

Abstract
One of the major challenges facing the long term survival of neurons is their requirement to maintain efficient axonal transport over long distances. In humans as large, long-lived vertebrates, the machinery maintaining neuronal transport must remain efficient despite the slow accumulation of cell damage during aging. Mutations in genes encoding proteins which function in the transport system feature prominently in neurologic disorders. Genes known to cause such disorders and showing traditional Mendelian inheritance have been more readily identified. It has been more difficult, however, to isolate factors underlying the complex genetics contributing to the more common idiopathic forms of neurodegenerative disease. At the heart of neuronal transport is the rail network or scaffolding provided by neuron specific microtubules (MTs). The importance of MT dynamics and stability is underscored by the critical role tau protein plays in MT-associated stabilization versus the dysfunction seen in Alzheimer’s disease, frontotemporal dementia and other tauopathies. Another example of the requirement for tight regulation of MT dynamics is the need to maintain balanced levels of post-translational modification of key MT building-blocks such as α-tubulin. Tubulins require extensive polyglutamylation at their carboxyl-terminus as part of a novel post-translational modification mechanism to signal MT growth versus destabilization. Dramatically, knock-out of a gene encoding a deglutamylation family member causes an extremely rapid cell death of Purkinje cells in the ataxic mouse model, pcd. This review will examine a range of neurodegenerative conditions where current molecular understanding points to defects in the stability of MTs and axonal transport to emphasize the central role of MTs in neuron survival.
INTRODUCTION
The focus of this review will be the role of microtubule (MT) stabilization and axonal transport as they relate to human pathology by looking at a series of inherited and sporadic conditions defined by neurodegeneration. In general, MTs form part of the cytoskeletal framework of all eukaryotes, and are primarily composed of tubular polymers of tubulin proteins with an outer diameter of ~ 25 nm (Nogales et al., 1998). Microtubules are formed by hetero-dimerization of α- and β-tubulin, the binding of GTP and subsequent incorporation of dimers into growing MT polymers (Figure 1). The dynamic stability of the MT will be favored or not depending upon β-tubulin’s binding of either GTP or GDP, respectively. For many years the tubulin protein family was thought to be specific to eukaryotes, but more recently a prokaryotic cell division protein FtsZ was uncovered as a ‘tubulin related protein’ (Nogales et al., 1998). Nonetheless, we now recognize that distinct tubulin post-translational modifications within the eukaryotic MT polymer, such as glutamylation, make it possible for the cell to differentiate axonal-MTs from other arrangements within the cell-body. With what appears to be increased neuronal MT complexity, comes the need for adequate regulation. Disease states will be discussed that range from rare, private gene mutations such as seen in the pcd mouse, to some of the most common human neuropathies such as Alzheimer’s disease, frontotemporal dementia & the tauopathies.
Figure 1. The microtubule is a dynamic structure essential to axonal transport and neuron survival.

The center panel depicts the basic neuron structure, highlighting the role of kinesin family proteins for anterograde transport from the cell body; and the dynein molecular motor, essential for retrograde transport back from the axon terminal. The upper panel represents and expansion from one isolated microtubule showing α-tubulin and β-tubulin as dynamic building blocks of the microtubule (side view and cross-section). The lower panel is an expansion of the axon in cross-section, underscoring the role of tau protein in microtubule stabilization.
Neuronal α-Tubulin post-translational modification defects
While various types of protein post-translational modification are well known, tubulin does require several additional forms that may not be well appreciated. For example, tubulin undergoes ligase mediated addition of glutamate and glycine residues (Edde et al., 1990, Redeker et al., 1994). However in neuronal MTs, tubulin glycylation is distinctly absent and glutamylation is of a specific form defined by the addition of one to six glutamate residues (Audebert et al., 1994). For both human and mouse, glutamylation and glycylation is orchestrated by a family of functional enzymes known as the ‘Tubulin Tyrosine Ligase – Like’ (TTLL) proteins. These range from TTLL1 through TTLL13 with varying specificities and expression profiles (van Dijk et al., 2007). Similarly, cleavage of glutamate side-chains is regulated by a family of six ‘Cytosolic Carboxy-Peptidases’ (CCPs), CCP1 through CCP6 (Kalinina et al., 2007, Rodriguez de la Vega et al., 2007). Also, for reasons not well understood, α-tubulin of neuronal MTs go through cleavage of the carboxyl-terminal tyrosine residue (Barra et al., 1988), followed by cleavage of the second to last gene encoded residue, a glutamate. The final product is referred to as delta 2-tubulin (Δ2-tubulin) and it is this form of the protein that accumulates in stable MT assemblies within neurons (Paturle-Lafanechere et al., 1994) (Figure 2).
Figure 2. The role of α-tubulin glutamate post-translational modification is depicted here, and can be appreciated by the rapid Purkinje Cell neurodegeneration which occurs in pcd mice via functional loss of the deglutamylation enzyme, CCP1.

At the top of the figure is the eleven carboxyl-terminal amino-acids of mouse α-tubulin. The terminal tyrosine reside, which is cleavable, is indicated in red. (A), to the left, detyrosination and polyglutamylation generate an α-tubulin isoform that can be acted upon by cytosolic carboxy-Peptidases, CCP1, CCP4, and CCP6. Each of these enzymes can cleave glutamate residues from the polyE side-chain down to the ‘branch point’, and cleave the second last gene-encoded glutamate, creating the Δ2-Tub isomer. The role of CCP5 alone is to specifically cleave the branch glutamate. (B), on the right side, addition of the branch point glutamate initiates these modification. Thereafter, the situation where CCP1 or Nna1 is absent (pink shading), hyperglutamylation – neurodegeneration; is contrasted with the normal CCP1 present situation (no shading), defined by equilibrated polyglutamylation. Redundant function of CCP4 and CCP6 does not compensate for CCP1 loss, as studies have shown CCP1 is the enzyme predominantly expressed in the neuron subset which undergoes degeneration in pcd mice. This illustration was modified from figure 7 and supplemental material 1st reported by Rogowski et al., (2010).
Proof of the need to maintain tight regulation of these neuronal MT modifications, are now emerging. TTLL knock-out mice lack the ability to correctly add glutamate residues and display respiratory problems due to abnormal cilia beating on airway epithelial (Ikegami et al., 2010). Cilia are structurally related to MTs as the cilia core consists of a MT-based cytoskeleton called the axoneme with nine outer microtubule doublets (Scholey, 2008). Conversely, the pcd mouse results from functional loss of the Nna1 gene which encodes the deglutamylation enzyme, CCP1 (Fernandez-Gonzalez et al., 2002). CCP1, as well as CCP4 and CCP6 cleave glutamate residues from tubulin side-chains as well as gene-encoded C-terminal glutamate residues. CCP5 is unique in its ability to remove the branch point glutamates (Figure 2). Pcd mice show overt ataxia beginning at 3 to 4 weeks of age, resulting from the rapid and dramatic degeneration of virtually all cerebellar Purkinje cells beginning at around P17 and is near complete by P45 (Figure 3). Degeneration of cerebellar granule neurons (CGNs), follows thereafter, as well as the subsequent loss of other neuron populations (Mullen et al., 1976). While there is redundancy in CCP enzyme function, Nna1 shows the highest expression in cell populations most susceptible in pcd mice, and α-tubulin polyglutamylation levels were increased in these same regions (Rogowski et al., 2010). Finally, partial rescue of the pcd phenotype was achieved by TTLL1 glutamylase knock-down, in an attempt to balance α-tubulin glutamylation, and as proof of principle. Motor coordination was improved by nearly 150% concomitant with the retention of a significant number Purkinje cells at day P40 (Rogowski et al., 2010).
Figure 3. Purkinje cell degeneration in pcd5J homozygous mice.
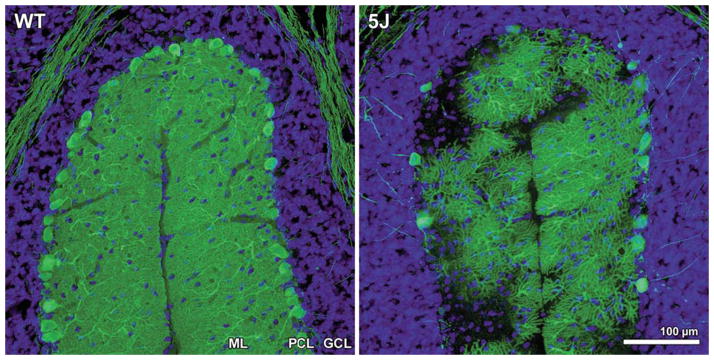
Here we see cerebellar sections for matched folia from P25 wild-type DBA/2J (WT) and P25 pcd5J homozygous mice (5J) immunostained with an anti-calbindin antibody and counterstained with DAPI. Although WT P25 cerebellum displays numerous Purkinje cell neurons with extensive dendritic arborization visible in the molecular layer (ML), the pcd5J P25 cerebellum reveals pronounced loss of Purkinje cells and dramatic degeneration of surviving Purkinje cells. The degree of Purkinje cell loss and degeneration observed in pcd5J homozygous mice is consistent with the disease’s natural history in pcd5J homozygous mice, suggesting that 5J is a severe allele. ML = molecular layer; PCL = Purkinje cell layer; GCL = granule cell layer. Reproduced with permission from the corresponding author Albert R. La Spada (Chakrabarti et al., 2006).
To date, mutations within the human CCP1-CCP6 protein orthologs have not been correlated with a specific ataxia syndrome reminiscent of the pcd mouse. Despite this, it is interesting to consider that hypomorphic mutations may underlie syndromes mapping to regions encompassing CCP deglutamylation gene loci. A non-exhaustive list of potential candidates may include the following conditions listed in Table 1.
Table 1.
Human conditions of unknown cause that map to defined genetic regions encompassing the six CCPs; CCP1 - CCP6.
| Condition | OMIM | Band Interval | Encoding gene |
|---|---|---|---|
| Spastic Paraplegia-19 | 607152 | 9q33-q34 | CCP1, AGTPBP1 |
| Spastic Paraplegia-41 | 613364 | 11p14.1-p11.2 | CCP2, AGBL2 |
| Spinocerebellar ataxia-32 | 613909 | 7q32-q33 | CCP3, AGBL3 |
| Primary ciliary dyskinesia-8 | 612274 | 15q24-q25 | CCP4, AGBL1 |
| Essential hereditary tremor-2 | 602134 | 2p25-p22 | CCP5, AGBL5 |
| Dystonia-9: episodic ataxia & spasticity | 601042 | 1p33 | CCP6, AGBL4 |
While a human correlate to the pcd mouse has not been reported, a suggestive condition was recently discovered in sheep (Ovis aries). Missense mutation of the sheep CCP1 ortholog was linked with a phenotype similar to pcd mice. Affected sheep (n=19) were normal at birth, then progressive weakness and quadriplegia developed after the first week of life (Zhao et al., 2012). The authors define the condition as a lower motor neuron disease and noted that the R970P mutation locates within the catalytically active site, and showed complete segregation with the recessively inherited condition.
Microtubule defects & motor dysfunction – Dynactin (retrograde)
Cytoplasmic dynein is a MT-based molecular motor protein that directly utilises the energy provided by ATP hydrolysis to allow retrograde transport of vesicles and organelles (Holzbaur and Vallee, 1994). But it is the coordinated action of the dynactin macromolecular complex in combination with the dynein-motor that facilitates and orchestrates retrograde transport back to the cell body (Figure 1). And of the many proteins within the complex, dynactin 1 is the largest at ~ 150 kDa, directly binding to both MT and dynein (Holzbaur and Tokito, 1996).
Early studies with the dynactin 1 Drosophila ortholog, p150/Glued, foreshadowed its critical role in neurons. Using a range of gene targeted and transgenic approaches, fly geneticists demonstrated that disruption of this protein results in a frequency increase of synaptic retraction events at the neuromuscular junction (Eaton et al., 2002). In 2003, Puls et al. were the first to link Dynactin-1 mutations with neural dysfunction. By studying a family showing dominant inheritance of a distal hereditary motor neuronopathy, (or lower motor neuron disease); they discovered that all nine affected family members harbored a Gly59Ser mutation. This substitution was predicted to “distort the folding of dynactin’s microtubule-binding domain” (Puls et al., 2003). Taken together these findings demonstrate a link between dynactin-1 mutation and neuronopathy.
While the inherited neuronopathy described above is debilitating, it is usually not fatal when only lower motor neurons are involved. However, true motor neuron disease or amyotrophic lateral sclerosis (ALS) is far more devastating and is not rare with an incidence of ~ 1 in 40,000 (Logroscino et al., 2010). Sporadic ALS (sALS) is a fatal, late-onset disorder characterized by rapid degeneration of upper and lower motor neurons (UMNs, LMNs), as well as bulbar neuron involvement (Hardiman et al., 2011). And thus, in a study of 250 cases of sALS, three patients were found to have substitution mutations of the dynactin 1 protein. But in each of these individual cases the patients were distinguished from the Gly59Ser family by the addition of UMN signs and symptoms (Munch et al., 2004). One of the three mutations described, Thr1249Ile, was subsequently identified in a range of control and neuropathy cohorts as follows: (i) 3 of 435 controls; (ii) 1 of 374 frontotemporal dementia (FTD) patients; and (iii) 5 of 372 ALS patients (Vilarino-Guell et al., 2009). As a consequence, it was concluded that the DCTN1 genetic loci likely represents a susceptibility gene to sALS as well as playing a direct role in rare, familial conditions. Partially overshadowing all of this may be the recent discovery that repeat expansions within the C9orf72 gene were found to be the most common cause of familial ALS (fALS), as well as a significant cause of sALS and FTD (see [OMIM: 614260], and discussed below). With C9orf72 being added to the list of about a dozen genes known to directly cause fALS, only about 40% of fALS cases remain of unknown genetic cause (Williams et al., 2013). What then may be the cause of the vast majority of all ALS cases where the cause(s) still remain undefined? ALS is a complex disease, where multiple genetic and environmental factors likely contribute to disease liability. Somatically acquired mutations within developing neurons, along with environmental insults and germ-line inheritance of hypomorphic mutations within a range of MT-based transport genes, are all likely to be involved and these may only be characterized with new, powerful, next-generation sequencing technologies.
Microtubule defects & motor dysfunction – Kinesin (anterograde)
For axonal transport in the anterograde direction, the family of proteins known as the kinesins are indispensable (Figure 1). The kinesins are essentially the equivalent to the dynein-dynactin complex in that they are a family of molecular motor proteins that also use the hydrolysis of ATP to enable direct walking along MTs. But in this case specific cargos are moved in the anterograde direction. Kinesins do vary in shape but the prototype is a heterotetramer where two motor subunits form a protein dimer and then bind with two light chains. In most cases transported cargo is harnessed to the light chains (Vale, 2003).
Potentially the best example of an axonal neuropathy stemming from a kinesin gene defect was first reported in the journal Cell, in 2001. Charcot-Marie-Tooth disease (CMT), also known as hereditary motor sensory neuropathy (HMSN), is the most common inherited peripheral neuropathy in humans with a prevalence of 1 in 2500. CMT (or HMSN) is clinically characterized by weakness and atrophy of distal muscles, depressed or absent deep tendon reflexes, and mild sensory loss (Shy et al., 1999). These disorders were traditionally classified into two forms differentiated only by electrophysiology testing. CMT type I, the demyelinating form showed reduced nerve conduction velocities (NCVs); and CMT type II, the axonal form showed normal NCVs, but reduced compound muscle action potentials. But in the current genomics era, more than 40 genes are known to cause CMT and the phenotypic boundaries are becoming blurred (http://neuromuscular.wustl.edu/time/hmsn.html). Genes that cause demyelinating CMT (or type 1) tend to directly afflict Schwann cells, while those that cause the axonal forms tend to affect neurons more directly. The most common axonal form is CMT2A, and thus its initial mapping to chromosome 1p35-36 was an important landmark (Ben Othmane et al., 1993). Then, as the kinesin encoding KIF1B gene was closely linked to the CMT2A genetic interval, it represented a strong candidate gene as it was known to transport mitochondria (Nangaku et al., 1994). Dramatically, a Q98L mutation of the KIF1B β isoform was reported to segregate with disease in a Japanese CMT2A pedigree (Zhao et al., 2001). In this report, data in support of the kinesis hypothesis for CMT2A was strong: (i) Q98L was located to the ATP binding consensus; (ii) Q98L was shown to decrease ATPase activity; and (iii) Q98L blocked migration of KIF1B to the cell membrane. The authors were led to suggest KIF1Bβ haploinsufficiency was responsible for CMT2A neuropathy and explained the autosomal dominant inheritance. Finally, heterozygous kif1B +/− mice showed symptoms reminiscent of human CMT2A (Zhao et al., 2001). But enigmatically, no further links of KIF1Bβ mutation with CMT2A were reported. Several years later the true or common cause of CMT2A was described and the causal mutation of several previously reported CMT2A pedigrees were isolated within the mitofusin-2 gene, MFN2 (Zuchner et al., 2004). Subsequent analysis has shown mitofusin-2 to be a mitochondrial membrane protein partially responsible for facilitating mitochondrial fusion and contributing to the maintenance and operation of the mitochondrial network (Gazaryan and Brown, 2007). And since the initial MFN2 mutation report, an array of gene mutations linked to CMT2A has been reported (OMIM: 609260).
The final point may be that mutation of another kinesin member, KIF5A (transporting a diverse range of cargos including mitochondria), causes autosomal dominant spastic paraplegia type 10 (SPG10) (Reid et al., 2002). Since this discovery, a significant number of KIF5A point mutations, all located within the highly conserved motor domain have been reported in support of the kinesin haploinsufficiency hypothesis in SPG10, in which upper motor neurons are primarily affected (OMIM: 604187).
Axonal Transport defects in motor neuron disease (MND, or ALS)
It is now accepted that just over a dozen genes are responsible for ~ 60% of all fALS cases. But it is not known what may be common to each of the pathogenic pathways represented by these seemingly divergent genes. Given the unique sensitivity large motor neurons show to mutations in genes that are often ubiquitously expressed, it is reasonable to assume that there is a length dependent and energy requirement dependency to this sensitivity. Direct evidence also suggests axonal transport defects play an important role in neurodegeneration (Jablonka et al., 2004). ALS1 results from mutations in the superoxide dismutase 1 gene (SOD1) (Rosen, 1993), and this form represents ~ 20% of fALS cases. But despite 2013 representing the 20 twenty year anniversary of the SOD1 discovery, the precise etiology of SOD1-mediated ALS still remains unclear. As a potential point of mechanistic convergence, it is important to note that defects in anterograde axonal transport are one of the earliest pathologies observed in SOD1 mice (Williamson and Cleveland, 1999). Mice with dominant mutations of the dynein heavy chain gene (Dnchc1), Phe580Tyr, are known as legs at odd angles (Loa) mice. Loa mice show defects in retrograde axonal transport and loss of motor neurons (Hafezparast et al., 2003). Intriguingly, while the Loa mice show an age-related progressive loss of muscle tone and locomotor ability, no major reduction in life-span results. The experiment was undertaken then to cross SOD1G93A mice with Loa/+ mice to produce double-heterozygotes (Loa/SOD1G93A) and observe the resultant motor phenotype. In essence, could the defect in ‘retrograde’ transport seen in Loa mice neutralize or partially neutralize the defect SOD1 mice harbor in ‘anterograde’ transport. SOD1G93A mice survive for an average of only 125 days, while Loa mice live a full life span. Thus, a conclusive result was found in that Loa/SOD1G93A mice survived for an average of 160 days, 28% greater than their SOD1G93A counterparts and disease onset was delayed (Kieran et al., 2005). In addition to the role of MTs, the related role of neurofilaments is clearly an overlapping aspect to the story. For example, an odd, over-accumulation of neurofilaments in parts of motor neurons is a hallmark of ALS, and mice with mutations in neurofilament genes have been found to have poorly functioning motor neurons (Xiao et al., 2006). But beyond this recognition, we will not focus on neurofilments in neurodegeneration, and consider it beyond the scope of this review.
Tau protein, Frontotemporal dementia and the Tauopathies
The microtubule-associated proteins (MAPs) bind directly to tubulin dimers via C-terminal domains (Figure 1), normally leading to stabilization. The extent to which MAP proteins bind MTs is normally regulated via phosphorylation levels (Drewes et al., 1998).
In 1989 it was established that six tau isoforms are produced by mRNA alternative splicing from the microtubule-associated protein tau gene, MAPT (Goedert et al., 1989). Critical to MT binding is a 31-amino acid repeat located in the carboxyl-terminus of tau that forms either three- or four-repeat isoforms dependent on the exclusion or inclusion of exon 10. Normal cerebral cortex contains similar levels of 3-repeat and 4-repeat tau. The repeats and adjoining sequences constitute the MT-binding domain (Goedert et al., 1989). Significant interest is centred on MAPT mutations, or abnormal functioning and processing of the tau protein in association with a range of neurodegenerative conditions (see OMIM: 157140). These include: Alzheimer’s disease (AD), Pick’s disease, FTD, cortico-basal degeneration and progressive supranuclear palsy.
The general tau hypothesis is that tau protein abnormalities initiate a pathologic cascade (Mudher and Lovestone, 2002). This is thought to begin when hyper-phosphorylated tau pairs with other tau threads, and this catalyst expands to form neurofibrillary tangles within nerve cell bodies (Goedert et al., 1991). It is posited that the ensuing MT destruction fatally halts the essential transport system of neurons, ultimately leading to cell death (Iqbal et al., 2005).
Frontotemporal dementia is underscored as just one possible model for an inherited neurodegenerative condition resulting from MAPT mutations or tau dysfunction. FTD is characterized by progressive deterioration of the brain’s ‘frontal’ lobe, gradually expanding on occasion to include degeneration of the ‘temporal’ lobe. FTD is second only to AD in prevalence, accounting for ~ 20% of pre-senile dementia cases (Snowden et al., 2002). In FTD patients, symptoms normally begin between 45 to 65 years of age, and commonly include behaviour changes and emotional dulling (Snowden et al., 2002). As with most of the better known neurodegenerative conditions, there is no cure, but new treatments for symptomatic relief are becoming available.
The genetics of FTD is complex, but differs significantly from AD in that a far higher percentage of FTD cases directly result from inherited mutations at ~ 30–50%, whereas only 2–5 % of AD is of the early-onset, inherited form. The three main genetic markers for FTD include the tau gene MAPT, as well as progranulin, PGRN, normally considered to function as a cell growth regulator; and together MAPT and PGRN account for almost half of all familial-FTD (Seelaar et al., 2011). More recently, the emergence of the C9orf72 gene (discussed above in reference to ALS), which is pathogenic by virtue of the presence of a hexanucleotide repeat expansion, was added to the list of important FTD genes. C9orf72, along with a range of other causal genes, account for the second half of all familial-FTD, which some subdivided as the FTLD-TDP43 forms (Seelaar et al., 2011).
FTDP-17 is an early-onset form of FTD caused by MAPT mutations and is characterized by the pathological aggregation of tau (OMIM: 600274). These mutations play an important role in the study of tau aggregation which closely correlate with progression and severity of brain atrophy. Most FTDP-17 mutations localize in or near the MT binding repeat regions. And by generating expression vectors for 12 different FTDP-17 mutants, it was demonstrated that many of these disease associated point mutations increased the propensity for in vitro aggregation and inhibited the MT stabilization ability of tau protein (Combs and Gamblin, 2012). Studies directly looking at the effects on the frontotemporal lobe function specifically examined the FTDP-17 associated mutation, V337M. Transgenic mice (Tg-mice) over-expressing this human tau mutation from the brain specific Thy-1 promoter were compared with wild type (wt) Tg-mice. Comparing wt and TauV337M Tg-mice, a series of goal-directed, decision making tests indicated the tauV337M mutation selectively disrupts a subset of processes related to executive brain function (Reichelt et al., 2013).
The development of neurodegenerative treatments and the regulatory environment
Globally neurodegenerative diseases are already a large burden on healthcare systems. To care for its existing population of neurodegenerative patients affected by Alzheimer’s disease and other related disorders, the European Union (EU) spends approximately €130 billion per annum and this figure will only increase as 25% of the population will reach the age of 65 or older in 2030 (EU Joint Programme - Neurodegenerative Disease Research, 2013). The Alzheimer’s Association estimate that in 2013 the cost to the United States of America government will reach US$203 billion dollars per annum with the number expected to rise to $1.2 trillion by 2050 (Thies and Bleiler, 2013).
The spiralling cost of these diseases is one of the reasons behind the Food and Drug Administration Safety and Innovation Act of 2012 (FDASIA), also known as the Prescription Drug User Fee Act V (PDUFA V) which has expanded two of the major bottle necks in the previous approval process. The Act intends to “facilitate and expedite development and review of new drugs to address unmet medical need in the treatment of a serious or life threatening condition” through the use of four programs: fast track designation, breakthrough therapy designation, accelerated approval and priority review designation (Center for Drug Evaluation, 2013). These programs have resulted in the average review time being shortened to only 6 months, with some receiving approval even faster (U.S. Food and Drug Administration). These changes should allow for more molecular therapies to become available for neurodegenerative diseases as they are intended to favour rare and/or life threatening conditions as well as those conditions affecting smaller numbers (Fox, 2012). However, out of the thirteen FDA approved molecular therapeutic entities in the first half of 2013 – only one Tecfidera (dimethyl fumarate) by Biogen Idec was for a neurodegenerative disorder – multiple sclerosis (FDA, 2013).
Currently, there are over 897 open clinical trials for novel neurodegenerative disease treatments with 76 trials specific to MND/ALS, 391 to Alzheimer’s disease and 28 to FTD (U.S. National Institutes of Health, 2013). Many of these trials will provide essential data towards demonstrating efficacy and safety within this new regulatory framework, especially as the use of meta-analysis on existing trial data is now recommended; especially between EU and Asian regulatory bodies (Fox, 2012, Byrne, 2013). The new FDA regulations will also allow for more personalised gene therapies to be developed against unique biomarkers of orphan diseases (Fox, 2012). This fast-track process is not without its risks as a single well designed trial with modified endpoints can be sufficient to gain approval; however that approval can be withdrawn if any product fails to meet ongoing approval criteria (Byrne, 2013). Within the next few years, we will see if any of those 897 trials yields an approved therapeutic under this new legislation.
SUMMARY
In this review we have focused on the microtubule and some of its important component building blocks such as α-tubulin and the stabilizing role played by MAP proteins such as tau. Many aspects of the review have been treated only briefly, examining the tip of the iceberg as it were; when in fact the pathogenic processes are extremely complex. For example, the dynamics of tau in its role of stabilizing MTs and the pathogenic onset of the tauopathies is very complex and its study could easily fill a successful career many times over. Our goal here was to draw together an overview of a range of primarily inherited disorders, where the mutations likely have a direct bearing on MT function within the neuron. We have looked at the role of glutamylation as one of the α-tubulin post-translation modifications used to maintain MT growth versus destruction. Yet few direct examples have been implicated in human disease showing Mendelian inheritance. Therefore, we highlighted the possibility that hypomorphic mutations of glutamylation or deglutamylation genes may be the cause of a range of human conditions where the genetic cause remains unknown. We have suggested hypomorphic mutations may be more likely as full knock-out of the human orthologs of these genes may cause embryonic lethality. Molecular motors responsible for either retrograde and anterograde were examined in light of a range of neurodegenerative conditions. These were found to result from mutation of important players in the dynein-dynactin complex as well as key kinesin family members. Throughout the review we have also tried to highlight the uniquely selective sensitivity neurons show in each condition, whether it is an ataxia, ALS, CMT, paraplegia or frontal lobe degenerative condition. And finally, we have included the latest changes to the regulatory environment which will facilitate the approval of next generation therapies for these often devastating conditions. This was looked at from the perspective of oversight bodies such as the FDA who now are actively working in concert with interested research bodies such as MDA and the ALS Association, as well as other global regulatory bodies such as the European Medicines Agency, in the hope that this group of patients may look forward to improved therapies in the near future.
References
- Audebert S, Koulakoff A, Berwald-Netter Y, Gros F, Denoulet P, Edde B. Developmental regulation of polyglutamylated alpha- and beta-tubulin in mouse brain neurons. J Cell Sci. 1994;107 (Pt 8):2313–2322. doi: 10.1242/jcs.107.8.2313. [DOI] [PubMed] [Google Scholar]
- Barra HS, Arce CA, Argarana CE. Posttranslational tyrosination/detyrosination of tubulin. Mol Neurobiol. 1988;2 (2):133–153. doi: 10.1007/BF02935343. [DOI] [PubMed] [Google Scholar]
- Ben Othmane K, Middleton LT, Loprest LJ, Wilkinson KM, Lennon F, Rozear MP, Stajich JM, Gaskell PC, Roses AD, Pericak-Vance MA, et al. Localization of a gene (CMT2A) for autosomal dominant Charcot-Marie-Tooth disease type 2 to chromosome 1p and evidence of genetic heterogeneity. Genomics. 1993;17 (2):370–375. doi: 10.1006/geno.1993.1334. [DOI] [PubMed] [Google Scholar]
- Byrne BJ. Pathway for approval of a gene therapy orphan product: treading new ground. Mol Ther. 2013;21 (8):1465–1466. doi: 10.1038/mt.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Drug Evaluation R, & Center for Biologics Evaluation and Research. Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics. F. a. D. A. U. S. A. U.S. Department of Health and Human Services; 2013. [Google Scholar]
- Chakrabarti L, Neal JT, Miles M, Martinez RA, Smith AC, Sopher BL, La Spada AR. The Purkinje cell degeneration 5J mutation is a single amino acid insertion that destabilizes Nna1 protein. Mamm Genome. 2006;17 (2):103–110. doi: 10.1007/s00335-005-0096-x. [DOI] [PubMed] [Google Scholar]
- Combs B, Gamblin TC. FTDP-17 tau mutations induce distinct effects on aggregation and microtubule interactions. Biochemistry. 2012;51 (43):8597–8607. doi: 10.1021/bi3010818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewes G, Ebneth A, Mandelkow EM. MAPs, MARKs and microtubule dynamics. Trends Biochem Sci. 1998;23 (8):307–311. doi: 10.1016/s0968-0004(98)01245-6. [DOI] [PubMed] [Google Scholar]
- Eaton BA, Fetter RD, Davis GW. Dynactin is necessary for synapse stabilization. Neuron. 2002;34 (5):729–741. doi: 10.1016/s0896-6273(02)00721-3. [DOI] [PubMed] [Google Scholar]
- Edde B, Rossier J, Le Caer JP, Desbruyeres E, Gros F, Denoulet P. Posttranslational glutamylation of alpha-tubulin. Science. 1990;247 (4938):83–85. doi: 10.1126/science.1967194. [DOI] [PubMed] [Google Scholar]
- Eu Joint Programme - Neurodegenerative Disease Research. Why choose Neurodegenerative diseases? 2013 Retrieved 03/08/2013, from http://www.neurodegenerationresearch.eu/about/why/
- Fda . FDA approvals for first half of 2013. Nat Rev Drug Discov. 2013;12 (8):568. [Google Scholar]
- Fernandez-Gonzalez A, La Spada AR, Treadaway J, Higdon JC, Harris BS, Sidman RL, Morgan JI, Zuo J. Purkinje cell degeneration (pcd) phenotypes caused by mutations in the axotomy-induced gene, Nna1. Science. 2002;295 (5561):1904–1906. doi: 10.1126/science.1068912. [DOI] [PubMed] [Google Scholar]
- Fox JL. Rare-disease drugs boosted by new Prescription Drug User Fee Act. Nat Biotechnol. 2012;30 (8):733–734. doi: 10.1038/nbt0812-733. [DOI] [PubMed] [Google Scholar]
- Fox JL. Rare-disease drugs boosted by new Prescription Drug User Fee Act. Nat Biotech. 2012;30 (8):733–734. doi: 10.1038/nbt0812-733. [DOI] [PubMed] [Google Scholar]
- Gazaryan IG, Brown AM. Intersection between mitochondrial permeability pores and mitochondrial fusion/fission. Neurochem Res. 2007;32 (4–5):917–929. doi: 10.1007/s11064-006-9252-2. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Crowther RA. Tau proteins and neurofibrillary degeneration. Brain Pathol. 1991;1 (4):279–286. doi: 10.1111/j.1750-3639.1991.tb00671.x. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3 (4):519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300 (5620):808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Hardiman O, Van Den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7 (11):639–649. doi: 10.1038/nrneurol.2011.153. [DOI] [PubMed] [Google Scholar]
- Holzbaur EL, Tokito MK. Localization of the DCTN1 gene encoding p150Glued to human chromosome 2p13 by fluorescence in situ hybridization. Genomics. 1996;31 (3):398–399. doi: 10.1006/geno.1996.0068. [DOI] [PubMed] [Google Scholar]
- Holzbaur EL, Vallee RB. DYNEINS: molecular structure and cellular function. Annu Rev Cell Biol. 1994;10:339–372. doi: 10.1146/annurev.cb.10.110194.002011. [DOI] [PubMed] [Google Scholar]
- Ikegami K, Sato S, Nakamura K, Ostrowski LE, Setou M. Tubulin polyglutamylation is essential for airway ciliary function through the regulation of beating asymmetry. Proc Natl Acad Sci U S A. 2010;107 (23):10490–10495. doi: 10.1073/pnas.1002128107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739 (2–3):198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Jablonka S, Wiese S, Sendtner M. Axonal defects in mouse models of motoneuron disease. J Neurobiol. 2004;58 (2):272–286. doi: 10.1002/neu.10313. [DOI] [PubMed] [Google Scholar]
- Kalinina E, Biswas R, Berezniuk I, Hermoso A, Aviles FX, Fricker LD. A novel subfamily of mouse cytosolic carboxypeptidases. FASEB J. 2007;21 (3):836–850. doi: 10.1096/fj.06-7329com. [DOI] [PubMed] [Google Scholar]
- Kieran D, Hafezparast M, Bohnert S, Dick JR, Martin J, Schiavo G, Fisher EM, Greensmith L. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J Cell Biol. 2005;169 (4):561–567. doi: 10.1083/jcb.200501085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logroscino G, Traynor BJ, Hardiman O, Chio A, Mitchell D, Swingler RJ, Millul A, Benn E, Beghi E. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2010;81 (4):385–390. doi: 10.1136/jnnp.2009.183525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudher A, Lovestone S. Alzheimer’s disease-do tauists and baptists finally shake hands? Trends Neurosci. 2002;25 (1):22–26. doi: 10.1016/s0166-2236(00)02031-2. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Eicher EM, Sidman RL. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc Natl Acad Sci U S A. 1976;73 (1):208–212. doi: 10.1073/pnas.73.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G, Hanemann CO, Stumm G, Ludolph AC. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63 (4):724–726. doi: 10.1212/01.wnl.0000134608.83927.b1. [DOI] [PubMed] [Google Scholar]
- Nangaku M, Sato-Yoshitake R, Okada Y, Noda Y, Takemura R, Yamazaki H, Hirokawa N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell. 1994;79 (7):1209–1220. doi: 10.1016/0092-8674(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Nogales E, Downing KH, Amos LA, Lowe J. Tubulin and FtsZ form a distinct family of GTPases. Nat Struct Biol. 1998;5 (6):451–458. doi: 10.1038/nsb0698-451. [DOI] [PubMed] [Google Scholar]
- Paturle-Lafanechere L, Manier M, Trigault N, Pirollet F, Mazarguil H, Job D. Accumulation of delta 2-tubulin, a major tubulin variant that cannot be tyrosinated, in neuronal tissues and in stable microtubule assemblies. J Cell Sci. 1994;107 (Pt 6):1529–1543. doi: 10.1242/jcs.107.6.1529. [DOI] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, Lamonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33 (4):455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Redeker V, Levilliers N, Schmitter JM, Le Caer JP, Rossier J, Adoutte A, Bre MH. Polyglycylation of tubulin: a posttranslational modification in axonemal microtubules. Science. 1994;266 (5191):1688–1691. doi: 10.1126/science.7992051. [DOI] [PubMed] [Google Scholar]
- Reichelt AC, Killcross S, Wilkinson LS, Humby T, Good MA. Transgenic expression of the FTDP-17 tau mutation in brain dissociates components of executive function in mice. Neurobiol Learn Mem. 2013;104C:73–81. doi: 10.1016/j.nlm.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, Graham FL, Gaskell PC, Dearlove A, Pericak-Vance MA, Rubinsztein DC, Marchuk DA. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10) Am J Hum Genet. 2002;71 (5):1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez De La Vega M, Sevilla RG, Hermoso A, Lorenzo J, Tanco S, Diez A, Fricker LD, Bautista JM, Aviles FX. Nna1-like proteins are active metallocarboxypeptidases of a new and diverse M14 subfamily. FASEB J. 2007;21 (3):851–865. doi: 10.1096/fj.06-7330com. [DOI] [PubMed] [Google Scholar]
- Rogowski K, Van Dijk J, Magiera MM, Bosc C, Deloulme JC, et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell. 2010;143 (4):564–578. doi: 10.1016/j.cell.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;364 (6435):362. doi: 10.1038/364362c0. [DOI] [PubMed] [Google Scholar]
- Scholey JM. Intraflagellar transport motors in cilia: moving along the cell’s antenna. J Cell Biol. 2008;180 (1):23–29. doi: 10.1083/jcb.200709133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelaar H, Rohrer JD, Pijnenburg YA, Fox NC, Van Swieten JC. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry. 2011;82 (5):476–486. doi: 10.1136/jnnp.2010.212225. [DOI] [PubMed] [Google Scholar]
- Shy ME, Kamholz J, Lovelace RE. Introduction to the Third International Symposium on Charcot-Marie-Tooth disorders. Ann N Y Acad Sci. 1999;883:xiii–xviii. [PubMed] [Google Scholar]
- Snowden JS, Neary D, Mann DM. Frontotemporal dementia. Br J Psychiatry. 2002;180:140–143. doi: 10.1192/bjp.180.2.140. [DOI] [PubMed] [Google Scholar]
- Thies W, Bleiler L. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 2013;9 (2):208–245. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration. About FDA Product Approval. 2009 Feb 26; Retrieved 03/08/2013 from http://www.fda.gov/NewsEvents/ProductsApprovals/ucm106288.htm.
- U.S. National Institutes of Health. Clinical Trials. 2013 Retrieved 03/08/2013, from http://clinicaltrials.gov.
- Vale RD. The molecular motor toolbox for intracellular transport. Cell. 2003;112 (4):467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- Van Dijk J, Rogowski K, Miro J, Lacroix B, Edde B, Janke C. A targeted multienzyme mechanism for selective microtubule polyglutamylation. Mol Cell. 2007;26 (3):437–448. doi: 10.1016/j.molcel.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Vilarino-Guell C, Wider C, Soto-Ortolaza AI, Cobb SA, Kachergus JM, et al. Characterization of DCTN1 genetic variability in neurodegeneration. Neurology. 2009;72 (23):2024–2028. doi: 10.1212/WNL.0b013e3181a92c4c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KL, Fifita JA, Vucic S, Durnall JC, Kiernan MC, Blair IP, Nicholson GA. Pathophysiological insights into ALS with C9ORF72 expansions. J Neurol Neurosurg Psychiatry. 2013;84 (8):931–935. doi: 10.1136/jnnp-2012-304529. [DOI] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2 (1):50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- Xiao S, Mclean J, Robertson J. Neuronal intermediate filaments and ALS: a new look at an old question. Biochim Biophys Acta. 2006;1762 (11–12):1001–1012. doi: 10.1016/j.bbadis.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y, Hirokawa N. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105 (5):587–597. doi: 10.1016/s0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]
- Zhao X, Onteru SK, Dittmer KE, Parton K, Blair HT, Rothschild MF, Garrick DJ. A missense mutation in AGTPBP1 was identified in sheep with a lower motor neuron disease. Heredity (Edinb) 2012;109 (3):156–162. doi: 10.1038/hdy.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36 (5):449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]