

Abstract
This review summarizes the current state of knowledge on neutrophil basic biology and discusses how the breakdown of neutrophil homeostasis affects periodontal health. The homeostasis of neutrophils is tightly regulated through coordinated bone marrow production, release into the circulation, transmigration to and activation in peripheral tissues, and clearance of senescent neutrophils. Dysregulation of any of these homeostatic mechanisms at any age can cause severe periodontitis in humans and animal models. Accordingly, both impaired and excessive neutrophil activity (in terms of numbers or immune function) can precipitate periodontitis. Neutrophil defects of congenital origin (e.g., congenital neutropenia, leukocyte adhesion deficiency, and Chediak-Higashi syndrome) are associated with cutaneous and systemic infections and early-onset forms of periodontitis affecting both the primary and permanent dentitions of children. However, the strong association between congenital neutrophil disorders and early-onset periodontitis is not currently adequately explained mechanistically. This suggests the operation of as-yet-unknown molecular mechanisms, although the available body of evidence leaves no doubt that neutrophils are integral to periodontal tissue homeostasis and health.
Keywords: bone marrow, congenital syndromes, periodontitis, leukocyte adhesion molecules, neutrophil infiltration, inflammation
Introduction
Neutrophils are terminally differentiated white blood cells, which are equipped with a plethora of microbicidal and pro-inflammatory mechanisms and form the first line of defense against pathogenic insults (Kobayashi and DeLeo, 2009; Amulic et al., 2012). They are produced in huge numbers in the bone marrow (≈ 109 cells per kg bodyweight per day), from where they are released into the circulation. However, neutrophils are short-lived (6- to 8-hour circulating half-life) and undergo apoptosis (programmed cell death) (von Vietinghoff and Ley, 2008).
In response to infection and/or inflammation, circulating neutrophils can migrate to peripheral tissues, such as the skin, gut, lungs, or the periodontium (Amulic et al., 2012; Hajishengallis and Chavakis, 2013). Defects in the function or numbers of neutrophils have been implicated in several forms of periodontal disease, ranging from early-onset forms in children to adult-type, chronic periodontitis (Deas et al., 2003; Darveau, 2010; Nussbaum and Shapira, 2011). Intriguingly, both insufficient and excessive neutrophil function or numbers can contribute to the pathogenesis of periodontitis (Deas et al., 2003; Ryder, 2010; Nussbaum and Shapira, 2011; Hasturk et al., 2012; Hajishengallis and Chavakis, 2013). The major focus of this review is to discuss established and emerging concepts that neutrophil homeostasis is essential to periodontal health for reasons that do not necessarily relate to the traditional defense functions of neutrophils. Understanding the mechanisms of neutrophil involvement in the various forms of periodontal disease can lead to more effective therapeutic interventions.
Neutrophil Biology and Homeostasis
To better understand the role of neutrophils in periodontal disease, it is instructive to first discuss basic concepts of neutrophil biology and homeostasis. To maintain a balance between the protective and potentially destructive effects of neutrophils, several homeostatic mechanisms regulate their production, trafficking, and clearance (von Vietinghoff and Ley, 2008; Summers et al., 2010). The granulocyte colony-stimulating factor (G-CSF) is the primary regulator of both granulopoiesis and neutrophil release from the bone marrow (Fig. 1). Mice or humans lacking a functional G-CSF receptor exhibit severe neutropenia (von Vietinghoff and Ley, 2008). The effects mediated by G-CSF include the commitment of progenitors to the myeloid lineage, proliferation of granulocytic precursors, and the release of mature neutrophils into the circulation (von Vietinghoff and Ley, 2008; Summers et al., 2010). The latter effect is mediated, at least in part, by the ability of G-CSF to interfere with the interaction between the CXC chemokine receptor 4 (CXCR4) and the stromal-derived factor-1 (SDF-1), a chemokine (also known as CXCL12) produced constitutively by bone-marrow stromal cells. The CXCR4–SDF-1 interaction is a major mechanism of neutrophil retention in the bone marrow (von Vietinghoff and Ley, 2008). In mice, the circulating pool of neutrophils represents < 2% of the mature neutrophils in the bone marrow, although this percentage is increased to about 10% following G-CSF treatment (Summers et al., 2010). Interleukin 17 (IL-17) promotes granulopoiesis and induces the recruitment, activation, and survival of neutrophils by acting mainly through up-regulation of G-CSF (Ye et al., 2001; von Vietinghoff and Ley, 2008) (Fig. 2).
Figure 1.

The life cycle and function of neutrophils: homeostatic perturbations in various syndromes or disorders. Key events in the life cycle of the neutrophil include granulopoiesis, release into the circulation, adhesion to the endothelium and chemotactic migration to tissues, antimicrobial function, and apoptotic cell clearance. Shown are congenital and other disorders that interfere with the indicated functions leading to development of severe forms of periodontitis (Deas et al., 2003; Kollner et al., 2006; Dotta et al., 2011; Ye et al., 2011; Hanna and Etzioni, 2012). LAD, leukocyte adhesion deficiency; SLE, systemic lupus erythematosus; WHIM, warts, hypogammaglobulinemia, infections, and myelokathexis (syndrome); G-CSF, granulocyte-colony stimulating factor.
Figure 2.
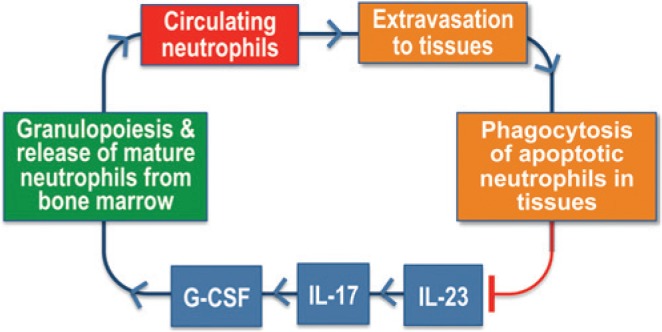
The neutrostat regulatory feedback loop. During infection or inflammation, IL-23-induced IL-17 promotes granulopoiesis and mobilization of mature neutrophils from the bone marrow by acting through up-regulation of G-CSF. Circulating neutrophils can normally extravasate into infected or inflamed tissues. Upon senescence, transmigrated neutrophils become apoptotic and undergo phagocytosis by tissue phagocytes, leading to suppression of IL-23 production, in turn, down-regulating the IL-17-G-CSF axis for maintaining steady-state neutrophil counts (Stark et al., 2005).
The process of neutrophil extravasation involves a cascade of low- and high-affinity adhesive interactions between the neutrophils and the vascular endothelium (Nourshargh et al., 2010). The first step involves transient rolling interactions mediated by endothelial cell-surface molecules (P- or E-selectin) and their glycoprotein ligands on neutrophils. This rolling-dependent deceleration of neutrophils is followed by β2 integrin-dependent firm adhesion and subsequent crawling on the endothelium, during which neutrophils seek an appropriate site for diapedesis through endothelial junctions (Fig. 1). Recently, a 52-kDa endothelial cell-secreted glycoprotein termed developmental endothelial locus-1 (Del-1; also known as EDIL3, for EGF-like repeats and discoidin I-like domains 3) was identified as a novel, perhaps the first, negative regulator of β2 integrin-dependent neutrophil recruitment (Choi et al., 2008; Hajishengallis and Chavakis, 2013). Specifically, Del-1 inhibits the interaction between the LFA-1 (CD11a/CD18) integrin on neutrophils and the intercellular adhesion molecule-1 (ICAM-1) on endothelial cells (Choi et al., 2008), thereby suppressing firm adhesion and consequently the transendothelial migration of human neutrophils (Eskan et al., 2012) (Fig. 3).
Figure 3.
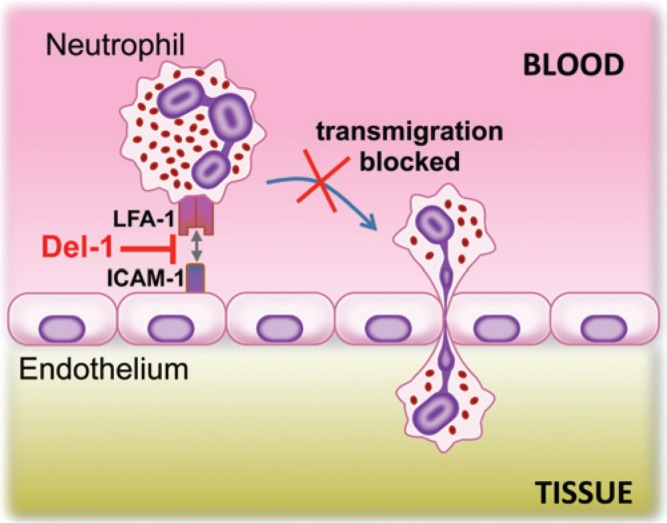
Del-1 acts as a local gatekeeper of normal neutrophil recruitment. Del-1 blocks the interaction between the LFA-1 integrin on neutrophils and ICAM-1 on endothelial cells. This interaction is required for firm neutrophil adhesion to the endothelium, which in turn is essential to subsequent transmigration (Choi et al., 2008; Eskan et al., 2012). As a consequence, Del-1 can suppress the migration of neutrophils from the circulation to peripheral tissues.
Neutrophil extravasation can be modulated by tissue-derived cytokines, which can up-regulate endothelial adhesion molecule expression, whereas tissue-derived chemokines decorating the apical endothelial cell surface can trigger conformational changes on leukocyte integrins, which thereby adopt their high-affinity state (Nourshargh et al., 2010; Kolaczkowska and Kubes, 2013). Although transmigrating neutrophils initially follow the chemokine gradient deposited by the endothelium, they then have to move toward a gradient existing in the infected or inflamed tissue. Such gradients could also involve chemoattractants derived from infecting bacteria (N-formyl-methionyl-leucyl-phenylalanine; fMLP) or from local complement activation (C5a fragment) (Kolaczkowska and Kubes, 2013).
Extravasated senescent neutrophils are cleared locally by tissue phagocytes, such as macrophages or dendritic cells, whereas senescent neutrophils in the blood home back to the bone marrow for clearance upon up-regulation of CXCR4 expression (Martin et al., 2003) (Fig. 1). Intriguingly, the clearance of transmigrated apoptotic neutrophils serves more than waste disposal, since it is crucial for the regulation of neutrophil production in the bone marrow (Stark et al., 2005) (Fig. 2). Indeed, the engulfment of apoptotic neutrophils by tissue phagocytes triggers anti-inflammatory signals; which decrease their production of IL-23, a key cytokine for induction of IL-17 by both innate and adaptive immune cells (Stark et al., 2005). The resulting inhibition of IL-17 production, in turn, leads to decreased production of G-CSF by cells such as fibroblasts, thereby limiting the stimulus for neutrophil production to maintain steady-state neutrophil counts (Stark et al., 2005). In essence, this is a neutrophil rheostat (‘neutrostat’) feedback mechanism (Fig. 2). For instance, the phagocytosis of apoptotic neutrophils associated with resolution of inflammation will signal the down-regulation of neutrophil production, since neutrophils are no longer needed in great numbers.
Neutrophil Recruitment to the Periodontium
Neutrophils constitute the overwhelming majority of cells recruited to the gingival crevice (≥ 95% of total leukocytes) and form a ‘defense wall’ against the tooth-associated subgingival biofilm (Delima and Van Dyke, 2003; Ryder, 2010). Extravasated neutrophils enter the crevice through the junctional epithelium, which, under inflammatory conditions, is largely occupied (≈ 60%) by trafficking neutrophils (Delima and Van Dyke, 2003).
Healthy human gingiva display coordinated gradients of chemokines and adhesion molecules that are thought to contribute to the directed migration of neutrophils to the gingival crevice. Specifically, gradients of interleukin (IL)-8 (CXCL8), ICAM-1, and E-selectin are topographically associated with the pathway of neutrophil migration, from the vasculature to the junctional epithelium and, ultimately, to the gingival crevice (Darveau, 2010). A recent study in mice demonstrated that neutrophil recruitment to the periodontium is entirely dependent upon the CXC chemokine receptor 2 (CXCR2), which responds to neutrophil-specific chemoattractants such as CXCL1 and CXCL2 (murine analogues of IL-8) (Zenobia et al., 2013). This finding probably reflects the essential role of CXC chemoattractants in the initial phase of the directed extravasation of neutrophils (Kolaczkowska and Kubes, 2013), and it should not necessarily be interpreted as evidence of lack of involvement of C5a or fMLP in neutrophil recruitment to the periodontium.
Intriguingly, the migration of neutrophils to the periodontium does not require commensal bacterial colonization, since recruited neutrophils were also observed in germ-free mice (Zenobia et al., 2013). This finding suggests that neutrophil recruitment may serve homeostatic function(s) that may not necessarily be related to infection control. In this respect, it should be noted that mechanisms sensing neutrophil recruitment to peripheral tissues also contribute to the homeostatic regulation of neutrophil numbers (Stark et al., 2005) (Fig. 2).
Neutrophil Homeostasis Breakdown and Periodontal Disease
The importance of neutrophil homeostasis for periodontal health is emphatically underscored by the development of periodontitis in conditions associated with defects in mechanisms that regulate the production and life cycle of neutrophils (Fig. 1). Many of these disorders are congenital and affect periodontal health early in life. These forms of early-onset periodontitis are generally unresponsive to antibiotics and/or mechanical removal of the tooth-associated biofilm (Deas et al., 2003; Nualart Grollmus et al., 2007), suggesting the involvement of mechanisms unrelated to or in addition to defective neutrophil control of the periodontal microbiota. Congenital or acquired conditions associated with neutrophil dysfunction and periodontitis are discussed below in sections organized according to the different stages of the neutrophil life cycle.
Defective Neutrophil Production
Defective granulopoiesis leads to neutropenia, which represents a persistent reduction in the absolute neutrophil count in the circulation (< 1,500 cells/µL) and is generally associated with susceptibility to infections. It is classified as congenital or acquired (i.e., autoimmune neutropenia, HIV-associated neutropenia, and neutropenia in cancer patients under chemotherapy or radiation therapy). Congenital neutropenias are further subdivided into those existing as the only phenotypic manifestations or those associated with other immunological or extra-hematopoietic abnormalities (Donadieu et al., 2011). Periodontal disease, often severe, involving both the primary and the permanent dentition, occurs in many of these neutropenic conditions (Hart and Atkinson, 2007). To restore the number of circulating neutrophils, G-CSF is frequently administered to neutropenic patients who typically show improvement in their periodontal condition, thus supporting the importance of neutrophils in periodontal health (Nussbaum and Shapira, 2011).
Mutations in the ELANE gene, which encodes neutrophil elastase, are involved in cyclic neutropenia and also in 40% to 55% of the cases of severe congenital neutropenia (Fig. 1). Elastase is a serine protease that is synthesized during the myeloblast-to-promyelocyte transition and stored in azurophilic granules. ELANE mutations cause the production of misfolded elastase protein molecules, which are thought to activate the unfolded protein response leading to accelerated apoptosis of neutrophil precursors in the bone marrow (Kollner et al., 2006). Mutations in the ELANE gene were recently associated with periodontitis in patients with severe congenital neutropenia (Ye et al., 2011).
Defective Neutrophil Release from the Bone Marrow
The syndrome known as WHIM is characterized by warts, hypogammaglobulinemia, infections, and myelokathexis (i.e., retention of neutrophils in the bone marrow) (Dotta et al., 2011). WHIM patients have mutations in the CXCR4, a chemokine receptor involved in the release of mature neutrophils from the bone marrow and also the homing of senescent neutrophils back to the bone marrow (Martin et al., 2003; von Vietinghoff and Ley, 2008). The CXCR4 mutations associated with the WHIM syndrome affect the desensitization of CXCR4 upon stimulation with its ligand (SDF-1), leading to altered responsiveness of WHIM neutrophils to SDF-1 (Lagane et al., 2008). How this defect relates to the clinical manifestations of the syndrome is not clear, although impaired cellular homeostasis and trafficking appear to be a factor. In this regard, WHIM patients cannot release neutrophils into the circulation (Fig. 1), leading to severe reduction of neutrophil counts in the blood and increased numbers of neutrophils in the bone marrow (Dotta et al., 2011). It should be noted that lymphocyte function and trafficking are also affected in WHIM patients. Periodontitis associated with premature tooth loss is common in children with WHIM syndrome, who generally suffer from recurrent superficial infections, such as cellulitis and cutaneous abscess, but also deeper tissue infections, including pneumonia, osteomyelitis, and meningitis (Gorlin et al., 2000; Mc Guire et al., 2010; Dotta et al., 2011).
Impaired or Excessive Neutrophil Recruitment to Peripheral Tissues
Leukocyte adhesion deficiency (LAD) represents a group of inherited disorders, which inhibit the normal extravasation of circulating neutrophils to sites of infection or inflammation (Hanna and Etzioni, 2012). In LAD patients, leukocytes have defects in the expression or function of β2 integrins or other adhesion molecules and consequently cannot adhere to vascular endothelial cells (Fig. 1). LAD type I (LAD-I) is caused by deficiency in β2 integrins, LAD-II is due to defective glycosylation of selectin ligands, and LAD-III involves dysfunction of signaling intermediates affecting integrin activation. The most common type is LAD-I, an autosomal-recessive immunodeficiency caused by mutations in the CD18-encoding ITGB2 gene (Hanna and Etzioni, 2012). Few, if any, neutrophils can be found in extravascular sites in these patients, who display neutrophilia (increased blood neutrophil counts) even in the absence of infection. The neutrophilia of LAD patients is explained by the disruption of the neutrostat mechanism, since the lack of neutrophils recruited to tissues – and hence the lack of apoptotic neutrophil phagocytosis – would lead to unrestrained expression of granulopoiesis factors (Fig. 2). LAD-I patients suffer from recurrent infections and develop severe periodontitis early in life, affecting both the primary and permanent dentitions (Waldrop et al., 1987; Deas et al., 2003; Hanna and Etzioni, 2012). Mice genetically deficient in CD18 or CD11a reproduce several aspects of the LAD-I phenotype, such as neutrophilia and defective neutrophil adhesion and extravasation (Stark et al., 2005; von Vietinghoff and Ley, 2008). More recently, LFA-1 (CD11a)-deficient mice were shown to develop spontaneous dysbiosis and alveolar bone loss early in life, mimicking the periodontal phenotype of LAD-I patients (Hajishengallis et al., 2011). A similar periodontal phenotype is seen in mice with combined P- and E-selectin deficiency, a model equivalent to LAD-II (Niederman et al., 2001). In either study, however, it is uncertain whether the dysbiosis of the microbiota was the cause or the consequence of inflammation, and further research is warranted to determine whether dysbiosis is a direct outcome of the neutrophil defects.
Other conditions associated with defective neutrophil recruitment are attributed to impaired neutrophil chemotaxis, such as the Chediak-Higashi syndrome and the Papillon-Lefèvre syndrome. Similar to LAD patients, individuals with defective chemotaxis develop rapidly advancing periodontal bone loss at a very young age (Deas et al., 2003; Hart and Atkinson, 2007; Hanna and Etzioni, 2012). These conditions are reproduced in CXCR2-deficient mice, which cannot recruit neutrophils to the periodontium and develop severe bone loss early in life (Hajishengallis et al., 2011; Zenobia et al., 2013).
The Chediak-Higashi syndrome is associated with mutations of the LYST gene, which encodes for a protein involved in the regulation of lysosomal trafficking (Kaplan et al., 2008). Phenotypically, this disorder is characterized by fusion of cytoplasmic granules, which, in the case of myelocytes, occurs early in myelopoiesis. As a consequence, many myeloid precursors die in the marrow, causing moderate neutropenia. In surviving neutrophils, the giant granules appear to interfere mechanically with the process of diapedesis (Clawson et al., 1978) (Fig. 1). Moreover, because the Chediak-Higashi syndrome neutrophils have reduced content of hydrolytic enzymes, they exhibit delayed intracellular killing of phagocytosed bacteria (Fig. 1). Affected individuals are susceptible to infections involving mucous membranes, the respiratory tract, and skin, whereas severe periodontitis and oral ulcerations are common oral manifestations of the syndrome (Delcourt-Debruyne et al., 2000; Nualart Grollmus et al., 2007).
The Papillon-Lefèvre syndrome is caused by deficiency in cathepsin C (dipeptidyl peptidase-I), a lysosomal exo-cysteine protease also involved in pro-enzyme activation (e.g., activation of neutrophil-derived serine proteases such as cathepsin G, elastase, and proteinase 3) (Dickinson, 2002; Kobayashi et al., 2013). Neutrophils from patients with Papillon-Lefèvre syndrome have defective chemotaxis (Van Dyke et al., 1984; Liu et al., 2000) but are not invariably affected in their bacterial killing capacity (Pham et al., 2004; de Haar et al., 2006) (Fig. 1). These defects could probably contribute to the clinical condition of Papillon-Lefèvre patients, who show dramatic susceptibility to periodontitis affecting both the primary and permanent dentitions, whereas a subset of patients is susceptible to cutaneous and systemic infections (Pham et al., 2004; Hart and Atkinson, 2007; Nualart Grollmus et al., 2007). Additionally or alternatively, the periodontal tissue destruction associated with the Papillon-Lefèvre syndrome could be related in part to inflammation resulting from defective degradation and hence excessive accumulation of the macrophage inflammatory protein-1α, due to the patients’ cathepsin C deficiency and thus failure to activate neutrophil-derived serine proteases (Ryu et al., 2005).
Apart from host-related factors, periodontal bacteria, such as the keystone pathogen Porphyromonas gingivalis (Hajishengallis et al., 2012; Hajishengallis and Lambris, 2011), can impair neutrophil recruitment by interfering with the coordinated expression of chemokines and cell adhesion molecules, such as IL-8 and E-selectin (Darveau et al., 1998; Darveau, 2010). The inhibitory effect on IL-8 was termed ‘local chemokine paralysis’ and depends on the capacity of P. gingivalis to invade the epithelial cells (Darveau et al., 1998) and secrete the serine phosphatase SerB (Takeuchi et al., 2013). P. gingivalis-invaded epithelial cells are prevented from eliciting IL-8 responses, even when exposed to bacteria like F. nucleatum that are potent inducers of IL-8 on their own (Darveau et al., 1998). In vivo mouse studies have shown that these subversive effects (inhibition of IL-8 and E-selectin expression) are transient (Hajishengallis et al., 2011), but, at least in principle, could allow adequate time for P. gingivalis to initiate colonization while delaying the influx of neutrophils. In this regard, a SerB-deficient isogenic mutant (hence, incapable of causing local chemokine paralysis) induces enhanced neutrophil recruitment to the periodontium and causes reduced bone loss compared with wild-type P. gingivalis (Bainbridge et al., 2010). Therefore, mechanisms by which bacteria delay neutrophil recruitment can contribute to periodontal pathogenesis.
Recent studies have suggested that the unrestrained recruitment of neutrophils to the periodontium is as problematic as the impaired neutrophil recruitment in LFA-1–deficient mice (Hajishengallis et al., 2011; Eskan et al., 2012). In line with the recent identification of Del-1 as an antagonist of LFA-1–dependent leukocyte adhesion (Hajishengallis and Chavakis, 2013) (Fig. 3), Del-1–deficient mice spontaneously exhibit excessive neutrophil recruitment to the gingiva, leading to destructive inflammation and alveolar bone loss (Eskan et al., 2012). Therefore, Del-1 appears to act as a gatekeeper for homeostatic recruitment of neutrophils to the periodontium. Consistent with this notion, inflamed human gingiva express lower levels of Del-1 than do healthy gingiva (Eskan et al., 2012). Whether loss-of-function or hypofunctional polymorphisms in the gene encoding Del-1 (EDIL3) exist and can account for increased susceptibility to periodontitis is yet to be investigated. Intriguingly, wild-type mice develop age-associated Del-1 deficiency, which correlates with heavy neutrophil infiltration and alveolar bone loss in old age, although both features are suppressed by local treatment with Del-1 (Eskan et al., 2012). Following induction of experimental gingivitis, the elderly exhibit increased recruitment of inflammatory cells to the gingiva as compared with young individuals, despite comparable dental plaque accumulation in the 2 age groups (Fransson et al., 1996). Although it is uncertain whether this heightened inflammatory state in old individuals is related to age-associated decline in periodontal Del-1 expression, as shown in mice (Eskan et al., 2012), it is nevertheless a testable hypothesis.
Excessive Neutrophil Activation
Because of their rich and potentially harmful assortment of antimicrobial and pro-inflammatory mechanisms, the activation of neutrophils at inflammatory sites should be tightly regulated to prevent unwarranted tissue damage. However, neutrophils from individuals with a specific polymorphism (131H/H) in the Fcγ receptor IIa (which mediates neutrophil activation and phagocytosis) exhibit a hyper-responsive phenotype, as compared with neutrophils from individuals with the more common (131R/R) Fcγ receptor IIa genotype (Nicu et al., 2007). Specifically, upon stimulation, the 131H/H neutrophils express higher levels of degranulation markers and release more elastase than do the 131R/R neutrophils, although no significant differences were observed regarding their oxidative burst. Importantly, periodontitis patients with the 131H/H genotype have deeper periodontal pockets and more bone loss than those with the 131H/R or 131R/R genotype (Wolf et al., 2006; Nicu et al., 2007). It should be noted, however, that longitudinal analysis failed to demonstrate an association between these polymorphisms and response to conventional periodontal therapy (Wolf et al., 2006). In general, whereas gene polymorphisms can play a contributory role in susceptibility or resistance to periodontitis, their effects may not always be readily detectable, given the multifactorial etiology of periodontitis and the redundancy and compensatory mechanisms of the immune system.
Peripheral neutrophil hyper-responsiveness associated with excessive production of reactive oxygen species has been observed in chronic periodontitis. Neutrophils from such patients exhibit a hyper-responsive phenotype, even in the absence of exogenous stimulation (Matthews et al., 2007). It is thought that the recruitment of hyper-responsive neutrophils to the periodontium could contribute to periodontitis by causing oxidative tissue damage (Chapple and Matthews, 2007).
Localized aggressive periodontitis (LAP), a rapidly advancing form of periodontitis that primarily affects young patients, has been traditionally associated with hypofunctional neutrophils (Kantarci et al., 2003; Ryder, 2010). However, more recent evidence suggests a rather hyperfunctional phenotype for LAP neutrophils, which display elevated secretion of inflammatory mediators and oxidative stress, combined with chemotactic and phagocytic abnormalities (Kantarci et al., 2003; Ryder, 2010). It is therefore thought that periodontal tissue destruction in LAP could result, at least in part, from an inherent incapacity of LAP neutrophils to restrain their immune and inflammatory responses, while failing to control the periodontal microbial challenge.
Defective Neutrophil Clearance
Although hyperactive neutrophils may contribute to the pathogenesis of periodontitis (Chapple and Matthews, 2007), even normally activated neutrophils could cause unwarranted collateral tissue damage if not cleared properly when they become apoptotic. In this context, the resolution of inflammation, which includes clearance of apoptotic cells and cellular debris, is essential to homeostasis and tissue repair (Serhan et al., 2008; Ricklin et al., 2010). The inappropriate persistence of neutrophils due to defective clearance mechanisms could lead to their necrosis and the release of toxic contents. The notion that periodontal tissue damage could result from eventual necrosis of non-cleared apoptotic neutrophils is consistent with findings that neutrophil necrosis predominates over apoptosis in the diseased periodontium (Crawford et al., 2000). In principle, therefore, this mechanism could contribute to the transition from gingivitis to periodontitis or the aggravation of existing periodontitis.
Inefficient removal and accumulation of apoptotic cells and cellular debris have been associated with autoimmune inflammatory disorders such as systemic lupus erythematosus, attributed to deficiencies in complement components involved in apoptotic cell removal (e.g., C1q) (Ricklin et al., 2010). The importance of defective apoptotic cell clearance in periodontal pathogenesis is currently uncertain, although such a mechanism may operate in periodontitis patients with co-morbid conditions associated with impaired apoptotic cell clearance (e.g., lupus) (Fig. 1). Interestingly, periodontitis and systemic lupus erythematosus are thought to share common risk factors associated with phagocytic receptor polymorphisms (Kobayashi et al., 2007). Independently of genetic deficiencies predisposing to ineffective apoptotic cell removal, aging could be associated with defective clearance of apoptotic neutrophils (Aprahamian et al., 2008) (Fig. 1), perhaps due to reduced expression of apoptotic cell uptake receptors in macrophages (Devitt and Marshall, 2011). Defective clearance of apoptotic neutrophils is a plausible mechanism that could account, at least in part, for the association of old age with increased susceptibility to periodontitis (Hajishengallis, 2010).
Conclusions and Future Directions
Neutrophil homeostasis is maintained by a fine balance among several functions, including granulopoiesis, retention vs. release of mature neutrophils from the bone marrow, trafficking and transmigration, and clearance of apoptotic neutrophils (Figs. 1-3). Neutrophil defects affecting these functions can all potentially lead to severe periodontitis in human patients and related animal models. Many of these defects are congenital and can initiate periodontitis early in life, often leading to premature loss of primary and permanent teeth, with adverse psychological and functional consequences. In contrast to the syndromes and disorders discussed (Fig. 1), patients with chronic granulomatous disease are not susceptible to periodontitis, although their neutrophils have defective bactericidal activity, and they suffer from recurrent or persistent infections (e.g., pneumonia and abscesses of the skin) (Nussbaum and Shapira, 2011). Combined with the evidence discussed above, that neutrophils are associated with important regulatory mechanisms, this suggests that the increased susceptibility to periodontitis of individuals with congenital neutrophil dysfunctions (Fig. 1) may not be explained adequately by defective immune control of the periodontal bacteria, especially since these conditions are generally refractory to standard periodontal therapy and antibiotics. Moreover, systems-biology-level approaches investigating the transcriptome, proteome, and metabolome of neutrophils have started providing a more comprehensive understanding of the role of neutrophils beyond pathogen phagocytosis and killing, including host response regulation and resolution of inflammation (Kobayashi and DeLeo, 2009). We thus suggest that neutrophil-associated diseases may not necessarily or exclusively be related to defective killing function by neutrophils but could, alternatively or additionally, involve breakdown of neutrophil-associated homeostatic mechanisms. Future research may uncover hitherto-unknown local regulatory defects forming a causal link between congenital neutrophil disorders and early-onset forms of periodontitis. A better molecular understanding of periodontitis-associated neutrophil dysfunctions, owing to congenital or acquired causes, could be exploited for targeted therapeutic interventions in affected individuals.
Supplementary Material
Footnotes
Research support came from the National Institutes of Health, National Institute of Dental and Craniofacial Research (grants DE015254, DE021580, DE017138, and DE021685).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. (2012). Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30:459-489. [DOI] [PubMed] [Google Scholar]
- Aprahamian T, Takemura Y, Goukassian D, Walsh K. (2008). Ageing is associated with diminished apoptotic cell clearance in vivo. Clin Exp Immunol 152:448-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge B, Verma RK, Eastman C, Yehia B, Rivera M, Moffatt C, et al. (2010). Role of Porphyromonas gingivalis phosphoserine phosphatase enzyme SerB in inflammation, immune response, and induction of alveolar bone resorption in rats. Infect Immun 78:4560-4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapple IL, Matthews JB. (2007). The role of reactive oxygen and antioxidant species in periodontal tissue destruction. Periodontol 2000. 43:160-232. [DOI] [PubMed] [Google Scholar]
- Choi EY, Chavakis E, Czabanka MA, Langer HF, Fraemohs L, Economopoulou M, et al. (2008). Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science 322:1101-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clawson CC, White JG, Repine JE. (1978). The Chediak-Higashi syndrome. Evidence that defective leukotaxis is primarily due to an impediment by giant granules. Am J Pathol 92:745-753. [PMC free article] [PubMed] [Google Scholar]
- Crawford JM, Wilton JM, Richardson P. (2000). Neutrophils die in the gingival crevice, periodontal pocket, and oral cavity by necrosis and not apoptosis. J Periodontol 71:1121-1129. [DOI] [PubMed] [Google Scholar]
- Darveau RP. (2010). Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol 8:481-490. [DOI] [PubMed] [Google Scholar]
- Darveau RP, Belton CM, Reife RA, Lamont RJ. (1998). Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun 66:1660-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haar SF, Hiemstra PS, van Steenbergen MT, Everts V, Beertsen W. (2006). Role of polymorphonuclear leukocyte-derived serine proteinases in defense against Actinobacillus actinomycetemcomitans. Infect Immun 74:5284-5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas DE, Mackey SA, McDonnell HT. (2003). Systemic disease and periodontitis: manifestations of neutrophil dysfunction. Periodontol 2000. 32:82-104. [DOI] [PubMed] [Google Scholar]
- Delcourt-Debruyne EM, Boutigny HR, Hildebrand HF. (2000). Features of severe periodontal disease in a teenager with Chediak-Higashi syndrome. J Periodontol 71:816-824. [DOI] [PubMed] [Google Scholar]
- Delima AJ, Van Dyke TE. (2003). Origin and function of the cellular components in gingival crevice fluid. Periodontol 2000. 31:55-76. [DOI] [PubMed] [Google Scholar]
- Devitt A, Marshall LJ. (2011). The innate immune system and the clearance of apoptotic cells. J Leukoc Biol 90:447-457. [DOI] [PubMed] [Google Scholar]
- Dickinson DP. (2002). Cysteine peptidases of mammals: their biological roles and potential effects in the oral cavity and other tissues in health and disease. Crit Rev Oral Biol Med 13:238-275. [DOI] [PubMed] [Google Scholar]
- Donadieu J, Fenneteau O, Beaupain B, Mahlaoui N, Chantelot CB. (2011). Congenital neutropenia: diagnosis, molecular bases and patient management. Orphanet J Rare Dis 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotta L, Tassone L, Badolato R. (2011). Clinical and genetic features of Warts, Hypogammaglobulinemia, Infections and Myelokathexis (WHIM) syndrome. Curr Mol Med 11:317-325. [DOI] [PubMed] [Google Scholar]
- Eskan MA, Jotwani R, Abe T, Chmelar J, Lim JH, Liang S, et al. (2012). The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol 13:465-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson C, Berglundh T, Lindhe J. (1996). The effect of age on the development of gingivitis. Clinical, microbiological and histological findings. J Clin Periodontol 23:379-385. [DOI] [PubMed] [Google Scholar]
- Gorlin RJ, Gelb B, Diaz GA, Lofsness KG, Pittelkow MR, Fenyk JR., Jr (2000). WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet 91:368-376. [PubMed] [Google Scholar]
- Hajishengallis G. (2010). Too old to fight? Aging and its toll on innate immunity. Mol Oral Microbiol 25:25-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Chavakis T. (2013). Endogenous modulators of inflammatory cell recruitment. Trends Immunol 34:1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Lambris JD. (2011). Microbial manipulation of receptor crosstalk in innate immunity. Nat Rev Immunol 11:187-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. (2011). Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10:497-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Darveau RP, Curtis MA. (2012). The keystone-pathogen hypothesis. Nat Rev Microbiol 10:717-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna S, Etzioni A. (2012). Leukocyte adhesion deficiencies. Ann NY Acad Sci 1250:50-55. [DOI] [PubMed] [Google Scholar]
- Hart TC, Atkinson JC. (2007). Mendelian forms of periodontitis. Periodontol 2000. 45:95-112. [DOI] [PubMed] [Google Scholar]
- Hasturk H, Kantarci A, Van Dyke TE. (2012). Paradigm shift in the pharmacological management of periodontal diseases. Front Oral Biol 15:160-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci A, Oyaizu K, Van Dyke TE. (2003). Neutrophil-mediated tissue injury in periodontal disease pathogenesis: findings from localized aggressive periodontitis. J Periodontol 74:66-75. [DOI] [PubMed] [Google Scholar]
- Kaplan J, De Domenico I, Ward DM. (2008). Chediak-Higashi syndrome. Curr Opin Hematol 15:22-29. [DOI] [PubMed] [Google Scholar]
- Kobayashi SD, DeLeo FR. (2009). Role of neutrophils in innate immunity: a systems biology-level approach. Wiley Interdiscip Rev Syst Biol Med 1:309-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Ito S, Yasuda K, Kuroda T, Yamamoto K, Sugita N, et al. (2007). The combined genotypes of stimulatory and inhibitory Fc gamma receptors associated with systemic lupus erythematosus and periodontitis in Japanese adults. J Periodontol 78:467-474. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Sugiura K, Takeichi T, Akiyama M. (2013). The novel CTSC homozygous nonsense mutation p.Lys106X in a patient with Papillon-Lefèvre syndrome with all permanent teeth remaining at over 40 years of age. Br J Dermatol [Epub ahead of print 5/11/2013] (in press). [DOI] [PubMed] [Google Scholar]
- Kolaczkowska E, Kubes P. (2013). Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13:159-175. [DOI] [PubMed] [Google Scholar]
- Kollner I, Sodeik B, Schreek S, Heyn H, von Neuhoff N, Germeshausen M, et al. (2006). Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood 108:493-500. [DOI] [PubMed] [Google Scholar]
- Lagane B, Chow KY, Balabanian K, Levoye A, Harriague J, Planchenault T, et al. (2008). CXCR4 dimerization and beta-arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood 112:34-44. [DOI] [PubMed] [Google Scholar]
- Liu R, Cao C, Meng H, Tang Z. (2000). Leukocyte functions in 2 cases of Papillon-Lefèvre syndrome. J Clin Periodontol 27:69-73. [DOI] [PubMed] [Google Scholar]
- Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. (2003). Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 19:583-593. [DOI] [PubMed] [Google Scholar]
- Matthews JB, Wright HJ, Roberts A, Ling-Mountford N, Cooper PR, Chapple IL. (2007). Neutrophil hyper-responsiveness in periodontitis. J Dent Res 86:718-722. [DOI] [PubMed] [Google Scholar]
- McGuire PJ, Cunningham-Rundles C, Ochs H, Diaz GA. (2010). Oligoclonality, impaired class switch and B-cell memory responses in WHIM syndrome. Clin Immunol 135:412-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicu EA, Van der Velden U, Everts V, Van Winkelhoff AJ, Roos D, Loos BG. (2007). Hyper-reactive PMNs in FcgammaRIIa 131 H/H genotype periodontitis patients. J Clin Periodontol 34:938-945. [DOI] [PubMed] [Google Scholar]
- Niederman R, Westernoff T, Lee C, Mark LL, Kawashima N, Ullman-Culler M, et al. (2001). Infection-mediated early-onset periodontal disease in P/E-selectin-deficient mice. J Clin Periodontol 28:569-575. [DOI] [PubMed] [Google Scholar]
- Nourshargh S, Hordijk PL, Sixt M. (2010). Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol 11:366-378. [DOI] [PubMed] [Google Scholar]
- Nualart Grollmus ZC, Morales Chavez MC, Silvestre Donat FJ. (2007). Periodontal disease associated to systemic genetic disorders. Med Oral Patol Oral Cir Bucal 12:E211-215. [PubMed] [Google Scholar]
- Nussbaum G, Shapira L. (2011). How has neutrophil research improved our understanding of periodontal pathogenesis? J Clin Periodontol 38(Suppl 11):49-59. [DOI] [PubMed] [Google Scholar]
- Pham CT, Ivanovich JL, Raptis SZ, Zehnbauer B, Ley TJ. (2004). Papillon-Lefèvre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin C/dipeptidyl peptidase I deficiency in humans. J Immunol 173:7277-7281. [DOI] [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. (2010). Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11:785-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder MI. (2010). Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontol 2000. 53:124-137. [DOI] [PubMed] [Google Scholar]
- Ryu OH, Choi SJ, Firatli E, Choi SW, Hart PS, Shen RF, et al. (2005). Proteolysis of macrophage inflammatory protein-1α isoforms LD78β and LD78α by neutrophil-derived serine proteases. J Biol Chem 280:17415-17421. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, Van Dyke TE. (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8:349-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. (2005). Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 22:285-294. [DOI] [PubMed] [Google Scholar]
- Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. (2010). Neutrophil kinetics in health and disease. Trends Immunol 31:318-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Hirano T, Whitmore SE, Morisaki I, Amano A, Lamont RJ. (2013). The serine phosphatase SerB of Porphyromonas gingivalis suppresses IL-8 production by dephosphorylation of NF-κB RelA/p65. PLoS Pathog 9:e1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke TE, Taubman MA, Ebersole JL, Haffajee AD, Socransky SS, Smith DJ, et al. (1984). The Papillon-Lefèvre syndrome: neutrophil dysfunction with severe periodontal disease. Clin Immunol Immunopathol 31:419-429. [DOI] [PubMed] [Google Scholar]
- von Vietinghoff S, Ley K. (2008). Homeostatic regulation of blood neutrophil counts. J Immunol 181:5183-5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldrop TC, Anderson DC, Hallmon WW, Schmalstieg FC, Jacobs RL. (1987). Periodontal manifestations of the heritable Mac-1, LFA-1, deficiency syndrome. Clinical, histopathologic and molecular characteristics. J Periodontol 58:400-416. [DOI] [PubMed] [Google Scholar]
- Wolf DL, Neiderud AM, Hinckley K, Dahlén G, van de Winkel JG, Papapanou PN. (2006). Fcgamma receptor polymorphisms and periodontal status: a prospective follow-up study. J Clin Periodontol 33:691-698. [DOI] [PubMed] [Google Scholar]
- Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. (2001). Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194:519-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Carlsson G, Wondimu B, Fahlen A, Karlsson-Sjoberg J, Andersson M, et al. (2011). Mutations in the ELANE gene are associated with development of periodontitis in patients with severe congenital neutropenia. J Clin Immunol 31:936-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenobia C, Luo XL, Hashim A, Abe T, Jin L, Chang Y, et al. (2013). Commensal bacteria-dependent select expression of CXCL2 contributes to periodontal tissue homeostasis. Cell Microbiol 15:1419-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.