

Abstract
Simple catalyst, high enantioselectivities. An easily prepared squaramide catalyst promotes highly enantioselective Michael addition reactions of diphenyl phosphite to a broad range of nitroalkenes, including those bearing acidic protons or sterically demanding aliphatic substituents. The methodology provides facile access to chiral β-nitro phosphonates, which are precursors to biologically active β-amino phosphonic acids.
Keywords: asymmetric catalysis, hydrogen bonds, Michael addition, organocatalysis, squaramides
Horiguchi and Kandatsu’s isolation of 2-amino ethylphosphonic acid from the rumen protozoa in 1959 demonstrated for the first time the occurrence of a C-P bond in nature.[1] Such β-amino phosphonic acids, as phosphorus analogs of β-amino acids, have been the subject of intense interest due to their diverse biological activities.[2] In contrast to the armamentarium of methods for the asymmetric synthesis of α-amino phosphonic acid derivatives,[3, 4] the enantioselective synthesis of β-amino phosphonic acids remains a considerable challenge.[5] A logical solution to this problem is through the enantioselective Michael addition of diaryl or dialkyl phosphites to nitroalkenes to provide β-nitro phosphonates, wherein reduction of the nitro group would produce chiral β-amino phosphonates.[6–8] To date, only two reports describe the use of metal-free catalysts for the conjugate addition reaction of phosphites.[9–11] Wang and coworkers showed in 2007 that quinine promotes this reaction and affords the addition products in modest to very good ee’s.[9] Significantly higher enantioselectivities were obtained by Terada and coworkers, who used an intricate, axially-chiral biaryl guanidine to promote this conjugate addition reaction.[10, 12] In connection with our interest in hydrogen-bond donor promoted enantioselective reactions,[13, 14] we have developed a new family of chiral catalysts based on the squaramide scaffold.[15] The modular nature of this scaffold allows quick access to a wide range of catalysts, tuned with regard to the chiral environment as well as the pKa of the donor hydrogens, and opens up opportunities for the exploration of new reactions and the development of highly effective catalysts for known reactions. We report here that a simple, easily prepared squaramide catalyst promotes the Michael addition reaction of diphenyl phosphite to a broad range of nitroalkenes, both aryl- and alkyl-substituted, affording the products in high yields and uniformly excellent enantioselectivities.
Despite the plethora of successful reactions promoted by various thiourea-based catalysts,[16] the only reported use of a chiral thiourea to promote the addition of diphenyl phosphite to trans-β-nitrostyrene is that by Wang, who obtained the addition product in 21% yield and 8% ee after a reaction time of 24 h.[9] Given the structural differences between thioureas and squaramides, particularly the spacing between the two donor hydrogen atoms,[15] we expected to see differences in their reactivity. Indeed, addition of the dimethyl-substituted squaramide 4a[17] to a solution of trans-β-nitrostyrene (1a) and diphenyl phosphite (2) at room temperature promoted a rapid reaction that went to 98% conversion after just 45 minutes and afforded the addition product in 81% ee (Table 1, entry 1).
Table 1.
Michael addition of diphenyl phosphite (2) to trans-β-nitrostyrene (1a) catalyzed by 4 or 5[a].
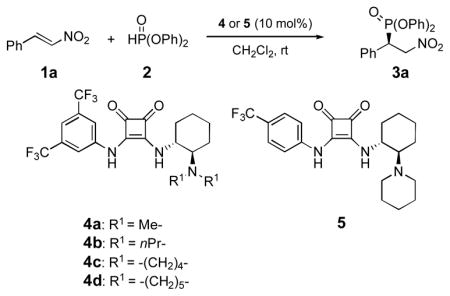
| ||||
|---|---|---|---|---|
| entry | catalyst | time | conv [%][b] | ee [%] |
| 1 | 4a | 45 min | 98 | 81 |
| 2 | 4b | 1 h | 96 | 84 |
| 3 | 4c | 30 min | 98 | 88 |
| 4 | 4d | 30 min | 97 | 95 |
| 5 | 5 | 15 min | 99 | 96 |
Reactions were carried out on 0.20 mmol of 1a with 1.25 equiv of 2 and 10 mol% 4 or 5 in 1.0 mL CH2Cl2 at room temperature.
Reaction conversions were determined by 1H-NMR.
In order to optimize the catalyst, a brief study of the structure-enantioselectivity relationship was carried out (Table 1). The effect of various substituents on the amino group of the catalyst was examined first (entries 2–4). Catalyst 4b bearing the bulkier n-propyl groups on the nitrogen improved the enantioselectivity slightly, with accompanying diminution in the reaction rate (entry 2). Higher enantioselectivities were observed when the amine substituents constitute a ring. Thus, the pyrrolidine-substituted catalyst 4c gave the product in 88% ee, and the corresponding piperidine-substituted catalyst 4d catalyst gave the product in 95% ee (entries 3, 4). The improved enantioselectivities from the cyclic substituents, particularly piperidine, are attributed to the relative conformational rigidity of these groups, which allows a more organized transition state, one that better differentiates the two sides of trans-β-nitrostyrene. After a brief survey of different substituents on the aryl moiety, we chose catalyst 5 for further studies, due to its ease of preparation and better catalytic selectivity (entry 5). Catalyst 5 is prepared in 3-steps from commercial starting materials (Scheme 1).
Scheme 1.
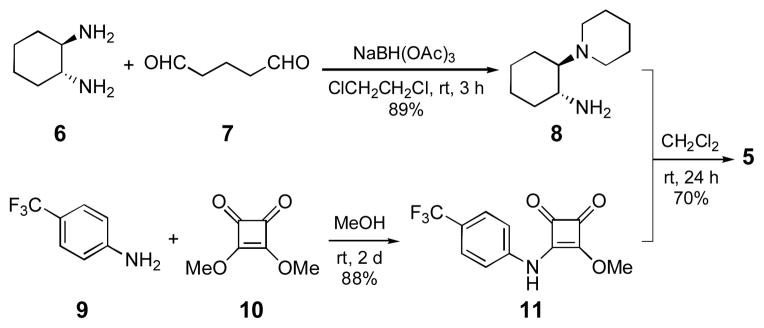
Synthesis of catalyst 5.
A survey of solvents showed the phosphite conjugate addition reaction to be relatively insensitive to the solvent used (Table 2). The enantioselectivity obtained in toluene was essentially the same as that obtained in ether solvents, including THF (entries 1–4).[18] Even very polar solvents such as acetonitrile and acetone afforded the addition product in 90% or greater ee (entries 5,6). The best solvent was found to be CH2Cl2, in which, as noted earlier, the room temperature reaction gave the product in 96% ee (entry 7). As expected, the enantioselectivity increased steadily as the reaction temperature was lowered (entries 8–10). Taking into account the practical advantages of carrying out the reaction at 0 °C, this temperature was used for evaluating the scope of the methodology.
Table 2.
Michael addition of diphenyl phosphite (2) to trans-β-nitrostyrene (1a) catalyzed by 5[a].
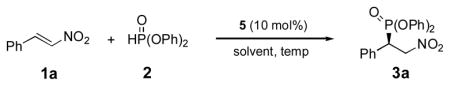
| |||||
|---|---|---|---|---|---|
| entry | solvent | temp [°C] | time | conv [%][b] | ee [%] |
| 1 | toluene | rt | 1 h | 90 | 94 |
| 2 | Et2O | rt | 1 h | 81 | 93 |
| 3 | tBuOMe | rt | 1 h | 71 | 94 |
| 4 | THF | rt | 1 h | 99 | 93 |
| 5 | MeCN | rt | 1 h | 89 | 90 |
| 6 | acetone | rt | 1 h | 64 | 93 |
| 7 | CH2Cl2 | rt | 15 min | 99 | 96 |
| 8 | CH2Cl2 | 0 | 30 min | 99 (95[c]) | 97 |
| 9 | CH2Cl2 | −10 | 1 h | 99 | 98 |
| 10 | CH2Cl2 | −20 | 2 h | 99 | 98.4 |
Reactions were carried out on 0.20 mmol of 1a with 1.25 equiv of 2 and 10 mol% 5 in 1.0 mL solvent.
Reaction conversions were determined by 1H-NMR.
Isolated yield.
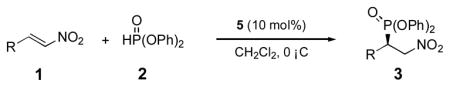
A diverse range of aryl-substituted nitroalkene substrates were selected to evaluate the scope of the squaramide catalyzed conjugate addition reaction. As shown in Table 3, the enantioselective conjugate addition reaction is remarkably general: under the optimized conditions, the full spectrum of substrates underwent the reaction in 30 minutes or less and afforded the products in good yields with 96–99% ee, regardless of the electronic properties and locations of substituents. The parent reaction can be scaled up without untoward effect on either the yield or enantioselectivity (entry 1). It is worth noting that even substrates with acidic protons capable of forming competing hydrogen bonds, such as 1l and 1q, were tolerated and afforded the expected products in 98% ee (entries 12, 17).
Table 3.
Enantioselective Michael addition reaction of diphenyl phosphite (2) to trans-nitroalkenes (1, R = aromatic substituent) catalyzed by 5[a].
| |||||
|---|---|---|---|---|---|
| entry | product | R | time [min] | yield [%][b] | ee [%] |
| 1[c] | 3a |
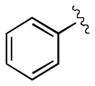
|
30 | 99 | 97 |
| 2 | 3b |
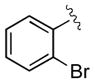
|
15 | 92 | 97 |
| 3 | 3c |
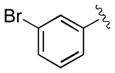
|
15 | 98 | 97 |
| 4 | 3d |
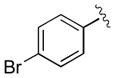
|
15 | 94 | 97 |
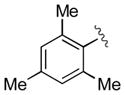
| 5 | 3e |
|
15 | 98 | 97 |
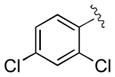
| 6 | 3f |
|
30 | 96 | 97 |
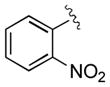
| 7 | 3g |
|
30 | 97 | 99 |
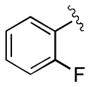
| 8 | 3h |
|
30 | 98 | 96 |
| 9 | 3i |
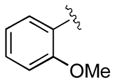
|
15 | 98 | 98 |
| 10 | 3j |
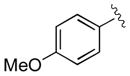
|
30 | 98 | 99 |
| 11 | 3k |
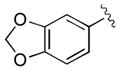
|
30 | 99 | 98 |
| 12 | 3l |
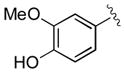
|
30 | 79 | 98 |
| 13 | 3m |
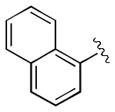
|
15 | 98 | 97 |
| 14 | 3n |
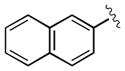
|
15 | 99 | 99 |
| 15[d] | 3o |
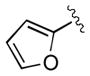
|
30 | 83 | 97 |
| 16 | 3p |
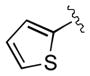
|
30 | 84 | 97 |
| 17 | 3q |
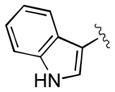
|
15 | 99 | 98 |
Unless otherwise noted, reactions were carried out on 0.20 mmol of 1 with 1.25 equiv of 2 and 10 mol% 5 in 1.0 mL CH2Cl2 at 0 °C.
Isolated yield.
1.0 mmol scale reaction.
Absolute configuration of 3o was assigned as S (see Supporting Information for details).
Among the most challenging substrates for the organocatalyzed phosphite conjugate addition reaction are alkyl-substituted nitroalkenes.[9, 10] The highest enantioselectivity recorded for such substrates is 87% ee.[19] To evaluate the effectiveness of catalyst 5 in these reactions, several alkyl-substituted nitroalkenes were subjected to the optimized conditions. As summarized in Table 4, excellent enantioselectivities were obtained even for aliphatic nitroalkenes (95–97% ee). Compared to aryl-substituted substrates, the reactions of alkyl substrates were slower, presumably due to steric and electronic factors. To circumvent the slow rate of substrates with secondary and tertiary alkyl groups, the catalyst loading was increased to 20 mol% (entries 4–6). With this modification, even the highly hindered t-butyl-containing substrate 3w reacted to completion, giving the phosphite addition product in 83% yield and 96% ee (entry 6).
Table 4.
Enantioselective Michael addition reaction of diphenyl phosphite (2) to trans-nitroalkenes (1, R = aliphatic substituent) catalyzed by 5[a].
| entry | product | R- | time [h] | yield [%][b] | ee [%] |
|---|---|---|---|---|---|
| 1 | 3r |
|
1.5 | 94 | 96 |
| 2 | 3s |
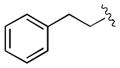
|
1.5 | 95 | 95 |
| 3 | 3t |
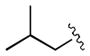
|
1.5 | 69 | 96 |
| 4[c] | 3u |
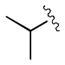
|
1.5 | 89 | 95 |
| 5[c] | 3v |
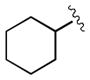
|
1.5 | 87 | 97 |
| 6[c] | 3w |
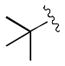
|
1.5 | 83 | 96 |
Unless otherwise noted, reactions were carried out on 0.20 mmol of 1 with 1.25 equiv of 2 and 10 mol% 5 in 1.0 mL CH2Cl2 at 0 °C.
Isolated yield.
Reaction was carried out using 20 mol% 5.
The results above show squaramide 5 to be a remarkably effective catalyst for the enantioselective Michael addition reactions of diphenyl phosphite to nitroalkenes. The reaction provides a simple, highly enantioselective synthesis of chiral β-nitro phosphonates, which are precursors to biologically active β-amino phosphonic acids. The high yields and uniformly excellent enantioselectivities obtained for both aryl- and alkyl-substituted nitroalkenes, including those bearing acidic protons or sterically-demanding substituents, point to the unique capability of the squaramide scaffold. Given the simple, modular assembly of squaramides, and the ready availability of its precursors, this scaffold is expected to provide many further opportunities in asymmetric catalysis.
Experimental Section
(R)-diphenyl 2-nitro-1-phenylethylphosphonate (3a)
Compound 1a (30 mg, 0.20 mmol) and catalyst (R, R)-5 (8.4 mg, 0.020 mmol) was added to CH2Cl2 (1.0 mL) at room temperature. The stirred mixture was cooled in an ice-water bath for 10 minutes then treated with 2 (48 μL, 0.25 mmol). After 30 minutes, the reaction mixture was loaded directly to the top of a flash chromatography column. Elution with hexanes:EtOAc (10:1 to 6:1 to 3:1) afforded the title compound as a white solid: 73 mg, 95% yield, 97% ee. On a 1 mmol scale, the product was obtained in 99% yield, 97% ee.
Supplementary Material
Footnotes
This work was funded by the National Institutes of Health, R01GM069990 and P50GM086145. We thank Professor Leo A. Paquette, Ohio State University, for a generous supply of squaric acid and dimethyl squarate.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Horiguchi M, Kandatsu M. Nature. 1959;184:901–902. doi: 10.1038/184901b0. [DOI] [PubMed] [Google Scholar]; b) Quin LD. A Guide to Organophosphorus Chemistry. Wiley; New York, N.Y: 2000. pp. 351–386. [Google Scholar]
- 2.For reviews on β-amino phosphonic acids and derivatives, see: Palacios F, Alonso C, de los Santos JM. Chem Rev. 2005;105:899–931. doi: 10.1021/cr040672y.Palacios F, Alonso C, de los Santos JM. In: Enantioselective Synthesis of β-Amino Acids. 2. Juaristi E, Soloshonok VA, editors. Wiley; Hoboken, N.J: 2005. pp. 277–317.
- 3.For biological activity of α-amino phosphonic acids, see: Kukhar VP, Hudson HR, editors. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity. Wiley; New York, N.Y: 2000. Metcalf WW, van der Donk WA. Annu Rev Biochem. 2009;78:65–94. doi: 10.1146/annurev.biochem.78.091707.100215. and references therein.
- 4.For reviews on stereoselective synthesis of α-amino phosphonic acids and derivatives, see: Ma JA. Chem Soc Rev. 2006;35:630–636. doi: 10.1039/b517100h.Ordonez M, Rojas-Cabrera H, Cativiela C. Tetrahedron. 2009;65:17–49. doi: 10.1016/j.tet.2008.09.083.
- 5.Examples of catalytic asymmetric synthesis of β-amino phosphonates and related compounds include: β-amino-α-hydroxy phosphonates by aminohydroxylations: Cravotto G, Giovenzana GB, Pagliarin R, Palmisano G, Sisti M. Tetrahedron: Asymmetry. 1998;9:745–748.Thomas AA, Sharpless KB. J Org Chem. 1999;64:8379–8385. doi: 10.1021/jo990060r.α-Hydroxy-β-nitro phosphonates by nitroaldol reactions: Mandal T, Samanta S, Zhao C. Org Lett. 2007;9:943–945. doi: 10.1021/ol070209i.Chen X, Wang J, Zhu Y, Shang D, Gao B, Liu X, Feng X, Su Z, Hu C. Chem --Eur J. 2008;14:10896–10899. doi: 10.1002/chem.200801958.α-Acyl-β-amino phosphonates by Mannich reactions: Kjaersgaard A, Jorgensen KA. Org Biomol Chem. 2005;3:804–808. doi: 10.1039/b416294c.Chen Z, Yakura K, Matsunaga S, Shibasaki M. Org Lett. 2008;10:3239–3242. doi: 10.1021/ol800965t.α-Diazo-β-amino phosphonates by Mannich reactions: Hashimoto T, Maruoka K. J Am Chem Soc. 2007;129:10054–10055. doi: 10.1021/ja0713375.β-Amino-α-nitro phosphonates by Mannich reaction: Wilt JC, Pink M, Johnston JN. Chem Commun. 2008:4177–4179. doi: 10.1039/b808393b.β-Substituted-β-amino phosphonates by hydrogenations: Kadyrov R, Holz J, Schaeffner B, Zayas O, Almena J, Boerner A. Tetrahedron: Asymmetry. 2008;19:1189–1192.Doherty S, Knight JG, Bell AL, El-Menabawey S, Vogels CM, Decken A, Westcott SA. Tetrahedron: Asymmetry. 2009;20:1437–1444.
- 6.For reviews on phospha-Michael reactions, see: Pudovik AN, Konovalova IV. Synthesis. 1979:81–96.Enders D, Saint-Dizier A, Lannou MI, Lenzen A. Eur J Org Chem. 2006:29–49.
- 7.Examples of diastereoselective Michael addition reactions of dialkyl phosphites to nitroalkenes include: chiral nitroalkenes: Paulsen H, Greve W. Chem Ber. 1973;106:2114–2123.Yamamoto H, Hanaya T, Kawamoto H, Inokawa S, Yamashita M, Armour MA, Nakashima TT. J Org Chem. 1985;50:3516–3521.Pachamuthu K, Figueroa-Perez I, Ali IAI, Schmidt RR. Eur J Org Chem. 2004:3959–3961.Chiral dialkyl phosphites: Enders D, Tedeschi L, Bats JW. Angew Chem. 2000;112:4774–4776.Angew Chem Int Ed. 2000;39:4605–4607.Enders D, Tedeschi L, Foerster D. Synthesis. 2006:1447–1460.
- 8.Enantioselective Michael addition reaction of dialkyl phosphites to nitroalkenes catalyzed by aluminum lithium bis(binaphthoxide): Rai V, Namboothiri INN. Tetrahedron: Asymmetry. 2008;19:2335–2338.
- 9.Wang J, Heikkinen LD, Li H, Zu L, Jiang W, Xie H, Wang W. Adv Synth Catal. 2007;349:1052–1056. [Google Scholar]
- 10.Terada M, Ikehara T, Ube H. J Am Chem Soc. 2007;129:14112–14113. doi: 10.1021/ja0746619. [DOI] [PubMed] [Google Scholar]
- 11.For reviews on organocatalytic enantioselective conjugate addition reactions, see: Almasi D, Alonso DA, Najera C. Tetrahedron: Asymmetry. 2007;18:299–365.Tsogoeva SB. Eur J Org Chem. 2007:1701–1716.Vicario JL, Badia D, Carrillo L. Synthesis. 2007:2065–2092.
- 12.Metal-free enantioselective Michael addition reactions of other phosphorous nucleophiles have been reported: Diaryl phosphine/phosphine oxide addition to nitroalkenes: Bartoli G, Bosco M, Carlone A, Locatelli M, Mazzanti A, Sambri L, Melchiorre P. Chem Commun. 2007:722–724. doi: 10.1039/b613477g.Fu X, Jiang Z, Tan C. Chem Commun. 2007:5058–5060. doi: 10.1039/b713151h.Hydrophosphination/phosphonylation of α, β-unsaturated aldehydes: Carlone A, Bartoli G, Bosco M, Sambri L, Melchiorre P. Angew Chem. 2007;119:4588–4590. doi: 10.1002/anie.200700754.Angew Chem Int Ed. 2007;46:4504–4506. doi: 10.1002/anie.200700754.Ibrahem I, Rios R, Vesely J, Hammar P, Eriksson L, Himo F, Cordova A. Angew Chem. 2007;119:4591–4594. doi: 10.1002/anie.200700916.Angew Chem Int Ed. 2007;46:4507–4510. doi: 10.1002/anie.200700916.Maerten, Cabrera S, Kjaersgaard A, Jorgensen KA. J Org Chem. 2007;72:8893–8903. doi: 10.1021/jo7018587.
- 13.a) Huang Y, Unni AK, Thadani AN, Rawal VH. Nature. 2003;424:146. doi: 10.1038/424146a. [DOI] [PubMed] [Google Scholar]; b) Thadani AN, Stankovic AR, Rawal VH. Proc Natl Acad Sci U S A. 2004;101:5846–5850. doi: 10.1073/pnas.0308545101. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McGilvra JD, Unni AK, Modi K, Rawal VH. Angew Chem. 2006;118:6276–6279. doi: 10.1002/anie.200601638. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:6130–6133. doi: 10.1002/anie.200601638. [DOI] [PubMed] [Google Scholar]; d) Gondi VB, Hagihara K, Rawal VH. Angew Chem. 2009;121:790–793. doi: 10.1002/anie.200804244. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:776–779. doi: 10.1002/anie.200804244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For reviews on hydrogen-bond donor catalysis, see: Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r.McGilvra JD, Gondi VB, Rawal VH. In: Enantioselective Organocatalysis. Dalko PI, editor. Wiley-VCH; Weinheim: 2007. pp. 189–254.Ting A, Schaus SE. Eur J Org Chem. 2007:5797–5815.Yu X, Wang W. Chem --Asian J. 2008;3:516–532. doi: 10.1002/asia.200700415.
- 15.Malerich JP, Hagihara K, Rawal VH. J Am Chem Soc. 2008;130:14416–14417. doi: 10.1021/ja805693p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For a review on thiourea-catalyzed asymmetric nucleophilic reactions, see: Takemoto Y, Miyabe H. Chimia. 2007;61:269–275.
- 17.Compound 4a was first synthesized by Koji Hagihara in our lab.
- 18.When the phosphite addition reaction was carried out in either Et2O or tBuOMe, a noticeable amount of a white precipitate formed, even at room temperature. The precipitate was separated by filtration and identified by NMR to be the desired product (3a). The precipitate had a lower ee than the product that remained in solution. These observations indicate that the product enriched in the racemate precipitates from the solution, leaving behind product that is even more enriched in the major enantiomer.
- 19.Wang et al. (ref. 9) reported four alkyl-substituted substrates (45–63% ee) and Terada et al. (ref. 10) reported three (80–87% ee).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.