

Abstract
Studies in recent years have shown a positive relationship between metabolic syndrome (MS) and periodontal disease (PD). Given that patients with MS take statins to reduce cholesterol, and statins also have anti-inflammatory effects, it is important to determine if statin intake hinders the progression of MS-associated PD. In this study, PD was induced in Zucker fat rats (ZFRs), an animal model for MS, and in control lean rats by periodontal injection of Aggregatibacter actinomycetemcomitans lipopolysaccharide (LPS), while simvastatin was given to some of the rats via gavage. After 4 wk of treatment, alveolar bone loss was determined by micro-computed tomography. To explore the underlying mechanisms, we determined the effect of simvastatin on tissue inflammation and the expression of molecules involved in osteoclastogenesis. Results showed that while bone loss was increased by LPS in both ZFRs and the control lean rats, it was significantly more in the former than the latter. Simvastatin effectively alleviated bone loss in both ZFRs and the control rats. Results also showed that LPS stimulated leukocyte tissue infiltration and expression of molecules for osteoclastogenesis, but simvastatin significantly modulated the stimulation. This study demonstrated that simvastatin inhibited LPS-induced alveolar bone loss and periodontal tissue inflammation in rats with MS.
Keywords: statin, periodontal disease, lipopolysaccharide, inflammation obesity, insulin resistance
Introduction
Statins reduce serum cholesterol by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, a rate-controlling enzyme for cholesterol biosynthesis (Gotto and Moon, 2012). In addition to lowering cholesterol, studies have well established that statins also have pleiotropic effects, including anti-inflammatory properties (Wang et al., 2008; Sadowitz et al., 2010). Clinical trials have shown that statins are effective in the treatment of not only hypercholesterolemia, atherosclerosis, and cardiovascular disease, but also other inflammation-associated diseases such as rheumatoid arthritis (Paraskevas, 2008). Since periodontal disease is an inflammation-associated disease in which lipopolysaccharide (LPS) from Gram-negative bacteria plays an important role in triggering periodontal inflammation and tissue destruction (Beck et al., 1998; Gu et al., 2011), it is important to determine if statin intake hinders the progression of periodontal disease.
It has been well established that periodontal disease is more prevalent and severe in patients with type 2 diabetes than in non-diabetic patients (Katz et al., 1991; Yalda et al., 1994). In addition to diabetes, cross-sectional and prospective studies have indicated a close correlation of obesity with periodontal disease (Pischon et al., 2007; Chaffee and Weston, 2010; Suvan et al., 2011, 2013; D’Aiuto and Suvan, 2012). Furthermore, a recent review of 20 clinical studies by meta-analysis has presented clear evidence that metabolic syndrome, which includes central obesity, dyslipidemia (high triglycerides and low high-density lipoprotein), impaired glucose tolerance, and hypertension, is associated with increased periodontal disease (Nibali et al., 2013).
Since patients with metabolic syndrome have dyslipidemia that may include hypercholesterolemia (Morabia and Costanza, 2005), statins have commonly been prescribed to these patients to lower cholesterol and prevent cardiovascular events (Towne and Thara, 2008; Wee et al., 2008). Given that patients with metabolic syndrome have an increased risk of periodontal disease and also receive statin treatment, it is essential to determine if statin intake exerts a beneficial effect on metabolic syndrome–related periodontal disease, in addition to its cholesterol-lowering action.
In the present study, we used Zucker fat rats (ZFRs), an animal model widely used for the study of metabolic syndrome (de Artinano and Castro, 2009), and induced periodontal disease in the rats by periodontal injection of Aggregatibacter actinomycetemcomitans lipopolysaccharide (LPS). We found that LPS induced alveolar bone loss in both ZFRs and the control lean rats, but simvastatin effectively alleviated the bone loss.
Materials & Methods
Animals
Twenty-two 12-week-old female Zucker fat rats (ZFRs, Zucker-Lepr-fa/fa) and 22 lean rats (Zucker-Lepr-+/+) purchased from Harlan Laboratories, Inc. (Haslett, MI, USA) were used for the study. ZFR is an animal model for metabolic syndrome and exhibits features of metabolic syndrome including obesity, insulin resistance, dyslipidemia, and hyperinsulinemia (de Artinano and Castro, 2009). Animals were housed with a 12-hour light/12-hour dark cycle, had free access to tap water, and were fed a regular rat chow. The Institutional Animal Care and Use Committee at the Medical University of South Carolina approved all experimental protocols. All animal-related work was performed in accordance with ARRIVE guidelines for preclinical studies and the National Institute of Health Guidelines.
For complete Materials & Methods, please see the online Appendix.
Results
The Effect of Simvastatin on Metabolic Parameters
Weight
As shown in Fig. 1A, the fat rats were 65% heavier than the lean rats before treatment (519 vs. 314 g). During the four-week treatment, either the fat or lean rats increased body weight by 27%, and thus the fat rats were still 65% heavier than the lean rats after treatment (660 vs. 400 g). Administration of LPS alone or LPS and simvastatin did not change the weight significantly (Fig. 1A).
Figure 1.

The effects of lipopolysaccharide (LPS) and simvastatin on weight, lipids, insulin, and insulin resistance in the Zucker fat rats (ZFRs) and the control lean rats. The ZFRs and the control lean rats were treated with phosphate-buffered saline (PBS) (n = 7), LPS (n = 7), or LPS plus simvastatin (n = 8) for 4 wk as described in “Materials & Methods”. Before and after the treatments, body weight (A), glucose (B), insulin (C), homeostasis model assessment of insulin resistance (HOMA-IR) (D), total cholesterol (E), triglycerides (F), and free fatty acids (G) were determined. The data are mean ± SD.
Glucose
As shown in Fig. 1B, there was no significant difference in plasma fasting glucose levels between the fat and lean rats before and after treatment. During the four-week treatment, glucose levels in the fat and the lean rats increased by 83% and 66%, respectively. Administration of LPS or LPS and simvastatin had no significant effect on glucose level.
Insulin and Insulin Resistance
The fasting insulin level in Zucker fat rats was about three-fold higher than that in the lean rats (Fig. 1C). Interestingly, LPS treatment reduced insulin levels in the fat rats, and the addition of simvastatin further reduced insulin levels. Insulin resistance (HOMA-IR) in the fat rats was markedly increased when compared with that in the lean rats (Fig. 1D). Similarly, LPS reduced HOMA-IR, and simvastatin further reduced it in the fat rats.
Cholesterol
The cholesterol level in fat rats was about two-fold that in the lean rats before treatment (Fig. 1E). LPS had no effect on cholesterol levels in both fat and lean rats. As expected, simvastatin markedly reduced cholesterol levels in the fat rats. Interestingly, simvastatin had no effect on the cholesterol level in lean rats.
Triglycerides
The triglycerides (TG) level in the fat rats was 5.7-fold that in the lean rats before treatment (Fig. 1F). After 4 wk, the TG levels in phosphate-buffered saline (PBS)-treated lean rats were increased, but LPS treatment reduced TG levels. In the fat rats, LPS alone did not change TG levels significantly, but the addition of simvastatin increased TG levels.
Free Fatty Acid
As shown in Fig. 1G, the free fatty acid (FFA) level in the fat rats was three-fold that in the lean rats before treatment. Similarly to TG, the FAA level in PBS-treated lean rats was increased after 4 wk, but LPS treatment reduced FAA level significantly. LPS and simvastatin did not reduce FAA levels in the fat rats.
Simvastatin Significantly Reduces LPS-stimulated Alveolar Bone Loss in Both Lean and Fat Rats
We have reported recently that simvastatin significantly reduces LPS-induced alveolar bone loss in non-obese Sprague Dawley rats (Jin et al., 2013a). In the present study, the effect of simvastatin on LPS-induced alveolar bone loss was confirmed in the control Zucker lean rats. Results from our µCT-based quantification of BVF (Figs. 2A, 2B) showed that LPS induced alveolar bone loss in the lean rats by 7.7% (0.366 vs. 0.397, p < .01), and simvastatin markedly attenuated the effect of LPS on bone loss. Interestingly, the PBS-injected ZFRs had greater BVF than the lean rats (0.421 vs. 0.397, p < .01), and LPS also significantly induced bone loss in ZFRs (0.384 vs. 0.421, p < .01). Statistical analysis indicates that the bone loss in ZFRs is greater than that in the control lean rats (8.9% vs. 7.7%, p < .05) (Fig. 2C). Consistently, simvastatin also effectively reduced LPS-induced bone loss in the ZFRs.
Figure 2.

Simvastatin inhibited lipopolysaccharide (LPS)-induced alveolar bone loss. The Zucker fat rats (ZFRs) and the control lean rats were treated with phosphate-buffered saline (PBS) (n = 7), LPS (n = 7), or LPS plus simvastatin (n = 8) for 4 wk as described in “Materials & Methods”. After treatment, the maxillae were scanned by mCT, and bone volume fractions were quantified. Representative µCT images (A) and bone volume fractions for 6 groups (B) are shown. The bone loss induced by LPS or LPS plus simvastatin is also presented as a percentage of the control (PBS-treated rats) (C). The data are mean ± SD.
Simvastatin Inhibited LPS-induced Periodontal Inflammation and Bone Resorption
Histological analysis of maxillae was conducted to determine the effect of simvastatin on periodontal inflammation and bone resorption. Results showed that LPS injection led to a marked increase in leukocyte infiltration and alveolar bone resorption in both fat and lean rats when compared with PBS injection, but administration of simvastatin significantly suppressed the effect of LPS (Fig. 3A). The mononuclear cells under the first molars were quantified, and results showed that LPS increased tissue infiltration of mononuclear cells in both ZFRs and the control lean rats, but simvastatin markedly reduced the increases (Fig. 3B). Although it appeared that, without simvastatin treatment, LPS induced tissue infiltration of more inflammatory cells in ZFRs than in the control lean rats, the difference did not reach statistical significance (Fig. 3B). Furthermore, the degree of leukocyte infiltration and bone resorption was also evaluated by the scoring system (McAbee et al., 2012). Results showed that LPS markedly increased inflammatory scores in both fat and lean rats, but simvastatin reduced it significantly (see Appendix Fig. 1).
Figure 3.

Simvastatin inhibited lipopolysaccharide (LPS)-induced leukocyte infiltration and alveolar bone resorption. (A) Histological evaluation of leukocyte infiltration and bone resorption in maxillae after treatment with phosphate-buffered saline (PBS), LPS, or LPS plus simvastatin. After the maxillae were examined by mCT as shown in Fig. 2, they were decalcified and sectioned. Tissue sections (7 µm) were stained with hematoxylin and eosin, and photomicrographs on the first molar of rat maxillae were taken at 40x magnification. The selected areas in the images were amplified to 400x and are presented as the insets in the lower right corner. The scale bar for the lower magnification (40x) images represents 50 µm and that for the higher magnification (400x) images represents 250 µm. (B) Quantification of mononuclear cells under the first molar for rats treated with PBS, LPS, or LPS plus simvastatin. The data are mean ± SD.
Simvastatin Inhibits the Stimulatory Effect of LPS on RANKL and CSF2 Expression
It is known that receptor activator of nuclear factor kappa-B ligand (RANKL) and colony-stimulating factor (CSF)2 play an important role in alveolar bone resorption (Taubman et al., 2005; Lee et al., 2009). To elucidate how simvastatin reduces LPS-induced alveolar bone resorption, we determined the effect of simvastatin on the expression of RANKL and CSF2 in the periodontal tissue. Results showed that LPS stimulated the expression of RANKL and CSF2, but simvastatin attenuated the stimulation in both fat and lean rats (Fig. 4).
Figure 4.
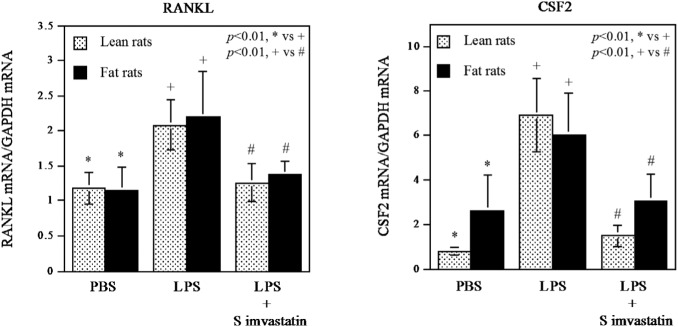
Simvastatin inhibited lipopolysaccharide (LPS)-stimulated receptor activator of nuclear factor kappa-B ligand (RANKL) and colony-stimulating factor (CSF)2 mRNA expression in periodontal tissue. RNA was isolated from periodontal tissue after treatments with vehicle (PBS) (n = 7), LPS (n = 7), or LPS plus simvastatin (n = 8), and real-time polymerase chain-reaction (PCR) was performed to quantify RANKL and CSF2 mRNA expression. The RANKL and CSF2 expression was normalized to GAPDH expression. The data are mean ± SD.
Simvastatin Inhibits the Stimulatory Effects of LPS and Palmitic Acid on IL-6 Secretion by Macrophages
Recently, we reported that LPS and palmitic acid (PA), the most abundant saturated fatty acid in obese patients (Weigert et al., 2004), synergistically stimulated IL-6 expression in murine macrophages (Jin et al., 2013a). Given that IL-6 is known to play an important role in periodontal disease (Javed et al., 2012), and that the fat rats in our current study had a markedly increased FAA as shown in Fig. 1G, we performed an in vitro experiment with murine macrophages to determine if simvastatin inhibits IL-6 secretion induced by LPS and PA. Results showed that LPS and PA exerted a synergy on IL-6 secretion, but simvastatin mitigated the effect of LPS and PA significantly (see Appendix Fig. 2).
Discussion
In the current study, analysis of our metabolic data clearly demonstrated that ZFRs had metabolic syndrome, since these rats showed increased body weight and levels of lipids including cholesterol, triglycerides, and free fatty acids, insulin, and HOMA-IR, an insulin resistance index. It is noteworthy that the fasting glucose in ZFRs was not increased (Fig. 1B), which is consistent with the previous report (de Artinano and Castro, 2009). Interestingly, our study showed that LPS-induced periodontal disease is associated with reduced fasting insulin in ZFRs, but not in the control lean rats (Fig. 1C). This observation may reveal the impact of periodontal disease on systemic insulin levels. Although the mechanism is largely unknown, it is possible that LPS-induced periodontal disease may lead to an increase in circulating pro-inflammatory cytokines such as tumor necrosis factor alpha (TNFα), which is known to inhibit insulin secretion from pancreatic beta cells (Zhang and Kim, 1995). Furthermore, it also remains unclear how simvastatin treatment further inhibited LPS-reduced fasting insulin in the fat rats (Fig. 1C).
Our current study showed that simvastatin increased TG in LPS-administrated Zucker fat rats, but not in the control lean rats (Fig. 1F). This is surprising, since it is known that simvastatin reduces TG in rats by increasing lipoprotein lipase activity and reducing triglyceride secretion rate (Sato et al., 1991). However, a recent study reported that atorvastatin, another statin drug, increased TG (391 vs. 297 mg/dL) in Zucker fat rats (Aguirre et al., 2013), which is in agreement with our findings. This study and our findings, as shown in Fig. 1F, suggest that the effects of statins on TG in the rats with metabolic syndrome, such as ZFRs, may be different from those in rats without metabolic syndrome, and more investigations are warranted to explore the underlying mechanisms.
Using µCT-based alveolar bone volume quantification, our study demonstrated that the vehicle-treated ZFRs had a higher alveolar BVF than the control lean rats (0.421 vs. 0.397). It is noteworthy that ZFRs have a genetic defect in leptin receptor that leads to hyperphagia and development of obesity at a young age (Shiota and Printz, 2012). The hyperphagia during body development is likely to contribute to increased alveolar bone in ZFRs. The finding that no significant reduction of BVF was observed in ZFRs without LPS injection indicates that metabolic syndrome per se as a co-morbidity of periodontal disease in ZFRs did not cause significant alveolar bone loss. Interestingly, this finding is different from that observed in animal models for rheumatoid arthritis, another co-morbidity of periodontal disease. Queiroz-Junior et al. reported that the C57BL/6 mouse model for experimental arthritis without bacterial inoculation developed periodontal disease and had alveolar bone loss spontaneously (Queiroz-Junior et al., 2011). They further found that arthritis-dependent release of TNFα played a critical role in arthritis-induced periodontal disease. It is possible that the circulating levels of inflammatory cytokines such as TNFα induced by metabolic syndrome may not be as high as those induced by experimental arthritis and thus are unable to induce significant alveolar bone loss.
Despite the fact that metabolic syndrome by itself in ZFRs did not increase alveolar bone loss, it significantly enhanced LPS-induced bone loss (Fig. 2C). Given that obesity and dyslipidemia are the major manifestations in metabolic syndrome, the role of obesity and dyslipidemia in the aggravated periodontal disease has been the focus of recent investigations (Chaffee and Weston, 2010; Jin et al., 2013a). We hypothesized that increased bioactive lipids associated with obesity and dyslipidemia play a crucial role in the increased periodontal inflammation and accelerated periodontal disease by amplifying LPS-triggered inflammatory signaling. Our recent study has shown that PA, the most abundant saturated fatty acid, potently augments LPS-initiated expression of pro-inflammatory cytokine IL-6 in macrophages (Jin et al., 2013a), a major type of leukocyte involved in periodontal disease, suggesting that dyslipidemia associated with metabolic syndrome may enhance the tissue inflammation triggered by LPS.
Impressively, this study showed that simvastatin treatment effectively reduced bone loss in ZFRs and the control lean rats by 48% and 66%, respectively. Given that LPS injection led to more bone loss in ZFRs than in the lean rats, it is conceivable that the treatment with simvastatin at the same dose would have less alleviating effect on ZFRs than on the control lean rats. Consistent with the findings from alveolar bone loss, histological analysis of periodontal tissues showed that LPS induced a remarkable leukocyte infiltration and alveolar bone resorption in both ZFRs and the control lean rats, but simvastatin inhibited the inflammatory infiltration and bone resorption effectively in both types of rats (Fig. 3). To further elucidate the mechanisms involved in the inhibition of LPS-stimulated bone resorption by simvastatin, we focused on the periodontal expression of RANKL and CSF2. It is known that the up-regulation of RANKL, a critical osteoclast differentiation factor, in T-lymphocytes by LPS plays an essential role in LPS-induced osteoclastogenesis and alveolar bone resorption (Taubman et al., 2005). In studies to elucidate how RANKL stimulates osteoclastogenesis, it was reported that CSF2 was responsible for the fusion of mononuclear osteoclasts into bone-resorbing osteoclasts to form multinucleated osteoclasts and thus enhanced the effect of RANKL on bone resorption (Lee et al., 2009). Analysis of data from our quantitative real-time PCR showed that LPS-stimulated RANKL and CSF2 expression, but simvastatin inhibited them in both ZFRs and the control lean rats (Fig. 4), indicating that simvastatin inhibited alveolar bone loss induced by LPS by targeting the key molecules involved in osteoclastogenesis.
In conclusion, this study demonstrated, for the first time, that LPS induced more alveolar bone loss in rats with metabolic syndrome than in the control lean rats, and administration of simvastatin significantly inhibited alveolar bone loss. In addition, this study also demonstrated that simvastatin inhibited expression of molecules involved in osteoclastogenesis in rats with metabolic syndrome.
Supplementary Material
Footnotes
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
This work was supported by National Institutes of Health grant DE016353 and by the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs (to Y.H.). Research reported in this publication was also supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P30103331.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Aguirre L, Hijona E, Macarulla MT, Gracia A, Larrechi I, Bujanda L, et al. (2013). Several statins increase body and liver fat accumulation in a model of metabolic syndrome. J Physiol Pharmacol 64:281-288. [PubMed] [Google Scholar]
- Beck JD, Offenbacher S, Williams R, Gibbs P, Garcia R. (1998). Periodontitis: a risk factor for coronary heart disease? Ann Periodontol 3:127-141. [DOI] [PubMed] [Google Scholar]
- Chaffee BW, Weston SJ. (2010). Association between chronic periodontal disease and obesity: a systematic review and meta-analysis. J Periodontol 81:1708-1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aiuto F, Suvan J. (2012). Obesity, inflammation, and oral infections: are microRNAs the missing link? J Dent Res 91:5-7. [DOI] [PubMed] [Google Scholar]
- de Artinano AA, Castro MM. (2009). Experimental rat models to study the metabolic syndrome. Br J Nutr 102:1246-1253. [DOI] [PubMed] [Google Scholar]
- Gotto AM, Jr, Moon JE. (2012). Management of cardiovascular risk: the importance of meeting lipid targets. Am J Cardiol 110(1 Suppl):3A-14A. [DOI] [PubMed] [Google Scholar]
- Gu Y, Lee HM, Sorsa T, Salminen A, Ryan ME, Slepian MJ, et al. (2011). Non-antibacterial tetracyclines modulate mediators of periodontitis and atherosclerotic cardiovascular disease: a mechanistic link between local and systemic inflammation. Pharmacol Res 64:573-579. [DOI] [PubMed] [Google Scholar]
- Javed F, Al-Askar M, Al-Hezaimi K. (2012). Cytokine profile in the gingival crevicular fluid of periodontitis patients with and without type 2 diabetes: a literature review. J Periodontol 83:156-161. [DOI] [PubMed] [Google Scholar]
- Jin J, Zhang X, Lu Z, Perry DM, Li Y, Russo SB, et al. (2013a). Acid sphingomyelinase plays a key role in palmitic acid-amplified inflammatory signaling triggered by lipopolysaccharide at low concentrations in macrophages. Am J Physiol Endocrinol Metab 305:E853-E867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Zhang X, Lu Z, Li Y, Lopes-Virella MF, Yu H, et al. (2013b). Simvastatin inhibits lipopolysaccharide-induced osteoclastogenesis and reduces alveolar bone loss in experimental periodontal disease. J Periodontal Res [Epub ahead of print 10/7/2013] (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz PP, Wirthlin MR, Jr, Szpunar SM, Selby JV, Sepe SJ, Showstack JA. (1991). Epidemiology and prevention of periodontal disease in individuals with diabetes. Diabetes Care 14:375-385. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kim HS, Yeon JT, Choi SW, Chun CH, Kwak HB, et al. (2009). GM-CSF regulates fusion of mononuclear osteoclasts into bone-resorbing osteoclasts by activating the Ras/ERK pathway. J Immunol 183:3390-3399. [DOI] [PubMed] [Google Scholar]
- McAbee J, Li Q, Yu H, Kirkwood KL. (2012). Sexual dimorphism in periapical inflammation and bone loss from mitogen-activated protein kinase phosphatase-1 deficient mice. J Endod 38:1097-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morabia A, Costanza MC. (2005). The obesity epidemic as harbinger of a metabolic disorder epidemic: trends in overweight, hypercholesterolemia, and diabetes treatment in Geneva, Switzerland, 1993-2003. Am J Public Health 95:632-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibali L, Tatarakis N, Needleman I, Tu YK, D’Aiuto F, Rizzo M, et al. (2013). Clinical review: Association between metabolic syndrome and periodontitis: a systematic review and meta-analysis. J Clin Endocrinol Metab 98:913-920. [DOI] [PubMed] [Google Scholar]
- Paraskevas KI. (2008). Statin treatment for rheumatoid arthritis: a promising novel indication. Clin Rheumatol 27:281-287. [DOI] [PubMed] [Google Scholar]
- Pischon N, Heng N, Bernimoulin JP, Kleber BM, Willich SN, Pischon T. (2007). Obesity, inflammation, and periodontal disease. J Dent Res 86:400-409. [DOI] [PubMed] [Google Scholar]
- Queiroz-Junior CM, Madeira MF, Coelho FM, Costa VV, Bessoni RL, Sousa LF, et al. (2011). Experimental arthritis triggers periodontal disease in mice: involvement of TNF-alpha and the oral microbiota. J Immunol 187:3821-3830. [DOI] [PubMed] [Google Scholar]
- Sadowitz B, Maier KG, Gahtan V. (2010). Basic science review: Statin therapy—Part I: The pleiotropic effects of statins in cardiovascular disease. Vasc Endovascular Surg 44:241-251. [DOI] [PubMed] [Google Scholar]
- Sato A, Watanabe K, Fukuzumi H, Hase K, Ishida F, Kamei T. (1991). Effect of simvastatin (MK-733) on plasma triacylglycerol levels in rats. Biochem Pharmacol 41:1163-1172. [DOI] [PubMed] [Google Scholar]
- Shiota M, Printz RL. (2012). Diabetes in Zucker diabetic fatty rat. Methods Mol Biol 933:103-123. [DOI] [PubMed] [Google Scholar]
- Suvan J, D’Aiuto F, Moles DR, Petrie A, Donos N. (2011). Association between overweight/obesity and periodontitis in adults. A systematic review. Obes Rev 12:e381-e404. [DOI] [PubMed] [Google Scholar]
- Suvan J, Petrie A, Moles DR, Nibali L, Patel K, Darbar U, et al. (2013). Body Mass Index as a predictive factor of periodontal therapy outcomes. J Dent Res [Epub ahead of print 10/28/2013] (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubman MA, Valverde P, Han X, Kawai T. (2005). Immune response: the key to bone resorption in periodontal disease. J Periodontol 76(11 Suppl):2033S-2041S. [DOI] [PubMed] [Google Scholar]
- Towne SP, Thara E. (2008). Do statins reduce events in patients with metabolic syndrome? Curr Atheroscler Rep 10:39-44. [DOI] [PubMed] [Google Scholar]
- Wang CY, Liu PY, Liao JK. (2008). Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med 14:37-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee CC, Girotra S, Weinstein AR, Mittleman MA, Mukamal KJ. (2008). The relationship between obesity and atherosclerotic progression and prognosis among patients with coronary artery bypass grafts the effect of aggressive statin therapy. J Am Coll Cardiol 52:620-625. [DOI] [PubMed] [Google Scholar]
- Weigert C, Brodbeck K, Staiger H, Kausch C, Machicao F, Haring HU, et al. (2004). Palmitate, but not unsaturated fatty acids, induces the expression of interleukin-6 in human myotubes through proteasome-dependent activation of nuclear factor-kappaB. J Biol Chem 279:23942-23952. [DOI] [PubMed] [Google Scholar]
- Yalda B, Offenbacher S, Collins JG. (1994). Diabetes as a modifier of periodontal disease expression. Periodontol 2000 6: 37-49. [DOI] [PubMed] [Google Scholar]
- Zhang S, Kim KH. (1995). TNF-alpha inhibits glucose-induced insulin secretion in a pancreatic beta-cell line (INS-1). FEBS Lett 377:237-239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.