1079–1081. doi: 10.1097/01.aids.0000060354.78202.fe. [DOI] [PubMed] [Google Scholar]
- 3.a) Cole ST, Alzari PM. Science. 2005;14:214–215. doi: 10.1126/science.1108379. [DOI] [PubMed] [Google Scholar]; b) Stover CK, Warrener P, van Devater DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. Nature. 2000;405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 4.a) Wainberg MA. Scientifica. 2012;2012:1–28. [Google Scholar]; b) Connolly LE, Edelstein PH, Ramakrishnan L. PLoS Med. 2007;4:435–441. doi: 10.1371/journal.pmed.0040120. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nachega JB, Hislop M, Dowdy DW, Chaisson RE, Regensberg L, Maartens G. Ann Int Med. 2007;146:564–573. doi: 10.7326/0003-4819-146-8-200704170-00007. [DOI] [PubMed] [Google Scholar]; d) Burman WJ, Jones BE. Am J Respir Crit Care Med. 2001;164:469–473. doi: 10.1164/ajrccm.164.1.2101133. [DOI] [PubMed] [Google Scholar]
- 5.a) Diacon AH, Donald PR, Pym A, Grobusch M, Patientia RF, Mahanyele R, Bantubani N, Narasimooloo R, De Marez T, van Heeswijk R, Lounis N, Meyvisch P, Andries K, McNeeley DF. Antimicrob Agents Chemother. 2012;56:3271–3276. doi: 10.1128/AAC.06126-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Matteelli A, Carvalho AC, Carvalho KE, Kritski A. Future Microbiol. 2010;5:849–858. doi: 10.2217/fmb.10.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wright GD. Science. 2007;315:1372–1373. doi: 10.1126/science.1140374. [DOI] [PubMed] [Google Scholar]
- 7.a) Cudic P, Behenna DC, Yu MK, Kruger RG, Szwczuk LM, McCafferty DG. Bioorg Med Chem Lett. 2001;11:3107–3110. doi: 10.1016/s0960-894x(01)00653-9. [DOI] [PubMed] [Google Scholar]; b) Helm JS, Hu Y, Chen L, Gross B, Walker S. J Am Chem Soc. 2003;125:11168–11169. doi: 10.1021/ja036494s. [DOI] [PubMed] [Google Scholar]; c) Bachelier A, Mayer R, Klein CD. Bioorg Med Chem Lett. 2006;16:5605–5609. doi: 10.1016/j.bmcl.2006.08.021. [DOI] [PubMed] [Google Scholar]; d) Antane S, Caufield CE, Hu W, Keeney D, Labthavikul P, Morris K, Naughton SM, Petersen PJ, Rasmussen BA, Singh G, Yang Y. Bioorg Med Chem. 2006;16:176–180. doi: 10.1016/j.bmcl.2005.09.021. [DOI] [PubMed] [Google Scholar]; e) Taha MO, Atallah N, Al-Bakri AG, Pradis-Bleau C, Yonis H, Levesque KS. Bioorg Med Chem. 2008;16:1218–1235. doi: 10.1016/j.bmc.2007.10.076. [DOI] [PubMed] [Google Scholar]; f) Bryskier A, Dini AC. Antimicr Agents: Antibacter Antifung. 2005:377–400. [Google Scholar]; g) Kotnik M, Humljan J, Contreras-Martel C, Oblak M, Kristan K, Hervé M, Blanot D, Urleb U, Gobec S, Dessen A, Solmajer T. J Mol Biol. 2007;370:107–115. doi: 10.1016/j.jmb.2007.04.048. [DOI] [PubMed] [Google Scholar]; h) Perdih A, Kovac A, Wolber G, Blanot D, Gobec S, Solmajer T. Bioorg Med Chem Lett. 2009;19:2668–2673. doi: 10.1016/j.bmcl.2009.03.141. [DOI] [PubMed] [Google Scholar]; i) Humljan J, Kotnik M, Contreras-Martel C, Blanot D, Urleb U, Dessen A, Solmajer T, Gobec S. J Med Chem. 2008;51:7486–7494. doi: 10.1021/jm800762u. [DOI] [PubMed] [Google Scholar]
- 8.Bugg TDH, Timothy DH, Lloyd AJ, Roper DI. Infect Disorders: Drug Targets. 2006;6:85–106. doi: 10.2174/187152606784112128. [DOI] [PubMed] [Google Scholar]
- 9.a) Kurosu M, Mahapatra S, Narayanasamy P, Crick DC. Tetrahedron Lett. 2007;48:799–803. [Google Scholar]; b) Kurosu M, Narayanasamy P, Crick DC. Heterocycles. 2007;72:339–352. [Google Scholar]; c) Kurosu M, Li K. J Org Chem. 2008;73:9767–9770. doi: 10.1021/jo801408x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mcdonald L, Barbieri L, Carter G, Lenoy E, Lotvin J, Petersen P, Siegel M, Singh G, Williamson R. J Am Chem Soc. 2002;124:10260–10261. doi: 10.1021/ja017748h. [DOI] [PubMed] [Google Scholar]
- 11.Ubukata M, Kimura K, Isono K, Nelson CC, Gregson JM, JA J Org Chem. 1992;57:6392–6403. [Google Scholar]
- 12.Igarashi M, Nakagawa N, Doi N, Hattori S, Naganawa H, Hamada M. J Antibiot. 2003;56:580–583. doi: 10.7164/antibiotics.56.580. [DOI] [PubMed] [Google Scholar]
- 13.Fernandes PB, Swanson RN, Hardy DJ, Hanson CW, Coen L, Rasmussen RR, Chen RH. J Antibiot. 1989;42:521–526. doi: 10.7164/antibiotics.42.521. [DOI] [PubMed] [Google Scholar]
- 14.Yamaguchi H, Sato S, Yoshida S, Takada K, Itoh M, Seto H, Otake N. J Antibiot. 1986;39:1047–1053. doi: 10.7164/antibiotics.39.1047. [DOI] [PubMed] [Google Scholar]
- 15.a) Reddy VM, Einck L, Nacy CA. Antimicro Agents Chemother. 2008;52:719–721. doi: 10.1128/AAC.01469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ichikawa S. Chem Pharm Bull. 2008;56:1059–1072. doi: 10.1248/cpb.56.1059. [DOI] [PubMed] [Google Scholar]; c) Dini C. Curr Top Med Chem. 2005;5:1221–1236. doi: 10.2174/156802605774463042. [DOI] [PubMed] [Google Scholar]; d) Dini C, Drochon N, Feteanu S, Guillot JC, Peixoto C, Aszodi J. Bioorg Med Chem Lett. 2001;11:529–5231. doi: 10.1016/s0960-894x(00)00715-0. [DOI] [PubMed] [Google Scholar]
- 16.a) Bogatcheva E, Dubuisson T, Protopopova M, Einck L, Nacy CA, Reddy VM. J Antimicro Chemother. 2011;66:578–587. doi: 10.1093/jac/dkq495. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reddy VM, Einck L, Nacy CA. J Antimicro Chemother. 2008;52:719–721. doi: 10.1128/AAC.01469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Koga T, Fukuoka T, Harasaki T, Inoue H, Hotoda H, Kakuta M, Muramatsu Y, Yamamura N, Hoshi M, Hirota T. J Antimicro Chemother. 2004;54:755–760. doi: 10.1093/jac/dkh417. [DOI] [PubMed] [Google Scholar]
- 17.Knapp S, Nandan SR. J Org Chem. 1994;59:281–283. [Google Scholar]
- 18.Kurosu M, Li K, Crick DC. Org Lett. 2009;11:2393–2396. doi: 10.1021/ol900458w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aleiwi BA, Kurosu M. Tetrahedron Lett. 2012;53:3758–3762. doi: 10.1016/j.tetlet.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Aleiwi BA, Kurosu M. unpublished data. [Google Scholar]
- 21.a) Wang Y, Kurosu M. Tetrahedron. 2012;68:4797–4804. doi: 10.1016/j.tet.2012.03.121. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kurosu M, Li K. Synthesis. 2009;21:3633–3641. [Google Scholar]; c) Kurosu M, Biswas K, Crick DC. Org Lett. 2007;9:1141–1144. doi: 10.1021/ol070150f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurosu M, Li K. Org Lett. 2009;11:911–914. doi: 10.1021/ol8028719. [DOI] [PubMed] [Google Scholar]
- 23.Kore AR, Parmar G, Reddy S. Nucleos Nucleot Nucleic Acids. 2006;25:307–314. doi: 10.1080/15257770500544529. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Aleiwi BA, Wang Q, Kurosu M. Org Lett. 2012;14:4910–4913. doi: 10.1021/ol3022337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.See: supporting information.
- 26.Lázár L, Bajza I, Jakab Z, Lipták A. Synlett. 2005;14:2242–2244. [Google Scholar]
- 27.a) Ranganathan RS, Jones GH, Moffatt JG. J Org Chem. 1974;39:290–302. doi: 10.1021/jo00917a003. [DOI] [PubMed] [Google Scholar]; b) Pfitzner KE, Moffatt JG. J Am Chem Soc. 1963;85:3026–3027. [Google Scholar]
- 28.a) North M. Tetrahedron: Asymmetry. 2003;14:147–288. [Google Scholar]; b) Krueger CA, Kuntz KW, Dzierba CD, Wirschun WG, Gleason JD, Snapper ML, Hoveyda AH. J Am Chem Soc. 1999;121:4284–4285. [Google Scholar]; c) Mori M, Imma H, Nakai T. Tetrahedron Lett. 1997;38:6229–6232. [Google Scholar]
- 29.Kurosu M, Lorca M. Synlett. 2005;7:1109–1112. [Google Scholar]
- 30.Watahiki T, Ohba S, Oriyama T. Org Lett. 2003;5:2679–2681. doi: 10.1021/ol0348295. [DOI] [PubMed] [Google Scholar]
- 31.Orita A, Hamada Y, Nakano T, Toyoshima S, Otera J. Chem Eur J. 2001;7:3321–3327. doi: 10.1002/1521-3765(20010803)7:15<3321::aid-chem3321>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt RR. Angew Chem Int Ed. 1986;25:212–235. [Google Scholar]
- 33.Kurosu M, Li K. J Org Chem. 2008;73:9767–9770. doi: 10.1021/jo801408x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harreus AH, Kunz H. Liebigs Ann Chem. 1986:717–730. [Google Scholar]
- 35.a) Fukase K, Hasuoka A, Kinoshita I, Aoki Y, Kusumoto S. Tetrahedron. 1995;51:4923–4932. [Google Scholar]; b) Yeeneman GH, van Leeuwen SH, van Boom JH. Tetrahedron Lett. 1990;31:1331–1334. [Google Scholar]
- 36.a) Carter MC, Alber DG, Baxter RC, Bithell SK, Budworth J, Chubb A, Cockerill GS, Dowdell VCL, Henderson EA, Keegan SJ, Kelsey RD, Lockyer MJ, Stables JN, Wilson LJ, Powell KL. J Med Chem. 2006;49:2311–2319. doi: 10.1021/jm051185t. [DOI] [PubMed] [Google Scholar]; b) Bock MG, DiPardo RM, Evans BE, Rittle KE, Veber DF, Freidinger RM. Tetrahedron Lett. 1987;28:939–942. [Google Scholar]
- 37.a) Park EY, Jin KB, Hyun MH. Chirality. 2012;24:427–431. doi: 10.1002/chir.22041. [DOI] [PubMed] [Google Scholar]; b) Henderson EA, Alber DG, Baxter RC, Bithell SK, Budworth J, Carter MC, Chubb A, Cockerill GS, Dowdell VCL, Fraser IJ, Harris RA, Keegan SJ, Kelsey RD, Lumley JA, Stables JN, Weerasekera N, Wilson LJ, Powell KL. J Med Chem. 2007;50:1685–1692. doi: 10.1021/jm060747l. [DOI] [PubMed] [Google Scholar]; c) Evans BE, Rittle KE, Veber DF, Freidinger RM, Hirshfield J, Springer JP. J Org Chem. 1987;52:3232–3232. [Google Scholar]
- 38.Sherrill RG, Sugg EE. J Org Chem. 1995;60:730–734. [Google Scholar]
- 39.Kim ES, Lee HS, Kim SH, Kim JN. Tetrahedron Lett. 2010;51:1589–1591. [Google Scholar]
- 40.Mackie DM, Perlin AS. Carbohydr Res. 1972;24:67–85. [Google Scholar]
- 41.a) Bal BS, Childers WE, Pinnick HW. Tetrahedron. 1981;37:2091–2096. [Google Scholar]; b) Bengt LO, Torsten N. Acta Chem Scand. 1973;27:888–890. [Google Scholar]
- 42.Wang Q, Wang Y, Kurosu M. Org Lett. 2012;14:3372–3375. doi: 10.1021/ol3013556. [DOI] [PMC free article] [PubMed] [Google Scholar]